

Abstract
Autosomal-recessive congenital ichthyoses represent a large and heterogeneous group of disorders of epidermal cornification. Recent data suggest that most of these disorders might result from defective lipid transport and metabolism. In the present study, we describe a late-onset form of recessive ichthyosis in a large consanguineous pedigree. By using a combination of homozygosity mapping and positional candidate-gene screening, we identified a 2 bp deletion in LIPN that segregated with the disease phenotype throughout the family. LIPN encodes one of six acid lipases known to be involved in triglyceride metabolism in mammals . LIPN was found to be exclusively expressed in the epidermis and to be strongly induced during keratinocyte differentiation.
Main Text
Autosomal-recessive congenital ichthyoses (ARCI) form a complex and heterogeneous group of nonsyndromic disorders of cornification (epidermal maturation) and have an approximate prevalence of 1:100,000.1 Recent data indicate that despite being rare, congenital ichthyoses represent a major economical burden,2 suggesting the need for improved preventive strategies as well as novel therapeutic interventions.
ARCI patients can display generalized dry scaling of the skin, known as lamellar ichthyosis (LI); alternatively, the skin surface of ARCI patients can be diffusely red and covered with fine whitish scales, a clinical presentation referred to as congenital ichthyosiform erythroderma (CIE). Some patients present with a particularly severe disease form known as Harlequin ichthyosis (MIM 242500), in which the skin is covered by thick and adherent scales;3 others display a phenotype that varies in severity over time and that was recently coined pleomorphic ichthyosis4. ARCIs not only feature an unusual degree of phenotypic heterogeneity, but they are also characterized by extensive genetic heterogeneity, and seven loci have been reported so far in various forms of ARCIs1 on 2q34 harboring ABCA12 (MIM 607800), 5q33.3 including NIPAL4 (MIM 609383), 14q11.2 harboring TGM1 (MIM 190195), 17p13.1 spanning ALOX12B (MIM 603741), and ALOXE3 (MIM 607206), 19p13.12 containing CYP4F22 (MIM 611495), as well as additional intervals in which no disease-associated variants have been identified so far.5–7
Despite these significant advances in our understanding of ARCIs pathogenesis, no causative mutations can be found in approximately one-fifth of ARCI cases.8 This is likely due to the existence of mutations located either in ARCI-associated gene regions that are not routinely sequenced or in genes not previously associated with ARCI. In the present study, we report on a ARCI-associated locus on 10q23.31 and identified a pathogenic mutation in LIPN, encoding lipase N, which was found to segregate with the disease phenotype in a large consanguineous family.
A 14-year-old girl of Arab Muslim origin was referred for evaluation because of generalized scaling of the skin. At birth, her skin looked normal; however, she developed widespread ichthyosis at age 5 years of age. Her parents were first-degree cousins. Six additional relatives were also affected by a similar disorder (Figure 1). On examination, the entire surface of her skin was covered with fine whitish scales (Figures 2A and 2B), whereas her face was slightly erythematous. Routine laboratory tests were normal. A skin biopsy revealed hyperkeratosis and acanthosis without evidence for epidermolytic changes or intracytoplasmic vacuoles (Figure 2C).
Figure 1.
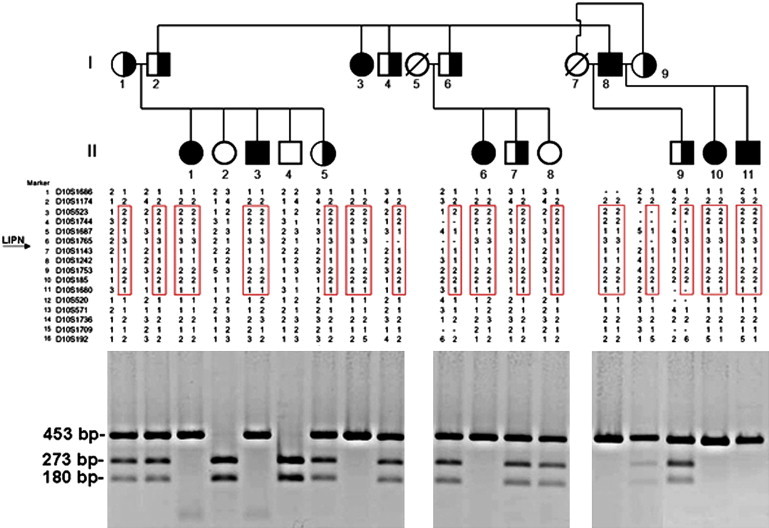
Family Pedigree, Haplotype Analysis, and PCR-RFLP Analysis
The family tree appears in the upper panel (black symbols denote affected individuals and half-filled symbols represent heterozygous carrier of mutation c.399_400delGA). Haplotype analysis using polymorphic markers on chromosome 10q23 is depicted in the middle panel and reveals a homozygous haplotype shared by all patients (boxed in red). The lower panel provides details of the PCR-RFLP analysis that confirm segregation of the mutation in the family: mutation c.399_400delGA abolishes a recognition site for BsmAI; thus, affected patients display a single fragment of 453 bp, healthy individuals show two fragments of 273 and 180 bp, and all fragments are found in heterozygous carriers of the mutation.
Figure 2.
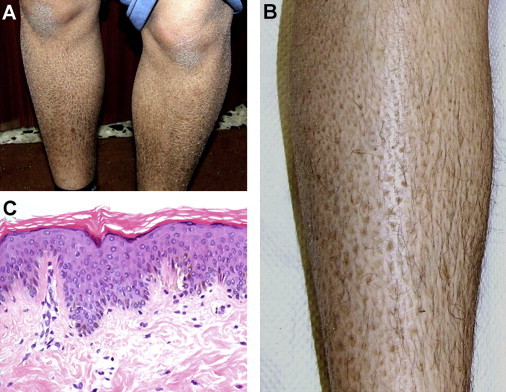
Clinical Features
(A and B) Diffuse lamellar ichthyosis over the legs of a 14-year-old affected individual (patient II-1).
(C) A skin biopsy shows hyperkeratosis, orthokeratosis, hypergranulosis, and acanthosis (hematoxylin and eosin staining, original magnification 100×).
A diagnosis of ARCI was posed. All affected and healthy family members or their legal guardian provided written and informed consent according to a protocol approved by the institutional review board at the Tel Aviv Medical Center and by the Israel National Committee for Human Genetic Studies in adherence to the Helsinki guidelines. DNA was extracted from peripheral blood lymphocytes. We initially excluded all ARCI-associated loci by using a panel of microsatellite markers, as previously reported.9,10 We therefore scrutinized the whole genome for regions of homozygosity shared by all affected individuals by using Illumina HumanLinkage-12 chip (Illumina Inc.) that contains ∼6000 tagged SNPs across the genome. Two hundred nanograms of DNA was hybridized according to the Infinium II assay (Illumina Inc.) and scanned with an Illumina BeadArray reader. The scanned images were imported into BeadStudio 3.1.3.0 (Illumina Inc.) for extraction and quality control and had an average call rate of 99.9%. The data were then scanned for homozygous regions with a MatLab (MathWorks)-based software script.
We identified five regions of homozygosity larger than 2 Mb on 10q23 (13 Mb; 19 consecutive SNPs), 4q12 (7 Mb; six consecutive SNPs), 19p11 (9 Mb; five consecutive SNPs), 4q31.3 (6 Mb; four consecutive SNPs), and 4q15.2 (2 Mb; four consecutive SNPs). Polymorphic microsatellite markers spanning those five areas were selected from the GeneLoc site. Genotypes were established by PCR amplification of genomic DNA as previously described11 by using the BigDye terminator system on an automated sequencer (ABI PRISM 3100 Genetic Analyzer; Applied Biosystems, Foster City, CA, USA), and allele sizes were determined with GeneMapper v4.0 software. Fine mapping of the disease-associated interval with microsatellite typing suggested linkage to 10q23 (Figure 1) and excluded all other candidate loci. Using the Superlink software,12 we obtained a maximal LOD score of 4.11 at marker D10S1687 (Figure 3). Haplotype analysis (Figure 1) revealed critical recombinations in individuals II-3, II-6, and II-11, setting the disease boundaries at markers D10S1174 and D10S520 (Figure 1). The 10.8 Mb interval was found to contain 124 genes of which 28 were sequenced (Table S1, available online).
Figure 3.
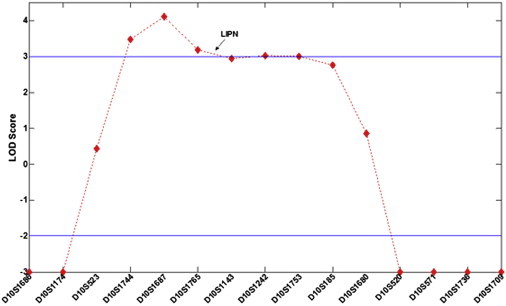
Multipoint Analysis of Lod Score
Lod scores are plotted against the 15 markers assessed.
Of particular interest was a group of three genes encoding epidermal acid lipases. At least six acid lipases are involved in triglyceride hydrolysis in mammals and are designated as lipase A (lysosomal; MIM 613497); lipase F (gastric; MIM 601980); lipase J (testis); and lipases K, M, and N, which are specifically expressed in the epidermis.13 Mammalian acid lipase genes have been shown to be clustered in all mammals examined to date, suggesting that they appeared through duplication during evolution.14 Although the National Center for Biotechnology Information database contains two SNPs in LIPN predicted to result in premature termination of translation (rs35519318 and rs75379216), we were unable to identify these variants in 496 healthy controls, including 250 Arab Muslims, 46 Druzes, and 200 Europeans, suggesting that these SNPs are either exceedingly rare or possibly nonexistant.
Genomic DNA was amplified by PCR by using primer pairs spanning the entire coding sequence and all intron-exon boundaries of LIPK, LIPM, and LIPN (Table S2) as well as 25 additional genes (Table S1). PCR amplification was performed with Taq polymerase. Cycling conditions were 94°C for 2 min followed by three sets of three cycles each (set 1: 94°C for 40 s, 61°C for 40 s, and 72°C for 40 s; set 2: 94°C for 40 s, 59°C for 40 s, and 72°C for 40 s; set 3: 94°C for 40 s, 57°C for 40 s, and 72°C for 40 s); by 33 cycles at 94°C for 40 s, 55°C for 40 s, and 72°C for 40 s; and then by a final extension step at 72°C for 10 min. DNA was extracted from gel, purified with QIAquick Gel Extraction kit (QIAGEN), and submitted to direct sequencing with the BigDye terminator system on an automated sequencer (ABI PRISM 3100 Genetic Analyzer; Applied Biosystems, Foster City, CA, USA).
Direct sequencing of the resulting PCR products revealed a homozygous 2 bp deletion in exon 3 of LIPN, encoding lipase member N, at position 399–400 of the cDNA sequence (c.399_400delGA; p.Glu133AspfsX3; accession numbers NM_001102469.1, NP_001095939.1) (Figure 4). To confirm the mutation, we amplified a 453 bp long DNA fragment with forward primer 5′- CTGTGATGTTTCCGACATGC-3′, and reverse primer 5′- GCTTCTTGCTGTTACCCTCC-3′ (Figure 1). The resulting amplicons were digested with BsmAI endonucleases and visualized with ethidium bromide. Using this assay, we confirmed segregation of the mutation throughout the extended family and also assessed 925 healthy individuals for the mutation, including 679 individuals of Arab Muslim origin, 46 Druzes, and 200 Ashkenazi Jews. One individual of Arab Muslim extraction was found to carry mutation c.399–400delGA in an heterozygous state. This individual was found to share a common haplotype with affected members of the family, possibly pointing to a founder effect (not shown). Taken together, these results indicate that p.Glu133AspfsX3 does not represent a common neutral polymorphism but rather is a disease-causing mutation.
Figure 4.
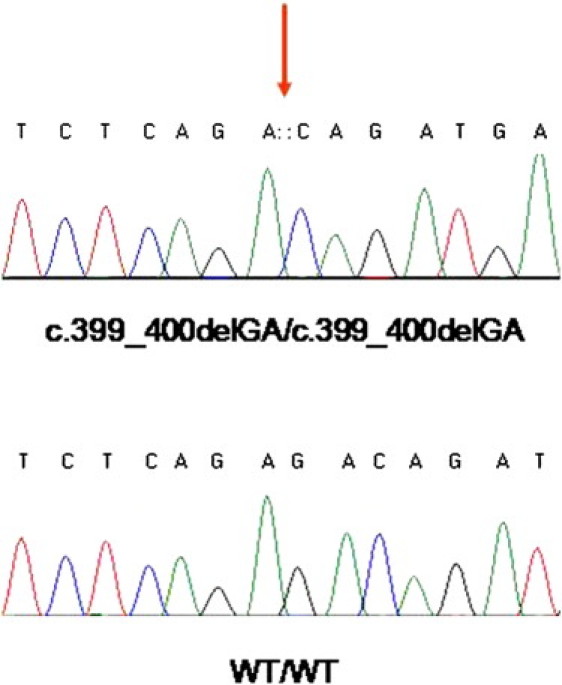
Mutation Analysis
Sequence analysis of LIPN reveals a homozygous deletion at cDNA position 399_400 (upper panel, red arrow). The wild-type sequence is given for comparison (lower panel).
Mutation c.399–400delGA is predicted to result in the generation of a stop codon two amino acids downstream from the mutation site (p.Glu133AspfsX3), which in turn is likely to lead to decreased LIPN mRNA levels because of nonsense mediated mRNA decay. To assess this possibility, we extracted total RNA from a patient's skin biopsy by using RNeasy Extraction Kit (QIAGEN). cDNA was synthesized (Thermo Scientific VersoTM cDNA Synthesis Kit, UK) and PCR amplified with exon-crossing primers located in LIPN exons 6 and 8, respectively (5′- TGAATCAGAGTCGAATGGATGTG-3′; 5′- GTGGTAAAGCTGTTTTATATGCAGAATG-3′). Figure 5A shows that mutation c.399–400delGA results in markedly decreased LIPN mRNA levels.
Figure 5.
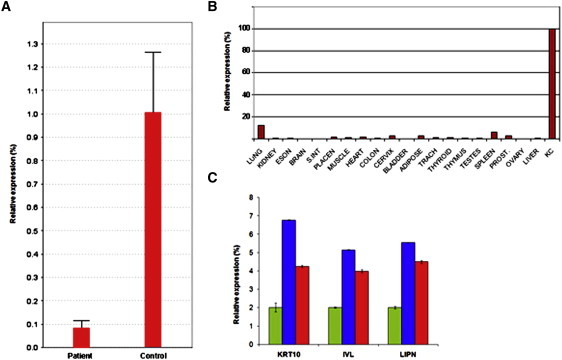
LIPN Expression Analysis
(A) Quantitative RT-PCR was performed on mRNA samples extracted from keratinocyte cultures derived from a patient and a nonaffected individual (control). Results are provided as a percentage of LIPN expression in control keratinocytes ± SD.
(B) LIPN expression was assessed via Clontech tissue blot cDNA array. Expression of LIPN was normalized to that of ACTB. Results are provided as a percentage of LIPN expression in control keratinocytes ± SD. The following abbreviations are used: ESON = esophagus; S.INT = small intestine; PLACEN = placenta; TRACH = trachea; PROST = prostate; KC = keratinocytes.
(C) Total RNA was extracted from organotypic cell cultures at day 0 (green), day 7 (blue), and day 10 (red). LIPN, IVL, and KRT10 expression were compared by using quantitative RT-PCR. Results are provided as a percentage of expression at day 0.
Acid lipases represent a special group of lipases that, unlike the neutral lipases such as liproprotein lipases, are capable of withstanding acid conditions14 within organelles such as lysosomes or in certain tissues, such as the gastric mucosa. For example, lipase A catalyzes the deacylation of triacylglycerols and cholesterol esters in lysosomal low-density lipoproteins and has been associated with the pathogenesis of several forms of lysosomal storage disease.15,16 Lipase F, in contrast, is critical for proper catabolism of triglycerides in the stomach.17 More limited information on the role of other acid lipases is available. Three of these lipases, K, M and N, have been shown to be expressed in the epidermis.13 All mammalian epidermal acid lipases seem to have been extremely well conserved over more than 100 million years of evolution,14 suggesting their importance for the normal maturation of stratified epithelia.
In line with previous semiquantitative RT-PCR experiments,13 LIPN mRNA levels were significantly higher by quantitative RT-PCR in epidermal cells compared with 20 different tissues (Figure 5B). Although this pattern of tissue-specific expression suggests involvement of lipase N in the process of cornification, which was obviously disturbed in our patients (Figure 2), we attempted to obtain more direct evidence for lipase N involvement in epidermal differentiation. At different time points during epidermal stratification, we extracted total RNA from organotypic cultures derived from primary human keratinocytes as previously described18 and compared the expression of LIPN to that of IVL and KRT10 (known markers of epidermal differentiation). Along with KRT10 and IVL, LIPN was found to be upregulated during epidermal differentiation (Figure 5C), suggesting that lipase N is part of the differentiation program of human keratinocytes.
In summary, the present data indicate that decreased expression of LIPN is associated with a late-onset form of ARCI and add to the steadily growing list of genes related to ARCI and encoding proteins involved in lipid metabolism. Abnormal epidermal lipid metabolism has been shown to underlie the pathogenesis of numerous forms of ichthyosis,19,20 including X-linked recessive ichthyosis (MIM 308100) due to deficient activity of steroid sulfatase;21 harlequin ichthyosis due to decreased function of the lipid transporter, ABCA12;22 ARCI caused by mutations in genes encoding enzymes belonging to the hepoxylin pathway;23–25 and ichthyosis prematurity syndrome (MIM 608649) caused by mutations in FATP4, encoding fatty acid transport protein 4.26 Despite the critical importance of triglyceride metabolism for proper epidermal maturation,25 the exact function of lipase N is unknown at the present. However, it is of interest to note that another syndromic form of congenital-recessive ichthyosis, Chanarin-Dorfman syndrome (MIM 275630), has been shown to be caused by mutations in ABHD5 (MIM 604780),27 which encodes a cofactor for adipose triglyceride lipase, also known as patatin-like phospholipase domain protein 2 (PNPLA2; MIM 609059).28,29 Recent data obtained in a mouse model for this disease30 indicate that ABHD5 targets epidermal lipases that are distinct from PNPLA2. It is tempting to speculate that LIPN may encode one of these enigmatic epidermal lipases.
Acknowledgments
We would like to acknowledge the participation of the family in this study. This study was supported in part by grants from the Israel General Trustee Fund.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
GeneLoc, http://genecards.weizmann.ac.il/geneloc/index.shtml
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Superlink, http://bioinfo.cs.technion.ac.il/superlink-online-twoloci/makeped/TwoLociMultiPoint.html
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Oji V., Tadini G., Akiyama M., Blanchet Bardon C., Bodemer C., Bourrat E., Coudiere P., DiGiovanna J.J., Elias P., Fischer J. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Dermatol. 2010;63:607–641. doi: 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 2.Styperek A.R., Rice Z.P., Kamalpour L., Pavlis M., Kuo J., Culler S., Spraker M.K., Chen S.C. Annual direct and indirect health costs of the congenital ichthyoses. Pediatr. Dermatol. 2010;27:325–336. doi: 10.1111/j.1525-1470.2010.01160.x. [DOI] [PubMed] [Google Scholar]
- 3.Akiyama M. ABCA12 mutations and autosomal recessive congenital ichthyosis: A review of genotype/phenotype correlations and of pathogenetic concepts. Hum. Mutat. 2010;31:1090–1096. doi: 10.1002/humu.21326. [DOI] [PubMed] [Google Scholar]
- 4.Vahlquist A. Pleomorphic ichthyosis: Proposed name for a heterogeneous group of congenital ichthyoses with phenotypic shifting and mild residual scaling. Acta Derm. Venereol. 2010;90:454–460. doi: 10.2340/00015555-0937. [DOI] [PubMed] [Google Scholar]
- 5.Mizrachi-Koren M., Geiger D., Indelman M., Bitterman-Deutsch O., Bergman R., Sprecher E. Identification of a novel locus associated with congenital recessive ichthyosis on 12p11.2-q13. J. Invest. Dermatol. 2005;125:456–462. doi: 10.1111/j.0022-202X.2005.23777.x. [DOI] [PubMed] [Google Scholar]
- 6.Hatsell S.J., Stevens H., Jackson A.P., Kelsell D.P., Zvulunov A. An autosomal recessive exfoliative ichthyosis with linkage to chromosome 12q13. Br. J. Dermatol. 2003;149:174–180. doi: 10.1046/j.1365-2133.2003.05386.x. [DOI] [PubMed] [Google Scholar]
- 7.Wu W.M., Lee Y.S. Autosomal recessive congenital ichthyosis maps to chromosome 15q26.3 in an isolated aboriginal population from southern Taiwan. J. Dermatol. Sci. 2011;61:62–64. doi: 10.1016/j.jdermsci.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 8.Fischer J. Autosomal recessive congenital ichthyosis. J. Invest. Dermatol. 2009;129:1319–1321. doi: 10.1038/jid.2009.57. [DOI] [PubMed] [Google Scholar]
- 9.Lugassy J., Hennies H.C., Indelman M., Khamaysi Z., Bergman R., Sprecher E. Rapid detection of homozygous mutations in congenital recessive ichthyosis. Arch. Dermatol. Res. 2008;300:81–85. doi: 10.1007/s00403-007-0815-0. [DOI] [PubMed] [Google Scholar]
- 10.Mizrachi-Koren M., Shemer S., Morgan M., Indelman M., Khamaysi Z., Petronius D., Bitterman-Deutsch O., Hennies H.C., Bergman R., Sprecher E. Homozygosity mapping as a screening tool for the molecular diagnosis of hereditary skin diseases in consanguineous populations. J. Am. Acad. Dermatol. 2006;55:393–401. doi: 10.1016/j.jaad.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 11.Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000;18:233–234. doi: 10.1038/72708. [DOI] [PubMed] [Google Scholar]
- 12.Fishelson M., Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18(Suppl 1):S189–S198. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 13.Toulza E., Mattiuzzo N.R., Galliano M.F., Jonca N., Dossat C., Jacob D., de Daruvar A., Wincker P., Serre G., Guerrin M. Large-scale identification of human genes implicated in epidermal barrier function. Genome Biol. 2007;8:R107. doi: 10.1186/gb-2007-8-6-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmes R.S., Cox L.A., VandeBerg J.L. Comparative studies of mammalian acid lipases: Evidence for a new gene family in mouse and rat (Lipo) Comp. Biochem. Physiol. Part D Genomics Proteomics. 2010;5:217–226. doi: 10.1016/j.cbd.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Assmann G., Krauss R.M., Fredrickson D.S., Levy R.I. Positional specificity of triglyceride lipases in post-heparin plasma. J. Biol. Chem. 1973;248:7184–7190. [PubMed] [Google Scholar]
- 16.Hoeg J.M., Demosky S.J., Jr., Pescovitz O.H., Brewer H.B., Jr. Cholesteryl ester storage disease and Wolman disease: Phenotypic variants of lysosomal acid cholesteryl ester hydrolase deficiency. Am. J. Hum. Genet. 1984;36:1190–1203. [PMC free article] [PubMed] [Google Scholar]
- 17.Bodmer M.W., Angal S., Yarranton G.T., Harris T.J., Lyons A., King D.J., Pieroni G., Riviere C., Verger R., Lowe P.A. Molecular cloning of a human gastric lipase and expression of the enzyme in yeast. Biochim. Biophys. Acta. 1987;909:237–244. doi: 10.1016/0167-4781(87)90083-2. [DOI] [PubMed] [Google Scholar]
- 18.Fuchs-Telem D., Stewart H., Rapaport D., Nousbeck J., Gat A., Gini M., Lugassy Y., Emmert S., Eckl K., Hennies H.C. CEDNIK syndrome results from loss-of-function mutations in SNAP29. Br. J. Dermatol. 2011;164:610–616. doi: 10.1111/j.1365-2133.2010.10133.x. [DOI] [PubMed] [Google Scholar]
- 19.Schmuth M., Gruber R., Elias P.M., Williams M.L. Ichthyosis update: Towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv. Dermatol. 2007;23:231–256. doi: 10.1016/j.yadr.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pilgram G.S., Vissers D.C., van der Meulen H., Pavel S., Lavrijsen S.P., Bouwstra J.A., Koerten H.K. Aberrant lipid organization in stratum corneum of patients with atopic dermatitis and lamellar ichthyosis. J. Invest. Dermatol. 2001;117:710–717. doi: 10.1046/j.0022-202x.2001.01455.x. [DOI] [PubMed] [Google Scholar]
- 21.Elias P.M., Crumrine D., Rassner U., Hachem J.P., Menon G.K., Man W., Choy M.H., Leypoldt L., Feingold K.R., Williams M.L. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J. Invest. Dermatol. 2004;122:314–319. doi: 10.1046/j.1523-1747.2003.22258.x. [DOI] [PubMed] [Google Scholar]
- 22.Hovnanian A. Harlequin ichthyosis unmasked: A defect of lipid transport. J. Clin. Invest. 2005;115:1708–1710. doi: 10.1172/JCI25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akiyama M., Shimizu H. An update on molecular aspects of the non-syndromic ichthyoses. Exp. Dermatol. 2008;17:373–382. doi: 10.1111/j.1600-0625.2007.00691.x. [DOI] [PubMed] [Google Scholar]
- 24.Fürstenberger G., Epp N., Eckl K.M., Hennies H.C., Jørgensen C., Hallenborg P., Kristiansen K., Krieg P. Role of epidermis-type lipoxygenases for skin barrier function and adipocyte differentiation. Prostaglandins Other Lipid Mediat. 2007;82:128–134. doi: 10.1016/j.prostaglandins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 25.Elias P.M., Williams M.L., Holleran W.M., Jiang Y.J., Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: Inherited disorders of lipid metabolism. J. Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klar J., Schweiger M., Zimmerman R., Zechner R., Li H., Törmä H., Vahlquist A., Bouadjar B., Dahl N., Fischer J. Mutations in the fatty acid transport protein 4 gene cause the ichthyosis prematurity syndrome. Am. J. Hum. Genet. 2009;85:248–253. doi: 10.1016/j.ajhg.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruno C., Bertini E., Di Rocco M., Cassandrini D., Ruffa G., De Toni T., Seri M., Spada M., Li Volti G., D'Amico A. Clinical and genetic characterization of Chanarin-Dorfman syndrome. Biochem. Biophys. Res. Commun. 2008;369:1125–1128. doi: 10.1016/j.bbrc.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 28.Montero-Moran G., Caviglia J.M., McMahon D., Rothenberg A., Subramanian V., Xu Z., Lara-Gonzalez S., Storch J., Carman G.M., Brasaemle D.L. CGI-58/ABHD5 is a coenzyme A-dependent lysophosphatidic acid acyltransferase. J. Lipid Res. 2010;51:709–719. doi: 10.1194/jlr.M001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown J.M., Chung S., Das A., Shelness G.S., Rudel L.L., Yu L. CGI-58 facilitates the mobilization of cytoplasmic triglyceride for lipoprotein secretion in hepatoma cells. J. Lipid Res. 2007;48:2295–2305. doi: 10.1194/jlr.M700279-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Radner F.P., Streith I.E., Schoiswohl G., Schweiger M., Kumari M., Eichmann T.O., Rechberger G., Koefeler H.C., Eder S., Schauer S. Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58) J. Biol. Chem. 2010;285:7300–7311. doi: 10.1074/jbc.M109.081877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.