

Abstract
Aims/hypothesis
We investigated the molecular mechanism by which the human glucagon-like peptide-1 analogue liraglutide preserves pancreatic beta cells in diabetic db/db mice.
Methods
Male db/db and m/m mice aged 10 weeks received liraglutide or vehicle for 2 days or 2 weeks. In addition to morphological and biochemical analysis of pancreatic islets, gene expression profiles in the islet core area were investigated by laser capture microdissection and real-time RT-PCR.
Results
Liraglutide treatment for 2 weeks improved metabolic variables and insulin sensitivity in db/db mice. Liraglutide also increased glucose-stimulated insulin secretion (GSIS) and islet insulin content in both mouse strains and reduced triacylglycerol content in db/db mice. Expression of genes involved in cell differentiation and proliferation in both mouse strains was regulated by liraglutide, which, in db/db mice, downregulated genes involved in pro-apoptosis, endoplasmic reticulum (ER) stress and lipid synthesis, and upregulated genes related to anti-apoptosis and anti-oxidative stress. In the 2 day experiment, liraglutide slightly improved metabolic variables in db/db mice, but GSIS, insulin and triacylglycerol content were not affected. In db/db mice, liraglutide increased gene expression associated with cell differentiation, proliferation and anti-apoptosis, and suppressed gene expression involved in pro-apoptosis; it had no effect on genes related to oxidative stress or ER stress. Morphometric results for cell proliferation, cell apoptosis and oxidative stress in db/db mice islets were consistent with the results of the gene expression analysis.
Conclusions/interpretation
Liraglutide increases beta cell mass not only by directly regulating cell kinetics, but also by suppressing oxidative and ER stress, secondary to amelioration of glucolipotoxicity.
Keywords: Apoptosis, Beta cell mass, Cellular differentiation, Cellular proliferation, ER stress, Glucagon-like peptide-1, Oxidative stress
Introduction
It is widely accepted that beta cell function progressively deteriorates in individuals with type 2 diabetes mellitus as described, for example, by the UK Prospective Diabetes Study [1]. To prevent diabetic complications, maintenance of strict glycaemic control is necessary, but this requires preservation of beta cell function. On the other hand, human diabetic patients exhibit an early defect in glucose-stimulated insulin secretion (GSIS) [2] and autopsy studies have demonstrated reduced beta cell mass in diabetic patients [3]. Thus, development of treatment strategies has focused on ways to improve beta cell function and to prevent beta cell death. Hence, there is increased interest in determining whether newer drugs such as insulin-sensitising agents produce sustained improvements in beta cell function [4, 5]. Likewise, agents associated with glucagon-like peptide-1 (GLP-1) have also attracted considerable attention because they may alter the natural history of type 2 diabetes by preserving pancreatic beta cell mass [6–8] and function [9–11].
GLP-1 has several beneficial effects that counteract the pathophysiology of diabetes mellitus. First, GLP-1 is a potent glucose-lowering polypeptide that induces glucose-dependent insulin secretion [12, 13] while suppressing glucagon secretion [14, 15]. Second, GLP-1 has extra-pancreatic effects, such as delayed gastric emptying [16, 17], appetite suppression [18] and improved insulin sensitivity [19]. Third and last, GLP-1 stimulates beta cell replication [20, 21], neogenesis [22] and differentiation [22], as well as inhibiting beta cell apoptosis via reduction of cellular stress [23–26]. Consequently, GLP-1-related agents are currently regarded as a powerful treatment option for type 2 diabetes [27, 28]. Liraglutide, a novel long-acting GLP-1 derivative, is resistant to dipeptidyl peptidase-IV. Its prolonged effects result from the substitution of Lys for Arg34 and the addition of a glutamic acid and a 16C NEFA to the Lys26 residue of native GLP-1 [29].
To investigate the molecular mechanism by which liraglutide preserves pancreatic beta cell mass, we treated obese diabetic db/db mice with liraglutide for 2 days or 2 weeks. We also treated normoglycaemic m/m mice with liraglutide for 2 weeks.
Methods
Animals
Male 9-week-old BKS.Cg-+ Lepr db/+ Lepr db/Jcl (db/db) mice and BKS. Cg-m+/m+/Jcl (m/m) mice were purchased (Clea, Tokyo, Japan). They were housed two to three animals per cage in all experiments under controlled ambient conditions and a 14:10 h light/dark cycle with lights on at 07:00 hours. Animals were given free access to drinking water and conventional food (Oriental Yeast, Tokyo, Japan). They received a subcutaneous injection of liraglutide (200 μg/kg) or vehicle (PBS) twice daily (09:00 and 16:00 hours). Doses of the peptides used in the study were based on a review of the literature [7]. Body weight and food intake were monitored weekly from 9 weeks of age. All experiments were approved by the Kawasaki Medical School Animal Experiment Committee (No. 06-062) and were performed in accordance with the Kawasaki Medical School guidelines for animal experiments.
Measurement of biochemical markers
Blood samples were collected from the tail vein once per week. Blood glucose was measured immediately using a commercially available enzyme electrode method (FreeStyle; Kissei Pharmaceutical, Nagano, Japan). Plasma was separated and stored at −80°C until use. The concentration of insulin in the plasma was measured using an ELISA kit (Morinaga Institute of Biological Science, Yokohama, Japan) and the plasma triacylglycerol concentration was determined enzymatically (E-Test; Wako, Osaka, Japan).
Insulin tolerance test
After the 2 day or 2 week liraglutide treatment in db/db mice, an insulin tolerance test was performed by a intraperitoneal injection of insulin (2 units/kg body weight) after an overnight fast. Blood samples were collected from the tail vein every 30 min and blood glucose was determined immediately as described above.
Measurement of insulin content in pancreatic islets
Pancreatic islets were isolated using the collagenase digestion method as previously described [30]. Briefly, Hanks’ balanced salt solution containing 1.5 mg/ml collagenase (Collagenase P; Roche, Basel, Switzerland) and 10% (vol./vol.) fetal calf serum was infused into the bile duct. The excised pancreas was transferred to Hanks’ balanced salt solution and centrifuged three times for 2 min each time at 1,100 rpm (200 g). The final pellet was passed through a metal filter and the filtrate was centrifuged for 22 min at 2,500 rpm (1,000 g) using Histopaque-1077 (Sigma, St Louis, MO, USA). The islet samples were stored at −80°C until measurement of insulin content.
Measurement of triacylglycerol content in pancreatic islets
Pancreatic islets were isolated as described above and 45–60 of the isolated pancreatic islets were washed twice in PBS, to which 50 μl of a high-salt buffer (2 mol/l NaCl, 2 mmol/l EDTA, 50 mmol/l sodium phosphate) was added, followed by sonication for 1 min to disrupt the pancreatic islets. After centrifugation for 5 min at 12,000 rpm (13,000 g), 10 μl of the supernatant fraction was mixed with 10 μl t-butanol plus 50 μl Triton X-100-methyl alcohol (1:1). Triacylglycerol content in the pancreatic islets was measured using E-Test (Wako) according to the manufacturer’s instructions.
Glucose-stimulated insulin secretion from isolated pancreatic islets
Size-matched pancreatic islets were prepared (five pancreatic islets per tube) and preincubated in KRB-HEPES buffer containing 5 mg/ml BSA, pH 7.4, and 95% O2/5% CO2 saturated at 37°C for 60 min. The supernatant fraction was replaced with a 3.0 or 16.7 mmol/l glucose solution, and the mixture was incubated for an additional 60 min. The supernatant fraction was recovered and stored at −80°C until the insulin assay was performed.
Immunohistochemistry
Pancreas tissue sections (4 μm) were stained with haematoxylin–eosin. For immunostaining, the sections were immersed for 15 min in methanol containing 3% (vol./vol.) hydrogen peroxide to block endogenous peroxidase activity. After rinsing with PBS (10 mmol/l, pH 7.0), the sections were incubated for 1 h with a mixture of antibodies (rabbit anti-glucagon antibody: anti-somatostatin antibody 1:1; Nichirei, Tokyo, Japan) at 25°C. Tissue sections were also incubated for 14 h at 4°C with either a mouse anti-insulin monoclonal antibody, an anti-proliferative cell nuclear antigen (PCNA) monoclonal antibody (Nichirei) or a mouse anti-4-hydroxy-2-noneal modified protein (4HNE) monoclonal antibody (25 μg/ml; Japan Institute for the Control of Aging, Shizuoka, Japan). After rinsing with PBS, simple stain diaminobenzidine solution (Nichirei) was added and the mixture was incubated for 7 min at 25°C. The sections were counterstained with haematoxylin. To investigate cell apoptosis, a TUNEL assay was performed using a colorimetric apoptotic detection system (DeadEnd; Promega, Madison, WI, USA), as described previously [31].
Morphometric analysis
The image analysis software NIH Image (Version 1.61; http://rsbweb.nih.gov/ij/) was used to calculate the entire pancreatic area, islet area and glucagon- and somatostatin-positive cells, and also to determine the relative islet area and the relative beta cell area. Using 15 sections (i.e. five sections from three different areas of the pancreas) for each group of mice, beta cell mass was estimated by the following formula: beta cell mass (mg) = the pancreas weight (mg) × per cent pancreatic islet area × percent beta cell count.
Laser capture microdissection
According to our previously established procedure [32], frozen, 8 μm thick tissue slices were immediately stained and subjected to laser capture microdissection. After tissue staining, the islets were irradiated with a laser using the PixCell system (Arcturus, Mountain View, CA, USA). The peripheral area was first removed and then the beta-cell-rich core area was collected.
Real-time PCR
RNA was extracted using an RNA isolation kit (PicoPure PN 12206-01; Arcturus). TaqMan reverse transcription reagents (N808-0234; Applied Biosystems, Foster, CA, USA) were used for reverse transcription and random hexamers were used as primers for cDNA synthesis. The primers were designed using Primer Express (Applied Biosystems) and were based on mRNA sequences downloaded from the GenBank nucleotide database (www.ncbi.nlm.nih.gov/nuccore).
A reaction solution was prepared by combining 0.5 μl of the sample, 1 μl of a 50 nmol/l primer solution, 5 μl Sybr Green PCR Master Mix (Applied Biosystems) solution and 3.5 μl diluent. Dissociation curve analysis was performed for each experiment to determine the dissociation temperature and the size of the PCR products was confirmed by agarose gel electrophoresis. We examined gene expression in the core area of the pancreatic islets, selecting primary genes for which a statistically significant difference between the groups in the first gene expression experiment had been observed. The genes analysed in the present study were those associated with cell differentiation (Hlxb-9 [also known as Mnx1], Hes1, Neurod [also known as Neurod1] and Pdx1), cell proliferation (CycD and Erk-1 [also known as Mapk3]), endoplasmic reticulum (ER) stress (Xbp1), antioxidative stress (Cat and Gpx), lipid synthesis (Srebp-1c [also known as Srebf1] and Fas) and cell apoptosis (Bcl2, Casp8, Casp3 and Cad). To quantify gene expression, 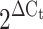 was calculated using 18S rRNA as an internal control.
was calculated using 18S rRNA as an internal control.
Statistical analysis
All data are presented as means ± SEM. A Mann–Whitney U test was used to test for differences among multiple groups, with p < 0.05 regarded as significant. The statistical analyses were performed with StatView (version 5; SAS, Cary, NC, USA).
Results
Metabolic variables in db/db mice with chronic liraglutide treatment
After 1 week of treatment, liraglutide significantly decreased food intake (23.7 ± 1.6 vs 39.8 ± 0.8 g per week, p < 0.0001; Fig. 1a) and suppressed body weight gain (41.3 ± 0.4 vs 43.3 ± 0.5 g, p < 0.05; Fig. 1b) in db/db mice compared with vehicle-treated mice. Liraglutide also reduced fasting glucose (7.0 ± 0.4 vs 9.4 ± 0.6 mmol/l, p < 0.05; Fig. 1c) and insulin concentrations (567.9 ± 51.6 vs 1,032 ± 86.1 pmol/l, p < 0.01; Fig. 1d) in db/db mice after 1 week of treatment. These effects were sustained by prolonged intervention with liraglutide until 2 weeks, at which time the liraglutide-treated group had a significant improvement in hypertriacylglycerolaemia (1.3 ± 0.03 vs 1.5 ± 0.07 mmol/l, p < 0.01; Fig. 1e). We also found that brief liraglutide treatment, i.e. for 2 days, resulted in similar metabolic improvements (Fig. 2a, b, d). The exception was fasting insulin, which was increased by 2 days of liraglutide treatment compared with vehicle (826.1 ± 86.1 vs 567.9 ± 34.4 pmol/l, p < 0.05; Fig. 2c). Insulin sensitivity in db/db mice was improved only after 2 weeks of liraglutide treatment (Figs 1f, 2e).
Fig. 1.

Metabolic variables in diabetic db/db mice treated for 2 weeks with vehicle (diamonds) or liraglutide (squares). a Food intake, (b) body weight, (c) Fasting blood glucose, (d) fasting plasma insulin concentration, (e) fasting plasma triacylglycerol concentration and (f) insulin sensitivity assessed by an i.p. insulin tolerance test. Results of the insulin tolerance test are expressed as a percentage relative to the basal blood glucose concentration before insulin administration. n = 7 for each group; *p < 0.05, **p < 0.01, ***p < 0.001; † p < 0.005
Fig. 2.

Metabolic variables in diabetic db/db mice treated for 2 days with vehicle (black bar, diamonds) or liraglutide (white bar, black squares). a Body weight (BW) change, (b) fasting blood glucose, (c) fasting plasma insulin concentration, (d) fasting plasma triacylglycerol concentration and (e) insulin sensitivity assessed by i.p. insulin tolerance test. Results of the tolerance test are expressed as a percentage relative to the basal blood glucose concentration before insulin administration. n = 11 for each group; *p < 0.05, ***p < 0.001
Islet function and morphology in db/db mice treated with liraglutide
To investigate whether 2 weeks of liraglutide treatment exerted beneficial effects on the pancreatic islets of db/db mice, we first subjected isolated pancreatic islets to morphological analysis and immunostaining with antibodies against glucagon and somatostatin. While liraglutide treatment in db/db mice conserved the normal morphology of pancreatic islets, in which glucagon- and somatostatin-positive cells were observed in the periphery, vehicle treatment disrupted these structures (Fig. 3a). In addition, beta cell mass was significantly increased in liraglutide-treated db/db mice, compared with vehicle-treated mice (7.7 ± 0.7 vs 4.9 ± 0.5 mg, p < 0.01; Fig. 3b).
Fig. 3.

Islet morphology and function in db/db mice treated with vehicle (black bars) or liraglutide (white bars) for 2 days or 2 weeks. a Haematoxylin–eosin staining, followed by double immunohistochemical staining with antibodies against glucagon and somatostatin. Scale bars 100 μm. b Beta cell mass, measured as described; n = 5 (a, b). c Immunohistochemical staining with antibodies against PCNA, quantified (d) as ratio. e, f 4-HNE and (g, h) TUNEL assays and quantification as indicated. c–h n = 3 for each assay, scale bars 100 μm. The proportion of cells positive for PCNA (c, d) and 4-HNE (e, f) was measured in each islet and expressed as average proportion for a minimum of 50 islets from each experimental animal. DNase-treated cells were used as a positive control in the TUNEL assay (g, h), with results of the TUNEL assay expressed as the average number of TUNEL-positive cells per islet by assessing a minimum of 50 islets from each experimental animal. i Islet insulin content and (j) islet triacylglycerol content; n = 5. k GSIS was assessed by use of lower and higher glucose concentrations (3 mmol/l and 16.7 mmol/l); n = 5. *p < 0.05, **p < 0.01
After the 2 week intervention, islet insulin content in liraglutide-treated db/db mice was greater than that in db/db mice treated with vehicle for 2 weeks (33.7 ± 2.3 vs 17.2 ± 2.1 ng/islet, p < 0.01; Fig. 3i). Although islet insulin content in vehicle-treated db/db mice tended to decrease with age (2 days 24.7 ± 2.7 vs 2 weeks 17.2 ± 2.1 ng/islet, p = 0.1; Fig. 3i), that in mice treated with liraglutide for 2 weeks was increased compared with mice treated with liraglutide for 2 days (2 weeks 33.7 ± 2.3 vs 2 days 27.0 ± 1.7 ng/islet, p < 0.05; Fig. 3i).
After 2 weeks of liraglutide treatment, insulin secretion was increased in response to 16.7 mmol/l glucose, but not to 3 mmol/l (1.5 ± 0.2 vs 0.9 ± 0.1 ng/ml per islet, p < 0.05; Fig. 3k). However, 2 days of liraglutide treatment had no effect on insulin secretion, irrespective of glucose concentration (Fig. 3k). These results suggest that chronic liraglutide treatment improved the reactivity of pancreatic beta cells in response to the higher glucose concentration.
Next, we examined the effect of liraglutide treatment on islet triacylglycerol content, which was not affected by 2 days of liraglutide treatment (Fig. 3j), although hypertriacylglycerolaemia was improved (Fig. 2d). However, the accumulation of triacylglycerol in the pancreatic islets was prevented by 2 weeks of liraglutide treatment (Fig. 3j), despite an increase in islet triacylglycerol content with ageing in vehicle-treated mice.
A morphological examination was used to study the mechanisms involved in the restoration of pancreatic beta cell mass by liraglutide in db/db mice. Histological sections of the pancreatic islet were stained by an antibody specific for PCNA and the proportion of antibody-positive cells was measured to evaluate the proliferative effect on pancreatic islets. Liraglutide-treated mice exhibited a significant increase in PCNA-positive cells in pancreatic islets (Fig. 3c, d). Oxidative stress, which was evaluated by scoring 4-HNE-positive cells in pancreatic islets, was reduced in the pancreatic islets of liraglutide-treated mice (Fig. 3e, f), while cellular apoptosis, analysed by a TUNEL assay, was suppressed (Fig. 3g, h).
Effect of liraglutide on gene expression in pancreatic beta cells of db/db mice
Both 2 weeks and 2 days of liraglutide treatment significantly augmented expression of genes involved in cellular differentiation (Hlxb-9, Neurod and Pdx1; Fig. 4a, c, d, g, i, j) and significantly reduced expression of an anti-differentiation gene (Hes1) in pancreatic beta cells of db/db mice (Fig. 4b, h). The expression of genes involved in cellular proliferation (CycD and Erk-1) was also increased significantly in db/db mice treated with liraglutide for 2 weeks, with similar results obtained in mice treated for 2 days (Fig. 4e, f, k, l). Liraglutide also significantly increased expression of the anti-apoptotic Bcl2 gene (Fig. 4m, q) and reduced expression of pro-apoptotic genes (Casp8, Casp3 and Cad) in both treatment regimens (Fig. 4n, o, p, r, s, t).
Fig. 4.

Gene expression in the core area of islets in db/db mice treated with vehicle (black bars) or liraglutide (white bars). a–f Expression of genes involved in cell differentiation and proliferation after 2 weeks and (g–l) 2 days of treatment. a, g Hlxb-9, (b, h) Hes1, (c, i) Neurod, (d, j) Pdx1, (e, k) Erk1, (f, l) CycD. m–t Expression of genes involved in pro-apoptosis and anti-apoptosis after 2 weeks (m–p) and 2 days (q–t) of treatment. m, q Bcl2, (n, r) Casp8, (o, s) Casp3, (p, t) Cad. u–dd Expression of genes involved in anti-oxidative stress, ER stress and lipid synthesis after 2 weeks (u–y) and 2 days (z–dd) of treatment. u, z Gpx, (v, aa) Cat, (w, bb) Xbp1, (x, cc) Fas, (y, dd) Srebp-1c. n = 4 for each group; *p < 0.05, **p < 0.01; † p < 0.005; ‡ p < 0.0001; § p = 0.08
In contrast, chronic but not acute liraglutide treatment significantly increased the expression of genes involved in anti-oxidative stress (Cat and Gpx; Fig. 4u, v, z, aa) and significantly reduced those related to ER stress (Xbp1; Fig. 4w, bb) and lipid synthesis (Srebp-1c and Fas) in beta cells of db/db mice (Fig. 4x, y, cc, dd). The expression profile of Srebp-1c and Fas (Fig. 4x, y, cc, dd), which was consistent with islet triacylglycerol content (Fig. 3j), indicated that liraglutide alleviates the lipotoxicity experienced by beta cells in a time-dependent fashion. Together, these results suggest that liraglutide directly accelerates cellular differentiation and proliferation, and inhibits apoptosis via a reduction in oxidative and ER stress mediated by improvements in glucolipotoxicity.
Impact of liraglutide on metabolism in normoglycaemic mice
We next investigated the effect of liraglutide in normoglycaemic m/m mice, which were treated with or without liraglutide for 1 week. Food intake in m/m mice, compared with the vehicle-treated group, was significantly reduced by liraglutide treatment (21.1 ± 0.8 vs 27.9 ± 1.0 g/week, p < 0.05; Fig. 5a), as was body weight gain (20.1 ± 0.2 vs 22.2 ± 0.9 g, p < 0.05; Fig. 5b). These effects were sustained by prolonged liraglutide treatment for up to 2 weeks, but were not correlated with metabolic variables, such as fasting concentrations of blood glucose, insulin and triacylglycerol, which did not differ statistically between liraglutide-treated and vehicle-treated m/m mice (Fig. 5c–e).
Fig. 5.

Metabolic variables in normoglycaemic m/m mice treated with vehicle (diamonds) or liraglutide (squares). a Food intake, (b) body weight, (c) fasting blood glucose, (d) fasting plasma insulin concentration and (e) fasting plasma triacylglycerol concentration. n = 5 for each group; *p < 0.05, **p < 0.01
Effect of liraglutide on morphology, function and gene expression of pancreatic beta cells in normoglycaemic mice
In m/m mice, beta cell mass and islet insulin content were significantly increased by liraglutide treatment (1.0 ± 0.1 vs 0.7 ± 0.1 mg, p < 0.01; 59.0 ± 3.7 vs 44.4 ± 3.9 ng/islet, p < 0.05; Fig. 6a, b). The increases in beta cell mass and islet insulin content were associated with improvements in GSIS in isolated islets in response to 16.7 mmol/l glucose (1.6 ± 0.1 vs 1.2 ± 0.03 ng/ml per islet, p < 0.05; Fig. 6c) in the liraglutide-treated group compared with the vehicle-treated group.
Fig. 6.

Islet morphology and islet function in m/m mice treated for 2 weeks with vehicle (black bars) or liraglutide (white bars). a Pancreatic beta cell mass, (b) islet insulin content and (c) GSIS. d Immunohistochemical staining of the pancreatic islet with antibodies against PCNA, (e) 4-HNE and (f) TUNEL. n = 4 for each group; *p < 0.05, **p < 0.01
The number of PCNA-positive cells was significantly increased (Fig. 6d), suggesting that the proliferation of pancreatic islets was upregulated in liraglutide-treated m/m mice. Nevertheless, the numbers of 4-HNE-positive and TUNEL-positive cells in the pancreatic islets did not differ between liraglutide- and vehicle-treated m/m mice (Fig. 6e, f). At the level of mRNA expression, m/m mice treated with liraglutide for 2 weeks experienced a significant increase in expression of genes involved in cellular differentiation (Hlxb-9, Neurod and Pdx1; Fig. 7a, c, d) and proliferation (Erk-1 and CycD; Fig. 7e, f), along with a significant reduction in expression of an anti-differentiation gene (Hes1; Fig. 7b). The expression of mRNA for genes associated with cell apoptosis, anti-oxidative stress, ER stress and lipid synthesis was not statistically different between liraglutide- and vehicle-treated mice (Fig. 7g–o). These results suggest that liraglutide affects beta cell mass by stimulating cellular differentiation and proliferation without affecting blood glucose, insulin or triacylglycerol concentrations.
Fig. 7.

Gene expression in the core area of islets in m/m mice treated with vehicle (black bars) or liraglutide (white bars) for 2 weeks. a–f Expression of genes involved in cell differentiation and proliferation, i.e. (a) Hlxb-9, (b) Hes1, (c) Neurod, (d) Pdx1, (e) Erk1 and (f) CycD. g–j Expression of genes involved in cell apoptosis, i.e. (g) Bcl2, (h) Casp8, (i) Casp3 and (j) Cad. k–o Expression of genes involved in anti-oxidative stress, ER stress and lipid synthesis, i.e. (k) Gpx, (l) Cat, (m) Xbp1, (n) Fas and (o) Srebp-1c. n = 4 for each group; *p < 0.05; † p < 0.005
Discussion
In the present study, we investigated the effects of liraglutide on obese diabetic mice. We found that the beta cell mass of db/db mice was increased by long-term liraglutide treatment. These results are essentially consistent with previous reports describing a stimulatory effect on beta cell mass of exendin-4, a potent GLP-1 receptor agonist, in a partial pancreatectomy rat model of type 2 diabetes [20] and of liraglutide, the long-acting GLP-1 derivative, in diabetic db/db mice [7].
The present results demonstrate that short- and long-term treatment with liraglutide affected mRNA expression of Hlxb-9, Hes1 and Neurod in the core of islets in diabetic db/db mice. The roles of several genes with well defined functions in pancreatic development, such as those mentioned above (Hlxb9, Hes-1 and Neurod) have not been previously studied in the adult pancreas. On the other hand, previous studies have suggested that the Notch signal through hairy and enhancer of split-1 (HES1) is activated even in the adult pancreas in conditions associated with cell regeneration, such as inflammation and neoplasia in the pancreas [33, 34]. In addition, a recent in vitro study also demonstrated that HES1 is involved in determining the beta cell fate of adult human beta cells [35]. These findings suggest the possibility that genes associated with an early stage of endocrine pancreas development are expressed in adult db/db mice and that liraglutide affects the expression of those genes. However, the present results cannot be regarded as conclusive, and further studies will be required to elucidate this issue.
In this study, cell proliferation related to CycD and Erk-1 gene expression was readily upregulated in diabetic and normoglycaemic mice treated with liraglutide for 2 days. Immunohistochemical analysis of the pancreatic islets suggested that enhancement of cellular proliferation might be an underlying mechanism that accounts for the restorative effects of GLP-1 on pancreatic beta cell mass. These results strongly support the hypothesis that liraglutide affects pancreatic beta cell mass by directly stimulating cellular proliferation. Interestingly, a previous study demonstrated that pancreatic beta cell mass in non-diabetic rats was significantly increased after 1 week of liraglutide treatment, but no different from that in control animals after 6 weeks of treatment, indicative of a temporary effect on beta cell mass [36]. This result suggests that the effect of liraglutide on beta cell kinetics is acute and temporary in non-diabetic conditions.
The binding of GLP-1 to its receptors activates adenylate cyclase and the cyclic AMP/protein kinase A signalling pathway. Additionally, GLP-1 activates phosphoinositide 3-kinase (PI3K), p42 mitogen-activated protein kinase (MAPK) and the epidermal growth factor receptor [37–39]. Furthermore, activation of the transcription factor for Pdx1 [38], p38 MAPK and protein kinase C-zeta [40] reportedly plays a role in GLP-1-induced DNA synthesis and replication. Thus there is emergent evidence for extensive cross-talk between the G-protein-coupled receptor and tyrosine kinase-coupled receptor signalling pathways in beta cells. The results of the present study show that modulation of the CycD gene is involved in the liraglutide-induced increase in beta cell mass, presumably through the MAPK pathway. We also found that cellular differentiation is affected by liraglutide via upregulation of Pdx1, which is located downstream of the PI3K–protein kinase B–forkhead box O1 pathway.
Liraglutide modified the expression of genes related to cell apoptosis such as Bcl2, Casp8, Casp3 and Cad in db/db mice during short- and long-term treatment, whereas mRNA levels of these genes were not altered in normoglycaemic m/m mice, suggesting that liraglutide directly suppressed beta cell apoptosis in mice under hyperglycaemic conditions. In a previous report, extracellular signal-regulated kinase (ERK)1/2 regulated Bcl2 gene expression and protein production, as well as the activity of caspase 3 through the regulation of cyclic AMP-responsive element-binding protein [41]. Furthermore, the extracellular matrix directly suppressed caspase 8 activity through regulation of the ERK but not the PI3K pathway in pancreatic beta cells [42]. These results indicate that liraglutide may have a direct anti-apoptotic effect, which is at least partly mediated by ERK.
With regard to the effect of GLP-1 on oxidative stress, Tews and co-workers [26] reported that exendin-4 directly reduces oxidative stress through counterregulation of the reduced abundance of electron transport chain proteins in INS-1 beta cells. In the present study, immunohistological analysis with anti-4-HNE staining and the TUNEL assay showed that 2 weeks of liraglutide treatment inhibited cellular oxidative stress and apoptosis of the pancreatic islets in diabetic db/db mice. We also showed that 2 weeks, but not 2 days of liraglutide treatment suppressed expression of Cat and Gpx. The expression of Srebp-1c and Fas mRNAs, as well as islet triacylglycerol content were also decreased only by the 2 week liraglutide treatment regimen. These results suggest that, in contrast to the action of exendin-4 in cultured cell lines, the improvements in oxidative stress observed with liraglutide treatment in db/db mice are secondary to improvements in glucolipotoxicity.
The inhibitory effect of long-term liraglutide treatment on expression of Xbp1 in the core area of pancreatic islets, which should reflect a decrease in the unfolded protein accumulated in the ER of pancreatic beta cells, is essentially consistent with the findings of Yusta et al., who reported that chronic administration of exendin-4 decreased expression of Chop (also known as Ddit3) and spliced Xbp1 in the pancreatic islets of db/db mice [43]. They also found that exendin-4 potentiated the induction of activating transcription factor 4, a transcription factor mediated by phosphorylated eukaryotic translation initiation factor 2 α (eIF2α), in the INS-1 beta cell in a manner that is dependent on protein kinase A, accelerating recovery from ER stress-mediated translational repression by growth arrest and DNA-damage-inducible 34, which promotes eIF2α dephosphorylation [43]. Contrary to the in vitro findings reported by Yusta et al. [43], the present in vivo study demonstrated that even short-term liraglutide treatment did not affect expression of Xbp1 in hyperglycaemic mice. The different study conditions, in vivo and in vitro experiments, may have led to these different results.
Fehmann and Habener demonstrated a stimulatory effect of GLP-1 on proinsulin biosynthesis in insulinoma beta TC-1 cells [9]. In the present study, we found that liraglutide enhanced the responsiveness of beta cells to a glucose challenge in diabetic and normoglycaemic mice. These results are essentially consistent with previous studies that have reported an improved responsiveness of the pancreatic beta cells to glucose when isolated mouse islets were incubated with liraglutide [10].
In conclusion, we have demonstrated that liraglutide restores pancreatic beta cell mass due to acute effects on cell kinetics and chronic effects on oxidative and ER stress that are secondary to improvements in glucolipotoxicity. These conclusions were mainly obtained by analysing the changes of gene expression in pancreatic islet cells. To fully understand the effect of liraglutide on pancreatic beta cell mass, further investigations, e.g. assessing the effect of liraglutide on peptide expression and function, as well as its role in neogenesis from the ducts, should be conducted.
Acknowledgements
This study was supported by a Grant-in-Aid from the Japan Society for the Promotion of Science (18591008, 21591153 to K. Kaku) and Research Project Grants from Kawasaki Medical School (18-501, 19-502, 20-505 to K. Kaku). We thank L. Bierre Knudsen, Novo Nordisk, for the generous gift of liraglutide and for valuable advice for this study. Abstracts of this report were presented at the 68th annual scientific session of the American Diabetes Association (San Francisco) and the 45th annual meeting of the European Association for the Study of Diabetes (Vienna).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- eIF2α
Eukaryotic translation initiation factor 2 α
- ER
Endoplasmic reticulum
- ERK
Extracellular signal-regulated kinase
- GLP-1
Glucagon-like peptide-1
- GSIS
Glucose-stimulated insulin secretion
- HES1
Hairy and enhancer of split-1
- 4HNE
4-Hydroxy-2-noneal modified protein
- MAPK
Mitogen-activated protein kinase
- PCNA
Proliferative cell nuclear antigen
- PI3K
Phosphoinositide 3-kinase
References
- 1.UK Prospective Diabetes Study (UKPDS) Group Overview of 6 years’ therapy of type II diabetes (UKPDS 16) Diabetes. 1995;44:1249–1258. doi: 10.2337/diabetes.44.11.1249. [DOI] [PubMed] [Google Scholar]
- 2.Bagdade JD, Bierman EL, Porte D., Jr The significance of basal insulin levels in the evaluation of the insulin response to glucose in diabetic and nondiabetic subjects. J Clin Invest. 1967;46:1549–1557. doi: 10.1172/JCI105646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 4.Buchanan TA, Xiang AH, Peters RK, et al. Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk Hispanic women. Diabetes. 2002;51:2796–2803. doi: 10.2337/diabetes.51.9.2796. [DOI] [PubMed] [Google Scholar]
- 5.Ovalle F, Bell DS. Effect of rosiglitazone versus insulin on the pancreatic beta-cell function of subjects with type 2 diabetes. Diab Care. 2004;27:2585–2589. doi: 10.2337/diacare.27.11.2585. [DOI] [PubMed] [Google Scholar]
- 6.Pospisilik JA, Martin J, Doty T, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes. 2003;52:741–750. doi: 10.2337/diabetes.52.3.741. [DOI] [PubMed] [Google Scholar]
- 7.Rolin B, Larsen MO, Gotfredsen CF, et al. The long-acting GLP-1 derivative NN2211 ameliorates glycemia and increases beta-cell mass in diabetic mice. Am J Physiol Endocrinol Metab. 2002;283:E745–E752. doi: 10.1152/ajpendo.00030.2002. [DOI] [PubMed] [Google Scholar]
- 8.Tourrel C, Bailbe D, Lacorne M, Meile MJ, Kergoat M, Portha B. Persistent improvement of type 2 diabetes in the Goto–Kakizaki rat model by expansion of the beta-cell mass during the prediabetic period with glucagon-like peptide-1 or exendin-4. Diabetes. 2002;51:1443–1452. doi: 10.2337/diabetes.51.5.1443. [DOI] [PubMed] [Google Scholar]
- 9.Fehmann HC, Habener JF. Insulinotropic hormone glucagon-like peptide-I (7-37) stimulation of proinsulin gene expression and proinsulin biosynthesis in insulinoma beta TC-1 cells. Endocrinology. 1992;130:159–166. doi: 10.1210/en.130.1.159. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Perfetti R, Greig NH, et al. Glucagon-like peptide-1 can reverse the age-related decline in glucose tolerance in rats. J Clin Invest. 1997;99:2883–2889. doi: 10.1172/JCI119482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Alessio DA, Kahn SE, Leusner CR, Ensinck JW. Glucagon-like peptide 1 enhances glucose tolerance both by stimulation of insulin release and by increasing insulin-independent glucose disposal. J Clin Invest. 1994;93:2263–2266. doi: 10.1172/JCI117225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7–37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. 1987;79:616–619. doi: 10.1172/JCI112855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet. 1987;2:1300–1304. doi: 10.1016/S0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- 14.Orskov C, Holst JJ, Nielsen OV. Effect of truncated glucagon-like peptide-1 [proglucagon-(78–107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology. 1988;123:2009–2013. doi: 10.1210/endo-123-4-2009. [DOI] [PubMed] [Google Scholar]
- 15.Nauck MA, Wollschlager D, Werner J, et al. Effects of subcutaneous glucagon-like peptide 1 (GLP-1 [7–36 amide]) in patients with NIDDM. Diabetologia. 1996;39:1546–1553. doi: 10.1007/s001250050613. [DOI] [PubMed] [Google Scholar]
- 16.Wettergren A, Schjoldager B, Mortensen PE, Myhre J, Christiansen J, Holst JJ. Truncated GLP-1 (proglucagon 72–107 amide) inhibits gastric and pancreatic function in man. Dig Dis Sci. 1993;38:665–673. doi: 10.1007/BF01316798. [DOI] [PubMed] [Google Scholar]
- 17.Näslund E, Gutniak MK, Skogar S, Rössner S, Hellström PM. GLP-1 increases the period of postprandial satiety and slows gastric emptying in obese humans. Am J Clin Nutr. 1998;68:525–530. doi: 10.1093/ajcn/68.3.525. [DOI] [PubMed] [Google Scholar]
- 18.Näslund E, Barkeling B, King N, et al. Energy intake and appetite are suppressed by glucagon-like-peptide 1 (GLP-1) in obese men. Int J Obes Relat Metab Disord. 1999;23:304–311. doi: 10.1038/sj.ijo.0800818. [DOI] [PubMed] [Google Scholar]
- 19.Lee YS, Shin S, Shigihara T, et al. Glucagon-like peptide-1 gene therapy in obese diabetic mice results in long-term cure of diabetes by improving insulin sensitivity and reducing hepatic gluconeogenesis. Diabetes. 2007;56:1671–1679. doi: 10.2337/db06-1182. [DOI] [PubMed] [Google Scholar]
- 20.Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270–2276. doi: 10.2337/diabetes.48.12.2270. [DOI] [PubMed] [Google Scholar]
- 21.Friedrichsen BN, Neubauer N, Lee YC, et al. Stimulation of pancreatic beta-cell replication by incretins involves transcriptional induction of cyclin D1 via multiple signalling pathways. J Endocrinol. 2006;188:481–492. doi: 10.1677/joe.1.06160. [DOI] [PubMed] [Google Scholar]
- 22.Paris M, Tourrel-Cuzin C, Plachot C, Ktorza A. Review: pancreatic beta-cell neogenesis revisited. Exp Diabesity Res. 2004;5:111–121. doi: 10.1080/15438600490455079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ. Glucagon-like peptide-1 receptor signaling modulates cell apoptosis. J Biol Chem. 2003;278:471–478. doi: 10.1074/jbc.M209423200. [DOI] [PubMed] [Google Scholar]
- 24.Bregenholt S, Møldrup A, Blume N, et al. The long-acting glucagon-like peptide-1 analogue, liraglutide, inhibits beta-cell apoptosis in vitro. Biochem Biophys Res Commun. 2005;330:577–584. doi: 10.1016/j.bbrc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 25.Hui H, Nourparvar A, Zhao X, Perfetti R. Glucagon-like peptide-1 inhibits apoptosis of insulin-secreting cells via a cyclic 5′-adenosine monophosphate-dependent protein kinase A- and a phosphatidylinositol 3-kinase-dependent pathway. Endocrinology. 2003;144:1444–1455. doi: 10.1210/en.2002-220897. [DOI] [PubMed] [Google Scholar]
- 26.Tews D, Lehr S, Hartwig S, Osmers A, Paslack W, Eckel J. Anti-apoptotic action of exendin-4 in INS-1 beta cells: comparative protein pattern analysis of isolated mitochondria. Horm Metab Res. 2009;41:294–301. doi: 10.1055/s-0028-1105911. [DOI] [PubMed] [Google Scholar]
- 27.Meier JJ, Nauck MA. The potential role of glucagon-like peptide 1 in diabetes. Curr Opin Investig Drugs. 2004;5:402–410. [PubMed] [Google Scholar]
- 28.Bjerre Knudsen L. Glucagon-like peptide I: the basis of a new class of treatment for type 2 diabetes. J Med Chem. 2004;47:4128–4134. doi: 10.1021/jm030630m. [DOI] [PubMed] [Google Scholar]
- 29.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyi peptidase-4 inhibitors in type 2diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 30.Kitamura T, Kido Y, Nef S, Merenmies J, Parada LF, Accili D. Preserved pancreatic beta-cell development and function in mice lacking the insulin receptor-related receptor. Mol Cell Biol. 2001;21:5624–5630. doi: 10.1128/MCB.21.16.5624-5630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farilla L, Hui H, Bertolotto C, et al. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143:4397–4408. doi: 10.1210/en.2002-220405. [DOI] [PubMed] [Google Scholar]
- 32.Kanda Y, Shimoda M, Hamamoto S, et al. Molecular mechanism by which pioglitazone preserves pancreatic β cells in obese diabetic mice: evidence for acute and chronic actions as a PPARγ agonist. Am J Physiol Endocrinol Metab. 2010;298:E278–E286. doi: 10.1152/ajpendo.00388.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jensen JN, Cameron E, Garay MV, et al. Replication of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–741. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Miyamoto Y, Maitra A, Ghosh B, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/S1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 35.Bar Y, Russ HA, Knoller S, et al. HES-1 is involved in adaptation of adult human beta-cells to proliferation in vitro. Diabetes. 2008;57:2413–2420. doi: 10.2337/db07-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bock T, Pakkenberg B, Buschard K. The endocrine pancreas in non-diabetic rats after short-term and long-term treatment with the long-acting GLP-1 derivative NN2211. APMIS. 2003;111:1117–1124. doi: 10.1111/j.1600-0463.2003.apm1111207.x. [DOI] [PubMed] [Google Scholar]
- 37.Frödin M, Sekine N, Roche E, et al. Glucose, other secretagogues, and nerve growth-factor stimulate mitogen- activated protein-kinase in the insulin-secreting beta-cell line, INS-1. J Biol Chem. 1995;270:7882–7889. doi: 10.1074/jbc.270.14.7882. [DOI] [PubMed] [Google Scholar]
- 38.Buteau J, Roduit R, Susini S, Prentki M. Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increases transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia. 1999;42:856–864. doi: 10.1007/s001250051238. [DOI] [PubMed] [Google Scholar]
- 39.Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. 2003;52:124–132. doi: 10.2337/diabetes.52.1.124. [DOI] [PubMed] [Google Scholar]
- 40.Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M. Protein kinase Cζ activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes. 2001;50:2237–2243. doi: 10.2337/diabetes.50.10.2237. [DOI] [PubMed] [Google Scholar]
- 41.Costes S, Broca C, Bertrand G, et al. ERK1/2 control phosphorylation and protein level of cAMP-responsive element-binding protein: a key role in glucose-mediated pancreatic beta-cell survival. Diabetes. 2006;55:2220–2230. doi: 10.2337/db05-1618. [DOI] [PubMed] [Google Scholar]
- 42.Hammar E, Parnaud G, Bosco D, et al. Extracellular matrix protects pancreatic beta-cells against apoptosis: role of short- and long-term signaling pathways. Diabetes. 2004;53:2034–2041. doi: 10.2337/diabetes.53.8.2034. [DOI] [PubMed] [Google Scholar]
- 43.Yusta B, Baggio LL, Estall JL, et al. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006;4:391–406. doi: 10.1016/j.cmet.2006.10.001. [DOI] [PubMed] [Google Scholar]