

Abstract
The ocean contains a host of macroscopic life in a great microbial soup. Unlike the terrestrial environment, an aqueous environment provides perpetual propinquity and blurs spatial distinctions. Marine organisms are under a persistent threat of infection by resident pathogenic microbes including bacteria, and in response they have engineered complex organic compounds with antibacterial activity from a diverse set of biological precursors. The diluting effect of the ocean drives the construction of potent molecules that are stable to harsh salty conditions. Members of each class of metabolite—ribosomal and non-ribosomal peptides, alkaloids, polyketides, and terpenes—have been shown to exhibit antibacterial activity. The sophistication and diversity of these metabolites points to the ingenuity and flexibility of biosynthetic processes in Nature. Compared with their terrestrial counterparts, antibacterial marine natural products have received much less attention. Thus, a concerted effort to discover new antibacterials from marine sources has the potential to contribute significantly to the treatment of the ever increasing drug-resistant infectious diseases.
Keywords: antibiotics, antimicrobials, metabolites, natural products
Introduction
The term “antibiotic,” defined in 1942 by Selman A. Waksman, originally referred to any microbial product antagonistic to the growth of another microorganism.[1] In common usage today, “antibiotic” describes any compound that kills (bactericidal) or inhibits the growth (bacteriostatic) of bacteria.[2] The naturally-occurring compounds discussed in this Review, which we refer to as “antibacterials,” fit into the latter definition. Those metabolites that act upon other microorganisms (fungi and protozoa) will not be considered. Taken together, the molecules featured herein are best defined as antibacterial marine natural products.
Most antibacterials used clinically are either naturally-produced or resemble natural products.[3] Seventy-four of the 98 antibacterial small molecules marketed between 1981 and 2005 contained discrete structural motifs that were taken from Nature.[3j] Of the 12 antibacterial classes, nine are derived from a natural product template. The molecular architectures of the β-lactams (penicillins, cephalosporins, carbapenems, monobactams), polyketides (tetracycline), phenylpropanoids (chloramphenicol), aminoglycosides (streptomycin), macrolides (erythromycin), glycopeptides (vancomycin), streptogramins (pristinamycin), and, most recently, the lipopeptides (daptomycin) and glycylcyclines (tegicycline) are borrowed from natural products. The other three classes—the sulfonamides, quinolones (ciprofloxacin), and oxazolidinones (linezolid)—have no precedence in Nature. Their design is purely synthetic and not influenced by structural elements of known metabolites.
Following the discovery of most of the antibacterial classes in the 1940s to 1960s, the so-called “Golden Age” of antibacterial research, the arsenal of compounds for the treatment of bacterial infections in humans was deemed sufficient. However, with the immediate development of antibacterial resistance in microbes, this belief was quickly dispelled. Application of antibacterials creates a selective pressure that culminates in certain random mutations, at a chromosomal level, rendering the bacterium more “fit” to survive in the presence of these antibacterial compounds.[4,5] The genetic material that encodes this fitness is rapidly propagated and/or dispersed. Improper or excessive use of antibacterial drugs dramatically accelerates the development of resistance. As a result, pathogenic strains of methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus (VRE), and fluoroquinolone-resistant Pseudomonas having emerged, bacterial infections have reached epidemic levels in 2010.
Modern pharmaceutical development of antibacterials has, by in large, relied upon incremental semisynthetic modifications of natural product templates validated more than half a century ago. In fact, 73% of the antibacterial drugs approved between 1981 and 2005 encompassed only three classes, the β-lactams, macrolides, and quinolones.[3j] This approach has produced molecules that narrowly, and temporarily, evade existing mechanisms of resistance. It seems obvious that only the discovery of new natural scaffolds that, by virtue of their chemical novelty, inhibit previously unknown bacterial targets, can satisfy long-term concerns over bacterial resistance.
Compared to the terrestrial environment, which was the focus of the pharmaceutical industry for more than 50 years, marine habitats have been virtually unexplored for their ability to yield antibacterial metabolites. We now know that species diversity in marine ecosystems dwarfs the biodiversity characteristic of tropical rainforests.[6] Life in the ocean encompasses a wide diversity of taxa, including protozoans, jellyfish, anemones, flatworms, roundworms, bryozoans, clams, squid, copepods, annelids, sea stars, sea cucumbers, corals, sponges, and algae.[7] In addition, open ocean seawater harbors 106 bacterial and 103 fungal cells per milliliter, and most marine organisms host specific populations of microbes on their surfaces or within the confines of their tissues. The diverse microbial population that exists in ocean sediments (108 bacterial cells per gram of wet mud),[8] like-wise, has been virtually overlooked as a source of novel antibiotic-producing microorganisms. Without a doubt, the pressing need for new antibacterials must be met via the exploration of under-explored habitats like the world’s oceans.
The literature concerning antibacterial natural products from the ocean has been reviewed before. Mayer and Hamann, in particular, have covered this area for the past 10 years with several concise reviews.[9] Other reviews of marine antibacterial natural products have been published,[10] though they often exclude discussions of an important class, a group of ribosomally-derived peptides referred to as the antimicrobial peptides (AMPs).[11] As stated earlier, the goal of this Review is to introduce several marine-derived natural products that possess significant antibacterial properties. A handful of metabolites have been selected that combine antibacterial potency with novelty and/or complexity in chemical structure. Some attention has also been paid to highlighting the vast variety of sources in the marine environment.
The 26 metabolites discussed herein are grouped into five classes based on their stuctures and/or biosynthetic origins: ribosomally-derived AMPs, nonribosomal peptides, polyketides, alkaloids, and terpenes.[12] Ribosomal peptides are often viewed as “primary metabolites” due to an apparent lack of structural complexity, although this distinction is quickly becoming obsolete.[13] Nonribosomal peptides, polyketides, alkaloids, and terpenes are formed from a series of enzymatic transformations employing a much more diverse set of precursors and biosynthetic reactions (Scheme 1). These compounds are classified as “secondary metabolites.” Despite this disparity in biosynthetic origin, we have included AMPs in this Review based on their prevalence in marine ecosystems and their common ability to exert antibacterial activity.
Scheme 1.

Building blocks for antibacterial natural product biosynthesis.
Testing for Antibacterial Activity
The bacterial strains employed in the testing laboratory can generally be divided into two classes based upon the distinct composition of their cell walls (Table 1). The cell walls of “Gram-positive” bacteria, and not those of “Gram-negative” bacteria, are vividly stained by a crystal violet-iodine dye. The distinction has broader implications for antibacterial efficacy since cell wall permeability is an essential criterion for the inhibition of an intracellular target. An antibacterial compound that is effectively transported across the thick peptidyglycan layer of Gram-positive bacteria (e.g., Staphylococcus aureus and Enterococcus faecium) lacks, in many cases, the appropriate chemical properties to cross the vastly different glycolipid layer and membrane characteristic of Gram-negative bacteria (e.g., Escherichia coli and Pseudomonas aeruginosa). In practice, the discovery of Gram-positive specific antibacterials has been successful, while the discovery of Gram-negative antibacterials has posed a greater challenge.
Table 1.
Common human pathogens used for laboratory assays
| Bacterium | Gram stain | Disease Pathology |
|---|---|---|
| Staphylococcus aureus[a] | + | staph infections |
| Streptococcus pneumoniae | + | Pneumonia |
| Streptococcus pyogenes | + | strep throat |
| Enterococcus faecium[b] | + | bacteraemia; endocarditis |
| Micrococcus luteus | + | non-pathogenic |
| Micrococcus nishinomiyaensis | non-pathogenic | |
| Listeria monocytogenes | + | listeriosis |
| Mycobacterium tuberculosis | + | tuberculosis |
| Escherichia coli | − | urinary tract infections; neonatal meningitis |
| Pseudomonas aeruginosa | − | pulmonary and urinary tract infections |
Including methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA).
Including vancomycin-resistant E. faecium (VRE).
Antibacterial activity can only be evaluated in the context of the bacterial strains that are selected. Traditionally, assessments of antibacterial activity in the marine natural products literature has focused on biomedically-relevant strains (see Table 1). Bacterial strains of biomedical importance have been screened in lieu of endogenous marine pathogens.[14] However, investigating the microbiological effects of secondary metabolites in an ecological context is extremely important if we are to develop a clear understanding of the extent to which microorganisms have influenced the evolution of secondary metabolites.[15] In one study, for example, extracts of several Atlantic and Indo-Pacific marine plants were shown to be active against Pseudoalter-monas bacteriolytica, an ecologically-relevant marine pathogenic bacterium.[16,17]
The study of antibacterial natural products is tied to the development of efficient assays to assess antibacterial activity. Two techniques are widely used in the laboratory. In the disc diffusion method,[18] a centralized paper disc is impregnated with the antibacterial compound and placed on a lawn of test bacteria. Antibiosis is measured by the diameter of bacterial growth inhibition on the agar plate. The diameter of the zone of inhibition is considered to correlate directly with antibacterial activity, although antibacterials will have inherently different diffusion coefficients in agar. In the microdilution method,[19] the compound of interest is serially diluted along each well of a microtiter plate and incubated with the test bacterium. The minimum inhibitory concentration (MIC) is determined from the change in optical density in the well. Notably, both methods offer a measure of antibacterial activity that makes no distinction (for better or for worse) regarding the exact bacterial protein target(s).
Ribosomal Peptides
Synthesized by the ribosome, antimicrobial peptides (AMPs) are the molecular agents of a non-specific innate immunity distinct from the adaptable lymphocyte-based immunity characteristic of higher vertebrates.[11] AMPs made by bacteria are called bacteriocins. In multicellular organisms, these natural products are found on external surfaces (lungs and skin) or in the granules of neutrophils. Invertebrate marine organisms, ascidians for example, harbor AMPs in neutrophile-like cells called hemocytes.
Although generally constructed of a simple set of amino acid precursors, AMPs adopt complex tertiary structures. The conformational properties of AMPs can be defined using circular dichrosim (CD), a technique that measures the differential absorption of left- and right-handed circularly polarized light.[20] In practice, β-sheet tertiary structures give a positive Cotton effect at 230 nm and a negative one near 215 nm. An α-helical structure yields a different profile with two negative Cotton effects at 222 and 208 nm. In combination with nuclear magnetic resonance spectroscopy, CD spectroscopy is a powerful tool for investigating the tertiary structure of ribosomal peptides.
Ribosomal peptides are generally translated as “prepro-peptides” composed of an N-terminal signal sequence, a “pro” segment, and a C-terminal peptide. Subsequent proteolytic processing and other post-translational modifications reveal the mature antibacterial peptide, which can be divided into six classes: 1) linear peptides that fold into α-helices, 2) cyclic peptides that form β-sheet structures, 3) peptides rich in one or more amino acid residues, 4) cyclic peptides with thio-ether bonds, 5) lipopeptides, and 6) macrocyclic knotted peptides.
Unlike secondary metabolites, for which no general structural features exist, AMPs are cationic, amphipathic molecules between 12 and 45 amino acids in length. At physiological pH, they are largely cationic (between +2 and +9) due to a high frequency of basic amino acid residues (lysine, arginine, histidine) and a low number of acidic and neutral residues. Their amphipathicity stems from an unequal distribution of neutral hydrophobic residues, comprising 30–50% of the peptide, and cationic hydrophilic residues.
The cationicity characteristic of AMPs is responsible for their specificity and selective toxicity against bacterial cells. Bacterial cell membranes predominantly contain the phospholipids phosphatidylglycerol (PG), phosphatidylserine (PS), cardiolipin (CL). These anionic phospholipids have electrostatic affinity for cationic AMPs. The phosphate groups of the lipopolysaccharide leaflet on the extracellular surface of all Gram-negative bacteria are also negatively-charged. Mammalian cell membranes, by contrast, contain phophatidylcholine (PC), phophatidylethanolamine (PE), and sphingomyelin (SM), zwitterionic phospholipids with no net charge. Other factors such as the differential transmembrane potential between bacterial cells in logarithmic phase growth (−130 to −150 mV) and mammalian cells (−90 to −110 mV) help account for the specificity of AMPs.
The amphipathic nature of AMPs is also linked to antibacterial activity. At some threshold concentration, the peptides aggregate, begin to traverse, and ultimately disrupt the cell membrane. α-Helical AMPs are often disordered in aqueous solution, but quickly adopt a rigid amphipathic helical structure when exposed to a lipid bilayer. β-sheet AMPs are much more ordered, even in aqueous solution, since they contain stabilizing intramolecular bonds formed from cyclization of the peptidyl backbone or oxidative coupling of internal cysteine residues.
For an antibacterial ribosomal peptide to find use as a therapeutic agent in humans depends upon its ability to function at physiological salt concentrations. The electrostatic attraction between a cationic AMP and the anionic bacterial cell membrane is crucial for antibacterial activity, and the presence of salt weakens this key interaction. Indeed, the loss in efficacy of salt-sensitive AMPs in the lungs has been shown to account for the persistent infections of P. aeruginosa in cystic fibrosis patients.[21] Intuitively, AMPs from the marine environment should be designed to sustain activity at elevated salt concentrations.[22–24] The frequent occurrence of intramolecular cystine bonds and cyclized peptidyl backbones in marine peptides, which decrease the sensitivity of AMPs toward the effects of salt, supports this notion.
Arenicin-1 (1), C127H193N41O25S2, was purified from coelomocytes (hemocytes) of the marine lugworm Arenicola marina, collected off the coast of Sredniy, White Sea, Russia (Scheme 2).[25] The peptide is translated as a 202-residue prepropeptide containing an N-terminal signal peptide (25 residues), a “pro” segment (156 residues), and a C-terminal arenicin segment (21 residues). The mature peptide is comprised of six basic arginine residues and several lipophilic residues. In solution, the peptide adopts a two-stranded antiparallel β-sheet structure as evidenced by its CD properties (positive cotton effect at 230 nm, negative at 216 nm).[25–27] The hairpin turn in 1 is stabilized by an internal disulfide bond, which contributes to the establishment and maintenance of its amphipathicity.[27] Supporting the notion that AMPs interact with the bacterial membrane, peptide 1 was shown to bind strongly to anionic (SDS) and zwitterionic (DPC) detergent micelles.[26] The specificity of these AMPs for bacterial membranes over mammalian cells, assessed by measuring the AMP’s hemolytic activity toward red blood cells, can be finely-tuned through the generation of analogues.[28,29]
Scheme 2.

Ribosomal (1–4) and NRPS-derived (5–8) antibacterial peptides.
Arenicin-1 (1) showed activity against the Gram-negative bacteria E. coli and Pseudomonas mirabilis (MIC=0.8 and 2 μgmL−1, respectively),[27] as well as Gram-positive bacteria such as S. aureus (MIC=6 μgmL−1).[26] Its antibacterial activity against E. coli was not greatly attenuated at elevated NaCl concentrations (up to 100–150 mM).[27] Linear derivatives of 1, in which the cysteine residues that contribute to the disulfide bond are replaced with serine residues, are two times less active than the parent molecule.[26] An analogue with two cystine bonds was two-fold more active owing to the increased rigidity of its tertiary structure.[29] Replacement of the arginine residues with lysine produced derivatives that were slightly less active and more susceptible to changes in salt concentration.[30]
Halocidin (2), C155H251N47O38S2, was isolated from hemocytes of the tunicate Halocynthia aurantium, purchased at a local seafood market in Sockcho, South Korea.[31] Before processing, the newly-translated peptide consists of an N-terminal signal peptide, a halocidin segment, a single glycine residue, and a C-terminal anionic extension.[32] The active molecule is composed of two peptides, one containing 15 amino acid residues and the other 18 residues, linked by a disulfide bond. Each mature subunit of 2 has two histidines and one lysine residue, interspersed with hydrophobic residues. A homodimer based on the longer 18 residue motif of halocidin (2) with C-terminal amidation showed little order in aqueous solution.[33] However, in the presence of SDS or membrane-like solvents (50% trifluoroethanol), the CD profile indicated a strong degree of α-helical character. Using the synthetic homodimer, antibacterial activities with MICs ranging from 2–4 μgmL−1 against clinical isolates of MRSA, VRE, and multi-drug resistant P. aeruginosa were measured.[32] Similar to arenicin-1 (1), the activity was maintained at salt concentrations as high as 300 mM. One derivative of 2 showed promising results in a mouse model of L. monocytogenes infection.[34]
Isolated from coelomocytes of the marine annelid Nereis diversicolor, hedistin (3), C112H166Br2N30O27, is another amphipathic antibacterial peptide.[35] Again, the prepropeptide is composed of a signal sequence, a hedistin segment, a glycine residue, and a C-terminal anionic extension. The molecule is composed of a mixture of basic and hydrophobic residues. Both tryptophan residues are decorated with bromine atoms, reflecting the common metabolism of bromine observed in marine organisms.[36] The incorporation of halogen into the peptide, however, is not required for antibacterial activity, as a synthetic hedistin molecule lacking bromine retained activity. The carboxy terminus of 3 is amidated, which in addition to halogenation, may make the compound more resistant to endogenous peptidases. Based upon interpretation of CD and NMR data, desbromo 3 was shown to adopt an amphipathic helix-turn-helix motif, though only upon introduction of SDS or DPC micelles.[37] Against Gram-positive bacteria, desbromo 3 showed excellent activity, giving an MIC=1–2 μgmL−1 against M. luteus and M. nishinomiyaensis. The synthetic peptide showed activity against S. aureus (MIC=8–15 μgmL−1) and other Staphylococcus species as well.[37]
Lastly, clavanin A (4), C131H185N35O26, isolated from hemocytes of the tunicate Styela clava, freshly-harvested at Long Beach, CA, is a 23 residue peptide.[38,39] Clavanin A shows structural and functional homology to magainin-1 from the skin of the frog Xenopus laevis,[40] but the tunicate peptide is unique due to the presence of C-terminal amidation and the replacement of lysines in magainin-1 with histidines in 4. The prepropeptide consists of a signal peptide, a “pro” region, a clavanin segment, a glycine residue, and an anionic extension.[41] CD spectra indicated an α-helical structure in trifluoroethanol. Glycine residues at position 6 and 13 were shown to act as essential hinges in the molecule, facilitating insertion of the hydrophobic N-terminal end of clavanin into the target membrane.[42] Other substitutions are less detrimental. The abundant phenylalanine residues can be replaced with residues with similar hydrophobic and conformational flexibility without a loss in antibacterial activity.[43] The molecules were demonstrated to interact strongly with PC bilayers.[44]
Clavanin (4) showed antibacterial activity against Gram-positive and Gram-negative bacteria.[45] Against S. aureus, including MRSA strains, clavanin (4) exhibited an MIC= 1.4–3.8 μgmL−1. MIC values of 0.1–1.1 μgmL−1 were measured against three strains of E. faecium. Strains of E. coli (MIC=0.4–2.3 μgmL−1) and three strains of P. aeruginosa (MIC=0.4–0.8 μgmL−1) were likewise susceptible to 4. Unlike magainin-1, but not unlike other marine AMPs, the peptide retained much of its activity in 100 mM NaCl. Notably, the histidine residues impart a pH-dependence on antibacterial activity. At pH 5.0–5.5 the molecule displayed significant activity against E. coli and L. monocytogenes, but at neutral pH its activity was greatly diminished.
In summary, ribosomally-produced AMPs are large, cationic, amphipathic molecules found among all classes of life. Ideally suited to target prokaryotic cell membranes, these peptides have broad-spectrum antibacterial properties. Their unique physical, chemical, and biological characteristics lend themselves well to academic pursuits, including, but not limited to, the study of their three-dimensional structure, biosynthesis, and mechanism-of-action. Unfortunately, to date, no AMP has been approved for systemic treatment of bacterial infection.[11g] Stitched together with amide and weak disulfide linkages, the molecules are susceptible to enzymatic degradation. Antibacterial secondary metabolites, which are covalently assembled via strong carbon bonds to nitrogen, oxygen, sulfur, and other carbon atoms, possess added stability that makes these molecules more likely effective as systemic drugs.
Nonribosomal Peptides
Nonribosomal peptides are constructed by large multi-functional protein complexes called nonribosomal peptide synthetases (NRPSs).[12] Adenylation (A), thiolation (T), and condensation (C) domains of NRPSs catalyze amide bond formation between amino acid residues. Nonproteinogenic amino acids, not encoded by DNA, are often incorporated into these peptides. Subsequent domains that catalyze epimerization of L-amino acids to D-amino acids, cyclization of cysteine and serine residues to thiazolines and oxazolines, N-methylation of amide functionalities, and a slew of other transformations may be scattered within the A-T-C arrangement.
A nonribosomal peptide, bogorol A (5), C80H142O16N16, was isolated from the marine bacterium Bacillus laterosporus PNG-276 collected near Lolata Island, Papau New Guinea (see Scheme 2).[46,47] The bacterial strain was obtained from the tissues of an unknown tube worm and identified by analyses of cellular fatty acids and 16S rRNA sequences. The natural product has structural features similar to the ribosomal peptides discussed earlier. Three basic lysine residues are interspersed with several hydrophobic residues. Complex “secondary” modifications—reduction of the C-terminal valine residue, conversion of the N-terminal isoleucine residue to 2-hydroxy-3-pentanoic acid, the presence amino acids in their unnatural D-configuration—distinguish peptides of this class from ribosomal peptides. Owing to its greater complexity and recent structural elucidation, no synthesis of 5 has yet been reported. Bogorol A (5) showed activity against MRSA with an MIC=2.5 μgmL−1 and VRE with an MIC=9 μgmL−1.[47] Like ribosomal antibacterial peptides, the cationicity of 5 may contribute toward targeting of the bacterial membrane. Whether 5 adopts an amphipathic tertiary structure like AMPs is not known.
Emericellamide A (6), C31H55N5O7, a 19-membered depsipeptide, was obtained from the marine-derived fungus Emericella sp. CNL-878.[48] The strain, identified using 18S rDNA sequence analysis, was collected from the surface of a green alga Halimeda sp. from Papua New Guinea. Production of the compound was greatly enhanced when the fungal strain was co-cultured with a marine actinomycete (Salinispora arenicola strain CNH-665), since, apparently, the fungus is then forced to compete for culture nutrients. The molecule is cyclized through condensation of the carboxy-terminus to a pendant secondary alcohol. The alcohol itself is part of a 3-hydroxy-2,4-dimethyldecanoic acid (HDMA) unit. Though the peptidyl portion of 6 is derived from an NRPS, the lipophilic HDMA unit originates from a polyketide synthase, the biosynthetic machinery responsible for the production of polyketides (see below). This mixed PKS-NRPS metabolite is much more difficult to synthesize than a typical ribosomal peptide, requiring between 8 and 16 synthetic steps to complete.[49–51] Furthermore, the biosynthetic pathway of the emercillamides was identified in Aspergillus nidulans.[52] The gene cluster was found to contain a single-module iterative type I polyketide synthase for the formation of the HDMA moiety. The polyketide intermediate is released as the free carboxylic acid and eventually loaded onto the free thiolation domain of an NRPS. The NRPS then adds five amino acids to the HDMA chain. The lack of a thioesterase (TE) domain at the end of the last module suggests that cyclization is not catalyzed by an enzyme. Emericellamide A (6) showed activity against MRSA (MIC=2.3 μgmL−1).[48] Exhibiting weak cytotoxicity against human colon carcinoma cell line HCT-116 (IC50 = 14 μgmL−1), this peptide may possess the selectivity against prokaryotic cells that is desirable of a drug.
Thiocoraline (7), C48H56N10O12S6, was isolated from a marine-derived actinomycete Micromonospora sp. strain L-13-ACM2-092. This strain, identified by analysis of its fatty acid components, was found in association with a soft coral in the Indian Ocean near the coast of Mozambique.[53,54] The natural product is a symmetric bicyclic octadepsipeptide. Reminiscent of halocidin (2), the molecule contains an intra-molecular disulfide bond. Thiocoraline is related to the quinaldic acid metabolite BE-22179[55] as well as the quinoxaline antibacterials triostin A[56] and echinomycin.[57] Though structural differences betweeen BE-22179 and 7 are subtle (the exocyclic olefin in BE-22179 is replaced with a methylated cysteine residue in 7), the molecules inhibit disparate cellular proteins involved in DNA synthesis (BE-22179 inhibits topoisomerase I and II, while 7 inhibits DNA polymerase α[58]). The total synthesis[59] and elucidation of the biosynthetic gene cluster[60] of thiocoraline have been reported. In addition to genes involved in the synthesis, loading, and transfer of the starter unit, the gene cluster contains a 2-gene 4-module NRPS. The function of the putative gene cluster was verified by heterologous expression in Streptomyces albus and S. lividans.[60]
Thiocoraline (7) showed exceptional activity against S. aureus (MIC=0.05 μgmL−1), B. subtilis (MIC= 0.05 μgmL−1), and M. luteus (MIC=0.03 μgmL−1).[53] However, the natural product was also highly cytotoxic to eukaryotic cells. Against P388 leukemia and A549 lung cancer cells, an IC50 = 0.002 μgmL−1 was determined. Against L1210 mouse lymphocyte cells, moreover, the molecule gave an IC50 = 230 pgmL−1.[59] The mechanism of anticancer action involves intercalation into the minor grove of double-stranded DNA.[61] Thus, the molecule is better classified as an anti-tumor-antibiotic and, because of its inherent cytotoxicity, may function as a better anticancer agent than as an antibacterial.
Compound YM-266183 (8), C48H47N13O10S6 was isolated from the broth of Bacillus cereus, strain QN03323, found in association with the marine sponge Halichondria japonica.[62,63] The peptide is closely related to the terrestrial thiocillins[64,65] and micrococcins.[66] Cyclization domains in the NRPS give rise to several thiazole rings, and an intramolecular Diels–Alder reaction forms the pyridine ring. Studies on thiocillin can be applied to 8. For instance, the YM-266183 biosynthetic gene cluster is likely analogous to the thiocillin cluster, a 22-kb cluster encoding 24 genes.[67] Though a synthesis of 8 has not been reported, a recent synthesis of micrococcin P1 could be adapted.[68] YM-266183 (8) is highly active against Gram-positive bacteria, including MRSA (MIC=0.68 μgmL−1) and VRE (MIC= 0.025 μgmL−1).[63] The compound lacked activity toward Gram-negative bacteria like E. coli. Drawing conclusions from studies on thiocillin, the molecule may inhibit ribosomal protein synthesis.[69,70]
Polyketides
Polyketide natural products are constructed by polyketide synthases (PKS) that are functionally and conceptually similar to nonribosomal peptide synthetases.[12] Instead of amino acids, however, the building blocks of the PKS machinery are acetate and propionate. An acyl transferase (AT) domain catalyzes thioester bond formation between an acyl carrier protein (ACP) domain and a coenzyme A (CoA)-bound starter unit. A ketosynthase (KS) domain then catalyzes the union of its cysteine-bound malonyl elongation unit with the growing ACP-bound polyketide. In effect, acetate units are loaded onto the cysteine residue of adjacent KS domains, and the chain is elongated via successive Claisen condensations. Ketoreductase (KS), dehydratase (DH), and enoyl reductase (ER) domains add complexity and diversity to the polyketide. PKS and NRPS modules can cooperate to form hybrid PKS-NRPS molecules like bogorol A (5).
Bisanthraquinone BE-43472B (9), C32H24O9, was isolated from Streptomyces sp. N1-78-1. The actinomycete strain was itself obtained from cultured cells of an unidentified cyano-bacterium (N36-11-10) that was, in turn, found in association with the tunicate Ecteinascidia turbinata in Puerto Rico (Scheme 3).[71,72] The species was identified using 16S rRNA gene sequence analysis, showing 97% similarity with its nearest neighbor (S. melanosporofaciens). This example illustrates the complexity that is often associated with determining the true source of a natural product. Here, a bacterial associate and not the tunicate nor cyanobacterium is responsible for production of the metabolite. Bisanthraquinone BE-43472B showed activity against several clinical isolates of methicillin-susceptible S. aureus (MSSA), MRSA, and tetracycline-resistant S. aureus (TRSA) with an MIC less than 0.13 μgmL−1.[72] Against a number of isolates of VRE, 9 showed an MIC=0.50 μgmL−1. Since the cytotoxicity of the bisanthraquinone against human colon cancer cells (HCT-116) was moderate (IC50 = 1.8 μgmL−1), an acceptable therapeutic window for in vivo studies may exist. Analogues of 9 were generated through semisynthesis[72] and total synthesis.[73,74] Structure–activity relationships showed that the substituents on the C-ring have a dramatic effect on antibacterial activity.[72] The C-1 hydroxy group is also essential for activity.[73]
Scheme 3.

Antibacterial polyketides 9–12.
The marine actinomycete Verrucosispora sp. AB 18-032, collected in the Japanese Sea, was shown to produce abyssomicin C (10), C19H22O6.[75] This apparent polyketide is a salient testament to the complex molecular architecture that is produced in Nature. Its bioactivity and structural complexity have elicited several prominent total syntheses.[76–78] Interestingly, an atropisomer of 10, which was later found in the culture extract,[79] is formed upon rotation of the O=C7–C8=C9 bond. An X-ray structure of natural 10 reveals a 145° dihedral angle between the olefin and ketone group. Atropo-abyssomicin C exhibits a much shorter dihedral angle (26°) wherein the carbonyl group and double bond are oriented toward the same plane. Abyssomicin C showed activity against MRSA (MIC=4 μgmL−1), and atropo-10 displayed even better antibacterial activity. Remarkably, 10 is the first natural inhibitor of p-aminobenzoate (pABA) biosynthesis, a pathway used by microorganisms but not found in humans. The molecule is thought to act as a substrate mimetic to chorismate, binding covalently to 4-amino-4-deoxy-chorismate (ADC) synthase at a cysteine residue near the active site of one of the synthase subunits.[80]
Pestalone (11), C21H20Cl2O6, a chlorinated tetra-ortho-substituted benzophenone, was produced by the marine fungus CNL-365, a Pestalotia species.[81] Similar to emericellamide A (6), the natural product was detected only when the fungus was co-cultured with a marine bacterium (strain CNJ-328). The X-ray structure of 11 showed that the two aromatic rings are out of plane, owing to the steric interaction between substituents ortho to the ketone functionality. A related C20 desmethyl compound was reported from the fungus Chrysosporium, but the biological activity of 11 is distinct.[82] Polyketide 11 has been fashioned through total synthesis,[83] in addition to one published approach.[84] Pestalone is active against MRSA with an MIC=37 ng/mL and VRE with an MIC=78 ng/mL.[81] Few marine natural products exhibit such potent antibacterial activity. The moderate cytotoxicity that 11 displayed against various eukaryotic cancer cell lines is promising in terms of antibacterial treatment.
Ariakemicin A (12), C32H38N4O7, was isolated from the culture of a gliding bacterium of the phylum Bacteriodetes HC35.[85] Unlike the previous bacterial metabolites, 12 is produced by a Gram-negative bacterium of the genus Rapidithrix.[86] The bacterial strain was isolated from muds collected in southwest Japan alongside Ariake Inland. The oxa-zole ring and the large number of unsaturated carbon-carbon bonds suggest the presence of ketoreductase and dehydratase domains in the ariakemicin polyketide gene cluster. Ariakemicin A showed significant activity against S. aureus (MIC=0.4 μgmL−1).[85] The metabolite was only slightly cytotoxic against A549 human lung cancer cells and BHK baby hamster kidney cells (IC50 = 25 and 15 μgmL−1, respectively).
Alkaloids
Alkaloids are nitrogen-containing natural products that are related more by common structural characteristics than by biosynthetic considerations.[12] The nitrogenous character of these metabolites, often derived from amino acids such as ornithine, lysine, tryosine, tryptophan, histidine, nicotinic acid, and anthranilic acid, often render these molecules basic or “alkaline.”
The dimeric tetra-brominated alkaloid nagelamide G (13), C22H22Br4N10O2, and several analogues were isolated from an Okinawan marine sponge of the genus Agelas (Scheme 4).[87] Agelas sponges have been shown to synthesize a slew of nagelamide-like compounds, including the closely related ageliferins.[88,89] The complex, highly halogenated, azacyclic structure of negalamide G (13) and other oroidin alkaloids has caught the attention of synthetic chemists.[90–92] Nagelamide G exhibited antibacterial activity against M. luteus with an MIC=1.8 μgmL−1. The compound was demonstrated to specifically inhibit the protein phosphatase 2A in vitro, a serine/threonine phosphatase required for cell growth with an IC50 = 8.4 μgmL−1.[87]
Scheme 4.

Antibacterial alkaloids 13–16.
8-Hydroxymanzamine (14), C36H44N4O2, is a β-carboline alkaloid isolated from the marine sponge Pachypellina sp.[93] The molecule, a derivative of manzamine A from an Okinawan sponge of the genus Haliclona,[94] contains 5-, 6-, 8-, and 13-membered heterocyclic rings with a β-carboline moiety. Alkaloid 14 has been a synthetic challenge due to its complex structure and remarkable activity.[95,96] The first total synthesis required no less than 31 steps. 8-Hydroxymanzamine showed an MIC=0.91 μgmL−1 against the tuberculosis-causing bacterium Mycobacterium tuberculosis.[97,98] With the opposite absolute configuration, the alkaloid was much less active (MIC=3.13 μgmL−1).[99] Manzamine A itself displayed antibacterial activity against M. tuberculosis with an MIC=1.53 μgmL−1.
Marinopyrrole A (15), C22H11BrCl4N2O4 is produced by a bacterium of the genus Streptomyces, collected from ocean sediment off the coast of La Jolla, California.[100] Like hedistin (3), pestalone (11), and nagelamide G (13), the natural product is decorated with multiple halogen substituents. The characteristic 1,3′-bipyrrole core of 15, formed from an enzyme-catalyzed union of two molecules of monodeoxy-pyoluteorin,[101] has no precedence among natural products. Marinopyrrole A showed activity against MRSA with an MIC=0.61 μgmL−1.[102] Against HCT-116 cells the compound showed moderate cytotoxicity (IC50 = 8.8 μgmL−1). As mentioned above, only those antibacterial agents which do not exhibit significant cytotoxicity against eukaryotic cells have the potential for clinical application.
Cribrostain 6 (16), C15H14N2O3, was isolated from the marine sponge Cribrochalina sp. collected in the Republic of Maldives.[103] This alkaloid, the first example of a imidazo-[5,1-a]isoquinoline, was synthesized shortly after its structural elucidation.[104–106] Originally pursued for its antineoplastic activity, cibrostatin 6 (16) was re-examined for its antibacterial properties.[107] This tricyclic quinone was active against a variety of Gram-positive bacteria, especially resistant strains of S. pneumoniae (MIC=0.5–2 μgmL−1). Expected of an antineoplastic agent, the compound showed cytotoxicity against MCF-7 breast-adenocarcinoma, SF-268 CNS glio-blastoma, DU-145 prostate, and P388 mouse leukemia cells with an IC50 = 0.21–0.38 μgmL−1.[103] Concerns about the toxic effects of cibrostatin 6 (16), however, may be underscored by its high tolerance (maximum tolerated dose= 750–1000 μgkg−1d−1) in mice.
Terpenes
Terpenes are a diverse class of natural products fashioned from isoprene units. The biosynthetic terpene building blocks, are dimethylallyl pyrophophate (DMAPP) and isopentenyl pyrophophate (IPP).[12] Both five-carbon compounds are derived either from the mevalonate pathway or the deoxyxylulose phosphate (non-mevalonate) pathway. The successive addition of one isoprene unit provides monoterpenes (C10), sesquiterpenes (C15), diterpenes (C20), sesterterpenes (C25), and so on. Skeletal rearrangements frequently occur which distort the regular head-to-tail arrangement of the isoprene units and add diversity to terpenoid structures.
From the Bahamian soft coral Pseudopterogorgia elisabethae, the tricylic diterpenoid pseudopterosin P (17), C22H38O6, was isolated (Scheme 5).[108,109] Although the pseudopterosins have been known for two decades,[110–112] their antibacterial activity against Gram-positive bacteria was only recently published.[113] The parent compound pseudopterosin A has inspired several synthetic studies, though 17 itself has not yet succumbed to total synthesis[114,115] For pseudopterosin P (17), MICs of 0.8 μgmL−1 against S. pyogenes, 2.0 μgmL−1 against S. aureus, and 3.5 μgmL−1 against E. faecalis were measured.[113] The finding that this potent anti-inflammatory metabolite also displays antibacterial properties highlights the biological relevance of these molecules.
Scheme 5.
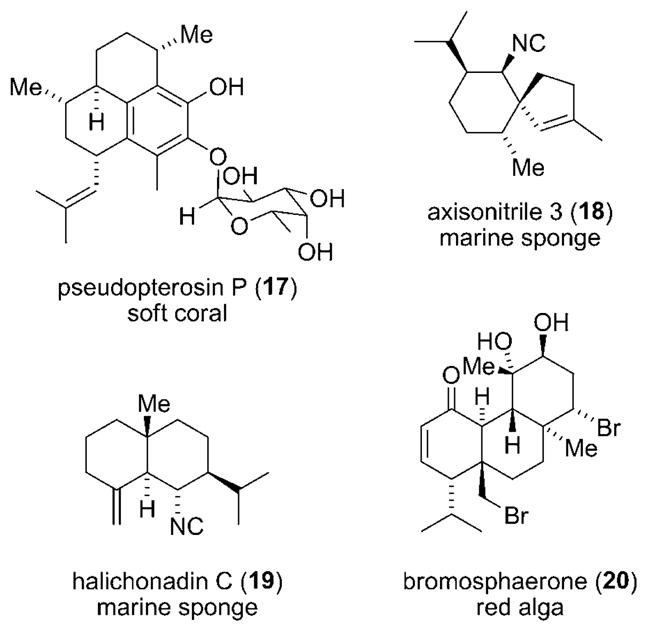
Antibacterial terpenes 17–20.
Axisonitrile 3 (18), C16H25N, was isolated from the marine sponge Axinella cannabina from the bay of Taranto, Italy,[116] and again from Acanthella klethra Pulitzer-Finali (Axinellidae) from the Great Barrier Reef, Australia.[117] This sesquiterpene features a spirocyclic quaternary center and an iso-nitrile substituent. Isocyano, isothiocyanato and formamide groups are a common feature of numerous marine sponge metabolites.[118] The synthesis of axisonitrile 3 has been completed.[119,120] Although discovered in the early 1990s, the activity of 18 against M. tuberculosis was only recently observed.[121] An MIC=2 μgmL−1 was measured. The lack of cytotoxicity of terpene 18 against human carcinoma (KB) cells is encouraging.
Haliconadin C (19), C16H25N, is a sesquiterpenoid isolated from a marine sponge Halichondria sp., which was collected in Okinawa, Japan.[122] The copper complex of haliconadin C, a CuI-coordinated trimer of the natural product, was described in a later publication.[123] The compound’s ability to tightly complex copper may be linked to its antibacterial activity. Like axisonitrile 3 (18), the metabolite features an iso-nitrile functionality. Derived from the same biosynthetic terpenoid pathway, and having an identical atomic constitution, the structures of 18 and 19 are very different. Haliconadin C showed considerable activity against several bacteria. Against M. luteus the compound showed antibacterial activity with an MIC=0.52 μgmL−1.[122] The isonitrile substituents of 18 and 19, specifically, may impart antibacterial properties to these metabolites.
Bromosphaerone (20), C20H30Br2O3, is a diterpenoid that was isolated from the red alga Sphaerococcus coronopifolius, collected along the Atlantic coast of Morocco.[124] The natural product is related to a metabolite previously isolated from Sphaerococcus, 12S-hydroxybromosphaerodiol.[125] Bromosphaerone showed striking activity against S. aureus with an MIC=0.048 μgmL−1.[124] The molecule potentially possesses an amphipathic structure: three polar alcohol functionalities reside on one side whilst hydrophobic aliphatic carbon and bromine atoms occupy the opposite side. Amphipathic structures like ribosomal antimicrobial peptides and possibly alkaloid 20 interact with bacterial membranes.
Targeting Bacterial Virulence
The use of bacteriocidal and bacteriostatic compounds is not the only means of combating infection. Targeting the pathways by which bacteria exhibit virulence is a promising approach.[126–128] When virulence is related to the production of a bacterial toxin, methods for thwarting virulence involve inhibiting the function or secretion of that toxin. Disrupting mechanisms of cell–cell adhesion also prevents the onset of virulence. A fourth method, targeting the regulation of virulence expression, includes interfering with quorum sensing in a bacterial population.
Quorum sensing is connected to virulence since the expression of virulence genes if often delayed, for the sake of reproduction and growth, until a minimum number or “quorum” is reached. In the case of Gram-negative bacteria, an accumulation of acyl-homoserine lactones (AHLs) above a certain threshold concentration activates a regulatory protein (LuxR or a LuxR homologue) responsible for transcription of virulence genes. Autoinducing peptides (AIPs) play a similar role in Gram-positive bacteria. Exogenous small molecules can mimic AHLs and bind to the regulatory protein.[129] These quorum sensing inhibitors (QSIs) prevent the expression of virulence genes by disrupting the AHL–LuxR interaction. A superb method of screening for new QSIs has been developed.[130]
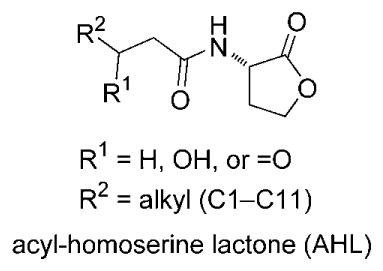
The marine environment is ideally suited to the discovery of QSIs since a multitude of epi- and endobiotic associations exist among marine prokaryotes and eukaryotes. The degree of bacterial epibiosis and endobiosis in the ocean, both in variety and magnitude, is unparalleled in terrestrial environments. This is true, in part, because “air rarely plays the role of nutrient (dissolved or particulate) vector in the same way that water does, while, for example, evaporation and dessication, are unknown problems in subtidal depths of the sea. As substrate-bound nutrient uptake is of secondary importance to most sessile hard-bottom organisms, all solid surfaces represented possible settlement sites for algae and sessile organisms.”[131] In fact, emercillamide (6), thiocoraline (7), YM-266183 (8), and BE-43472B (9), subjects of this Review, were isolated from microbial strains in association with a green alga, soft coral, marine sponge, and tunicate. Indeed, many of the compounds attributed to marine invertebrates may actually be produced by bacterial symbionts, though studies of this sort are complex and mostly inconclusive.[132–136]
The fimbrolides (21–24) are a family of brominated fura-nones isolated from the Australian red algae Delisea fimbriata[137] and D. pulchra (Scheme 6).[138] In accordance with the discussion above, these marine metabolites interfere with AHL-mediated processes by competitively binding to the AHL receptor (a LuxR homologue). In this way, the polyketide blocks AHL-dependent phenotypes like swarming and bioluminescence without inhibiting the growth of the bacteria.[139,140] In one ecologically relevant study, surface colonization by marine bacteria was examined. It was determined that both attachment and swarming—phenotypes linked to colonization—were inhibited by the presence of the fimbrolides.[141]
Scheme 6.

Marine-derived quorum sensing inhibitors 21–26.
Lastly, manoalides 25 and 26 are sesterterpenoids extracted from the sponge Luffariella variabilis collected in Palau.[142] Like the fimbrolides, the natural products are potent quorum sensing inhibitors that have a key structural element related to the acyl-homoserine lactones.[143]
Summary and Outlook
There are two fundamentally different approaches to natural product drug discovery. According to the “reverse chemogenomics” approach to drug discovery,[144–148] metabolically-essential proteins are cloned and screened in vitro with libraries of compounds. This technique has the potential to identify mechanistically-novel antibacterials, however, it often yields protein inhibitors that lack whole cell antibacterial activity.[149–150] Certain features of the in vitro lead limit its drug applicability, such as an inability to cross the bacterial cell membrane. Medicinal chemists can try to optimize the lead, but this work is itself a tedious art that requires the integration of many simultaneous structural parameters.[3i] The “forward chemogenomics” approach, by contrast, involves the identification of a lead structure through consideration of the molecule’s in vivo antibacterial activity.[144–148] Once a compound with activity is found, target and mechanism-of-action studies can be immediately undertaken.[151] Molecules selected in this way have built-in characteristics, such as membrane permeability, that make them more suitable as drugs.
The success of the “forward chemogenomics” approach to antibacterial discovery is dependent upon the quality of the chemical library. The track record of naturally-occurring compounds functioning as antibacterial agents is long,[3] and only a fraction of the Earth’s biota (especially marine biota) have been examined.[144] Thus, provided new avenues are developed to access phylogenetically-novel sources, natural products will continue to be the principal resource for new antibacterial agents. The finding of new pharmacophores resulting from collection or cultivation of novel biological sources enables, following a forward chemogenomics approach, the discovery of new therapeutic targets.
Developing new techniques in microbiology and molecular biology is vital to the discovery of new antibacterials from Nature.[152–158] For instance, novel methods for the collection and cultivation of new species, including the bacterial symbionts of invertebrates, must be conceived to realize the full biosynthetic potential of the oceans. As a testament to this wasted biodiversity, it is predicted that 5% or less of marine bacterial species can be cultured under standard conditions. New approaches, which divert from the standard paradigm of microbial culture, such as the manipulation of regulatory genes or chromatin remodeling, must be exploited. With no prior knowledge of metabolite structure, the products of cryptic gene clusters can be visualized through gene inactivation (“knockouts”) and heterologous gene expression followed, in both cases, by comparative metabolic profiling.
Although no significant efforts have been reported, the potential to discover new antibacterial natural product drugs from marine organisms is great. Relative to terrestrial environments, the ocean remains an under-explored habitat with unparalleled biodiversity. Regarding animal life, for instance, 32 of the 36 phyla are represented in the ocean, and 15 of these are exclusively marine.[159] In this brief review, a small subset of antibacterial compounds were presented that support increasing discovery programs for marine-derived antibacterial agents. It is without question that life in the sea represents a logical and under-examined source, which has the potential to fill the multidecade void in antibacterial drug discovery.
Biographies

William Fenical received his Ph.D. with Prof. P. Radlick (University of California, Riverside). He remained at Riverside to work with Prof. D. R. Kearns as an ACS postdoctoral fellow until 1969. Beginning as a lecturer at the Scripps Institution of Oceanography in 1973, he became Professor and Director of the Marine Research Division in 1989. He is now Distinguished Professor and Director of the Center for Marine Biotechnology and Biomedicine at Scripps Institution of Oceanography, University of California, San Diego (UCSD).

Chambers Hughes is a postdoctoral researcher with Prof. W. Fenical (Scripps Institution of Oceanography, UCSD). He obtained a Ph.D. with Prof. D. Trauner in 2004 (then at the University of California, Berkeley) focusing on the total synthesis of both terrestrial and marine natural products. Currently, he is pursuing the isolation, structural elucidation, total synthesis, and target identification of natural products from marine actinomycetes.
References
- 1.Waksman SA. Mycologia. 1947;39:565–569. [PubMed] [Google Scholar]
- 2.Davey PG. In: Concise Oxford Textbook of Medicine. Ledingham JGG, Warrell DA, editors. Oxford University Press; Oxford: 2000. p. 1475. [Google Scholar]
- 3.a) Bush K, Macielag M, Weidner-Wells M. Curr Opin Microbiol. 2004;7:466–476. doi: 10.1016/j.mib.2004.08.013. [DOI] [PubMed] [Google Scholar]; b) Appelbaum PC, Jacobs MR. Curr Opin Microbiol. 2005;8:510–517. doi: 10.1016/j.mib.2005.07.001. [DOI] [PubMed] [Google Scholar]; c) Wang G, Tang W, Bidigare RR. In: Natural Products: Drug Discovery and Therapeutic Medicine. Zhang L, Demain AL, editors. Humana Press; Totowa: 2005. pp. 198–227. [Google Scholar]; d) Butler MS. Nat Prod Rep. 2005;22:162–195. doi: 10.1039/b402985m. [DOI] [PubMed] [Google Scholar]; e) Alekshun MN. Expert Opin Invest Drugs. 2005;14:117–134. doi: 10.1517/13543784.14.2.117. [DOI] [PubMed] [Google Scholar]; f) Butler MS, Buss AD. Biochem Pharmacol. 2006;71:919–929. doi: 10.1016/j.bcp.2005.10.012. [DOI] [PubMed] [Google Scholar]; g) Paláez F. Biochem Pharmacol. 2006;71:981–990. doi: 10.1016/j.bcp.2005.10.010. [DOI] [PubMed] [Google Scholar]; h) Clardy J, Fischbach MA, Walsh CT. Nat Biotechnol. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]; i) von Nussbaum F, Brands M, Hinzen B, Weigand S, Häbich D. Angew Chem. 2006;118:5194–5254. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:5072–5129. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]; j) Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; k) Demain AL, Sanchez S. J Antibiot. 2009;62:5–16. doi: 10.1038/ja.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Fischbach MA, Walsh CT. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stone MJ, Williams DH. Mol Microbiol. 1992;6:29–34. doi: 10.1111/j.1365-2958.1992.tb00834.x. [DOI] [PubMed] [Google Scholar]
- 5.Walsh C. Antibiotics: Actions, Origins, Resistance. ASM Press; Washington: 2003. pp. 89–156. [Google Scholar]
- 6.Allsopp M, Page R, Johnston P, Santillo D. State of the World’s Oceans. Springer; Dordrecht: 2008. pp. 1–31. [Google Scholar]
- 7.a) Sverdrup HU, Johnson MW, Fleming RH. The Oceans: Their Physics, Chemistry and General Biology. Prentice Hall; New York: 1942. [Google Scholar]; b) Rheinheimer G. Aquatic Microbiology. 3. Wiley; New York: 1992. [Google Scholar]
- 8.ZoBell CE, Anderson DQ. Am Assoc Petrol Geol Bull. 1936;20:258–269. [Google Scholar]
- 9.a) Mayer AMS, Lehmann VKB. Pharmacologist. 2000;42:62–69. [Google Scholar]; b) Mayer AMS, Hamann MT. Comp Biochem Physiol. 2002;132:315–339. doi: 10.1016/s1532-0456(02)00094-7. [DOI] [PubMed] [Google Scholar]; c) Mayer AMS, Hamann MT. Mar Biotechnol. 2004;6:37–52. doi: 10.1007/s10126-003-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mayer AMS, Hamann MT. Comp Biochem Physiol. 2005;140:265–286. doi: 10.1016/j.cca.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mayer AMS, Rodríguez AD, Berlinck RGS, Hamann MT. Comp Biochem Physiol. 2007;145:553–581. doi: 10.1016/j.cbpc.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Mayer AMS, Rodríguez AD, Berlinck RGS, Hamann MT. Biochim Biophys Acta. 2009;1790:283–308. doi: 10.1016/j.bbagen.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) El Sayed KA, Bartyzel P, Shen X, Perry TL, Zjawiony JK, Hamann MT. Tetrahedron. 2000;56:949–953. [Google Scholar]; b) Donia M, Hamann MT. Lancet Infect Dis. 2003;3:338–348. doi: 10.1016/S1473-3099(03)00655-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kasanah N, Hamann MT. Curr Opin Invest Drugs. 2004;5:827–837. [PMC free article] [PubMed] [Google Scholar]; d) Bernan VS, Greenstein M, Carter GT. Curr Med Chem: Anti-Infect Agents. 2004;3:181–195. [Google Scholar]; e) Fiedler HP, Bruntner C, Bull AT, Ward AC, Goodfellow M, Potterat O, Puder C, Mihm G. Antonie van Leeuwenhoek. 2005;87:37–42. doi: 10.1007/s10482-004-6538-8. [DOI] [PubMed] [Google Scholar]; f) Zhang L, An R, Wang J, Sun N, Zhang S, Hu J, Kuai J. Curr Opin Microbiol. 2005;8:276–281. doi: 10.1016/j.mib.2005.04.008. [DOI] [PubMed] [Google Scholar]; g) Sipkema D, Franssen MCR, Osinga R, Tramper J, Wijffels RH. Mar Biotechnol. 2005;7:142–162. doi: 10.1007/s10126-004-0405-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Laatsch H. In: Frontiers in Marine Biotechnology. Proksch P, Müller WEG, editors. Horizon Bioscience; Norfolk: 2006. pp. 225–288. [Google Scholar]; i) Lam KS. Curr Opin Microbiol. 2006;9:245–251. doi: 10.1016/j.mib.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 11.a) Hancock REW, Lehrer R. Trends Biotechnol. 1998;16:82–88. doi: 10.1016/s0167-7799(97)01156-6. [DOI] [PubMed] [Google Scholar]; b) Epand RM, Vogel HJ. Biochim Biophys Acta Biomembr. 1999;1462:11–28. doi: 10.1016/s0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]; c) Hancock REW, Chapple DS. Antimicrob Agents Chemother. 1999;43:1317–1323. doi: 10.1128/aac.43.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zasloff M. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]; e) Yeaman MR, Yount NY. Pharmacol Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]; f) Boman HG. J Intern Med. 2003;254:197–215. doi: 10.1046/j.1365-2796.2003.01228.x. [DOI] [PubMed] [Google Scholar]; g) Andrès E, Dimarco JL. J Intern Med. 2004;255:519–520. doi: 10.1046/j.1365-2796.2003.01278.x. [DOI] [PubMed] [Google Scholar]; h) Tincu JA, Taylor SW. Antimicrob Agents Chemother. 2004;48:3645–3654. doi: 10.1128/AAC.48.10.3645-3654.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Zhang L, Falla TJ. Expert Opin Invest Drugs. 2004;13:97–106. doi: 10.1517/13543784.13.2.97. [DOI] [PubMed] [Google Scholar]; j) Reddy RVR, Yedery RD, Aranha C. Int J Antimicrob Agents. 2004;24:536–547. doi: 10.1016/j.ijantimicag.2004.09.005. [DOI] [PubMed] [Google Scholar]; k) Bulet P, Stöcklin R, Menin L. Immunol Rev. 2004;198:169–184. doi: 10.1111/j.0105-2896.2004.0124.x. [DOI] [PubMed] [Google Scholar]; l) Hancock RE, Brown KL, Mookherjee N. Immunology. 2006;211:315–322. doi: 10.1016/j.imbio.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 12.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. Wiley; New York: 2001. [Google Scholar]
- 13.McIntosh JA, Donia MS, Schmidt EW. Nat Prod Rep. 2009;26:537–559. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faulkner DJ, Hay ME, Paul VJ, Pawlik JR, Steinberg PD. In: Ecological Roles of Marine Natural Products. Paul VJ, editor. Comstock Press; Ithaca: 1992. [Google Scholar]
- 15.Engel S, Jensen PR, Fenical W. J Chem Ecol. 2002;28:1971–1985. doi: 10.1023/a:1020793726898. [DOI] [PubMed] [Google Scholar]
- 16.Engel S, Puglisi MP, Jensen PR, Fenical W. Mar Biol. 2006;149:991–1002. [Google Scholar]
- 17.Puglisi MP, Engel S, Jensen PR, Fenical W. Mar Biol. 2006;150:531–540. [Google Scholar]
- 18.Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Disk Susceptibility Tests, Approved Standard. 10. CLSI document M2A10; Wayne, PA (USA): 2009. [Google Scholar]
- 19.Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Approved Standard. 8. CLSI document M7A8; Wayne, PA (USA): 2009. [Google Scholar]
- 20.Woody RW. In: The Peptides. Undenfriend S, Meienhofer J, Hruby VJ, editors. Academic Press; New York: 1985. pp. 15–114. [Google Scholar]
- 21.Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Cell. 1997;88:553–560. doi: 10.1016/s0092-8674(00)81895-4. [DOI] [PubMed] [Google Scholar]
- 22.Yu Q, Lehrer RI, Tam JP. J Biol Chem. 2000;275:3943–3949. doi: 10.1074/jbc.275.6.3943. [DOI] [PubMed] [Google Scholar]
- 23.Tam JP, Lu YA, Yang JL. J Biol Chem. 2002;277:50450–50456. doi: 10.1074/jbc.M208429200. [DOI] [PubMed] [Google Scholar]
- 24.Kandasamy SK, Larson RG. Biochim Biophys Acta. 2000;1758:1274–1284. doi: 10.1016/j.bbamem.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 25.Ovchinnikova TV, Aleshina GM, Balandin SV, Krasnosdembskaya AD, Markelov ML, Frolova EI, Leonova YF, Tagaev AA, Krasnodemsky EG, Kokryakov VN. FEBS Lett. 2004;577:209–214. doi: 10.1016/j.febslet.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 26.Ovchinnikova TV, Shenkarev ZO, Nadezhdin KD, Balandin SV, Zhmak MN, Kudelina IA, Finkina EI, Kokryakov VN, Arseniev AS. Biochem Biophys Res Commun. 2007;360:156–162. doi: 10.1016/j.bbrc.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 27.Lee JU, Kang DI, Zhu WL, Shin SY, Hahm KS, Kim Y. Biopolymers. 2007;88:208–216. doi: 10.1002/bip.20700. [DOI] [PubMed] [Google Scholar]
- 28.Andrä J, Monreal D, Martinez de Tejada G, Olak C, Brezesinski G, Sanchez Gomez S, Goldmann T, Bartels R, Brandenburg K, Moriyon I. Biochem J. 2008;410:113–122. [Google Scholar]
- 29.Lee JU, Park KH, Lee JY, Kim J, Shin SY, Park Y, Hahm KS, Kim Y. Bull Korean Chem Soc. 2008;29:1190–1194. [Google Scholar]
- 30.Andrä J, Hammer MU, Grötzinger J, Jakovin I, Lindner B, Vollmer E, Fedders H, Leippe M, Gutsmann T. Biol Chem. 2009;390:337–349. doi: 10.1515/BC.2009.039. [DOI] [PubMed] [Google Scholar]
- 31.Jang WS, Kim KN, Lee YS, Nam MH, Lee IH. FEBS Lett. 2002;521:81–86. doi: 10.1016/s0014-5793(02)02827-2. [DOI] [PubMed] [Google Scholar]
- 32.Jang WS, Kim CH, Kang MS, Chae HJ, Son SM, Seo SJ, Lee IH. Peptides. 2005;26:2360–2367. doi: 10.1016/j.peptides.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Jang WS, Kim CH, Kim KN, Park SY, Lee JH, Son SM, Lee IH. Antimicrob Agents Chemother. 2003;47:2481–2486. doi: 10.1128/AAC.47.8.2481-2486.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jang WS, Lee SC, Lee YS, Shin YP, Shin KH, Sung BH, Kim BS, Lee SH, Lee IH. Antimicrob Agents Chemother. 2007;51:4148–4156. doi: 10.1128/AAC.00635-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tasiemski A, Schikorski D, Marrec-Croq FL, Pontoire-Van Camp C, Boidin-Wichlacz C, Sautiére P-E. Dev Comp Immunol. 2007;31:749–762. doi: 10.1016/j.dci.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Dittmar W. Challenger Repts Phys Chem. 1884;1:1–251. [Google Scholar]
- 37.Xu G, Wu M, Wang L, Zhang X, Cao S, Liu M, Cui Y. Biochim Biophys Acta. 2009;1788:2497–2508. doi: 10.1016/j.bbamem.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Lee IH, Zhao C, Cho Y, Harwig SSL, Cooper EL, Lehrer RI. FEBS Lett. 1997;400:158–162. doi: 10.1016/s0014-5793(96)01374-9. [DOI] [PubMed] [Google Scholar]
- 39.For a review of AMPs from tunicates, see: Lehrer RI, Tincu JA, Taylor SW, Menzel LP, Waring AJ. Integr Comp Biol. 2003;43:313–322. doi: 10.1093/icb/43.2.313.
- 40.Zasloff M. Proc Natl Acad Sci USA. 1987;84:5449–5453. doi: 10.1073/pnas.84.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao C, Liaw L, Lee IH, Lehrer RI. FEBS Lett. 1997;410:490–492. doi: 10.1016/s0014-5793(97)00646-7. [DOI] [PubMed] [Google Scholar]
- 42.van Kan EJM, van der Bent A, Demel RA, de Kruijff B. Biochemistry. 2001;40:6398–6405. doi: 10.1021/bi0028136. [DOI] [PubMed] [Google Scholar]
- 43.van Kan EJM, Demel RA, van der Bent A, de Kruijff B. Biochim Biophys Acta Biomembr. 2003;1615:84–92. doi: 10.1016/s0005-2736(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 44.van Kan EJM, Ganchev DN, Snel MME, Chupin V, van der Bent A, de Kruijff B. Biochemistry. 2003;42:11366–11372. doi: 10.1021/bi0349017. [DOI] [PubMed] [Google Scholar]
- 45.Lee IH, Cho Y, Lehrer RI. Infect Immun. 1997;65:2898–2903. doi: 10.1128/iai.65.7.2898-2903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barsby T, Kelly MT, Gagné SM, Andersen RJ. Org Lett. 2001;3:437–440. doi: 10.1021/ol006942q. [DOI] [PubMed] [Google Scholar]
- 47.Barsby T, Warabi K, Sørensen D, Zimmerman WT, Kelly MT, Andersen RJ. J Org Chem. 2006;71:6031–6037. doi: 10.1021/jo060667p. [DOI] [PubMed] [Google Scholar]
- 48.Oh DC, Kauffman CA, Jensen PR, Fenical W. J Nat Prod. 2007;70:515–520. doi: 10.1021/np060381f. [DOI] [PubMed] [Google Scholar]
- 49.Ghosh S, Pradhan TK. Tetrahedron Lett. 2008;49:3697–3700. [Google Scholar]
- 50.Li S, Liang S, Tan W, Xu Z, Ye T. Tetrahedron. 2009;65:2695–2702. [Google Scholar]
- 51.Ma JY, Xu LF, Huang WF, Wei BG, Lin GQ. Synlett. 2009;1307–1310 [Google Scholar]
- 52.Chiang YM, Szewczyk E, Nayak T, Davidson AD, Sanchez JF, Lo H-C, Ho W-Y, Simityan H, Kuo E, Praseuth A, Watanabe K, Oakley BR, Wang CCC. Chem Biol. 2008;15:527–532. doi: 10.1016/j.chembiol.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Romero F, Espliego F, Baz JP, de Quesada TG, Grávalos D, de La Calle F, Fernández-Puentes JL. J Antibiot. 1997;50:734–737. doi: 10.7164/antibiotics.50.734. [DOI] [PubMed] [Google Scholar]
- 54.Baz JP, Cañedo LM, Fernández-Puentes JL, Silva Elipe MV. J Antibiot. 1997;50:738–741. doi: 10.7164/antibiotics.50.738. [DOI] [PubMed] [Google Scholar]
- 55.Okada H, Suzuki H, Yoshinari T, Arakawa H, Okura A, Suda H. J Antibiot. 1994;47:129–135. doi: 10.7164/antibiotics.47.129. [DOI] [PubMed] [Google Scholar]
- 56.Shoji J, Katagiri K. J Antibiot. 1961;14:335–339. [PubMed] [Google Scholar]
- 57.Corbaz R, Ettlinger L, Gäumann E, Keller-Schierlein W, Kradolfer F, Neipp L, Prelog V, Reusser P, Zähner H. Helv Chim Acta. 1957;40:199–204. [Google Scholar]
- 58.Erba E, Bergamaschi D, Ronzoni S, Taverna S, Bonfanti M, Catapano CV, Faircloth G, Jimeno J, D’Incalci M. Br J Cancer. 1999;80:971–980. doi: 10.1038/sj.bjc.6690451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boger DL, Ichikawa S, Tse WC, Hedrick MP, Jin Q. J Am Chem Soc. 2001;123:561–568. doi: 10.1021/ja003602r. [DOI] [PubMed] [Google Scholar]
- 60.Lombó F, Velasco A, Castro A, de La Calle F, Braña AF, Sánchez-Puelles JM, Méndez C, Salas JA. ChemBioChem. 2006;7:366–376. doi: 10.1002/cbic.200500325. [DOI] [PubMed] [Google Scholar]
- 61.Negri A, Marco E, García-Hernández V, Domingo A, Llamas-Saiz AL, Porto-Sandá S, Riguera R, Laine W, David-Cordonnier MH, Bailly C, García-Fernández LF, Vaquero JJ, Gago F. J Med Chem. 2007;50:3322–3333. doi: 10.1021/jm070381s. [DOI] [PubMed] [Google Scholar]
- 62.Nagai K, Kamigiri K, Arao N, Suzumura K, Kawano Y, Yamaoka M, Zhang H, Watanabe M, Suzuki K. J Antibiot. 2003;56:123–128. doi: 10.7164/antibiotics.56.123. [DOI] [PubMed] [Google Scholar]
- 63.Suzumura KI, Yokoi T, Funatsu M, Nagai K, Tanaka K, Zhang H, Suzuki K. J Antibiot. 2003;56:129–134. doi: 10.7164/antibiotics.56.129. [DOI] [PubMed] [Google Scholar]
- 64.Shoji J, Hinoo H, Wakisaka Y, Koizumi K, Mayama M, Matsuura S, Matsumoto K. J Antibiot. 1976;29:366–374. doi: 10.7164/antibiotics.29.366. [DOI] [PubMed] [Google Scholar]
- 65.Shoji J, Kato T, Yoshimura Y, Tori K. J Antibiot. 1981;34:1126–1136. doi: 10.7164/antibiotics.34.1126. [DOI] [PubMed] [Google Scholar]
- 66.Walker J, Olesker A, Valente L, Rabanal R, Lukacs G. J Chem Soc Chem Commun. 1977;706–708 [Google Scholar]
- 67.Wieland Brown LC, Acker MG, Clardy J, Walsh CT, Fischbach MA. Proc Natl Acad Sci USA. 2009;106:2549–2553. doi: 10.1073/pnas.0900008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lefranc D, Ciufolini MA. Angew Chem. 2009;121:4262–4265. doi: 10.1002/anie.200900621. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:4198–4201. doi: 10.1002/anie.200900621. [DOI] [PubMed] [Google Scholar]
- 69.Pestka S, Brot N. J Biol Chem. 1971;246:7715–7722. [PubMed] [Google Scholar]
- 70.Kushida H, Nakajima S, Koyama T, Suzuki K, Ojiri K, Suda H. 08143569. JP Patent. 1996
- 71.Socha AM, Garcia D, Sheffer R, Rowley DC. J Nat Prod. 2006;69:1070–1073. doi: 10.1021/np050449b. [DOI] [PubMed] [Google Scholar]
- 72.Socha AM, LaPlante KL, Rowley DC. Bioorg Med Chem. 2006;14:8446–8454. doi: 10.1016/j.bmc.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 73.Nicolaou KC, Lim YH, Becker J. Angew Chem. 2009;121:3496–3500. [Google Scholar]; Angew Chem Int Ed. 2009;48:3444–3448. doi: 10.1002/anie.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nicolaou KC, Becker J, Lim YH, Lemire A, Neubauer T, Montero A. J Am Chem Soc. 2009;131:14812–14826. doi: 10.1021/ja9073694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bister B, Bischoff D, Ströbele M, Riedlinger J, Reicke A, Wolter F, Bull AT, Zähner H, Fiedler HP, Süssmuth RD. Angew Chem. 2004;116:2628–2630. doi: 10.1002/anie.200353160. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:2574–2576. doi: 10.1002/anie.200353160. [DOI] [PubMed] [Google Scholar]
- 76.Zapf CW, Harrison BA, Drahl C, Sorensen EJ. Angew Chem. 2005;117:6691–6695. doi: 10.1002/anie.200502119. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:6533–6537. doi: 10.1002/anie.200502119. [DOI] [PubMed] [Google Scholar]
- 77.Nicolaou KC, Harrison ST. Angew Chem. 2006;118:3334–3338. [Google Scholar]; Angew Chem Int Ed. 2006;45:3256–3260. [Google Scholar]
- 78.Nicolaou KC, Harrison ST. J Am Chem Soc. 2007;129:429–440. doi: 10.1021/ja067083p. [DOI] [PubMed] [Google Scholar]
- 79.Keller S, Nicholson G, Drahl C, Sorensen E, Fiedler HP, Süssmuth RD. J Antibiot. 2007;60:391–394. doi: 10.1038/ja.2007.54. [DOI] [PubMed] [Google Scholar]
- 80.Keller S, Schadt HS, Ortel I, Süssmuth RD. Angew Chem. 2007;119:8433–8435. doi: 10.1002/anie.200701836. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:8284–8286. doi: 10.1002/anie.200701836. [DOI] [PubMed] [Google Scholar]
- 81.Cueto M, Jensen PR, Kauffman C, Fenical W, Lobkovsky E, Clardy J. J Nat Prod. 2001;64:1444–1446. doi: 10.1021/np0102713. [DOI] [PubMed] [Google Scholar]
- 82.Wachi Y, Yamashita T, Komatsu K, Yoshida S. 07061950A2 19950307. JP Patent. 1995
- 83.Iijima D, Tanaka D, Hamada M, Ogamino T, Ishikawa Y, Nishiyama S. Tetrahedron Lett. 2004;45:5469–5471. [Google Scholar]
- 84.Kaiser F, Schmalz HG. Tetrahedron. 2003;59:7345–7355. [Google Scholar]
- 85.Oku N, Adachi K, Matsuda S, Kasai H, Takatsuki A, Shizuri Y. Org Lett. 2008;10:2481–2484. doi: 10.1021/ol8007292. [DOI] [PubMed] [Google Scholar]
- 86.Srisukchayakul P, Suwanachart C, Sangnoi Y, Kanjana-Opas A, Hosoya S, Yokota A, Arunpairojana V. Int J Syst Evol Microbiol. 2007;57:2275–2279. doi: 10.1099/ijs.0.65087-0. [DOI] [PubMed] [Google Scholar]
- 87.Endo T, Tsuda M, Okada T, Mitsuhashi S, Shima H, Kikucki K, Mikami Y, Fromont J, Kobayashi J. J Nat Prod. 2004;67:1262–1267. doi: 10.1021/np034077n. [DOI] [PubMed] [Google Scholar]
- 88.Kobayashi J, Tsuda M. Tetrahedron. 1990;46:5579–5586. [Google Scholar]
- 89.Keifer PA, Schwartz RE, Koker MES, Hughes RG, Jr, Rittschof D, Rinehart KL. J Org Chem. 1991;56:2965–2975. [Google Scholar]
- 90.Baran PS, O’Malley DP, Zografos AL. Angew Chem. 2004;116:2728–2731. [Google Scholar]; Angew Chem Int Ed. 2004;43:2674–2677. doi: 10.1002/anie.200453937. [DOI] [PubMed] [Google Scholar]
- 91.O’Malley DP, Li K, Maue M, Zografos AL, Baran PS. J Am Chem Soc. 2007;129:4762–4775. doi: 10.1021/ja069035a. [DOI] [PubMed] [Google Scholar]
- 92.For a review, see: Arndt H-D, Riedrich M. Angew Chem. 2008;120:4864–4867.Angew Chem Int Ed. 2008;47:4785–4788. doi: 10.1002/anie.200801793.
- 93.Ishiba T, Corgiat JM, Scheuer PJ, Kelly-Borges M. J Nat Prod. 1994;57:168–170. doi: 10.1021/np50103a027. [DOI] [PubMed] [Google Scholar]
- 94.Sakai R, Higa T, Jefford CW, Bernardinelli G. J Am Chem Soc. 1986;108:6404–6405. [Google Scholar]
- 95.Winkler JD, Axten JM. J Am Chem Soc. 1998;120:6425–6426. doi: 10.1021/ja981303k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Humphrey JM, Liao Y, Ali A, Rein T, Wong YL, Chen HJ, Courtney AK, Martin SF. J Am Chem Soc. 2002;124:8584–8592. doi: 10.1021/ja0202964. [DOI] [PubMed] [Google Scholar]
- 97.Rao KV, Santarsiero BD, Mesecar AD, Schinzi RF, Tekwani BL, Hamann MT. J Nat Prod. 2003;66:823–828. doi: 10.1021/np020592u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peng J, Kudrimoti S, Prasanna S, Odde S, Doerksen RJ, Pennaka HK, Choo YM, Rao KV, Tekwani BL, Madgula V, Khan SI, Wang B, Mayer AMS, Jacob MR, Tu LC, Gertsch J, Hamann MT. J Med Chem. 2010;53:61–76. doi: 10.1021/jm900672t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.El Sayed KA, Kelly M, Kara UAK, Ang KKH, Katsuyama I, Dunbar DC, Khan AA, Hamann MT. J Am Chem Soc. 2001;123:1804–1808. doi: 10.1021/ja002073o. [DOI] [PubMed] [Google Scholar]
- 100.Hughes CC, Prieto-Davo A, Jensen PR, Fenical W. Org Lett. 2008;10:629–631. doi: 10.1021/ol702952n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Durham DG, Hughes CG, Rees AH. Can J Chem. 1972;50:3223–3228. [Google Scholar]
- 102.Hughes CC, Yang YL, Liu WT, Dorrestein PC, La Clair JJ, Fenical W. J Am Chem Soc. 2009;131:12094–12096. doi: 10.1021/ja903149u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pettit GR, Collins JC, Knight JC, Herald DL, Nieman RA, Williams MD, Pettit RK. J Nat Prod. 2003;66:544–547. doi: 10.1021/np020012t. [DOI] [PubMed] [Google Scholar]
- 104.Nakahara S, Kubo A. Heterocycles. 2004;63:2355–2362. [Google Scholar]
- 105.Nakahara S, Kubo A, Mikami Y, Ito J. Heterocycles. 2006;68:515–520. [Google Scholar]
- 106.Markey MD, Kelly TR. J Org Chem. 2008;73:7441–7443. doi: 10.1021/jo801694w. [DOI] [PubMed] [Google Scholar]
- 107.Pettit RK, Fakoury BR, Knight JC, Weber CA, Pettit GR, Cage GD, Pon S. J Med Microbiol. 2004;53:61–65. doi: 10.1099/jmm.0.05250-0. [DOI] [PubMed] [Google Scholar]
- 108.Duque C, Puyana M, Narváez G, Osorno O, Hara N, Fujimoto Y. Tetrahedron. 2004;60:10627–10635. [Google Scholar]
- 109.Rodríguez HI, Shi Y-P, García OJ, Rodriguez AD, Mayer AMS, Sánchez JA, Ortega-Barria E, González J. J Nat Prod. 2004;67:1672–1680. doi: 10.1021/np049802o. [DOI] [PubMed] [Google Scholar]
- 110.Look SA, Fenical W, Jacob RS, Clardy J. Proc Natl Acad Sci USA. 1986;83:6238–6240. doi: 10.1073/pnas.83.17.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Look SA, Fenical W, Matsumoto GK, Clardy J. J Org Chem. 1986;51:5140–5145. [Google Scholar]
- 112.Roussis V, Wu Z, Fenical W. J Org Chem. 1990;55:4916–4922. [Google Scholar]
- 113.Ata A, Win HY, Holt D, Holloway P, Segstro EP, Jayatilake GS. Helv Chim Acta. 2004;87:1090–1098. [Google Scholar]
- 114.Broka CA, Chan S, Peterson B. J Org Chem. 1988;53:1586–1587. [Google Scholar]
- 115.Corey EJ, Carpino P. J Am Chem Soc. 1989;111:5472–5474. [Google Scholar]
- 116.Di Blasio B, Fattorusso E, Magno S, Mayol L, Pedone C, Santacroce C, Sica D. Tetrahedron. 1976;32:473–478. [Google Scholar]
- 117.König GK, Wright AD, Sticher O, Fronczek FR. J Nat Prod. 1992;55:633–638. doi: 10.1021/np50080a004. [DOI] [PubMed] [Google Scholar]
- 118.Scheuer PJ. Acc Chem Res. 1992;25:433–439. [Google Scholar]
- 119.Caine D, Deutsch H. J Am Chem Soc. 1978;100:8030–8031. [Google Scholar]
- 120.Tamura K, Nakasaki A, Kobayashi S. Synlett. 2009;2449–2452 [Google Scholar]
- 121.König M, Wright AD, Franzblau SG. Planta Med. 2000;66:337–342. doi: 10.1055/s-2000-8534. [DOI] [PubMed] [Google Scholar]
- 122.Ishiyama H, Hashimoto A, Fromont J, Hoshino Y, Mikami Y, Kobayashi J. Tetrahedron. 2005;61:1101–1105. [Google Scholar]
- 123.Ishiyama H, Kozawa S, Aoyama K, Mikami Y, Fromont J, Kobayashi J. J Nat Prod. 2008;71:1301–1303. doi: 10.1021/np800164s. [DOI] [PubMed] [Google Scholar]
- 124.Etahiri S, Bultel-Poncé V, Caux C, Guyot M. J Nat Prod. 2001;64:1024–1027. doi: 10.1021/np0002684. [DOI] [PubMed] [Google Scholar]
- 125.Cafieri F, De Napoli L, Fattorusso E, Santacroce C. Phytochemistry. 1987;26:471–473. [Google Scholar]
- 126.Rasmussen TB, Givskov M. Int J Med Microbiol. 2006;296:149–161. doi: 10.1016/j.ijmm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 127.Clatworthy AE, Pierson E, Hung DT. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 128.Baron C, Coombes B. Infect Dis Drug Targets. 2007;7:19–27. doi: 10.2174/187152607780090685. [DOI] [PubMed] [Google Scholar]
- 129.Pan J, Ren D. Expert Opin Ther Pat. 2009;19:1581–1601. doi: 10.1517/13543770903222293. [DOI] [PubMed] [Google Scholar]
- 130.Bjarnsholt T, van Gennip M, Jakobsen TH, Christensen LD, Jensen PO, Givskov M. Nat Protoc. 2010;5:282. doi: 10.1038/nprot.2009.205. [DOI] [PubMed] [Google Scholar]
- 131.Wahl M. Mar Ecol Prog Ser. 1989;58:175–189. [Google Scholar]
- 132.Haygood MG, Schmidt EW, Davidson SK, Faulkner DJ. J Mol Microbiol Biotechnol. 1999;1:33–43. [PubMed] [Google Scholar]
- 133.Hentschel U, Usher KM, Taylor MW. FEMS Microbiol Ecol. 2006;55:167–177. doi: 10.1111/j.1574-6941.2005.00046.x. [DOI] [PubMed] [Google Scholar]
- 134.Taylor MW, Radax R, Steger D, Wagner M. Micobiol Mol Biol Rev. 2007;71:295–347. doi: 10.1128/MMBR.00040-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Thomas TRA, Kavlekar DP, LokaBharathi PA. Mar Drugs. 2010;8:1417–1468. doi: 10.3390/md8041417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abdelmohsen UR, Pimentel-Elardo SM, Hanora A, Radwan M, Abou-El-Ela SH, Ahmed S, Hentschel U. Mar Drugs. 2010;8:399–412. doi: 10.3390/md8030399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kazlauskas R, Murphy PT, Quinn RJ, Wells RJ. Tetrahedron Lett. 1977;18:37–40. [Google Scholar]
- 138.de Nys R, Wright AD, König GM, Sticher O. Tetrahedron. 1993;49:11213–11220. [Google Scholar]
- 139.Givskov M, de Nys R, Manefield M, Gram L, Maximilien R, Eberl L, Molin S, Steinberg PD, Kjelleberg S. J Bacteriol. 1996;178:6618–6622. doi: 10.1128/jb.178.22.6618-6622.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kjelleberg S, Steinberg P, Givskov M, Gram L, Manefield M, de Nys R. Aquat Microb Ecol. 1997;13:85–93. [Google Scholar]
- 141.Maximilien R, de Nys R, Holmström C, Gram L, Givskov M, Crass K, Kjelleberg S, Steinberg PD. Aquat Microb Ecol. 1998;15:233–246. [Google Scholar]
- 142.de Silva ED, Scheuer PJ. Tetrahedron Lett. 1980;21:1611–1614. [Google Scholar]
- 143.Skindersoe ME, Ettinger-Epstein P, Rasmussen TB, Bjarnsholt T, de Nys R, Givskov M. Mar Biotechnol. 2008;10:56–63. doi: 10.1007/s10126-007-9036-y. [DOI] [PubMed] [Google Scholar]
- 144.Bull AT, Ward AC, Goodfellow M. Microbiol Mol Biol Rev. 2000;64:573–606. doi: 10.1128/mmbr.64.3.573-606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Salemme FR. Pharmacogenomics. 2003;4:257–267. doi: 10.1517/phgs.4.3.257.22692. [DOI] [PubMed] [Google Scholar]
- 146.Bredel M, Jacoby E. Nat Rev Genet. 2004;5:262–275. doi: 10.1038/nrg1317. [DOI] [PubMed] [Google Scholar]
- 147.Singh SB, Barrett JF. Biochem Pharmacol. 2006;71:1006–1015. doi: 10.1016/j.bcp.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 148.Puri AW, Bogyo M. ACS Chem Biol. 2009;4:603–616. doi: 10.1021/cb9001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hammond GG, Huber JL, Greenlee ML, Laub JB, Young K, Silver LL, Balkovec JM, Pryor KD, Wu JK, Leiting B, Pompliano DL, Toney JH. FEMS Microbiol Lett. 1999;459:289–296. doi: 10.1111/j.1574-6968.1999.tb08740.x. [DOI] [PubMed] [Google Scholar]
- 150.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. Nature. 2009;458:83–86. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 151.La Clair JJ. Nat Prod Rep. 2010;27:969–995. doi: 10.1039/b909989c. [DOI] [PubMed] [Google Scholar]
- 152.Chopra I, Hesse L, O’Neill AJ. J Appl Microbiol. 2002;92:4S–15S. [PubMed] [Google Scholar]
- 153.Miesel L, Greene J, Black TA. Nat Rev Genet. 2003;4:442–456. doi: 10.1038/nrg1086. [DOI] [PubMed] [Google Scholar]
- 154.O’Neill AJ, Chopra I. Expert Opin Invest Drugs. 2004;13:1045–1063. doi: 10.1517/13543784.13.8.1045. [DOI] [PubMed] [Google Scholar]
- 155.Freiberg C, Brötz-Oesterhelt H, Labischinski H. Curr Opin Microbiol. 2004;7:451–459. doi: 10.1016/j.mib.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 156.Bode HB, Müller R. Angew Chem. 2005;117:6988–7007. [Google Scholar]; Angew Chem Int Ed. 2005;44:6828–6846. doi: 10.1002/anie.200501080. [DOI] [PubMed] [Google Scholar]
- 157.Challis GL. J Med Chem. 2008;51:2618–2628. doi: 10.1021/jm700948z. [DOI] [PubMed] [Google Scholar]
- 158.Zerikly M, Challis GL. ChemBioChem. 2009;10:625–633. doi: 10.1002/cbic.200800389. [DOI] [PubMed] [Google Scholar]
- 159.Norse E. Global Marine Biological Diversity: A Strategy for Building Conservation into Decision Making. Island Press; Washington: 1993. [Google Scholar]