

Abstract
The last 20 years have witnessed major advances in the understanding of muscle diseases and significant inroads are being made to treat muscular dystrophy. However, no curative therapy is currently available for any of the muscular dystrophies, despite the immense progress made using several approaches and only palliative and symptomatic treatment is available for patients. The discovery of miRNAs as new and important regulators of gene expression is expected to broaden our biological understanding of the regulatory mechanism in muscle by adding another dimension of regulation to the diversity and complexity of gene-regulatory networks. As important regulators of muscle development, unravelling the regulatory circuits involved may be challenging, given that a single miRNA can regulate the expression of many mRNA targets. Although the identification of the regulatory targets of miRNAs in muscle is a challenge, it will be critical for placing them in genetic pathways and biological contexts. Therefore, combining informatics, biochemical and genetic approaches will not only expected to reveal the elucidation of the miRNA regulatory network in skeletal muscle and to bring a better knowledge on muscle tissue regulation but will also raise new opportunities for therapeutic intervention in muscular dystrophies by identifying candidate miRNAs as potential targets for clinical application.
Keywords: muscular dystrophy, microRNA, skeletal muscle
Introduction
The muscular dystrophies are a heterogeneous group of over 30 different inherited disorders all involving progressive weakness and degeneration of skeletal muscle with variable distribution and severity, resulting in significant morbidity and disability. Characteristic features of dystrophic muscle include central nuclei, small regenerating fibres and accumulation of connective tissue and fatty tissue. More than two decades after the cloning of dystrophin as the disease-causing gene in Duchenne muscular dystrophy [1], additional forms of muscular dystrophy have been associated with numerous gene defects. Mutations were identified in structural proteins, signalling molecules and enzymes as well as mutations that result in aberrant processing of mRNA or alterations in post-translational modifications of proteins [2]. These findings have not only revealed important insights into the protein network of skeletal muscle fibres and led to a better understanding of skeletal muscle function through the analysis of muscle dysfunction but also provided new approaches for therapy. However, although considerable progress has been made in the understanding of muscle biology, revealing the overall complexity of the pathogenesis of the various muscular disorders, the underlying molecular pathways remain poorly understood and still mainly unknown.
There are substantial underlying muscle disease mechanisms, which remain to be elucidated. A detailed knowledge of the signalling pathways and transcriptional networks that regulate cellular and molecular processes of skeletal muscle function is necessary to understanding the pathology of muscle.
Recently, another layer of transcriptional regulation has begun to emerge. In a genetic screen for molecules that control the timing of larval development in C. elegans, lin-4, the founding member of the miRNA gene class, was discovered and later identified as a gene that does not encode a protein but instead produces a pair of small RNAs, with the longer one predicted to be the precursor of the shorter one [3]. A second miRNA gene, let-7, was found in 2000, in worms [4, 5]. miRNAs have now emerged as widely acting regulatory molecules that silence their cognate target genes, either by degrading mRNA molecules or, more frequently in mammalian cells, by inhibiting their translation [6]. Computational and biological evidence suggests that miRNA-mediated gene regulation represents a fundamental mechanism of post-transcriptional regulation with diverse functional effects [7].
miRNAs have been implicated in a range of biological processes including cancer [8, 9], development of the limb [10, 11], lung [12], and haematopoetic system [13]. In addition, a number of miRNAs have been characterized as modulators of myogenic differentiation [14] and there is increasing evidence for miRNA involvement in myopathies and muscular dystrophies [15–17]. Here we review recent progress that addresses the functions of skeletal muscle-mediating miRNAs. These findings are expected to advance our understanding of the molecular mechanisms underlying muscle pathology.
Mechanisms of miRNA-mediated repression
miRNAs as an endogenously expressed small RNAs are transcribed as larger transcripts (pri-miRNAs) that are processed by Drosha into stem-loop precursors (pre-miRNAs) [6]. The pre-miRNAs are transported from the nucleus to the cytoplasm by Exportin-5 and are subsequently cleaved by the Dicer RNase to yield 20–22 nucleotide mature miRNA. The resulting miRNAs bind, largely through their ‘seed’ regions (nucleotides 2–8 at the 5′ end of an miRNA), to partially complementary sequences at the 3′UTRs of specific target mRNA and are loaded into an RNA-induced silencing complex (RISC), which directs silencing of target genes [6]. The most important factor governing miRNA binding efficacy is perfect base pairing between the target site and the miRNA seed region, ∼7 nucleotides at the 5′-end of the miRNA. Further determinants of probable mRNA targets, such as target site flanking sequence, accessibility caused by local secondary structure, number and spacing of target sites, base paring between the target site and the 3′-end of the miRNA and expression patterns, have been identified and used in computational prediction algorithms [18, 19]. Combined computational-experimental analyses suggest that at least ∼30% of human mRNAs are miRNA targets [20]. Once a miRNA binds to a target site, the associated RISC is the primary mediator of miRNA function, which is usually either translational repression or direct mRNA cleavage.
miRNA regulatory network in muscle
During embryonic development, specification of mesodermal precursor cells to the myogenic lineage is a multi-step process highly regulated by positive and negative signals from surrounding tissues. Specification to the myogenic lineage requires the up-regulation of MYOD and MYF5, basic helix-loop-helix transcriptional activators of the myogenic regulatory factor family (MRF). Proliferating myoblasts withdraw from the cell cycle to become terminally differentiated myocytes that express the late MRFs, Myogeninand and MRF4, and subsequently muscle-specific genes such as myosin heavy chain (MHC) and muscle creatine kinase (MCK) [21]. Finally, through complex pathways regulated at both transcriptional and post-transcriptional levels and multiple regulatory factors, including transcription factors and cellular signalling molecules, mononucleated myocytes specifically fuse to each other to form multinucleated syncytium, which eventually mature into contracting muscle fibres (Fig. 1). Recent evidence supports a role for miRNAs as integral components of the regulatory circuitry for muscle development. Characterization of regulatory mechanisms involving miRNA expression and activity is providing novel clues for the identification of genes and complex regulatory circuits and collectively these studies indicate that miRNAs function as regulators of gene expression important for myoblast proliferation and differentiation and may play decisive roles in specifying cell types during development.
1.
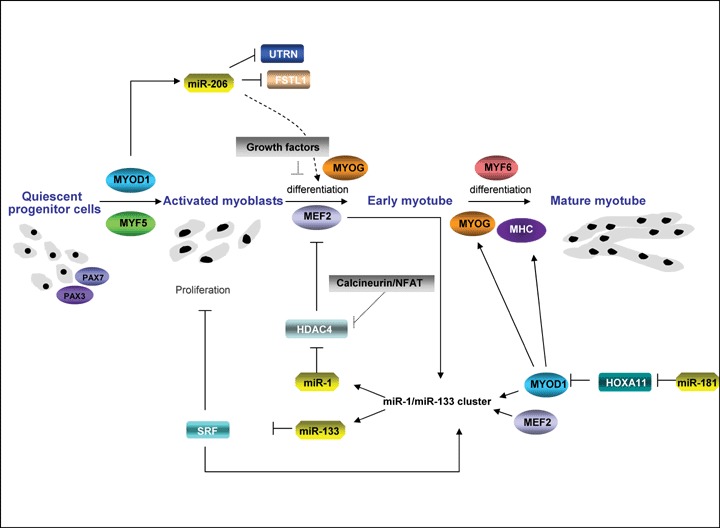
miRNA in muscle development. miR-1 and miR-133 along with miR-206 and miR-181 function at the centre of a network of transcription factors to regulate skeletal myoblast proliferation and differentiation. Myocyte enhancer factor-2 (MEF2) and myogenic basic helix loop helix (bHLH) proteins, including myogenic transcription factor MYOD1, regulate their own expression, as well as the expression of downstream muscle structural genes. Additionally, these transcription factors use upstream and intragenic enhancers to activate transcription of bicistronic miR-1/133 clusters encoding miR-1 and miR-133 in differentiated skeletal muscle. miR-1 represses expression of HDAC4 (histone deacetylase 4), a signal-dependent repressor of MEF2 activity, thereby establishing a negative feedback loop to modulate miR-1 and miR-133 expression and promoting myoblast differentiation. As myogenesis progresses from the myoblast stage to the myotube stage, the level of the muscle-specific miR-133 increases and miR-133 represses expression of serum response factor (SRF), a positive regulator of miR-1/133 expression and repressor of myoblast proliferation. Upon differentiation, miR-181 is also up-regulated, resulting in down-regulation of Hox-A11 and in the release of MYOD1 expression. As a result, myogenin (MYOG) and muscle marker proteins including MHC (myosin heavy chain) are up-regulated. In parallel, activation of MYOD1 increases the expression of the primary miR-206 transcript which in turn lead to down-regulation of Follistatin-like 1 (FSTL1) and to the repression of Utrophin (Utrn) expression and through a mechanism that is not yet known promotes muscle differentiation.
Current data on the roles of miRNAs in myogenesis have been obtained largely from studies on muscle-specific miR-1, miR-133 and miR-206 [15, 16, 22–31]. These findings have generated more detailed insights into the mechanisms underlying the myogenesis process and have uncovered different pathways that lead to myofibre proliferation and differentiation. However, the complete roles of miRNAs in muscle growth and development still remain to be elucidated and the roles of non-muscle-specific miRNAs in myogenesis have not been thoroughly examined.
Muscle-specific miRNAs
The functional characterization of miR-1 and miR-133 has been an important step in our understanding of miRNA-mediated muscle development (summarized in Fig. 1). miR-1 is highly expressed in skeletal and heart muscle across species from D. melanogaster to humans. Investigation of a loss-of-function phenotype of Drosophila miR-1 showed that miR-1 is not required for the formation or physiological function of the larval musculature, but is required for the post-mitotic growth of larval muscle [24]. miR-1 and miR-133 are transcribed in a muscle-specific manner during development from a common polycistronic gene [25] and modulate muscle growth and differentiation by regulating SRF and MEF2 activity, thereby establishing negative feedbacks loops within muscle cell lineages [25]. Recent studies showed that miR-1 promotes myogenesis by targeting histone deacetylase 4 (HDAC4), a transcriptional repressor of muscle gene expression. miR-1 represses the expression of histone deacetylase 4 (HDAC4) [25], which acts as a signal-dependent repressor of muscle differentiation together with MEF2 [32]. Thus, miR-1 up-regulation during differentiation is a mechanism to reduce HDAC4 expression and to potentiate MEF2 pro-myogenic activity. Among its many predicted targets, miR-1 also represses the translation of Hand2 [26] a bHLH transcription factor that is required for cardiac growth during embryogenesis.
Consistent with these findings, similar to miR-1, another miRNA, miR-206, has also been characterized as a muscle regulator in recent studies and has also been shown to promote myoblast differentiation [27–29]. Gap junction protein connexin43 (Cx43) and the p180 subunit (Pola1) of DNA polymerase alpha have been identified as regulatory targets of miR-206. Although Cx43 is required for the initial phase of myogenesis, it is rapidly down-regulated post-transcriptionally after the induction of differentiation [33], thus miR-206 is suggested to decrease communication between developing muscle fibres by decreasing Cx43 expression [29]. Down-regulation of Pola1 by miR-206 during early differentiation reduces DNA synthesis and contributes to the suppression of cell proliferation during myotube formation [16]. miR-206 is also suggested to mediate MyoD-dependent inhibition of follistatin-like 1 (FSTL1) and Utrophin (Utrn) genes in myoblasts [30]. In this case, MYOD1 activates the expression of miR-206, which in turn represses FSTL1 and Utrn gene expression post-transcriptionally. This mechanism could explain some of the previous observations in which MYOD1, known as a transcrip-tional activator, repressed FSTL1 and Utrn gene expression. Although Utrn expression was repressed by miR-206 during myoblast differentiation [30], its expression was up-regulated in mdx diaphragm muscle [15]. This phenomenon might reflect decreased efficiency of miRNA-mediated translational repression during a diseased state.
While experiments in cell culture suggested that miR-1 and miR-206 promote differentiation of myoblasts, miR-133 has been proposed to promote myoblast proliferation, a role opposite to that of miR-1 [25] through down-regulation of different target genes [25, 28]. The ability of miR-133 to promote proliferation has been ascribed to the repression of SRF, an essential regulator of muscle differentiation. miR-133 also represses translation of the polypyrimidine tract-binding protein (nPTB), which promotes differential splicing of a variety of transcripts that influence the muscle differentiation program [22]. In addition, ChIP on CHIP analysis also indicated that the myogenic regulatory factors, MYOD1 and Myogenin, bind to sequences upstream of miR-1 and miR-133 [27]. It seems as miR-1 and miR-133 that are encoded by the same MEF2-regulated bicistronic transcripts would exert opposing effects on muscle growth and differentiation. However, both miR-1 and miR-133 fine-tune key regulatory pathways in an antagonistic manner with the balance being tipped one way zor the other by additional transcription factors and regulatory pathways.
Interestingly, different evidence for the involvement of muscle-specific miRs in the control of skeletal muscle growth comes from a spontaneous mutation that causes dramatic muscularity in the Texel strain of sheep. The particular mutation has been mapped to a single G-to-A mutation within the 3′ UTR of the mRNA encoding Myostatin, a member of the transforming growth factor β (TGF-β) family of growth factors that represses muscle growth. This mutation creates a target site for miR-1 and miR-206, resulting in Myostatin translational repression [34]. This results in a pheno-type that matches Myostatin loss of function mutations in mice, cattle and humans [35].
Non-muscle-specific miRNAs
In contrast to miRNAs discussed above, which are specifically expressed in a tissue-restricted manner, miR-181 is broadly expressed. The expression of miR-181 was found to be increased in the regenerating muscle from a mouse model of muscle injury [36] and further analysis using C2C12 cells demonstrated that miR-181 depletion reduced MYOD1 expression and inhibited myoblast differentiation. One of the genes targeted by miR-181 is homeobox protein Hox-A11, which in turn represses MYOD1 expression. The proposed mechanism underlying miR-181 function is that miR-181 is up-regulated upon differentiation and targets a repressor (Hox-A11) of the differentiation process to allow new muscle growth (Fig. 1). However, although miR-181 is required for skeletal myoblast terminal differentiation, neither up-regulation of miR-181 nor down-regulation of Hox-A11 triggered terminal differentiation of proliferating myoblasts. Thus, miR-181 is necessary, but not sufficient, for differentiation. In addition to myogenesis, miR-181 was shown to modulate haematopoietic lineage differentiation in another study [13], which suggests that individual miRNAs may play very diverse biological roles depending upon their cellular context.
miR-214 is expressed in skeletal muscle cell progenitors during zebrafish development and was shown to specify muscle cell type during somitogenesis by modulating the response of muscle progenitors to Hedgehog signalling [37]. Blocking miR-214 activity by chemically modified antisense oligonucleotides decreased the number of slow-muscle cell types present in the developing somites and distinctly changed the gross morphology of the somites in manner previously associated with attenuated Hedgehog signalling. This phenotype was attributed to relief of miR-214-mediated inhibition of suppressor of fused (su(fu)) expression [37], a fine-tuner of Hedgehog signalling essential for proper specification of muscle cell types during somitogenesis [38].
Another example for a non-muscle-specific miRNA involved in myogenesis is miR-24. Members of TGF-β have been shown to potently inhibit terminal differentiation of cultured myoblasts by down-regulating MRFs [39, 40]. Myostatin, a member of the TGF-β family, has been shown to be a negative regulator of muscle differentiation and growth [41, 42]. Recent studies revealed that SMAD3 mediates the suppression of myogenesis by TGF-β through suppressing the expression of E-box muscle genes and MEF2 proteins [43, 44], whereas SMAD7 has been implicated in promoting and enhancing myogenesis by interacting with MYOD1 and abrogating of Myostatin signalling [45].
Recently, miR-24 was shown as an essential miRNA for the modulation of TGF-β-inhibited myogenesis [46] thus providing new clues for better understanding the molecular mechanisms underlying the physiological roles of TGF-β during myogenesis. Because changed expression of miR-24 affects both early and late myogenic markers, the underlying mechanism might be that miR-24 regulates an unknown upstream signal, which then affects the expression of both early and late myogenic markers [46]. It is also possible that miR-24 first regulates the expression of early genes like Myogenin and MEF2, which then affects the expression of late genes including MHC.
In addition to being strongly induced during myogenesis [46], miR-24 expression is maintained at high levels in terminally differentiated muscle tissues including heart and skeletal muscle. These observations suggest that miR-24 might function during both differentiation and homeostatic maintenance of cardiac and skeletal muscle tissues. Indeed, previous studies have shown that miR-24 is up-regulated during cardiac hypertrophy and is able to induce hypertrophic growth when overexpressed in primary cardiomy-ocytes [47]. Considering that miR-24 is broadly expressed in multiple tissues, it is speculated that miR-24 might play different roles in the homeostasis of these tissues.
miRNA signatures as biomarkers of myopathies
Several recent studies have championed the possibilities for the use of miRNA signatures as biomarkers for the detection of cancer, pregnancy and disease [48–50]. Presently, the most frequent method for detection and diagnosis of genetic mutations as a cause of myopathies is through large-scale PCR sequencing assays [51]. Because many of the limb girdle muscular dystrophies (LGMD) have similar symptoms, it can be difficult to diagnose the exact form of the disease without genomic screening. Although the results of these myopathic sequencing assays are highly accurate, they often involve high costs and can take several weeks to obtain conclusive results. Given the changes in protein expression and miRNA expression diversity, the usage of miRNAs as biomarkers present an attractive alternative to large-scale genome sequencing at reduced cost and time to the patient.
The usage of miRNAs in the detection of various cancers is one of the most promising areas of biomarker identification (reviewed in [52]). Cancer in general terms remains problematic in its diagnoses that rely primarily on the detection of overexpressed levels of various antigens such as prostate-screening antigen (PSA) for prostate cancer detection. Often cancers are not detected until they have metastasized and few treatment options for the patient remain. Several recent studies have highlighted the usage of specific miRNAs in various arrays of large-scale cancer studies. A recent study by Szafranska and colleagues identified miR-217 as significantly down-regulated and miR-196a significantly overex-pressed in patients with the difficult to diagnose pancreatic ductal adenocarcinoma (PDAC) [53]. Interestingly, the rs11614913 SNP in miRNA-196a was recently reported as an indicator of non-small cell lung cancer (NSCLC) [54]. Individuals who were homozygous for the CC mutation had statistically significantly higher mortality compared with individuals whom had either TT or CT at the locus. It is hypothesized that cancer in general may have a set of miRNAs that are dysregulated during uncontrolled cell proliferation. The miRNA polycistron miR-17–92 locus has been characterized as an oncogene and is highly overexpressed in B-cell lymphoma [55]. Indeed, the miR-17–92 locus is activated upon the overexpression of c-myc a hallmark of B-cell lymphomas diagnoses. Thus, it is logical to assume that many disease states involving cellular proliferation might have a unique miRNA signature that functions in concordance with various cell cycle genes to promote a non-native microenvironment.
Recently, our laboratory has characterized the unique miRNA signatures found in several of the limb girdle myopathies as well as Duchenne muscular dystrophy (DMD) [17]. Many of the miRNAs expressed in the muscle of specific LGMD were unique to each separate disorder. A strategy to first detect the miRNA signature using miRNA arrays on muscle biopsies followed by subsequent genome sequencing could greatly enhance the speed of myopathic diagnosis at reduced time and cost. For example, one who wishes to make the diagnosis based on the symptoms associated with Duchenne's muscular dystrophy (DMD) and distinguish it from the milder Beck muscular dystrophy (BMD) would be able make an accurate diagnosis based on the miRNA that would reveal a significant up-regulation of miRNAs -299-5p, -487b, and -362 present only in DMD [17]. More difficult to diagnosis myopathies such as nemaline myopathy (NM), which can be detected by mutations in six filamental genes, can be distinguished from the inflammatory myopathies such as inclusion body myositis (IBM) or dermatomyositis (DM) based on the expression levels of specific inflammatory miRNAs-155 and miR-146b among others [17, 56]. A positive Facioscapulohumeral muscular dystrophy (FSHD) diagnosis could be distinguished from DMD based on the lack of dysregualtion of miRNAs-381 and -382 found in FSHD patients [17]. Similarly, nearly all of the LGMD myopathies have a significant up-regulation of miRNAs-100, -103, -107; whereas DMD patients do not [17]. As the cost of arrays subsequently decreases as the technology becomes more mainstream and high-throughput, the usage of miRNAs as biomarkers in diagnostic laboratories should vastly increase.
miRNA therapeutic promise
Locked nucleic acids (LNAs) and antagomiRs
The ability to inhibit miRNA function through the use of complimentary sequences makes miRNAs an attractive candidate for therapeutic treatments. As noted, several miRNAs are significantly up-regulated in muscular dystrophies and other myopathies [17]. The ability to inhibit these up-regulated miRNAs using locked nucleic acids (LNAs) or antagomirs (cholesterol-modified) single stranded nucleic acids consisting of the complementary miRNA sequence thus offers a molecular strategy for miRNA inhibition.
Locked nucleic acids are more stable than single-stranded RNA molecules due to a modification of the ribose moiety of the oligonucleotide that results in enhanced stability and greater resistance to increases in temperature (reviewed in [57]). LNAs have been used successfully as qualitative markers to measure miRNA levels through in situ hybridization, northern blot analysis and real-time PCR [58, 59]. A recent study by Elmén and colleagues demonstrated that a miRNA could be successfully targeted in a living organism using LNA-antimiR conjugate [60]. Elmén and colleagues delivered LNA-antimiR targeting miR-122, a liver-specific miRNA previously shown to regulate lipid metabolism and the hepatitis C hepatic response, to the livers of African-green monkeys through a catheter in the saphenous vein. The monkeys that received the miR-122 LNA-antimiR had lower plasma cholesterol levels, no significant changes in essential liver metabolic enzymes, and importantly no adverse side effects.
The skeletal-muscle-specific miR-206 miRNA has been shown to be up-regulated in the diaphragm muscle of the dystrophin-mutant (mdx) mouse [15]. Rosenberg and colleagues recently identified two muscle-enriched miR-206 target genes, Utrophin (Utrn) and follistatin-like 1 (FSTL1), which were down-regulated following miR-206 overexpression in vitro[30]. Overexpression of miRNA in mouse myoblasts resulted in increased differentiation, regardless of the levels of serum in the media [28]. Likewise, inhibition of miR-206 by antagomirs resulted in the inhibition of myogenic differentiation. These findings would suggest that the overexpression of miR-206 observed in the diaphragm muscle of mdx mice might also explain the accelerated differentiation and cell cycle kinetics in the muscle satellite cells isolated from the diaphragm muscle of mdx mice [61]. Delivery of LNAs targeting miR-206 to the diaphragm muscles of patients might reverse the accelerated differentiation observed in DMD patients and increase life-span. Given the restrictive expression of miR-206 to skeletal muscle, intravenous or direct intramuscular injection of LNAs targeting miR-206 would most likely have relatively minor side effects.
Overexpression of miRNAs in myopathies
A modest cluster of miRNAs, both skeletal muscle-specific and non-skeletal muscle-specific, are significantly down-regulated in various myopathies of patients [17]. In both cardiac and skeletal muscle hypertrophy, the cardiac/skeletal muscle-enriched miRNA, miR-133a, was strongly down-regulated [16, 62]. Thus, the option of correcting the lower levels of specific miRNAs becomes another tool for treatment of myopathies. Overexpression of miRNAs in vivo would most likely involve injection of a stable oligonucleotide that would be cleaved to a 21–24 nucleotide strand by endogenous mechanisms or the injection of an miRNA overexpression virus. Recent advances in the viral infection levels and decreased toxicity using adeno-asso-ciated viruses (AAV) have made this system an attractive delivery method for gene therapy [63, 64]. Intramuscular injections of either the 21–24 nucleotide miRNA or stem-loop structure over-expression virus would ensure successful delivery of the miRNA to the affected muscle.
Specificity of miRNA treatment
Given the close proximity and cross-talk communication between skeletal muscle, immune cells (i.e. macrophages, neutrophils, etc.) and vascular smooth muscle cells, it is essential that any treatment involving the modification of the levels of a specific or group of miRNAs have a high degree of specificity for the affected muscle and the neighbouring cells. The role of miRNAs as essential regulators of the immune system and the inflammatory response has been well documented (reviewed in [65]. Many of the miRNAs that were dysregulated in different myopathies have essential functions in both myogenesis and the inflammatory response [17]. miRNA-181a/b is an upstream repressor of Hox-A11 transcription, a validated repressor of MYOD1 levels and is significantly up-regulated during skeletal muscle regeneration [36]. However, miR-181a/b also plays an essential role in the maturation of T cells and their affinity for antigen recognition [66]. Additional miRNAs, such as miR-1, whereas substantially expressed in skeletal muscle, have broad functions as regulators of stem cell differentiation, cell cycle progression, and cardiac conduction [67, 68]. The ability to achieve specificity and selectivity in targeting miRNAs of a specific cell type is a significant barrier that must be explored if non-skeletal muscle-specific miRNAs are to be used as a treatment for myopathies.
miRNAs as regulators of myogenic stem cells
Cellular therapies toward the treatment of myopathies have shown considerable promise in their ability to expand and repopulate the damaged muscle architecture [69]. Recent studies have identified myogenic and vascular-derived stem cells as capable of restoring physiological function and amelioration of skeletal muscle degeneration resulting from the lack of a functional dystrophin protein [70–72].
miRNAs are essential for normal myoblast proliferation and function. Deletion of the miRNA processing component Dicer using the MyoD-cre transgene resulted in abnormal mouse limb development and increased apoptosis of cultured embryonic myoblasts, thus demonstrating that miRNAs have an essential function in myoblast proliferation and maintenance [73]. Genetic and/or pharmacological manipulation of specific miRNA levels in cultured myoblasts may enhance engraftment in myopathic muscles. Chen and colleagues recently undertook a survey of miRNAs in mouse myoblasts and identified several essential miRNAs that were both enriched and depleted following the removal of serum [25]. MiR-1 overexpression accelerated myogenic differentiation through the repression of the histone deactylase HDAC4; whereas miR-133 overexpression accelerated the proliferative capacity of the myoblasts by repressing the SRFgene.
MiR-221 and miR-222 are two miRNAs, which are expressed in skeletal muscle, are validated regulators of cell proliferation through the repression of cell cyclins and pleuripotency markers [74, 75]. MiR-221 and miR-222 cluster together on the human and mouse genomic loci, are evolutionarily conserved among vertebrates and have similar seed sequences. MiR-221/222 target the 3’ UTR of the kit receptor and impairs erythropoietic and myeloid proliferation and enhance lineage differentiation in isolated haematopoietic stem cells [74]. Additionally, miR-221 and miR-222 down-regulate the cyclin-dependent kinase inhibitors p27 (Cdkn1b/Kip1) and p57 (Cdkn1c/Kip2) and are considered to be essential for the maintenance of the proliferative state of the cell cycle [75]. Several current studies are underway to inhibit miR-221 and miR-222 as treatment to inhibit cancer cell proliferation. Both miR-221 and miR-222 are significantly up-regulated in DMD and other muscular dystrophies [17]. Inhibiting these miRNAs using LNAs or antogomirs might correct the dysregulated cell cycle kinetics and restore normal function to the skeletal muscle stem cells of myopathic patients.
Perspective
miRNAs are novel regulators of gene expression, and have a dynamic expression in skeletal muscle. Several miRNAs are enriched in skeletal muscle during normal growth and regeneration, whereas others have essential functions in skeletal muscle but are ubiquitously expressed. Additionally, many miRNAs are either induced or repressed in muscular disorders, making them ideal candidates as drug targets as well as useful diagnostic bio-markers. Newer technologies, such as locked nucleic acids and antagomirs open up new techniques and potential therapies for miRNA-associated myopathies (Table 1). Similarly, overexpression of candidate miRNAs in cultured myoblasts for injection or using direct intramuscular injections also have great potential as therapeutic agents
1.
Potential miRNA-based therapies for various human diseases
| Disease/Disorder | Disease Symptoms | miRNA(s) dysregulated in disease | miRNA(s) Target(s) | Type of Therapy Proposed | Reference | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypercholesterolemia | Elevated blood pressure, high risk for heart failure/attack, high risk for stroke | miR-122 (overex-pressed) | Aldoa | Locked Nucleic Acid (LNA) intravenous injections | 60 | ||||||||||||
| Duchenne Muscular Dystrophy (DMD) | Muscular degeneration, respiratory failure, death before early 20's | miR-206 (overex-pressed) | Utrn | LNA delivery into diaphragm | 15 | ||||||||||||
| miR-133 (down-regulated) | Runx2 | overexpression in cultured myoblasts using Adeno-Associated Virus (AAV) | 16,62 | ||||||||||||||
| miR-221/222 (overexpressed) | Cdkn1b (p27), Cdkn1c (p57), Kit | LNA delivery in cultured muscle stem cells followed by intramuscular injection | 31 75 | ||||||||||||||
| Skeletal Muscle | Muscle weakness, | miR-1 and miR-133 | Hdac4 | Overexpression in cultured | 25 | ||||||||||||
| Atrophy | myofibre degeneration, immobility | (down-regulated) | myoblasts using Adeno-Associated Virus (AAV) | ||||||||||||||
| B-cell lymphoma | Immunodeficiency, tumor formation and metastasis potential, death if not treated | miR-17–92 cluster (overexpressed) | E2f1 | LNA intravenous delivery | 55 | ||||||||||||
| non-small cell lung cancer (NSCLC) | Respiratory failure, chronic obstructive pulmonary disease (COPD), death | miR-196a (overex-pressed) | Hoxb8, Ccna2 (cyclin A2),Lsp1,Myc, Casp3 | LNA intravenous delivery | 54 | ||||||||||||
However, despite the enormous potential of miRNAs as therapeutic agents, many questions still remain. Two recent loss-of-function studies revealed that miRNAs have an essential function as rheostats for making adjustments in global protein levels and not dramatic alterations in proteomic output [76, 77]. Baek and colleagues analysed global protein output using proteomic arrays in enriched neutrophils from mice that lacked miR-223 compared to their wild-type littermates. Surprisingly, very few specific genes were significantly induced or repressed at significant levels in mouse neutrophils lacking miR-223 at the proteomic level.
Although there were both slight decreases and increases in a large number of specific proteins resulting from the loss of miR-223, the overall protein output for many genes remained unchanged. Similarly, when Selbach and colleagues overexpressed several miRNAs in HeLa cells and analysed global protein output, they noticed very few genes that were significantly dysregulated at the protein level.
These proteomic studies raise the question as to whether or not the observed changes in specific miRNAs in myopathies have a significant effect on their proteomic output. It is likely that each specific myopathy has a separate degree of proteomic dysregulation resulting from gain- or loss-of-function of specific genes (reviewed in [1]). For example, the loss-of-function of an enzymatic transferase such as fukutin related protein (Fkrp) in patients with LGMD2I would have a different effect on global protein output in skeletal muscle compared to that of a myopathy involving a structural protein, such as loss-of-function mutations in components of the sarcoglycan complex in patients with LGMD2C-F. Likewise, the modulation of the expression of miRNAs in LGMD2A, which is caused by mutations in the CAPN3gene (a regulator of calcium signalling), might have a different effect than miRNA modulation in a structural protein-based myopathy, such as LGMD2B (resulting from mutations in the DYSF gene) [17]. Additional questions in regards to which specific miRNAs, or a possible combination would be the best drug targets.
Current treatments of patients with myopathies usually involve the use of various drugs to maintain muscle strength and prevent muscle atrophy. Depletion of myostatin, a natural muscle inhibitor, through the use of monoclonal antibodies has shown some efficacy in its long-term use to rebuild muscle dystrophin-mutant mice [78]. Recent advances in gene therapy have produced promising results using a technique referred to as ‘exon skipping’ in which cellular infection of myoblasts with antisense sequences designed to skip over exon 51 of the DMDgene results in translation of a functional dystrophin protein [79]. The most promising field of treatment involves muscle-derived stem cell (MDSC) therapies to restore the function and physiology of degenerate muscle in myopathic muscle (reviewed in [80]. Although recent studies using donor-derived MDSCs for treatment of myopathies have shown remarkable promise, the ability to correct a myopathy with a patient's autologous MDSCs and using gene therapy might have the best approach. Knowledge of miRNA function in myopathic MDSCs could greatly enhance myopathic engraphment in skeletal muscle and minimize any inflammatory response.
miRNAs serve important roles as modulators of protein output and determinants of cellular fate during critical periods of human growth and development. Many skeletal muscle-specific and nonspecific miRNAs are dysregulated in several myopathies. These dysregulated miRNAs can be utilized as diagnostic biomarkers for disease-specific myopathies based on their unique expression patterns. Additionally, these myopathic-specific miRNA signatures offer another category of potential drug targets through manipulation of their expression levels using LNA/antagomiR miRNA inhibitors and adenoviral-associated viruses (AAV) for muscle-specific overexpression. In combination with recent breakthroughs in cellular therapies, miRNA-based therapies offer a powerful addition to the amelioration of myopathies and eventual cure for these debilitating diseases.
Acknowledgments
We specially thank Emanuela Gussoni and Genri Kawahara for helpful discussion and members of the Kunkel lab for their support and useful suggestions. L.M.K. is an Investigator of the Howard Hughes Medical Institute.
References
- 1.Dalkilic I, Kunkel LM. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev. 2003;13:231–8. doi: 10.1016/s0959-437x(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 2.Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;7:762–73. doi: 10.1038/nrm2024. [DOI] [PubMed] [Google Scholar]
- 3.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 4.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–6. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 5.Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. The lin-41 RBCC gene acts in the Celegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell. 2000;5:659–69. doi: 10.1016/s1097-2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 8.Dalmay T, Edwards DR. MicroRNAs and the hallmarks of cancer. Oncogene. 2006;25:6170–5. doi: 10.1038/sj.onc.1209911. [DOI] [PubMed] [Google Scholar]
- 9.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 10.Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci USA. 2005;102:10898–903. doi: 10.1073/pnas.0504834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hornstein E, Mansfield JH, Yekta S, Hu JK, Harfe BD, McManus MT, Baskerville S, Bartel DP, Tabin CJ. The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature. 2005;438:671–4. doi: 10.1038/nature04138. [DOI] [PubMed] [Google Scholar]
- 12.Harris KS, Zhang Z, McManus MT, Harfe BD, Sun X. Dicer function is essential for lung epithelium morphogenesis. Proc Natl Acad Sci USA. 2006;103:2208–13. doi: 10.1073/pnas.0510839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 14.Callis TE, Deng Z, Chen JF, Wang DZ. Muscling through the microRNA world. Exp Biol Med. 2008;233:131–8. doi: 10.3181/0709-MR-237. [DOI] [PubMed] [Google Scholar]
- 15.McCarthy JJ, Esser KA, Andrade FH. MicroRNA-206 is overexpressed in the diaphragm but not the hindlimb muscle of mdx mouse. Am J Physiol. 2007;293:C451–7. doi: 10.1152/ajpcell.00077.2007. [DOI] [PubMed] [Google Scholar]
- 16.McCarthy JJ, Esser KA. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol. 2007;102:306–13. doi: 10.1152/japplphysiol.00932.2006. [DOI] [PubMed] [Google Scholar]
- 17.Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S, Flanigan KM, Neely LA, Whitney D, Beggs AH, Kohane IS, Kunkel LM. Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci USA. 2007;104:17016–21. doi: 10.1073/pnas.0708115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Fortin K, Mourelatos Z. MicroRNAs: biogenesis and molecular functions. Brain Pathol. 2008;18:113–21. doi: 10.1111/j.1750-3639.2007.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38:S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 20.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 21.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–38. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 22.Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M, Jr, Black DL. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–52. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakajima N, Takahashi T, Kitamura R, Isodono K, Asada S, Ueyama T, Matsubara H, Oh H. MicroRNA-1 facilitates skeletal myogenic differentiation without affecting osteoblastic and adipogenic differentiation. Biochem Biophys Res Commun. 2006;350:1006–12. doi: 10.1016/j.bbrc.2006.09.153. [DOI] [PubMed] [Google Scholar]
- 24.Sokol NS, Ambros V. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev. 2005;19:2343–54. doi: 10.1101/gad.1356105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38:228–33. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–20. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 27.Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci USA. 2006;103:8721–6. doi: 10.1073/pnas.0602831103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol. 2006;174:677–87. doi: 10.1083/jcb.200603008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson C, Catoe H, Werner R. MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006;34:5863–71. doi: 10.1093/nar/gkl743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg MI, Georges SA, Asawachaicharn A, Analau E, Tapscott SJ. MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J Cell Biol. 2006;175:77–85. doi: 10.1083/jcb.200603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Hassan MQ, Volinia S, Van Wijnen AJ, Stein JL, Croce CM, Lian JB, Stein GS. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc Natl Acad Sci USA. 2008;105:13906–11. doi: 10.1073/pnas.0804438105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–40. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 33.Proulx A, Merrifield PA, Naus CC. Blocking gap junctional intercellular communication in myoblasts inhibits myogenin and MRF4 expression. Dev Genet. 1997;20:133–44. doi: 10.1002/(SICI)1520-6408(1997)20:2<133::AID-DVG6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 34.Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibé B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38:813–8. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- 35.Tobin JF, Celeste AJ. Myostatin, a negative regulator of muscle mass: implications for muscle degenerative diseases. Curr Opin Pharmacol. 2005;5:328–32. doi: 10.1016/j.coph.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Naguibneva I, Ameyar-Zazoua M, Polesskaya A, Ait-Si-Ali S, Groisman R, Souidi M, Cuvellier S, Harel-Bellan A. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat Cell Biol. 2006;8:278–84. doi: 10.1038/ncb1373. [DOI] [PubMed] [Google Scholar]
- 37.Flynt AS, Li N, Thatcher EJ, Solnica-Krezel L, Patton JG. Zebrafish miR-214 modulates Hedgehog signaling to specify muscle cell fate. Nat Genet. 2007;39:259–63. doi: 10.1038/ng1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolff C, Roy S, Ingham PW. Multiple muscle cell identities induced by distinct levels and timing of hedgehog activity in the zebrafish embryo. Curr Biol. 2003;13:1169–81. doi: 10.1016/s0960-9822(03)00461-5. [DOI] [PubMed] [Google Scholar]
- 39.Olson EN, Sternberg E, Hu JS, Spizz G, Wilcox C. Regulation of myogenic differentiation by type beta transforming growth factor. J Cell Biol. 1986;103:1799–805. doi: 10.1083/jcb.103.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brennan TJ, Edmondson DG, Li L, Olson EN. Transforming growth factor beta represses the actions of myogenin through a mechanism independent of DNA binding. Proc Natl Acad Sci USA. 1991;88:3822–6. doi: 10.1073/pnas.88.9.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 42.Amthor H, Huang R, McKinnell I, Christ B, Kambadur R, Sharma M, Patel K. The regulation and action of myostatin as a negative regulator of muscle development during avian embryogenesis. Dev Biol. 2002;251:241–57. doi: 10.1006/dbio.2002.0812. [DOI] [PubMed] [Google Scholar]
- 43.Liu D, Black BL, Derynck R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001;15:2950–66. doi: 10.1101/gad.925901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu D, Kang JS, Derynck R. TGF-beta-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J. 2004;23:1557–66. doi: 10.1038/sj.emboj.7600179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kollias HD, Perry RL, Miyake T, Aziz A, McDermott JC. Smad7 promotes and enhances skeletal muscle differentiation. Mol Cell Biol. 2006;26:6248–60. doi: 10.1128/MCB.00384-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Q, Zhang Y, Yang G, Chen X, Zhang Y, Cao G, Wang J, Sun Y, Zhang P, Fan M, Shao N, Yang X. Transforming growth factor-beta-regulated miR-24 promotes skeletal muscle differentiation. Nucleic Acids Res. 2008;36:2690–9. doi: 10.1093/nar/gkn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006;103:18255–60. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen SY, Wang Y, Telen MJ, Chi JT. The genomic analysis of erythrocyte microRNA expression in sickle cell diseases. PLoS ONE. 2008;3:e2360. doi: 10.1371/journal.pone.0002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, Benjamin H, Kushnir M, Cholakh H, Melamed N, Bentwich Z, Hod M, Goren Y, Chajut A. Serum microRNAs are promising novel biomarkers. PLoS ONE. 2008;3:e3148. doi: 10.1371/journal.pone.0003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513–8. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dent KM, Dunn DM, Von Niederhausern AC, Aoyagi AT, Kerr L, Bromberg MB, Hart KJ, Tuohy T, White S, Den Dunnen JT, Weiss RB, Flanigan KM. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet. 2005;134:295–8. doi: 10.1002/ajmg.a.30617. [DOI] [PubMed] [Google Scholar]
- 52.Jay C, Nemunaitis J, Chen P, Fulgham P, Tong AW. miRNA profiling for diagnosis and prognosis of human cancer. DNA Cell Biol. 2007;26:293–300. doi: 10.1089/dna.2006.0554. [DOI] [PubMed] [Google Scholar]
- 53.Szafranska AE, Davison TS, John J, Cannon T, Sipos B, Maghnouj A, Labourier E, Hahn SA. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26:4442–52. doi: 10.1038/sj.onc.1210228. [DOI] [PubMed] [Google Scholar]
- 54.Hu Z, Chen J, Tian T, Zhou X, Gu H, Xu L, Zeng Y, Miao R, Jin G, Ma H, Chen Y, Shen H. Genetic variants of miRNA sequences and non-small cell lung cancer survival. J Clin Invest. 2008;118:2600–8. doi: 10.1172/JCI34934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–33. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agrawal PB, Greenleaf RS, Tomczak KK, et al. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am J Hum Genet. 2007;80:162–7. doi: 10.1086/510402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vester B, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233–41. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 58.Varallyay E, Burgyan J, Havelda Z. MicroRNA detection by northern blotting using locked nucleic acid probes. Nat Protoc. 2008;3:190–6. doi: 10.1038/nprot.2007.528. [DOI] [PubMed] [Google Scholar]
- 59.Castoldi M, Schmidt S, Benes V, Hentze MW, Muckenthaler MU. miChip: an arraybased method for microRNA expression profiling using locked nucleic acid capture probes. Nat Protoc. 2008;3:321–9. doi: 10.1038/nprot.2008.4. [DOI] [PubMed] [Google Scholar]
- 60.Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjärn M, Hansen HF, Berger U, Gullans S, Kearney P, Sarnow P, Straarup EM, Kauppinen S. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–9. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 61.Yablonka-Reuveni Z, Anderson JE. Satellite cells from dystrophic (mdx) mice display accelerated differentiation in primary cultures and in isolated myofibers. Dev Dyn. 2006;235:203–12. doi: 10.1002/dvdy.20602. [DOI] [PubMed] [Google Scholar]
- 62.Car A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW, 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–8. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 63.Rodino-Klapac LR, Lee JS, Mulligan RC, Clark KR, Mendell JR. Lack of toxicity of alpha-sarcoglycan overexpression supports clinical gene transfer trial in LGMD2D. Neurology. 2008;71:240–7. doi: 10.1212/01.wnl.0000306309.85301.e2. [DOI] [PubMed] [Google Scholar]
- 64.Pacak CA, Walter GA, Gaidosh G, Bryant N, Lewis MA, Germain S, Mah CS, Campbell KP, Byrne BJ. Long-term skeletal muscle protection after gene transfer in a mouse model of LGMD-2D. Mol Ther. 2007;15:1775–81. doi: 10.1038/sj.mt.6300246. [DOI] [PubMed] [Google Scholar]
- 65.Sonkoly E, Stahle M, Pivarcsi A. MicroRNAs and immunity: novel players in the regulation of normal immune function and inflammation. Semin Cancer Biol. 2008;18:131–40. doi: 10.1016/j.semcancer.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 66.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–61. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 67.Zhao Y, Ransom JF, Li A, Vedantham V, Von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell. 2007;129:303–17. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 68.Ivey KN, Muth A, Arnold J, King FW, Yeh RF, Fish JE, Hsiao EC, Schwartz RJ, Conklin BR, Bernstein HS, Srivastava D. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell. 2008;2:219–29. doi: 10.1016/j.stem.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kunkel LM, Bachrach E, Bennett RR, Guyon J, Steffen L. Diagnosis and cell-based therapy for Duchenne muscular dystrophy in humans, mice, and zebrafish. J Hum Genet. 2006;51:397–406. doi: 10.1007/s10038-006-0374-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–7. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- 71.Sampaolesi M, Blot S, D'Antona G, Granger N, Tonlorenzi R, Innocenzi A, Mognol P, Thibaud JL, Galvez BG, Barthélémy I, Perani L, Mantero S, Guttinger M, Pansarasa O, Rinaldi C, Cusella De Angelis MG, Torrente Y, Bordignon C, Bottinelli R, Cossu G. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2006;444:574–9. doi: 10.1038/nature05282. [DOI] [PubMed] [Google Scholar]
- 72.Cerletti M, Jurga S, Witczak CA, Hirshman MF, Shadrach JL, Goodyear LJ, Wagers AJ. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell. 2008;134:37–47. doi: 10.1016/j.cell.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O'Rourke JR, Georges SA, Seay HR, Tapscott SJ, McManus MT, Goldhamer DJ, Swanson MS, Harfe BD. Essential role for Dicer during skeletal muscle development. Dev Biol. 2007;311:359–68. doi: 10.1016/j.ydbio.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Felli N, Fontana L, Pelosi E, Botta R, Bonci D, Facchiano F, Liuzzi F, Lulli V, Morsilli O, Santoro S, Valtieri M, Calin GA, Liu CG, Sorrentino A, Croce CM, Peschle C. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci USA. 2005;102:18081–6. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafrè SA, Farace MG, Agami R. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 77.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bogdanovich S, Perkins KJ, Krag TO, Khurana TS. Therapeutics for Duchenne muscular dystrophy: current approaches and future directions. J Mol Med. 2004;82:102–15. doi: 10.1007/s00109-003-0484-1. [DOI] [PubMed] [Google Scholar]
- 79.Benchaouir R, Meregalli M, Farini A, D'Antona G, Belicchi M, Goyenvalle A, Battistelli M, Bresolin N, Bottinelli R, Garcia L, Torrente Y. Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell. 2007;1:646–57. doi: 10.1016/j.stem.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 80.Péult B, Rudnicki M, Torrente Y, Cossu G, Tremblay JP, Partridge T, Gussoni E, Kunkel LM, Huard J. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007;15:867–77. doi: 10.1038/mt.sj.6300145. [DOI] [PubMed] [Google Scholar]