

Abstract
MVM NS2 is essential for viral DNA amplification, but its mechanism of action is unknown. A classification scheme for autonomous parvovirus-associated replication (APAR) center development, based on NS1 distribution, was used to characterize abnormal APAR body maturation in NS2null mutant infections, and their organization examined for defects in host protein recruitment. Since acquisition of known replication factors appeared normal, we looked for differences in invoked DNA damage responses. We observed widespread association of H2AX/MDC1 damage response foci with viral replication centers, and sequestration and complex hyperphosphorylation of RPA32, which occurred in wildtype and mutant infections. Quantifying these responses by western transfer indicated that both wildtype and NS2 mutant MVM elicited ATM activation, while phosphorylation of ATR, already basally activated in asynchronous A9 cells, was downregulated. We conclude that MVM infection invokes multiple damage responses that influence the APAR environment, but that NS2 does not modify the recruitment of cellular proteins.
Keywords: Parvovirus, Minute virus of Mice (MVM), viral replication centers, DNA damage responses
Introduction
Minute Virus of Mice (MVM), a member of the genus Parvovirus, has a linear single-stranded negative-sense DNA genome of ~5kb, with small imperfect palindromes at each end that fold into hairpin telomeres. Since incoming virions do not provide the cell with duplex transcription templates, viral gene expression is not activated upon nuclear entry, but instead the virus appears to remain silent until its host cell enters S-phase under its own cell cycle control (reviewed in Cotmore and Tattersall, 2006a, 2007). At this time, the hairpin structure at the viral 3’ (left-end) telomere primes complementary strand synthesis by DNA polymerase-delta (Bashir et al., 2000; Christensen and Tattersall, 2002), creating a duplex template that can support rapid synthesis of mRNAs encoding the first viral proteins, NS1 and NS2 (Clemens and Pintel, 1988). Of these, the multifunctional 83kDa NS1 polypeptide is absolutely required for viral replication in all cell types, since it carries essential site-specific duplex-DNA binding, single-strand endonuclease, and helicase elements required for genome amplification and progeny excision (reviewed in Cotmore and Tattersall, 2006a, 2006b; Nuesch, 2006). By comparison, the functions of the 25kDa NS2 polypeptides are poorly understood.
NS2 is the major viral protein synthesized early in S-phase, but its accumulation relative to NS1 then declines, in part due to its relatively short half-life (Cotmore and Tattersall, 1990; Miller and Pintel, 2001). Although not required for infection in some transformed human cell lines, it is absolutely required in cells from the virus’ natural murine host (Cater et al., 1992; Naeger et al., 1990). MVM mutants that fail to accumulate NS2 show slightly delayed expression of the viral NS1 protein in mouse cells, which becomes detectable in western blots 6 hours into S-phase rather than after 3 hours as found for the wildtype virus, and NS1 levels remain impaired relative to wildtype throughout the course of infection. NS2null mutants also show an early and extreme defect in viral DNA amplification (Cater et al., 1992; Cotmore et al., 1997; Naeger et al., 1990; Ruiz et al., 2006). The molecular basis for this block is unknown, but there is no evidence for direct involvement of NS2 in genome replication in vitro, and the block must presumably occur after complementary strand synthesis, since this is required to support the observed NS1 expression. Mutants that express low levels of NS2 also exhibit a late defect in A9 cells, which manifests as a reduction in progeny virus production resulting from impaired capsid assembly (Cotmore et al., 1997; Ruiz et al., 2006). This defect appears linked to the ability of NS2 to bind the nuclear export factor Crm1 at high affinity (Bodendorf et al., 1999; Choi et al., 2005; Eichwald et al., 2002; Miller and Pintel, 2002). NS2 is also known to bind several members of the 14-3-3 family members, but the role of these interactions has yet to be determined (Brockhaus et al., 1996).
During infection, replicating MVM DNA is first detected in nuclear sub-domains called autonomous parvovirus-associated replication (APAR) bodies, which contain most of the available NS1 and a variety of cellular proteins involved in viral DNA amplification (Bashir et al., 2001; Cziepluch et al., 2000). These foci are initially separate and distinct from previously-described subnuclear compartments such as promyelocytic leukemia (PML) oncogenic domains (PODs), Cajal bodies, or speckled domains (Cziepluch et al., 2000; Young et al., 2002), although they subsequently interact and sometimes merge with these structures (Ihalainen et al., 2007; Young et al., 2002; Young et al., 2005), as accumulating NS1 and viral DNA expand to occupy most of the enlarged nucleus, leaving the cellular chromatin compacted and marginated (Ihalainen et al., 2009). Thus, there appears to be initial separation of nuclear domains, and differential segregation of cellular proteins into the APAR compartment, which provides a window of time when is possible to explore the cellular environment required to support viral DNA amplification by immunofluorescent staining and microscopy. Using a two-step (isoleucine deprivation/aphidicolin block) synchronization procedure, in which virus is allowed to penetrate into the cell nucleus prior to S-phase release, we have previously characterized sequential changes in the rates of cellular and viral DNA synthesis with time in S-phase (Cotmore and Tattersall, 1987). The current experiments were initially designed to correlate these phases in DNA metabolism with progressive changes in intranuclear NS1 distribution, and thus establish a developmental sequence for productive infection by the wildtype virus that reflects time in S-phase.
A difference in the pattern of NS1 distribution was previously observed in A9 cells infected with MVM wildtype and NS2null mutants (Ruiz, et al., 2006). Specifically, while comparable numbers of APAR-like structures were detected in both infections, these structures were consistently smaller in NS2null infections, perhaps suggesting that NS2 expression might modulate the organization and/or development of these viral replication centers. We used the classification scheme for NS1 progression to characterize and quantify the defect in APAR body maturation during NS2null infections, and then explored their organization, looking for defects in the recruitment of specific constituent proteins in the absence of NS2.
The speed and severity of the DNA replication block seen for NS2null mutants in A9 cells suggested either a profound inability to recruit compatible replication machinery, or perhaps the induction of an antiviral state, such as might accompany activation of a DNA damage response (DDR). MVM replication mechanisms have been studied extensively in vitro (reviewed in Cotmore and Tattersall, 2006a, 2006b), but such studies have relied on cellular proteins derived from uninfected cells or generated from expression vectors, so that the potential effects of induced damage responses have not been explored. However, the alien structure of incoming, uncoated, viral genomes could potentially evoke such responses, and viral DNA is replicated via a unidirectional, leading-strand specific, displacement mechanism called “rolling hairpin replication” that displaces long stretches of single-stranded DNA and generates a series of blunt-ended duplex intermediates, either of which would be expected to elicit DNA repair. Since this quasi-circular mechanism relies predominantly on cellular synthetic machinery, aided and orchestrated by the viral NS1 protein, it would be potentially susceptible to such changes in replication control, and the virus would likely have evolved to combat or suborn whatever cellular responses it induces. There is a growing body of evidence that DNA viruses typically do evoke, and then counter, cellular damage responses, although specific details vary greatly between viruses (reviewed in Lilley et al., 2010; Weitzman et al, 2010). Accordingly, we looked for evidence of induced damage responses, assessed whether activated proteins were recruited to the viral replication compartment, and determined if NS2 expression influenced this process.
Results and Discussion
Distinct patterns of NS1 organization emerge with time in S-phase nuclei
Our first study was designed to correlate phases in DNA metabolism previously observed in MVM infected cells during S-phase, with changes in nuclear NS1 morphology. Accordingly, we used a two-step cell synchronization procedure that allows virus to enter the cell and penetrate to the cell nucleus before release of the DNA replication block. Using this same procedure, we had previously shown that DNA synthesis rates in MVM infected A9 cells parallel those in uninfected cells for the first two hours in S phase, but then progressively drop to almost pre-release levels over the next four hours. Viral NS protein expression becomes apparent by around 3 hours post-release under these conditions, but a new wave of DNA synthesis, reflecting the amplification of duplex viral DNA, is not observed until 5–6 hours into S, peaks at 10 hours, and then remains relatively steady following the onset of progeny virus release (Cotmore and Tattersall, 1987). In the current experiments NS1 expression was monitored by indirect immuno-fluorescence and confocal microscopy in cells infected with wild-type MVMp at a multiplicity of 3,000 genome-containing particles per cell (g/cell), and fixed at 3, 6, 12 and 24 hours after release into S-phase. During this period the pattern of nuclear NS1 staining progressed through characteristic stages, allowing us to derive a classification scheme that tracked its redistribution with time in a productive infection. Fig 1 shows representative cells belonging to each class.
Figure 1. Changes in the nuclear morphology of NS1 with time in S-phase.
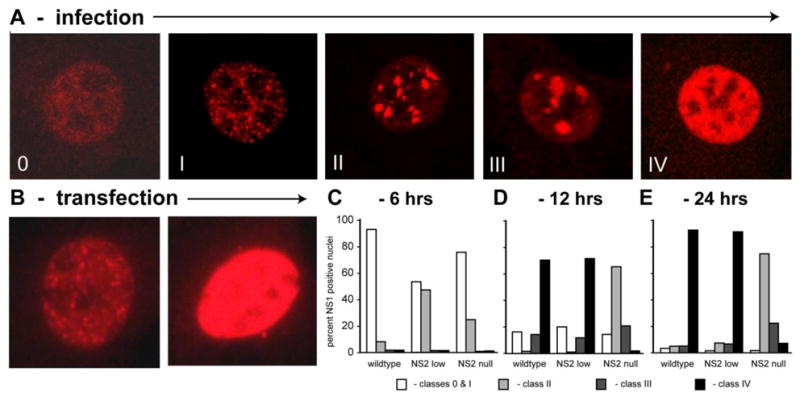
A. A classification scheme, with Classes 0 – IV, was developed to describe the progressive changes in NS1 morphology that emerge with time after synchronized A9 cells infected with wild type MVMp are released into S-phase. Overall classes 0 and I were first observed 3 h into S-phase, class II at 6 h, and classes III and IV at 12 hours, although the number of cells exhibiting the later class morphologies progressively increased with time. B. When expressed by transfection, NS1 adopts the Class 0–I, or Class IV distribution, with no intermediate forms. C–E. Kinetic analysis of NS1 redistribution in A9 cells infected with wildtype or NS2 mutant viruses at 6 h (C), 12 h (D), and 24 h (E) after release into S-phase.
At the 3h time point, NS1 was most commonly observed as a cloud of tiny puncta spread throughout the nucleus, but absent from nucleoli, as shown in the first panel of Fig 1A (Class 0). However, even at this early time, in some cells individual foci were larger and stained more intensely, creating a series of distinct speckles, which we termed developmental Class I foci (Fig 1A). By 6 hours into S-phase, the intensity of NS1 staining in the nuclei increased, and it became sequestered in a limited number of progressively larger distinct foci (Class II), although such nuclei also contained a population of smaller puncta that resembled those seen in the earlier Class I nuclei. This Class II distribution matured to a pattern containing fewer and larger, but still discrete, NS1 bodies (Class III), while at later times a staining pattern in which NS1 occupied the entire nucleoplasm predominated (Class IV). Classes II and III in this scheme correspond to the previously characterized APAR foci. This time frame indicates that APAR bodies are relatively short-lived, emerging as the cell embarks on the major phase of viral DNA amplification at around 6 hours into S-phase, but are lost within a few hours during a productive infection, as the nucleus rapidly fills with viral products. A very similar developmental sequence was observed in NLFK cells infected with canine parvovirus (Ihalainen et al, 2007), suggesting that this rapid progression is characteristic for members of the genus Parvovirus. In contrast, when NS1 was expressed by transfection, all NS1-positive nuclei belonged to either Class I, showing dozens of distinct small speckles, or Class IV, where the entire nucleoplasm was filled with NS1 (Fig 1B), but the intermediate APAR clustering patterns were absent. Thus, maturation through APAR-like intermediates only occurs during infection, and correlates in time with the onset of rapid viral duplex DNA expansion.
APAR centers are established in NS2null infections, but fail to mature
To document the effects of NS2 on this sequence of events, we carried out a kinetic analysis in synchronized cells, using wildtype and two NS2 mutant MVMp viruses, as shown in Fig 1C, D & E. One of the mutants expressed no stable NS2 due to a mutation at residue 86 (NS2-am, NS2null) that effectively prevents expression and/or accumulation of the truncated product (Cotmore et al., 1997; Gersappe et al., 1999; Naeger et al, 1992; Ruiz et al., 2006), while the other expressed approximately one sixth of the total wild-type NS2 level (NS2low). Since NS1 expression cannot be detected until 6 hours post-release in cells infected with NS2null viruses (Ruiz et al., 2006), cells were fixed at 6, 12 and 24 hours after release from aphidicolin, stained for NS1, and blind-scored according to the classes identified in Fig 1A. At 6 hours into S-phase NS1-positive cells predominantly exhibited the Class I distribution pattern, although some class II nuclei were apparent in all infections (Fig 1C). However, at later times two distinct developmental patterns emerged. In cells infected with either the wildtype or NS2low viruses, the NS1-staining pattern progressed to the Class IV stage in almost 80% of infected cells by 12 hours post-release (Fig 1D). In contrast, a similar percentage of cells infected with NS2null viruses showed evidence of NS1 expression, but staining generally failed to progress beyond the Class II stage by 12 hours post-release, and this defect persisted through 24 hours after release (Fig 1E). This indicates that in NS2null infections NS1 foci are established and develop normally during early S-phase, but the NS2null phenotype rapidly emerges at around 6 hours post-release, with the onset of viral DNA amplification. It also suggests that APAR progression is not merely retarded, but is effectively blocked in all but a small percentage of cells infected with NS2null viruses, even though cells with class II/III nuclei have been reported to survive for several days in culture (Young et al., 2005). We conclude that the presence of NS2 had a major impact on APAR development in MVM-infected cells, although only relatively low levels of the protein are required since even one sixth of the wildtype concentration, expressed from the NS2low mutant, was compatible with normal maturation and progression. This data highlights the possibility that the APAR defect, and the failure of NS2null mutants to replicate viral DNA effectively, may reflect critical abnormalities in the organization of the early viral replication compartment.
NS2 is not required for recruitment of replication factors to APAR foci
To explore whether the accumulation of replication factors known to be recruited to wildtype APAR bodies was dependent upon NS2, asynchronous populations of A9 cells were infected with wildtype and NS2null virions (3,000 g/cell) under single round infection conditions, fixed and processed for immunofluorescence 24 hours post infection using antibodies directed against a range of known APAR body constituents. Cellular replication factors known to be essential for MVM replication, exemplified here by RPA and PCNA, co-localized with NS1 in APAR bodies as previously reported (Bashir et al., 2001; Cziepluch et al., 2000) in cells infected with both wildtype and NS2null viruses, as shown in Fig 2. The lagging strand DNA polymerase pol-α is also known to be recruited to APAR foci in wild-type infections, even though this enzyme is not required for MVM DNA synthesis in vitro (Bashir et al., 2001; Christensen and Tattersall, 2002). Recruitment of this seemingly irrelevant factor suggests that parvoviruses may usurp pre-existing cellular replication complexes, rather than accumulate individual components (Bashir, et al., 2001), as discussed later. However, as shown in Fig 2, pol-α was detected in APAR bodies in both wildtype and NS2null infections. Normally pol-α exists as a complex with primase, a DNA-dependent RNA polymerase, but published data suggests that this interaction may be disrupted in MVM infected cells, leading to the accumulation of a primase-free form of pol-α, which could potentially impede bidirectional cellular DNA synthesis and thus explain its cessation following viral infection (Gupta and Faust, 1993; Ho et al., 1989). However, in the current study the primase component was readily detected and similarly sequestered in APAR foci in both wildtype and NS2null infections, suggesting that this holo-enzyme is recruited in an NS2-independent fashion. Likewise, cyclin A is recruited into APAR foci in both wildtype and NS2null infections (Fig 2). Cyclin A is essential for the duplex conversion of single-stranded MVM genomes by cellular replication extracts in vitro, via a mechanism that requires its associated cyclin dependent kinase activity (Bashir et al., PNAS 2000). Its precise role(s) in the APAR compartment remains uncertain, but it has been suggested that this sequestration might effectively render it inaccessible for cdk1 activation in G2, thus preventing cell cycle progression (Bashir et al., 2001). If so, the constrained size of APAR foci in NS2null infections could render cyclin A sequestration less efficient, even though it is effectively recruited. However, the control of cell cycle progression during MVM infection is likely complex, since expression of NS1 alone has been shown to impede cellular DNA replication in some cell types, leading to arrest in S and G2 phases of the cell cycle (Op de Beeck et al., 1997,Op de Beeck et al., 2001).
Figure 2.

Co-localization of cellular replication proteins with NS1 in APAR foci during infection with wild type and NS2 mutant MVM
Overall this analysis shows that recruitment of a range of normal replication factors into the APAR compartment proceeds similarly in wildtype and NS2null infections, arguing against the possibility that the defect underlying APAR maturation and the DNA replication block seen in NS2null infections, results from failure in the organization of nascent APAR compartments.
RPA 32kDa subunit is hyper-phosphorylated during MVM infection
Finding no differences in the qualitative re-localization of known cellular replication factors in wildtype and NS2null infections, we extended this analysis to include modified proteins that might be induced in response to DNA damage (reviewed in Huen and Chen, 2010; Jackson, 2009). Displacement synthesis from the unidirectional MVM fork is totally dependent upon the availability of high concentrations of RPA (Christensen and Tattersall, 2002), so that damage-mediated phosphorylation of this complex might severely impact its synthesis. RPA is a heterotrimeric complex, composed of RPA70, RPA32 and RPA14 subunits. The RPA32 subunit is phosphorylated at specific sites by cyclin-dependent kinases when cells enter S-phase, but there are also seven residues in its extreme N-terminus that can become phosphorylated in response to DNA damage (Nuss et al., 2005; Olson et al., 2006).
Accordingly, we asked if the RPA32 subunit in wildtype and NS2null infections carried DDR-induced PI3K-like kinase concensus and non-consensus phosphorylations, and if so whether such complexes were still recruited to APAR foci. As shown in the top two panels of Fig 3A, indirect immunofluorescence with phospho-specific antibodies revealed accumulation of RPA32 phosphorylated at the non-consensus serine 4 and 8 sites and at the consensus serine 33 site in both wildtype and NS2null infected cells, which substantially colocalized with NS1 in APAR foci. This pattern of induction was confirmed by Western analysis, discussed later, and supported the suggestion that uncoated viral genomes and/or viral replication could elicit response from the cell’s damage surveillance and repair machinery. This prompted us to broaden our investigation to look for additional evidence of damage responses.
Figure 3.

Co-localization of cellular DNA damage response proteins with NS1 in cells infected with wildtype and NS2 mutant viruses
γH2AX/MDC1 DNA damage foci associate with APAR bodies
A wave of histone H2AX phosphorylation at Ser-139, generating a product referred to as γH2AX, can spread very rapidly for many thousands of bases on either side of double-strand breaks in cellular DNA, creating DNA damage foci (DDF) that are large enough to be observed by immunofluorescence microscopy (Fernandez-Capetollo et al., 2004; Rogaku et al., 1998). As seen in the third panel of Fig 3A, and at higher magnification in Fig 3B, γH2AX foci developed in close association with the APAR compartment during both wildtype and NS2null infections, at first seeming to surround, rather than colocalize with, the accumulated NS1. However DDF and APAR foci then merged, and γH2AX-staining lost its previous punctate appearance as it accumulated to high concentration in the developing APAR compartment. Ultimately, in cells with a class IV pattern of NS1 staining, viral replication was associated with major overall intensification of the γH2AX signal, as seen, for example, in the two cells at the top of the wildtype sample in the third panel of Fig 3A, but co-localization even extended to some of the very smallest, class I, NS1 foci seen in the lower cell in this panel, where overall signal amplification appeared as yet minimal. We conclude that γH2AX damage foci are highly enriched in the immediate vicinity of developing APAR bodies in both wildtype and NS2null mutant infections, and that the modified histone becomes assimilated into the APAR compartment where it effectively co-localizes with accumulated NS1. It is hard to envisage how γH2AX foci of this size could be organized without a DNA core, but individual viral genomes in the process of being trafficked to the APAR compartment would not be distinguishable at this magnification. Thus, it seems possible that these γH2AX foci became organized on cellular DNA, which in turn suggests that damaged host DNA may be specifically relocated to developing MVM replication centers and/or that the virus induces damage responses in surrounding normal cellular DNA, and is then able to organize transfer of the modified histone into the MVM compartment. Both scenarios suggest close juxtaposition of APAR foci and cellular DNA in this early phase of amplification, allowing directional transfer of components.
During cellular damage responses H2AX phosphorylation creates binding sites that recruit Mediator of DNA Damage Checkpoint 1 (MDC1), a multi-domain molecular platform that, in turn, binds and stably accumulates a variety of effector and transducer molecules. These serve multiple essential roles in relaxing chromatin structure, and in amplifying and propagating various arms of the damage response (reviewed in Huen and Chen., 2010). Thus MDC1 plays a central role in the development of cellular γH2AX repair foci. DDR foci containing MDC1 were also commonly observed clustered around developing class II APAR bodies (as in the NS2null sample shown in Fig 3A), but, unlike γH2AX, MDC1 was not conspicuously sequestered inside the APAR compartment in either wildtype or NS2null infections. Instead, late stage (NS1 type III–IV) nuclei contained very few discrete MDC1 foci, but did stain diffusely for this protein, and Western analysis (detailed below) suggested that the overall MDC1 concentration in the cell did not change appreciably within the first 24 hours of infection.
Thus in A9 cells γH2AX colocalized with even the smallest MVM APAR foci in both wildtype and NS2null infections, and then remained associated with the developing viral replication compartment, whereas MDC1 foci segregated around type II viral bodies but did not conspicuously accumulate within the APAR compartment. This contrasting sequence of events was even more apparent in mouse embryonic fibroblasts (MEFs), as illustrated in Fig 4A. In these wildtype MEFs large clusters of MDC1 foci commonly surrounded type II/III NS1 foci, as seen at higher magnification in the bottom row of panels, whereas cells with a type IV NS1 distribution (as seen in the second row) showed no sign of MDC1 accumulation, in sharp contrast to the major overall γH2AX accumulation seen in type IV nuclei (upper panel). If this reflects specific inclusion of γH2AX within the viral replication compartment, but the exclusion of MDC1, such disruption may imply a sorting mechanism that could potentially limit aspects of the normal damage cascade. As a control, we show, in Fig 4B, that APAR bodies are generated during MVM infection of H2AX knock-out MEFs, so that APAR center development is not dependent upon factors delivered via recruitment of these damage foci. However, as would be predicted, such cells failed to show induction or APAR body recruitment of MDC1-containing foci. Whether or not sequestration of γH2AX, or associated damage response proteins, in the viciniy of replicating viral DNA is beneficial or detrimental to virus replication remains to be explored. Fig 4C serves as a control for 4B, and documents that MDC1 staining is distributed throughout the nucleus of these wildtype and H2AX−/− MEFs whereas, as would be expected, H2AX staining is absent from H2AX−/− nuclei. Overall, this data indicates that at the start of the major phase of viral DNA amplification, cellular γH2AX/MDC1 DNA damage foci are specifically induced and/or recruited to the vicinity of the viral replication compartment, where they lose their focal appearance, individual components appear to dissociate, and at least some components become sequestered in close apposition to replicating viral DNA.
Figure 4. Accumulation of γH2AX and MDC1 DNA damage response foci around APAR bodies in mouse embryonic fibroblasts.

A, B.WT and H2AX MEFs were stained as indicated. Stippled vertical bars in the middle panel of section A and in section B indicate the juxtaposition of two separate images. The lower panel in section A shows NS1 and MDC1 staining at a higher magnification. Panel C confirms the loss of H2AX expression in the H2AX-KO MEFs by differential staining with antibodies against H2AX and MDC1.
Quantitative assessment of damage responses during MVM infection
In order to assess which damage response pathways were activated during MVM infection, and to ask whether they were conspicuously implicated in the NS2null defect, we probed Western transfers with a series of diagnostic antibodies, and quantified the signals using indirect immunofluorescence. Cellular responses to DNA double-strand breaks or collapsed replication forks involve a complex series of signal transduction cascades in which lesions are detected by sensor proteins that in turn activate members of the phosphoinositide 3 kinase-like protein kinase (PI3KK) family, specifically ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3-related) and DNA-PK (DNA-dependent protein kinase). Which particular kinase is activated depends on several factors, predominantly the nature of the DNA lesion and the stage of the cell cycle. Thus, double-strand breaks in S-phase typically activate ATM, while single-stranded DNA, often associated with collapsed cellular replication forks, leads to activation of ATR. However, these cascades are complex and overlapping, with certain transducers and effectors involved in multiple pathways. As a positive control for these experiments uninfected A9 cells were exposed to the topoisomerase I inhibitor camptothecin. We also used a specific inhibitor af ATM activation (2-morpholin-4-yl-6-thianthren-1-yl-pyran-4-one, Kudos 55933) that is at least 100-fold more active against ATM than against related kinases (reviewed in O’Connor et al., 2007), to probe the complexity of virus induced repsonses. This inhibitor was first screened for cytotoxicity in A9 cells at drug concentrations from 2.5–10μM, using a “Live/Dead” assayed based on differential calcein AM/ethidium homodimer staining, and showed little evidence of inducing cell death over the time course used for the Western assays (Fig 5A). However, parvoviruses can only initiate infection in cells that enter S-phase under normal cell cycle control. Thus, inhibitors that do not kill the cell but do impede cell cycle progression can be erroneously scored as inhibiting specific aspects of viral infection. To assess the effect of Kudos 55933 on the rate of entry into S-phase, uninfected A9 cells were incubated under Western assay conditions, and pulsed for 1 hour with the nucleotide analogue Edu immediately prior to fixation. Incorporated Edu was visualized using “Click-it” chemistry and fluorescence microscopy. As seen in Fig 5B, the percentage of cells actively engaged in DNA synthesis remained constant for drug concentrations up to 5μM, but was suppressed by approximately 40 percent at higher doses. This assay was performed multiple times, and consistently gave results comparable to those illustrated in Fig 5B, indicating that in uninfected asynchronous A9 cells exposure to concentrations of inhibitor in excess of 5μM for 24 hours slowed cell cycle progression. We therefore selected 5μM as our standard working concentration.
Figure 5. Infection with wildtype and NS2 mutant MVM induces similar DNA damage responses in asynchronous A9 cells.

A. Effects of Kudos 55933 on A9 viability
Uninfected A9 cells were screened for cell viability in the presence of variable doses of Kudos 55933 using a fluorescence based Live/dead cell assay (Biotium).
B. Effects of Kudos 55933 on cell entry into S-phase
Uninfected A9 cells were assessed, by “Click-It” chemistry and fluorescence microscopy, for their ability to enter S-phase and incorporate EdU after exposure to variable doses of Kudos 55933.
C. Western quantitation of DNA damage response following MVM infection
Lanes 1 and 2 contain extract from camptothecin (camp)-treated cells; lanes 3 and 4 from uninfected (NCC) cells; lanes 5 and 6 from cells infected with wild type MVM; lanes 7 and 8 from NS2-am infected cells; and lanes 9 and 10 from NS2-sam infected cells. Extracts in lanes, 2, 4, 6, 8 and 10 also received the ATM-inhibitor, Kudos 55933 (ATMi) at 5μM. The percent of NS1 positive cells in each infection (lanes 5–10) is shown at the bottom of the figure, with standard deviations. Band intensities, plotted under certain panels, are expressed as fluorescence units × 105. Band intensities for upper and lower bands of RPA32 ser4/8 were quantified separately.
ATM kinase is activated in response to MVM infection
Asynchronous cells were infected under single cycle conditions, harvested at 24 hours post infection, and probed with antibodies directed against a variety of candidate factors. In the experiment illustrated in Fig 5C, cells were infected for 4 hours at 10,000 g/cell with wild-type MVMp and two related NS2null mutants, NS2−am and NS2−sam, the latter being the NS2−am mutant with an additional major splice acceptor site mutation (Cotmore et al., 1997). To assess infection initiation frequency, parallel samples were analysed for NS1 expression by indirect immunofluorescence microscopy. These results, tabulated at the bottom of Fig 5C, indicate that the infections were matched at the level of overt initiation, with around ~50% of cells in each sample showing conspicuous NS1 accumulation by 24 hours post infection. As expected, neither of the NS2null mutants gave evidence of NS2 accumulation, and both showed similar levels of NS1, which were approximately 3-fold lower than levels seen in the wildtype infection (Fig 5C, panels a and b). For each virus, exposure to the ATM-inhibitor impaired NS1 accumulation somewhat (by 17–35%), and generally had a more marked effect on NS2 expression in the wildtype infection (53% impaired). This pattern of inhibitor-mediated supression of viral protein accumulation was seen in multiple experiments, perhaps suggesting that ATM activation may promote the efficiency of early stages in viral replication that are required for the generation of transcription templates or some aspect of viral gene expression.
Probing extracts with an antibody specific for the ATM autophosphorylation site (generally referred to as ATM-p-ser1981, although this specifies the equivalent position in human ATM) suggested that ATM was activated in all infections (Fig 5C, panel c). By comparison with uninfected control cells phosphorylated ATM was found to be induced approximately 11-fold in the positive, camptothecin-treated, control sample and this was readily inhibited by Kudos. In contrast, both wild-type and mutant MVM infections induced ATM activation to somewhat lower levels (2.5 to 5.5-fold induction), but this could not be totally abrogated by the ATM-inhibitor (29–36% inhibition). As a control, we show that total ATM accumulation remained essentially similar in all samples (Fig 5C, panel d). Why virus-induced activation resists this inhibitor remains uncertain. We explored the effects of adding Kudos 55933 at higher doses (10uM), or 4 hours earlier (i.e. 4 hours prior to infection), but without quantitatively inhibiting ATM autophosphorylation (data not shown). We also considered the possibility that using lower virus inocula might render the response easier to dissect, but similar results were obtained with virus input multiplicities of 2,000 and 400 genomes per cell (resulting in NS1 positive initiation frequencies of around 30% and 10%, respectively, data not shown). However, at present we have been unable to consistently suppress ATM phosphorylation at its autocatalytic site during asynchronous infections using inhibitor doses that are compatible with normal entry into S-phase, and suggest that this might, at least in part, reflect an unusual mode of phosphorylation or longevity of the activated kinase, as recently demonstrated during SV40 infection of CV1 cells (Rohaly et al., 2010).
Overall, we conclude that both wildtype and NS2null viruses induce conspicuous phosphorylation at a site in the ATM molecule which critically regulates its oligomerization and thus its activity, and that this phosphorylation is at least partially inhibited by a highly specific ATM kinase inhibitor. Since viral DNA amplification in NS2null infections is minimal, and NS1 accumulation is reduced, it is remarkable that the mutant viruses activated ATM so effectively, again suggesting that early events in the viral life cycle are particularly prone to induce this damage response.
ATR phosphorylation at serine 428 is downregulated in response to MVM infection
In contrast to ATM, the ATR kinase appeared to be phosphorylated at its activating site (serine 428) in asynchronous A9 cells (Fig 5C, panel e), although accumulation of this form could still be enhanced by exposure to camptothecin. Thus, it appears that ATR activation may be somewhat atypical in these cells, but the pathway remains functional. Significantly, the anti-phospho-ATR antibody used in these blots gave diffuse staining in both uninfected and infected A9 cells, but in wild-type MEFs staining with the same antibody co-localized with NS1 in APAR bodies (data not shown), supporting the suggestion that ATR regulation may be somewhat atypical in A9 cells. When assessed by western blotting 24 hours postinfection in asynchronous A9 cells, both wild-type and NS2null MVM caused its downregulation by ~2-fold, while overall levels of total ATR remained relatively constant (Fig 5C, panel f). This downregulation was observed repeatedly, correlated inversely with virus input (data not shown), and was not influenced by the ATM inhibitor. Since NS1 expression was observed in ~50% of cells, and there was an ~2-fold reduction in ATR-phospho-ser428 accumulation, it is theoretically possible that infection totally suppressed ATR phosphorylation in the infected cell population.
H2AX phosphorylation is reduced in NS2null compared to wildtype infection
As expected from the confocal analysis, H2AX phosphorylation was markedly induced in all infected samples relative to uninfected control cells (Fig 5C, panel g), but this was strongest for the wildtype virus (~11-fold), while NS2null viruses only upregulated its phosphorylation ~6-fold. Camptothecin-treated positive control cells also showed H2AX activation (~6-fold), which was again reduced to background levels by addition of the ATM inhibitor. Although, Kudos 55933 only partially suppressed H2AX phosphorylation in infected cells, this did parallel levels of residual ATM-p-ser1981, suggesting that ATM was likely responsible for much of the γH2AX induction. In contrast, total levels of both H2AX and MDC1 remained similar in all samples (Fig 5C, panels h & n. Thus, overall we find clear evidence of virus induced γH2AX induction, and this response is somewhat tempered, but not either eliminated or potentiated, in infections with NS2null mutants.
RPA32 is hyper-phosphorylated at multiple residues in infected cells
Confocal analysis indicated that complexes containing RPA32 phosphorylated at the PI3kinase-like-kinase consensus (Ser-33) and nonconsensus (Ser-4 and Ser-8) sites were both relocated to viral replication centers. RPA32 phosphorylation at non-consensus sites (e.g. at residues 4, 8, 11, 12 and 13) has been shown to generate a slower migrating form of RPA32, perhaps indicating induction of a conformational change in the protein, while phosphorylation of the consensus Thr-21 (TQ) and Ser-33 (SQ) sites has only minor effects on protein migration (Olson et al., 2006). Thus, by western analysis we were able to detect both individual and complex phosphorylation events. In camptothecin-treated control samples, a single, retarded, band was detected with the RPA32 Ser-4/8 antibody (Fig 5C, panel k), which was substantially enhanced by addition of the ATM kinase inhibitor (2.4-fold), suggesting complex cross-talk between damage response pathways in this situation. In contrast, the RPA32 Ser-33 antibody detected two distinct species following exposure to camptothecin, a predominant lower band indicating the presence of molecules that did not have additional non-consensus modifications, and a slower band indicating the presence of multiple damage-induced phosphate additions (Fig 5C, panel l). In this case, the ATM inhibitor only enhanced overall Ser-33 accumulation by around 25 percent, but again drove it into the slower form, confirming that in this case ATM inhibition allowed the subunit to be phosphorylated at additional non-consensus sites.
While neither antibody reacted with extracts from uninfected cells, all virus infections led to potent RPA32 phosphorylation at both consensus and non-consensus sites. Moreover, because the Ser-33 specific antibody only detected species with retarded migration, these must carry additional non-consensus phosphorylations. Unlike the camptothecin control, in infected samples addition of Kudos 55933 somewhat inhibited RPA32 phosphorylation, likely suggesting a role for the ATM kinase in its accumulation. Strong induction of phosphorylation at non-consensus sites was also apparent when samples were probed with an antibody that recognized all forms of RPA32 (Figure 5C, panel m). In this case extracts from uninfected and camptothecin-treated cells exhibited relatively low levels of the upper band (1.5% and 1.8% of the total RPA32, respectively), as did a hydroxyurea-treated control (data not shown), whereas all MVM infected samples showed more extensive conversion to the slower form (4.1 – 7.4% of the total RPA32). Thus MVM infection induces unusually high levels of non-consensus RPA32 phosphorylation, but these levels were remarkably similar in wild-type and NS2null infections. This suggests that critical inducing events occur early in the viral life cycle, and that the presence of NS2 may have minimal effects on the extent and/or specificity of these modifications.
Overall the western data supports our confocal analysis, showing that by 24 hours postinfection MVM induces major DNA damage responses in its host that must markedly impact the available replication machinery, and which the virus must exploit and/or circumvent. However, these studies do not suggest a major role for NS2 in the induction or evasion of these modifications, or in the organization of the viral replication compartment. We originally chose to avoid synchronization procedures in the APAR organization studies, since aphidicolin alone would be expected to induce ATR responses, but such analyses will likely be required to help distinguish early and late effects during, and preceding, viral duplex amplification, and to avoid potential complexity from responses evoked in cells that initiate replication late in S-phase. Since we observed no dislocation in the organization of cellular proteins in the APAR compartment during NS2null infections, failure to routinely develop beyond class II in our NS1 classification scheme may indicate that such progression is predominantly driven by the onset of rapid duplex DNA amplification. If so, class I and II stages in this scheme represent an early phase in viral infection when essential factors are recruited and the composition of the replication compartment established. How H2AX damage foci, and presumably cellular DNA, become organized around developing APAR foci, and whether the retained histone serves any specific purpose, or must merely be removed from the cellular environment at this time, pose interesting questions for future analysis.
Materials and Methods
Cells and viruses
The prototype strain of MVMp and its mutant derivatives NS2−am and NS2−sam (previously called NS2-F86 and NS2-SA86, respectively, Cotmore et al., 1997), and NS2wam and NS2low (previously called NS2P-/Y+ and Plo/Y- respectively, Ruiz et al., 2006) were propagated in the SV40-transformed newborn human kidney cell line 324K. Viral stocks were purified by centrifugation through iodixanol (Optiprep, Axis-Shield, Oslo, Norway) step gradients, as previously described (D’Abramo et al, 2005) and quantified by Southern blot against known amounts of genome-length viral DNA. For experiments, specified virion multiplicities expressed as genome equivalents per cell (g/cell), were used to infect the mouse L cell derivative A9 ouabr11 or embryonic fibroblasts from normal or H2AX knock-out C57Bl/6 mice (MEFs, the gift of Dr Junjie Chen, generated as in Minter-Dykhouse et al., 2008) cultured in Dulbecco’s modified Eagle’s minimal medium (DMEM) containing 5% and 10% fetal bovine serum, for A9s and MEFs respectively. Although we did not titer the viral stocks used in this paper by assessing their plaque-forming ability, we estimate that, for wildtype MVMp, an input multiplicity of 10,000 genomes would correlate to between 2 and 5 PFU per cell, as determined on A9 cell monlayers. Experiments were conducted under asynchrononous conditions or, in A9s, after synchronization using an isoleucine deprivation/aphidocolin double block schedule described previously (Cotmore et al., 1997). For transfection experiments, NS1 was expressed under the control of a CMV promoter in pCDNA3.1 (Invitrogen; MVM nucleotides 240-2291), using Superfect (Qiagen).
Confocal imaging
For confocal microscopy, cells were seeded onto coverslips in 35mm dishes at a density of 2.9 × 105 cells per dish and synchronized, or not, as detailed in text. At the indicated times post infection, cells were fixed in 2.5% paraformaldehyde, permeabilized with PBS containing 1% NP-40 and processed for indirect immunofluorescence. NS1 was detected using CE10, a mouse monoclonal antibody directed against the MVM NS1 C-terminus (Yeung et al., 1991) or with a rabbit polyclonal serum directed against the C-terminal 16 amino acids of NS1 (Cotmore and Tattersall, 1988), and cells were counterstained with 4-6-diamidino-2-phenylindole (DAPI) and the following commercial antibodies: PCNA (FL-261, Santa Cruz); Pol-alpha (N19, Santa Cruz); DNA primase (p49 Biomeda); Cyclin A (H432, Santa Cruz); total RPA32, Phospho RPA32 S4/S8, and Phospho RPA32 S33, Bethyl Laboratories; γH2AX clone JBW 301 (sci 139, Millipore). Mouse monoclonal antibodies against total H2AX and MDC1, and a rabbit polyclonal antibody against MDC1 were the gift of Dr Junjie Chen. After immunostaining and mounting, specimens were analyzed by confocal laser microscopy (LSM 510) using a 63x objective, and were detected using the 488- and 568-nm excitation lines of an argon-krypton laser. Images were acquired and modified with the LSM 510 software version 2.8 and processed in Adobe Photoshop.
Western blots
A9 cells, seeded at 5 × 105 per 60-mm-diameter dish, were infected with 10,000 g/cell of wild type or mutant MVMp for 4 h at 37°C in DMEM containing 1% fetal bovine serum and 20 mM HEPES, pH 7.3, before the inoculum was removed and cultures transferred to fresh medium containing 0.04 units per ml of neuraminidase (Clostridium perfrigens, Type V, Sigma) to remove surface virus and prevent re-infection. Where specified, the ATM-inhibitor Kudos 55933 (Calbiochem) was added (at 5μM, as determined below) to cultures at the time of infection (from a 2000x stock in DMSO), and replenished following the medium change at 4 h post infection. As a positive control for DNA damage, uninfected cultures were exposed to Camptothecin at 1μM for 3h prior to harvest. At 24h post infection all cells were washed twice with ice cold PBS and lysed in 1X lysis buffer (2% SDS, 10% glycerol, 62.5 mM TrisHCI, pH 6.8) containing 1x Complete, EDTA-free, Protease inhibitor cocktail (Roche) and 1x PhosSTOP, phosphatase inhibitor cocktail (Roche). Cell lysates were collected, boiled for 10 min, aliquoted and stored at −80°C.
Protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Scientific) and 20 μg aliquots (with added dyes and 100mM DTT) loaded onto 4–15 % Mini-Protean TGX gels (Bio-Rad). After separation proteins were transferred to Hybond ECL membranes (GE healthcare). These were stained with Ponceau Red to confirm sample uniformity, and probed by indirect immunofluorescence using antibodies against the following: MVM NS1/2 N-terminal peptide (Cotmore and Tattersall, 1986); phospho-H2AX S139 (Cell Signaling); phospho-ATM S1981 (Cell Signaling); phospho-ATR S428 (Cell Signaling), total ATR (Abcam), total ATM (GeneTex), total RPA32, Phospho RPA32 S4/8 and Phospho RPA32 S33 (Bethyl Laboratories). Signal was developed using an ECLTM Plex Western blotting system (GE Healthcare), including ECL Plex goat-α-mouse IgG Cy3 and goat-α-rabbit IgG Cy5 secondary antibodies, and detected using a Typhoon Trio Variable Mode Imager (GE Healthcare). ImageQuant TL 7.0 (GE Healthcare) image Analysis Software was used for proteins quantitation.
Kudos 55933 toxicity and effects on cell entry into S-phase
Prior to use in the above experiment, the ATM inhibitor Kudos 55933 was screened for toxicity in A9ouabr11 cells using a Viability/Cytotoxicity Assay kit for Animal Live & Dead Cells (Biotium, Inc.). Uninfected cells were seeded onto glass spots on Teflon coated “spot-slides” (Cell-Line Associates, Inc., Newfield, NJ) at 20% confluence (3000 cells per spot), and exposed to a range of Kudos concentrations over the time course, and under the culture conditions, used for western assays. Viable cells were detected by their ability to convert non-fluorescent calcein AM to fluorescent calcein, producing intense green fluorescence (excitation/emission ~495nm/~515nm). Dead cells were identified by their ability to take up of ethidium homodimers through their damaged membranes, causing a 40-fold enhancement of red fluorescence in dead cells (excitation/emission ~ 530nm/~635nm). Green and red fluorescent cells were counted in six fields per Kudos dose, and the percent of live cells plotted, with standard deviations.
Kudos 55933 was also screened (at 2.5, 5, 7.5 and 10 μM) for possible effects on the rate at which A9 cells entered S-phase under the standard experimental regime used for western assays. Cells were seeded at 3000 cells per spot on microscope slides. At 23 h post infection 10 μM EdU (5-ethynyl-2’-deoxyuridine) was added, and cells were fixed with 2.5% paraformaldehyde 1 hour later and processed using Click-iT chemistry (Click-iT EdU Alexa Fluor High-Throughput Imaging Assay) according to the manufacturer’s instructions (Invitrogen). Percent EdU positive cells were counted in ten fields, and plotted with standard deviations.
Determining the initiation efficiency of viral infection
A9 cells were seeded at 20% confluence on Teflon coated spot-slides, infected and cultured under the experimental regime used for western assays, and fixed at 24 h post infection with 2.5% paraformaldehyde. Following permeabilization in 0.1% Triton X-100, cells were stained with the α-NS1 CE10 antibody, followed by AlexaFuor 594-conjugated goat anti-mouse IgG (Invitrogen) and Dapi. Images were acquired using standardized exposures on a Nikon OptiPhot epi-fluorescence microscope fitted with a Kodak digital camera driven by MDS 290 software. Ten fields were counted per sample, using Adobe Photoshop 6.0, and the percent of infected cells calculated by comparing NS1-positive nuclei with the total number of DAPI-stained cells.
Acknowledgments
We thank Drs. Junjie Chen and Michael Huen for providing antibodies and MEF cell lines, Dr. David Pintel for exchanging information on DDR induction by MVM prior to publication, and Tony D’Abramo, Jr., for his valuable assistance in many phases of this study. Aspects of this work relating to virally induced DNA damage responses, including RPA32 and H2AX phosphorylation, and the recruitment of modified RPA and γH2AX/MDC1 DNA damage foci to viral replication centers, was previously presented at the XIIth Parvovirus Workshop, Cordoba, Spain, June, 2008. This work was supported by Public Health Service grant numbers CA029303 and AI26109 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Bashir T, Horlein R, Rommelaere J, Willwand K. Cyclin A activates the DNA polymerase delta -dependent elongation machinery in vitro: A parvovirus DNA replication model. Proc Natl Acad Sci U S A. 2000;97 (10):5522–5527. doi: 10.1073/pnas.090485297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir T, Rommelaere J, Cziepluch C. In vivo accumulation of cyclin A and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J Virol. 2001;75 (9):4394–4398. doi: 10.1128/JVI.75.9.4394-4398.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhaus K, Plaza S, Pintel DJ, Rommelaere J, Salome N. Nonstructural proteins NS2 of minute virus of mice associate in vivo with 14-3-3 protein family members. J Virol. 1996;70 (11):7527–7534. doi: 10.1128/jvi.70.11.7527-7534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodendorf U, Cziepluch C, Jauniaux JC, Rommelaere J, Salome N. Nuclear export factor CRM1 interacts with nonstructural protein NS2 from parvovirus minute virus of mice. J Virol. 1999;73 (9):7769–7779. doi: 10.1128/jvi.73.9.7769-7779.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater JE, Pintel DJ. The small non-structural protein NS2 of the autonomous parvovirus minute virus of mice is required for virus growth in murine cells. J Gen Virol. 1992;73 (Pt 7):1839–1843. doi: 10.1099/0022-1317-73-7-1839. [DOI] [PubMed] [Google Scholar]
- Choi EY, Newman AE, Burger L, Pintel D. Replication of minute virus of mice DNA is critically dependent on accumulated levels of NS2. J Virol. 2005;79 (19):12375–12381. doi: 10.1128/JVI.79.19.12375-12381.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Tattersall P. Parvovirus initiator protein NS1 and RPA coordinate replication fork progression in a reconstituted DNA replication system. J Virol. 2002;76 (13):6518–6531. doi: 10.1128/JVI.76.13.6518-6531.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens KE, Pintel DJ. The two transcription units of the autonomous parvovirus minute virus of mice are transcribed in a temporal order. J Virol. 1988;62 (4):1448–1451. doi: 10.1128/jvi.62.4.1448-1451.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotmore SF, Tattersall P. Organisation of nonstructural genes of the autonomous parvovirus minute virus of mice. J Virol. 1986;58 (3):724–732. doi: 10.1128/jvi.58.3.724-732.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotmore SF, Tattersall P. The autonomously replicating parvoviruses of vertebrates. Adv Virus Res. 1987;33:91–174. doi: 10.1016/s0065-3527(08)60317-6. [DOI] [PubMed] [Google Scholar]
- Cotmore SF, Tattersall P. The NS-1 polypeptide of minute virus of mice is covalently attached to the 5' termini of duplex replicative-form DNA and progeny single strands. J Virol. 1988;62 (3):851–860. doi: 10.1128/jvi.62.3.851-860.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotmore SF, D'Abramo AM, Jr, Carbonell LF, Bratton J, Tattersall P. The NS2 polypeptide of parvovirus MVM is required for capsid assembly in murine cells. Virology. 1997;231 (2):267–280. doi: 10.1006/viro.1997.8545. [DOI] [PubMed] [Google Scholar]
- Cotmore SF, Tattersall P. A rolling hairpin strategy: basic mechanisms of DNA replication in the parvoviruses. In: Kerr JR, Cotmore SF, Bloom ME, Linden RM, Parrish CR, editors. Parvoviruses. Edward Arnold Publishers Ltd; London: 2006a. pp. 171–188. [Google Scholar]
- Cotmore SF, Tattersall P. Parvoviruses. In: DePamphilis M, editor. DNA replication and Human Disease. Ch 29. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2006b. pp. 593–608. [Google Scholar]
- Cotmore SF, Tattersall P. Parvoviral host range and cell entry mechanisms. Adv Virus Res. 2007;70:183–232. doi: 10.1016/S0065-3527(07)70005-2. [DOI] [PubMed] [Google Scholar]
- Cziepluch C, Lampel S, Grewenig A, Grund C, Lichter P, Rommelaere J. H-1 parvovirus-associated replication bodies: a distinct virus-induced nuclear structure. J Virol. 2000;74 (10):4807–15. doi: 10.1128/jvi.74.10.4807-4815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Abramo AM, Jr, Ali AA, Wang F, et al. Host range mutants of Minute Virus of Mice with a single VP2 amino acid change require additional silent mutations that regulate NS2 accumulation. Virology. 2005;340 (1):143–54. doi: 10.1016/j.virol.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Eichwald V, Daeffler L, Klein M, Rommelaere J, Salome N. The NS2 proteins of parvovirus minute virus of mice are required for efficient nuclear egress of progeny virions in mouse cells. J Virol. 2002;76 (20):10307–10319. doi: 10.1128/JVI.76.20.10307-10319.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3 (8–9):959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Gersappe A, Burger L, Pintel DJ. A premature termination codon in either exon of minute virus of mice P4 promoter-generated pre-mRNA can inhibit nuclear splicing of the intervening intron in an open reading frame-dependent manner. J Biol Chem. 1999;274 (32):22452–22458. doi: 10.1074/jbc.274.32.22452. [DOI] [PubMed] [Google Scholar]
- Ho TF, Gupta JS, Faust EA. A novel primase-free form of murine DNA polymerase alpha induced by infection with minute virus of mice. Biochemistry. 1989;28 (11):4622–8. doi: 10.1021/bi00437a017. [DOI] [PubMed] [Google Scholar]
- Gupta JS, Faust EA. Minute virus of mice-induced modification of the murine DNA polymerase alpha-primase complex permits the salt-induced dissociation of 12S DNA primase and 10S DNA polymerase alpha components. Biochem Mol Biol Int. 1993;31 (4):599–611. [PubMed] [Google Scholar]
- Huen MS, Chen J. Assembly of checkpoint and repair machineries at DNA damage sites. Trends Biochem Sci. 2010;35 (2):101–108. doi: 10.1016/j.tibs.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihalainen TO, Niskanen EA, Jylhava J, Paloheimo O, Dross N, Smolander H, Langowski J, Timonen J, Vihinen-Ranta M. Parvovirus induced alterations in nuclear architecture and dynamics. PLoS One. 2009;4 (6):e5948. doi: 10.1371/journal.pone.0005948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihalainen TO, Niskanen EA, Jylhava J, Turpeinen T, Rinne J, Timonen J, Vihinen-Ranta M. Dynamics and interactions of parvoviral NS1 protein in the nucleus. Cell Microbiol. 2007;9 (8):1946–59. doi: 10.1111/j.1462-5822.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461 (7267):1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Chaurushiya MS, Weitzman MD. Chromatin at the intersection of viral infection and DNA damage. Biochim Biophys Acta. 2010;1799 (3–4):319–327. doi: 10.1016/j.bbagrm.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Pintel DJ. The NS2 protein generated by the parvovirus minute virus of mice is degraded by the proteasome in a manner independent of ubiquitin chain elongation or activation. Virology. 2001;285 (2):346–55. doi: 10.1006/viro.2001.0966. [DOI] [PubMed] [Google Scholar]
- Miller CL, Pintel DJ. Interaction between parvovirus NS2 protein and nuclear export factor Crm1 is important for viral egress from the nucleus of murine cells. J Virol. 2002;76 (7):3257–3266. doi: 10.1128/JVI.76.7.3257-3266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter-Dykhouse K, Ward I, Huen MS, Chen J, Lou Z. Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J Cell Biol. 2008;181 (5):727–735. doi: 10.1083/jcb.200801083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeger LK, Cater J, Pintel DJ. The small nonstructural protein (NS2) of the parvovirus minute virus of mice is required for efficient DNA replication and infectious virus production in a cell-type-specific manner. J Virol. 1990;64 (12):6166–6175. doi: 10.1128/jvi.64.12.6166-6175.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeger LK, Schoborg RV, Zhao Q, Tullis GE, Pintel DJ. Nonsense mutations inhibit splicing of MVM RNA in cis when they interrupt the reading frame of either exon of the final spliced product. Genes Dev. 1992;6 (6):1107–19. doi: 10.1101/gad.6.6.1107. [DOI] [PubMed] [Google Scholar]
- Nuesch JPF. Regulation of non-structural protein functions by differential synthesis, modification and trafficking. In: Kerr JR, Cotmore SF, Bloom ME, Linden RM, Parrish CR, editors. Parvoviruses. Edward Arnold Publishers Ltd; London: 2006. pp. 275–289. [Google Scholar]
- Nuss JE, Patrick SM, Oakley GG, Alter GM, Robison JG, Dixon K, Turchi JJ. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry. 2005;44 (23):8428–8437. doi: 10.1021/bi0480584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor MJ, Martin NM, Smith GC. Targeted cancer therapies based on inhibition of DNA strand break Repair. Oncogene. 2007;26 (56):7816–7824. doi: 10.1038/sj.onc.1210879. [DOI] [PubMed] [Google Scholar]
- Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J Biol Chem. 2006;281 (51):39517–39533. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- Op De Beeck A, Sobczak-Thepot J, Sirma H, Bourgain F, Brechot C, Caillet-Fauquet P. NS1- and minute virus of mice-induced cell cycle arrest: involvement of p53 and p21(cip1) J Virol. 2001;75 (22):11071–11078. doi: 10.1128/JVI.75.22.11071-11078.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Op De Beeck A, Caillet-Fauquet P. The NS1 protein of the autonomous parvovirus minute virus of mice blocks cellular DNA replication: a consequence of lesions to the chromatin? J Virol. 1997;71 (7):5323–5329. doi: 10.1128/jvi.71.7.5323-5329.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohaly G, Korf K, Dehde S, Dornreiter I. Simian Virus 40 activates ATR-Delta p53 signaling to override cell cycle and DNA replication control. J Virol. 2010;84 (20):10727–10747. doi: 10.1128/JVI.00122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273 (10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Ruiz Z, D'Abramo A, Jr, Tattersall P. Differential roles for the C-terminal hexapeptide domains of NS2 splice variants during MVM infection of murine cells. Virology. 2006;349 (2):382–395. doi: 10.1016/j.virol.2006.01.039. [DOI] [PubMed] [Google Scholar]
- Yeung DE, Brown GW, Tam P, Russnak RH, Wilson G, Clark-Lewis I, Astell CR. Monoclonal antibodies to the major nonstructural nuclear protein of minute virus of mice. Virology. 1991;181 (1):35–45. doi: 10.1016/0042-6822(91)90467-p. [DOI] [PubMed] [Google Scholar]
- Young PJ, Jensen KT, Burger LR, Pintel DJ, Lorson CL. Minute virus of mice NS1 interacts with the SMN protein, and they colocalize in novel nuclear bodies induced by parvovirus infection. J Virol. 2002;76 (8):3892–3894. doi: 10.1128/JVI.76.8.3892-3904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young PJ, Newman A, Jensen KT, Burger LR, Pintel DJ, Lorson CL. Minute virus of mice small non-structural protein NS2 localizes within, but is not required for the formation of, Smn-associated autonomous parvovirus-associated replication bodies. J Gen Virol. 2005;86 (Pt 4):1009–1014. doi: 10.1099/vir.0.80564-0. [DOI] [PubMed] [Google Scholar]
- Weitzman MD, Lilley CE, Chaurushiya MS. Genomes in conflict: maintaining genome integrity during virus infection. Annu Rev Microbiol. 2010;64:61–81. doi: 10.1146/annurev.micro.112408.134016. [DOI] [PubMed] [Google Scholar]