

Abstract
During middle G1 of the cell cycle origins of replication orchestrate the ordered assembly of the pre-replication complex (pre-RC), allowing licensing of DNA required for DNA replication. Cyclin-dependent kinase activation of the pre-RC facilitates the recruitment of additional signaling factors, which triggers DNA unwinding and replication, while limiting such DNA replication to once and only once per cell cycle. For both the normal and malignant prostate, androgen is the major stimulator of cell proliferation and thus DNA replication. In both cases, the binding of androgen to the androgen receptor (AR) is required. However, the biochemical cascade involved in such AR-stimulated cell proliferation and DNA synthesis is dramatically different in normal versus malignant prostate cells. In normal prostate, AR-stimulated stromal cell paracrine secretion of andromedins stimulates DNA replication within prostatic epithelial cells, in which AR functions as a tumor suppressor gene by inducing proliferative quiescence and terminal differentiation. By direct contrast, nuclear AR in prostate cancer cells autonomously stimulates continuous growth via incorporation of AR into the pre-RC. Such a gain of function by AR-expressing prostate cancer cells requires that AR be efficiently degraded during mitosis since lack of such degradation leads to re-licensing problems, resulting in S-phase arrest during the subsequent cell cycle. Thus, acquisition of AR as part of the licensing complex for DNA replication represents a paradigm shift in how we view the role of AR in prostate cancer biology, and introduces a novel vulnerability in AR-expressing prostate cancer cells apt for therapeutic intervention.
Introduction
Over the last several decades, there have been improvements in prostate cancer diagnosis and treatment primarily due to the diagnostic/prognostic use of prostate specific antigen, the advent of nerve sparing radical prostatectomy, conformal external beam radiation, and better hormonal and taxane based chemotherapies (Denmeade & Isaacs 2002). With these advances, prostate cancer annual mortality has begun to decrease, but despite this, over 28 000 men will still die this year as a result of prostate cancer in the USA (Jemal et al. 2008). To accelerate the development of more effective therapies to combat this devastating disease, novel targets need to be identified and validated. Since the lethality of prostate cancer is dependent upon its ability to grow continuously, the basis for one approach to discover and validate novel targets is to identify molecular processes that restrict growth of normal prostate epithelial cells and determine how this restriction is subverted during prostatic carcinogenesis and cancer progression. For its development and maintenance, normal prostatic epithelium absolutely requires the expression and appropriate degree of androgen receptor (AR) occupancy by its cognate ligand. This is based upon the following: 1) the AR gene is located on the X-chromosome and therefore males are hemizygous for AR; and 2) patients harboring truncation mutations in the first exon express no AR protein and therefore exhibit complete androgen insensitivity and failed prostatic development (Gottlieb et al. 1999). In non-mutated hosts, a sufficient degree of ligand-bound AR is required for both initial development of the prostate and maintenance of the homeostatic balance between proliferation and survival signaling in adult prostatic epithelial cells. This homeostasis involves a balance in which AR-dependent stromal cell-induced paracrine growth stimulation of the prostatic epithelium is countered by AR-dependent growth suppression within prostate epithelial cells (Litvinov et al. 2003; Fig. 1).
Figure 1.

Androgen receptor signaling in normal and malignant prostate cells. (top) In the normal prostate, growth and maintenance of prostatic epithelium depends on paracrine signaling of andromedins (growth and survival factors) produced by supporting stromal cells (smooth muscle and fibroblasts). Andromedins are secreted due to androgen signaling through AR, a nuclear hormone receptor expressed by prostate luminal epithelia but not by basal epithelia. (bottom) During prostate cancer transformation, the paracrine AR signaling mechanism is replaced by an emergent autocrine mechanism, whereby cancer cells exhibit less dependency on stromally-derived factors. Androgens acting through AR can directly stimulate production of growth and survival factors by the cancer cells.
During prostate carcinogenesis changes in the molecular ‘hard wiring’ of AR signaling pathways obviate the requirement for AR-dependent stromal cell-induced paracrine growth signaling. Diminished stromal dependence coupled with AR acquiring ‘gain of function’ growth stimulation as an autocrine tumor oncogene in prostate cancer cells (Gao et al. 2001, Litvinov et al. 2003, Isaacs & Isaacs 2004; Fig. 1) provides the mechanistic basis for androgen-ablation therapy and validates AR signaling pathways as therapeutic targets (Denmeade & Isaacs 2002). Presently, many efforts focus on developing better small molecule antiandrogen antagonists that compete for AR ligand binding as well as metabolic inhibitors for androgen synthesis. An alternative approach is to target the ability of AR to partner with other proteins uniquely involved in DNA licensing and replication, and thus interrupt proliferation of prostate cancer cells.
Licensing of DNA replication
In 1963, initial studies working with Escherichia coli first established that DNA is replicated bi-directionally from one initiation point (Cairns 1963) and that transacting initiator proteins regulate DNA replication at a specific region of DNA characterized as the origin of replication (Jacob & Brenner 1963). A few years later, it was discovered that replication of mammalian chromosomal DNA occurs simultaneously at many origin of replication sites and that such DNA replication is bi-directional (Huberman & Riggs 1968). The earliest clues to DNA licensing came about via cell fusion experiments where fusion of a G1 cell to an S-phase cell prematurely initiates DNA replication in the G1 cell nucleus, indicating that a G1 cell is capable of DNA replication but lacks necessary factors expressed during S-phase (Rao & Johnson 1970). By contrast, fusion of a G2 cell to an S-phase cell fails to initiate DNA replication in the G2 nucleus, demonstrating that the G2 cell is not able to re-license its DNA. Thus, cells in G1 are capable of ‘licensing’ DNA for replication followed by a removal of this license sometime during or after DNA replication in order to prevent re-replication.
Since these seminal studies it has been established that, in mammalian cells, the rate of replication at any one replication fork is ~50 nucleotides per second. This means that in order for DNA (i.e. ~3×109 bps in human genome) to be replicated during the 8–12 h of S-phase, DNA synthesis must occur simultaneously and bi-directionally starting from one-to-two thousand origins of replication sites spaced 30 000–300 000 bps apart within the genome. Critically, such DNA replication must be restricted so that only a single duplication of the genome occurs per S-phase. These two conditions are maintained by the process known as DNA ‘licensing’. Following mitosis, the genome of the daughter cells is ‘un-licensed’ for DNA replication. With the appropriate signaling, such post mitotic cells re-enter the cell cycle and progress into G1. During a critical period in early-to-middle G1, the temporally-coordinated binding of a series of ‘licensing factors’, which include origin recognition complex (Orc1–6), cell division cycle 6 homolog (Cdc6), chromatin licensing and DNA replication factor 1 (Cdt1), and mini chromosome maintenance (Mcm2–7) proteins, forms pre-replication complexes (pre-RC) at specific origins of replication sites in the DNA (Bell & Dutta 2002, Tabancay & Forsburg 2006). Origin sites are sequence specific in Saccharomyces cerevisiae but metazoans, including Drosophila, Xenopus, and humans, lack canonically defined origin consensus sequences (Bell 2002, Tabancay & Forsburg 2006).
Distributed stochastically throughout the eukaryotic genome, origins are first bound by the highly conserved Orc1–6 ATPase protein complex. ATP hydrolysis by ORC is required for loading of subsequent pre-RC factors Cdc6, Cdt1, and Mcm2–7 (Blow & Dutta 2005; Fig. 2). In human cells, Orc1 is chromatin-bound in late-M to G1 as the pre-RC forms; but, as cells progress from G1 to S-phase and cyclin A/cyclin-dependent kinase 2 (cdk2) levels accumulate Orc1 dissociates from ORC, is phosphorylated and subsequently recognized by the E3 ubiquitin ligase, SCFSKP2, polyubiquitinated and degraded via the proteasome (Mendez et al. 2002). By contrast, Orc2–6 levels remain stable throughout the cell cycle in human cells (Mendez et al. 2002, DePamphilis 2005).
Figure 2.

Mammalian pre-RC formation and DNA licensing in the context of cell cycle progression. In early G1, cyclin D-cdk4/6 phosphorylation of Rb, which permits E2F transcriptional activity, triggers entry into the cell cycle. Pre-RCs form via stepwise binding of licensing factors Orc1–6, Cdc6, Cdt1, and Mcm2–7, required for DNA licensing. Progressing into S-phase, CDK, and DDK phosphorylation of pre-RC constituents initiates Orc1 dissociation, and the removal and subsequent nuclear export of Cdc6, while geminin, along with CDK and DDK activity, trigger the removal and degradation of Cdt1. This regulation serves to ‘un-license’ the DNA and prevents re-licensing before completion of the initiated cell cycle. E2F target genes: Cdc6, MCM, Dbf4 (DDK), Cyclins E, and A. SCFSKP2, Skp1-cullin-F box E3 ubiquitin ligase; CUL4, CUL4-DDB1-CDT2 ubiquitin ligase; R, restriction point; G, geminin; U, ubiquitin; P, phosphate.
Human Cdc6 binds ORC and through ATPase nucleotide hydrolysis recruits Cdt1 to origins of replication (Bell & Dutta 2002). In mammalian cells, cellular Cdc6 levels remain stable throughout the active cell cycle; however, studies show cell cycle-specific variation in Cdc6 localization. In human cells, Cdc6 is primarily localized to the nucleus during G1 but as cells enter S-phase Cdc6 is phosphorylated by cyclin A/cdk2 and exported to the cytoplasm (Jiang et al. 1999, Petersen et al. 1999; Fig. 2), however, evidence shows residual chromatin-bound Cdc6 resistant to nuclear export (Coverley et al. 2000).
Recruited by Cdc6, Cdt1 binds to pre-RCs in mid-G1 and serves in recruiting Mcm2–7 helicase complexes to the DNA. In human cells, Cdt1 expression is under very tight, yet complex, cell cycle-dependent regulation. Owing to APC/CCdh1 repression of cyclins and the Cdt1 negative regulator geminin (Fujita 2006), Cdt1 is stably bound to chromatin in late M through G1 (Nishitani et al. 2001, Woo & Poon 2003). After DNA becomes fully licensed and cells transition from G1 into S-phase, geminin expression increases and inhibits Cdt1 by inducing Cdt1 ubiquitination and degradation (Fig. 2), however, after successful DNA replication and progression through G2 into mitosis, geminin appears to protect Cdt1 from ubiquitin-associated proteolysis (Ballabeni et al. 2004). These steps ensure timely and accurate pre-RC formation in G1 while preventing re-licensing in S-phase. It has also been demonstrated that cdk2 and cdk4 interact with and phosphorylate human Cdt1, leading to ubiquitination by SCFSKP2 and proteosomal degradation (Liu et al. 2004). However, the predominant mechanism responsible for CDT1 degradation, which ultimately prevents premature re-licensing, is the ubiquitin ligase CUL4-DDB1-CDT2 (Fig. 2). Recruited to the DNA polymerase processivity factor, PCNA, CUL4-DDB1-CDT2-dependent ubiquitination of Cdt1 effectively links Cdt1 degradation to initiated DNA replication, therefore reducing the chances of aberrant re-licensing during DNA replication (Arias & Walter 2006, Jin et al. 2006, Nishitani et al. 2006, Senga et al. 2006).
The recruitment of MCM proteins to origins of replication in G1 completes pre-RC formation, thereby fully licensing DNA for replication. Origin-bound Mcm2–7 has been described as the putative DNA helicase that unwinds DNA for replication in S-phase, therefore functioning in DNA synthesis as well as DNA licensing (Tabancay & Forsburg 2006). However, as DNA is unwound and replicated in a bi-directional manner, one would expect no more than two MCM complexes bound at each origin, but studies have detected roughly 10–40 MCM bound per origin site, suggesting additional functions associated with MCM-DNA association (Edwards et al. 2002, Blow & Dutta 2005). In terms of licensing control, several studies suggest that certain MCM proteins undergo phosphorylation-associated regulation, in turn switching off helicase activity (Woo & Poon 2003); however, there exists no experimental evidence that Mcm2–7 is subjected to direct negative regulation in human cells (Arias & Walter 2007).
A key mechanism for inhibition of aberrant re-replication involves CDK-induced degradation of Orc1 and Cdt1, inactivation and nuclear export of Cdc6, and inactivation of MCM proteins following G1 (Ballabeni et al. 2004, Takeda & Dutta 2005, Cvetic & Walter 2006, Arias & Walter 2007). A few members of the pre-RC remain associated with origins of replication during G2 to prevent DNA re-licensing and then are removed to permit re-licensing of the DNA, in late-M to early-G1, thus allowing for only one round of DNA replication per cycle (Takeda & Dutta 2005). In carcinogenesis, recent studies discovered altered expression of pre-RC proteins in a variety of cancers, including breast, prostate, oral, colorectal, ovarian, renal, and hematological cancers (Gonzalez et al. 2005, Williams & Stoeber 2007). Specifically, altered levels of Mcm2–7 may serve as a marker of increased proliferative capacity, whereas Cdc6 and Cdt1 may function more so as oncogenes ultimately effecting genome instability in cells (Blow & Gillespie 2008). Additionally, studies focusing on oncogenes such as cyclin D1 and v-Ha-ras Harvey rat sarcoma viral oncogene homolog (HRAS) have uncovered that oncogene induction can disrupt DNA licensing resulting in DNA re-replication and genomic instability (Blow & Gillespie 2008). Having discussed the role of AR as an oncogene in prostate cancer, these findings suggest a link between AR function and DNA licensing in cancer development and progression.
AR as a licensing factor for AR-expressing prostate cancer cells
Proliferation of AR-expressing prostate cancer cells can be inhibited by treatment with AR antagonists if administered during early G1 prior to pre-RC assembly; however, if AR antagonism is initiated after pre-RC formation later in G1, cells successfully enter S-phase (Cifuentes et al. 2003, Bai et al. 2005). These observations propose that the timing of androgen-induced cell proliferation in AR-expressing prostate cancer cells might be closely linked with the assembly of the pre-RC machinery. This correlation raised the issue of whether nuclear AR associates with the DNA licensing complex in AR-expressing prostate cancer cells. To test this possibility, nuclei were isolated from AR-expressing LNCaP and LAPC-4 human prostate cancer cells, where the primary function of AR is to promote cell growth and thus DNA replication, and incubated with AR antibodies to immunoprecipitate proteins associated with nuclear AR proteins. The immunoprecipitated proteins were analyzed by western blotting to determine whether AR associates with proteins of the pre-RC. These studies document that, in AR-expressing human prostate cancer lines, nuclear AR is associated with licensing proteins Orc2, Cdc-6, Cdt-1 (Fig. 3), and Mcm2 (data not shown). These results were reproducibly obtained using either N- or C-terminal AR specific antibodies (Vander Griend & Isaacs 2008).
Figure 3.
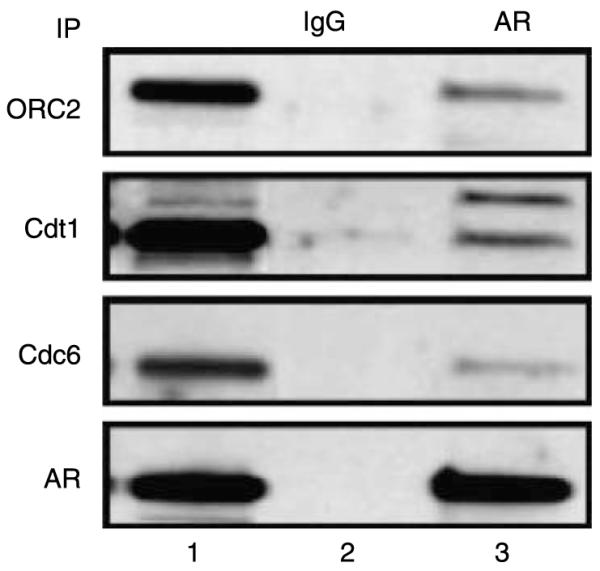
AR interacts with members of the DNA licensing complex. Androgen-sensitive LNCaP prostate cancer cells express AR as well as members of the DNA licensing complex, Orc2, Cdt1, and Cdc6 (lane 1). Immunoprecipitation (lane 2 – IgG control, lane 3 – anti-AR antibody) of AR from LNCaP cells followed by western blotting reveal that Orc2, Cdt1, and Cdc6 co-immunoprecipitated with AR. These data demonstrate AR presence at pre-RCs and strongly support a role for AR in modulating DNA replication licensing to drive prostate cancer cell proliferation. Figure from Vander Griend & Isaacs (2008) (LAPC4 data not shown).
These observations further establish a potential role for AR in DNA licensing for AR-expressing prostate cancer cells and suggest that as a licensing factor, AR might need to be removed from the DNA and degraded in mitosis or early-G1 for effective DNA re-licensing. To test this hypothesis, AR protein levels in LNCaP, CWR22Rv1, and LAPC-4 cells were examined throughout the cell cycle and found to vary both within and between different phases of the cell cycle. Additionally, it was learned that AR protein is degraded via the proteasome sometime in mitosis in each of the AR-expressing prostate cancer cell lines tested (Litvinov et al. 2006). Distinguishing from these prostate cancer cells, in which AR stimulates DNA replication, proliferation of non-malignant normal human prostate stromal cells (PrSCs), which express lower levels of AR, is not controlled by AR. As stated earlier, the primary function of AR in PrSCs is to drive expression of secreted paracrine growth factors (i.e. andromedins) that support the growth of normal prostate epithelial cells. Experiments looking at cell cycle dependent AR expression in PrSCs found that AR is not degraded during mitosis in PrSCs (Litvinov et al. 2003). These combined results provide a strong link between AR and DNA licensing in prostate cancer cells, which unlike PrSCs, completely degrade AR to allow re-licensing and re-entry into the next cell cycle.
Therapeutic vulnerability of AR as a licensing factor
Defining the mechanism(s) by which AR promotes DNA licensing and cell division is critical in identifying new targets for therapy. Many of the genomic areas of active transcription also serve as the earliest sites of DNA replication (Tabancay & Forsburg 2006). Therefore, AR binding to AREs in the promoters of androgen-regulated genes may serve to recruit licensing factors to AR-target genes. The limited number of AR-regulated genes (i.e. hundreds/genome; Wang et al. 2007), however, is not able to account for the larger number of origins of replication (i.e. thousands/genome). Regardless of the mechanism for how AR is captured as part of the DNA replication licensing process, such ‘gain of function’ requires that AR is degraded during mitosis since lack of such degradation results in subsequent S-phase arrest of androgen-sensitive prostate cancer cells (Vander Griend et al. 2007). This requirement is coordinated by an auto-regulatory ability of AR-expressing prostate cancer cells to maintain AR protein at a level optimal for the particular ligand concentration. This auto-regulation results in a decrease in AR protein in the presence of elevated ligand and an increase when androgen levels are reduced (Langeler et al. 1993, Kokontis et al. 1998). However, such auto-regulation requires a critical time frame.
The realization that AR functions in DNA replication licensing in malignant prostate cells provides an explanation for a long standing experimental paradox with regard to the biphasic acute dose–response of AR-expressing prostate cancer cells to androgen. When prostate cancers are placed in an androgen-depleted environment, the most consistent molecular change is a two to fourfold up-regulation of AR expression (Chen et al. 2004, D’Antonio et al. 2008). Paradoxically, however, prostate cancer cells expressing high levels of AR protein are growth inhibited when the level of androgen is raised acutely above a critical threshold (Langeler et al. 1993, Kokontis et al. 1998). These observations are consistent with the fact that androgen ligand stabilizes AR protein from degradation but also down-regulates AR transcription (Quarmby et al. 1990, Krongrad et al. 1991, Kemppainen et al. 1992), thus when androgen levels are raised acutely, AR protein is stabilized to a point where it is not degraded sufficiently during mitosis. Excessive ligand-dependent stabilization thus results in a fraction of AR protein remaining associated with origins of replication sites, which were licensed and used during the previous cell cycle, such that these origins of replication cannot re-license in G1 of the subsequent cell cycle in the daughter cells. Although this situation allows the daughter cells to progress into S-phase, it prevents them from completely replicating the full content of their genomic DNA, which induces early S-phase growth arrest (Vander Griend et al. 2007).
Several novel therapeutic approaches for prostate cancer can be imagined based upon this new revelation concerning the role and consequence of AR as a licensing factor. For example, the time dependence for AR adaptation provides a theoretical rationale for improving intermittent androgen deprivation therapy (IADT). Presently, such intermittent therapy involves giving ADT for a limited time followed by a relief period to allow recovery to a physiological level of testosterone (Mottet et al. 2005). From this, we hypothesize that during each cycle of ADT, AR protein is slowly up-regulated to compensate for diminished ligand, and when ADT is stopped, the rate at which tissue androgen levels return determines whether adaptive changes have sufficient time to down-regulate the elevated level of AR to prevent re-licensing problems. Theoretically, therefore, the efficacy of IADT can be enhanced by maintaining the patient on androgen ablation with a long acting depot (i.e. 6 months–1 year) of an LHRH analog interrupted by episodic (e.g. every 3–4 months) administration of pharmacologically high doses of androgen given acutely for only a limited period (i.e. 1 week). In this way, there is insufficient time to completely down-regulate AR during each androgen restoration cycle resulting in DNA replication re-licensing problems that inhibit the growth of the prostate cancer cells.
Additional ways exist to maximize the re-licensing problems in prostate cancer cells. One alternative is to take advantage of the fact that mitotic degradation of AR protein (Litvinov et al. 2006) and the growth of AR-expressing prostate cancer cell lines (Williams et al. 2003, Papandreou et al. 2004) are inhibited with proteasome inhibitors. A novel approach would combine proteasome inhibitors with high dose androgen in patients who have failed previous ADT treatments.
A further approach is not to abnormally stabilize the AR/licensing to produce re-licensing problems in AR-expressing prostate cancer cells, but to screen for compounds that down-regulate the levels of AR protein needed for DNA replication, proliferation, and cancer cell survival. Along these lines, we have identified the SERCA pump inhibitor, Thapsigargin, which induces an ER-stress response (Sohoel et al. 2006) resulting in the down-regulation of AR protein and apoptotic death of prostate cancer cells. Presently, Thapsigargin analogs covalently linked to carrier peptides to make ‘Smart Bomb’ prodrugs are in drug development based upon the fact that they can be given systemically since they are water soluble and therefore impermeable to cells until hydrolyzed to cell-permeable toxic moieties by prostate cancer-restricted proteases (Denmeade et al. 2003).
Concluding remarks
Based upon the understanding of the genomic actions of AR signaling in the prostate, various therapeutic approaches for disrupting AR signaling have yielded improvements in the treatment of prostate cancer. However, the benefits of these therapies are limited due to numerous adaptive modifications in the AR signaling cascade that ultimately circumvent present therapeutic modalities. The development of novel therapeutic strategies is much needed. Targeting AR’s ability to stimulate DNA licensing, which promotes cancer cell proliferation, provides a novel anti-AR approach that can be combined with the present prostate cancer therapies to hopefully maximize therapeutic benefit.
Acknowledgements
We would like to thank Lizamma Antony for her expertise in protein co-immunoprecipitation and Western blotting techniques.
Funding
This work was funded by NIH grant CA091409. JMD is supported by the Johns Hopkins Urology Training Grant (NIH T32DK07552); DVG was supported by the Urology Training Grant and is now supported by a Department of Defense Postdoctoral Training Award (PC060843).
Footnotes
Declaration of interest
There is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Arias EE, Walter JC. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nature Cell Biology. 2006;8:84–90. doi: 10.1038/ncb1346. [DOI] [PubMed] [Google Scholar]
- Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes and Development. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- Bai VU, Cifuentes E, Menon M, Barrack ER, Reddy GP. Androgen receptor regulates Cdc6 in synchronized LNCaP cells progressing from G1 to S phase. Journal of Celluar Physiology. 2005;204:381–387. doi: 10.1002/jcp.20422. [DOI] [PubMed] [Google Scholar]
- Ballabeni A, Melixetian M, Zamponi R, Masiero L, Marinoni F, Helin K. Human geminin promotes pre-RC formation and DNA replication by stabilizing CDT1 in mitosis. EMBO Journal. 2004;23:3122–3132. doi: 10.1038/sj.emboj.7600314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP. The origin recognition complex: from simple origins to complex functions. Genes and Development. 2002;16:659–672. doi: 10.1101/gad.969602. [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annual Review of Biochemistry. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nature Reviews. Molecular Cell Biology. 2005;6:476–486. doi: 10.1038/nrm1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow JJ, Gillespie PJ. Replication licensing and cancer – a fatal entanglement? Nature Reviews. Cancer. 2008;8:799–806. doi: 10.1038/nrc2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J. The bacterial chromosome and its manner of replication as seen by autoradiography. Journal of Molecular Biology. 1963;6:208–213. doi: 10.1016/s0022-2836(63)80070-4. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nature Medicine. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Cifuentes E, Croxen R, Menon M, Barrack ER, Reddy GP. Synchronized prostate cancer cells for studying androgen regulated events in cell cycle progression from G1 into S phase. Journal of Celluar Physiology. 2003;195:337–345. doi: 10.1002/jcp.10317. [DOI] [PubMed] [Google Scholar]
- Coverley D, Pelizon C, Trewick S, Laskey RA. Chromatin-bound Cdc6 persists in S and G2 phases in human cells, while soluble Cdc6 is destroyed in a cyclin A-cdk2 dependent process. Journal of Cell Science. 2000;113:1929–1938. doi: 10.1242/jcs.113.11.1929. [DOI] [PubMed] [Google Scholar]
- Cvetic CA, Walter JC. Getting a grip on licensing: mechanism of stable Mcm2–7 loading onto replication origins. Molecular Cell. 2006;21:143–144. doi: 10.1016/j.molcel.2006.01.003. [DOI] [PubMed] [Google Scholar]
- D’Antonio JM, Ma C, Monzon FA, Pflug BR. Longitudinal analysis of androgen deprivation of prostate cancer cells identifies pathways to androgen independence. Prostate. 2008;68:698–714. doi: 10.1002/pros.20677. [DOI] [PubMed] [Google Scholar]
- Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nature Reviews. Cancer. 2002;2:389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H, Christensen SB, Isaacs JT. Prostate-specific antigen-activated thapsigargin prodrug as trageted therapy for prostate cancer. Journal of the National Cancer Institutes. 2003;95:990–1000. doi: 10.1093/jnci/95.13.990. [DOI] [PubMed] [Google Scholar]
- DePamphilis ML. Cell cycle dependent regulation of the origin recognition complex. Cell Cycle. 2005;4:70–79. doi: 10.4161/cc.4.1.1333. [DOI] [PubMed] [Google Scholar]
- Edwards MC, Tutter AV, Cvetic C, Gilbert CH, Prokhorova TA, Walter JC. MCM2–7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. Journal of Biological Chemistry. 2002;277:33049–33057. doi: 10.1074/jbc.M204438200. [DOI] [PubMed] [Google Scholar]
- Fujita M. Cdt1 revisited: complex and tight regulation during the cell cycle and consequences of deregulation in mammalian cells. Cell Division. 2006;1:22. doi: 10.1186/1747-1028-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Research. 2001;61:5038–5044. [PubMed] [Google Scholar]
- Gonzalez MA, Tachibana KE, Laskey RA, Coleman N. Control of DNA replication and its potential clinical exploitation. Nature Reviews. Cancer. 2005;5:135–141. doi: 10.1038/nrc1548. [DOI] [PubMed] [Google Scholar]
- Gottlieb B, Vasiliou DM, Lumbroso R, Beitel LK, Pinsky L, Trifiro MA. Analysis of exon 1 mutations in the androgen receptor gene. Human Mutation. 1999;14:527–539. doi: 10.1002/(SICI)1098-1004(199912)14:6<527::AID-HUMU12>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Vander Griend DJ, Isaacs JT. Androgen receptor as a licensing factor for DNA replication. In: Tindall DJ, Mohler JL, editors. Androgen Action in Prostate Cancer. Springer; New York, NY, USA: 2008. [Google Scholar]
- Vander Griend DJ, Litvinov IV, Isaacs JT. Stabilizing androgen receptor in mitosis inhibits prostate cancer proliferation. Cell Cycle. 2007;6:647–651. doi: 10.4161/cc.6.6.4028. [DOI] [PubMed] [Google Scholar]
- Huberman JA, Riggs AD. On the mechanism of DNA replication in mammalian chromosomes. Journal of Molecular Biology. 1968;32:327–341. doi: 10.1016/0022-2836(68)90013-2. [DOI] [PubMed] [Google Scholar]
- Isaacs JT, Isaacs WB. Androgen receptor outwits prostate cancer drugs. Nature Medicine. 2004;10:26–27. doi: 10.1038/nm0104-26. [DOI] [PubMed] [Google Scholar]
- Jacob F, Brenner S. On the regulation of DNA synthesis in bacteria: the hypothesis of the replicon. Comptes Rendus Des Séances de l’Académie Des Sciences. 1963;256:298–300. [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA: A Cancer Journal for Clinicians. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Jiang W, Wells NJ, Hunter T. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. PNAS. 1999;96:6193–6198. doi: 10.1073/pnas.96.11.6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Molecular Cell. 2006;23:709–721. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Kemppainen JA, Lane MV, Sar M, Wilson EM. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. Journal of Biological Chemistry. 1992;267:968–974. [PubMed] [Google Scholar]
- Kokontis JM, Hay N, Liao S. Progression of LNCaP prostate tumor cells during androgen deprivation: hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Molecular Endocrinology. 1998;12:941–953. doi: 10.1210/mend.12.7.0136. [DOI] [PubMed] [Google Scholar]
- Krongrad A, Wilson CM, Wilson JD, Allman DR, McPhaul MJ. Androgen increases androgen receptor protein while decreasing receptor mRNA in LNCaP cells. Molecular and Cellular Endocrinology. 1991;76:79–88. doi: 10.1016/0303-7207(91)90262-q. [DOI] [PubMed] [Google Scholar]
- Langeler EG, van Uffelen CJ, Blankenstein MA, van Steenbrugge GJ, Mulder E. Effect of culture conditions on androgen sensitivity of the human prostatic cancer cell line LNCaP. Prostate. 1993;23:213–223. doi: 10.1002/pros.2990230304. [DOI] [PubMed] [Google Scholar]
- Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? Journal of Clinical Endocrinology and Metabolism. 2003;88:2972–2982. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- Litvinov IV, Vander Griend DJ, Antony L, Dalrymple S, De Marzo AM, Drake CG, Isaacs JT. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. PNAS. 2006;103:15085–15090. doi: 10.1073/pnas.0603057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E, Li X, Yan F, Zhao Q, Wu X. Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. Journal of Biological Chemistry. 2004;279:17283–17288. doi: 10.1074/jbc.C300549200. [DOI] [PubMed] [Google Scholar]
- Mendez J, Zou-Yang XH, Kim SY, Hidaka M, Tansey WP, Stillman B. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Molecular Cell. 2002;9:481–491. doi: 10.1016/s1097-2765(02)00467-7. [DOI] [PubMed] [Google Scholar]
- Mottet N, Lucas C, Sene E, Avances C, Maubach L, Wolff JM. Intermittent androgen castration: a biological reality during intermittent treatment in metastatic prostate cancer? Urologia Internationalis. 2005;75:204–208. doi: 10.1159/000087794. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Taraviras S, Lygerou Z, Nishimoto T. The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. Journal of Biological Chemistry. 2001;276:44905–44911. doi: 10.1074/jbc.M105406200. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Sugimoto N, Roukos V, Nakanishi Y, Saijo M, Obuse C, Tsurimoto T, Nakayama KI, Nakayama K, Fujita M, et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO Journal. 2006;25:1126–1136. doi: 10.1038/sj.emboj.7601002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, et al. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. Journal of Clinical Oncology. 2004;22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- Petersen BO, Lukas J, Sorensen CS, Bartek J, Helin K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO Journal. 1999;18:396–410. doi: 10.1093/emboj/18.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarmby VE, Yarbrough WG, Lubahn DB, French FS, Wilson EM. Autologous down-regulation of androgen receptor messenger ribonucleic acid. Molecular Endocrinology. 1990;4:22–28. doi: 10.1210/mend-4-1-22. [DOI] [PubMed] [Google Scholar]
- Rao PN, Johnson RT. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature. 1970;225:159–164. doi: 10.1038/225159a0. [DOI] [PubMed] [Google Scholar]
- Senga T, Sivaprasad U, Zhu W, Park JH, Arias EE, Walter JC, Dutta A. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. Journal of Biological Chemistry. 2006;281:6246–6252. doi: 10.1074/jbc.M512705200. [DOI] [PubMed] [Google Scholar]
- Sohoel H, Jensen AM, Moller JV, Nissen P, Denmeade SR, Isaacs JT, Olsen CE, Christensen SB. Natural products as starting materials for development of second-generation SERCA inhibitors targeted towards prostate cancer cells. Bioorganic & Medicinal Chemistry. 2006;14:2810–2815. doi: 10.1016/j.bmc.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Tabancay AP, Jr, Forsburg SL. Eukaryotic DNA replication in a chromatin context. Current Topics in Developmental Biology. 2006;76:129–184. doi: 10.1016/S0070-2153(06)76005-7. [DOI] [PubMed] [Google Scholar]
- Takeda DY, Dutta A. DNA replication and progression through S phase. Oncogene. 2005;24:2827–2843. doi: 10.1038/sj.onc.1208616. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Molecular Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GH, Stoeber K. Cell cycle markers in clinical oncology. Current Opinion in Cell Biology. 2007;19:672–679. doi: 10.1016/j.ceb.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ. Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Molecular Cancer Therapeutics. 2003;2:835–843. [PubMed] [Google Scholar]
- Woo RA, Poon RY. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle. 2003;2:316–324. [PubMed] [Google Scholar]