

Abstract
Preclinical data suggest that PIK3CA mutations predict response to PI3K/AKT/mTOR inhibitors. Concomitant KRAS or BRAF mutations may mediate resistance. Therefore tumors from patients referred to the Phase I Program for targeted therapy starting in October 2008 were analyzed for PIK3CA mutations using PCR-based DNA sequencing of exons 9 and 20. Consecutive patients with diverse tumor types and PIK3CA mutations were treated whenever possible with agents targeting the PI3K/AKT/mTOR pathway. Overall, PIK3CA mutations were detected in 25 of 217 patients (11.5%) (exon 9, n=11; exon 20, n=14). In tumor types with >10 patients tested, PIK3CA mutations were most frequent in endometrial (3/14, 21%), ovarian (5/30, 17%), colorectal (9/54, 17%), breast (2/14, 14%), cervical (2/15, 13%), and squamous cell cancer of head and neck (1/11, 9%). Seventeen of the 25 patients (68%) with PIK3CA mutations were treated on a protocol that included a PI3K/AKT/mTOR pathway inhibitor, and 6 (35%) achieved a partial response. In contrast, only 15 of 241 patients (6%) without documented PIK3CA mutations treated on the same protocols responded (p=0.001). Six of the 17 (35%) patients with PIK3CA mutations had simultaneous KRAS or BRAF mutations (colorectal, n=4; ovarian, n=2). Colorectal cancer patients with PIK3CA and KRAS mutations did not respond to therapy, while both ovarian cancer patients with PIK3CA and KRAS or BRAF mutations did. In conclusion, PIK3CA mutations were detected in 11.5% of patients with diverse solid tumors. The response rate was significantly higher for patients with PIK3CA mutations treated with PI3K/AKT/mTOR pathway inhibitors than for those without documented mutations.
Keywords: PIK3CA mutation, RAS mutation, RAF mutation, Cancer, Clinical trial
Introduction
Recently, major therapeutic advances have been made in tumors with druggable targets (1-4). These include the highly successful use of KIT kinase inhibitors in KIT mutation-positive gastrointestinal stromal tumors (GIST), ABL kinase inhibitors in BCR-ABL-positive chronic myelogenous leukemia (CML), and BRAF inhibitors in BRAF mutation-positive melanoma (1, 2, 4). Common solid tumors, such as breast, lung, and colorectal cancer remain difficult to treat, perhaps in part because they are heterogeneous, with each subset of patients having different molecular abnormalities (3). Identifying relevant molecular subtypes within heterogeneous diseases, and matching patients with appropriate targeted agents or combinations of them is crucial to future therapeutic progress (5).
The phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR signaling pathway is activated in many different cancers (Supplementary Figure 1) (6). Activation is frequently mediated by mutations in the p110α subunit of PI3K called PIK3CA, with most mutations (>80%) occurring either in exon 9, which codes for the helical domain, or exon 20, which codes for the kinase domain (Supplementary Figure 2) (7). Preclinical studies suggested that PIK3CA mutations may predict for response to PI3K inhibitors (8).
We investigated the PIK3CA mutation status of patients referred to the Phase I Clinical Trials Program clinic (known as the Clinical Center for Targeted Therapy). Whenever possible, patients with PIK3CA mutations were offered treatment targeting the PI3K/AKT/mTOR pathway and their clinical outcomes were analyzed.
Patients and Methods
Patients
We investigated the PIK3CA mutation status of patients with advanced tumors and available tissue referred to the Department of Investigational Cancer Therapeutics (Phase I Clinical Trials Program) at The University of Texas M. D. Anderson Cancer Center (M. D. Anderson) starting in October 2008. The registration of patients in the database, pathology assessment, and mutation analysis were performed at M. D. Anderson. Eligible patients were those referred for clinical trials of targeted therapeutic agents. The study and all treatments were conducted in accordance with the guidelines of the M. D. Anderson Institutional Review Board.
Tissue samples and mutation analyses
PIK3CA mutations were investigated in archival formalin-fixed, paraffin-embedded tissue blocks or material from fine needle aspiration biopsy obtained from diagnostic and/or therapeutic procedures. All histologies were centrally reviewed at M. D. Anderson. PIK3CA mutation testing was performed in the Clinical Laboratory Improvement Amendment–certified Molecular Diagnostic Laboratory (MDL) within the Division of Pathology and Laboratory Medicine at M. D. Anderson. DNA was extracted from microdissected paraffin-embedded tumor sections and analyzed using a polymerase chain reaction (PCR)-based DNA sequencing method for PIK3CA mutations in codons [c]532-554 of exon 9 (helical domain) and c1011-1062 of exon 20 (kinase domain), which included the mutation hot spot region of the PIK3CA proto-oncogene by Sanger sequencing following amplification of 276 bp and 198 bp amplicons, respectively, utilizing primers designed by the M.D. Anderson MDL. Whenever possible, in addition to PIK3CA, mutation analysis was done for KRAS and NRAS c12, c13, and c61 mutations of exon 2; and BRAF codon 595-600 mutations of exon 15 by pyrosequencing as previously described (9).
Treatment and evaluation
Consecutive patients with underlying PIK3CA mutations were enrolled whenever possible in clinical trials containing inhibitors of the PI3K/AKT/mTOR pathway, particularly protocols with anti-mTORC1 (rapalog)-based regimens or regimens containing PI3K inhibitors. Treatment continued until disease progression or unacceptable toxicity occurred.
Treatment was carried out according to the specific requisites in the treatment protocols selected.
Assessments, including history, physical examination, and laboratory evaluations, were performed as specified in each protocol, typically before the initiation of therapy, weekly during the first cycle, and then, at a minimum, at the beginning of each new treatment cycle. Efficacy was assessed from computed tomography (CT) scans and/or magnetic resonance imaging (MRI) at baseline before treatment initiation and then every 2 cycles (6-8 weeks). All radiographs were read in the Department of Radiology at M. D. Anderson and reviewed in the Department of Investigational Cancer Therapeutics tumor measurement clinic. Responses were categorized per RECIST 1.0 criteria and were reported as best response (10). In brief, complete response (CR) was defined as the disappearance of all measurable and non-measurable disease; partial response (PR) was defined as at least a 30% decrease in the sum of the longest diameter of measurable target lesions; progressive disease (PD) was defined as at least a 20% increase in the sum of the longest diameter of measurable target lesions, or unequivocal progression of a non-target lesion, or the appearance of a new lesion; and stable disease (SD) was defined as neither sufficient shrinkage to qualify for partial response nor sufficient increase to qualify for progressive disease.
Statistical analysis
Statistical analysis was verified by our statistician (XW). Fisher's exact test was used to assess the association among categorical variables and PIK3CA mutation status. The Wilcoxon rank-sum test assessed the association between age and PIK3CA mutation status. Time to progression (TTP) was defined as the time interval from the start of therapy to the first observation of disease progression or death, whichever occurred first. All tests were two-sided, and a P value less than 0.05 was considered statistically significant. All statistical analyses were carried out using SAS 9.1 software (SAS Institute, Cary, NC).
Results
Patients
A total of 217 patients with advanced tumors were analyzed for the presence of PIK3CA mutations. One-hundred-and-thirty-one (60%) patients were women and 86 (40%) were men (Table 1). The median age was 56 years (range 13 to 91 years). One-hundred-and-seventy-six (81%) were Caucasians, 19 (9%) African Americans, 12 (6%) Hispanic, and 10 (4%) Asians. Fifty-four (25%) had colorectal cancer, 30 (14%) ovarian cancer, 18 (8%) melanoma, 14 (6%) breast cancer, 14 (6%) endometrial cancer, 8 (4%) squamous cell cervical cancer, 7 (3%) cervical adenocarcinoma, 8 (4%) soft tissue sarcoma (excluding GIST), and 11 (5%) squamous cell cancer of head and neck. A variety of other tumors made up the rest of the patients (Table 1).
Table 1. Patients characteristics and distribution of PIK3CA mutations.
| Variable | Patients (n=217) | Patients with PIK3CA mutations in the category | ||
|---|---|---|---|---|
| N | % | N | % | |
| Sex | ||||
| Male | 86 | 40 | 11 | 13 |
| Female | 131 | 60 | 14 | 11 |
| Age | ||||
| < 50 years | 66 | 30 | 8 | 12 |
| 50-70 years | 127 | 59 | 17 | 13 |
| > 70 years | 24 | 11 | 0 | 0 |
| Ethnicity | ||||
| Caucasian | 176 | 81 | 17 | 10 |
| African American | 19 | 9 | 4 | 21 |
| Hispanic | 12 | 6 | 1 | 8 |
| Asian | 10 | 4 | 3 | 30 |
| Tumor type | ||||
| Colorectal | 54 | 25 | 9 | 17 |
| Ovarian | 30 | 14 | 5 | 17 |
| Melanoma | 18 | 8 | 0 | 0 |
| Breast | 14 | 6 | 2 | 14 |
| Endometrial | 14 | 6 | 3 | 21 |
| Cervix | 15 | 7 | 2 | 13 |
| Soft tissue sarcomas | 8 | 4 | 0 | 0 |
| Non-small cell lung cancer | 7 | 3 | 1 | 14 |
| Small cell lung cancer | 2 | <1 | 0 | 0 |
| Head & Neck: squamous | 11 | 5 | 1 | 9 |
| Head & Neck: adenoid cystic | 3 | 1 | 0 | 0 |
| Renal | 4 | 2 | 0 | 0 |
| Parotid | 4 | 2 | 0 | 0 |
| Thymoma | 3 | 1 | 0 | 0 |
| Pancreatic | 3 | 1 | 1 | 33 |
| Neuroendocrine | 3 | 1 | 0 | 0 |
| Vulvar: squamous | 2 | <1 | 0 | 0 |
| Adrenocortical | 2 | <1 | 0 | 0 |
| Small intestine | 1 | <1 | 1 | 100 |
| Other cancers | 19 | 9 | 0 | 0 |
PIK3CA mutations
PIK3CA proto-oncogene mutations were detected in 25 of the 217 patients (11.5%). In 11 patients, a mutation in exon 9 was detected: 8 in c545, 1 in c542, 1 in c546, and 1 in c545/c549. Exon 20 mutations were found in the 14 remaining individuals: 10 in c1047, 3 in c1049, and 1 in c1043 (Table 2). In tumor types with more than three patients tested, PIK3CA mutations were most frequent in endometrial cancer (3 of 14 patients [21%]). Mutations were also present in 5 of 30 patients (17%) with ovarian cancer, in 9 of 54 patients (17%) with colorectal cancer, in 2 of 14 patients (14%) with breast cancer, in 2 of 15 patients (13%) with cervical cancer (both of whom had squamous histology), in 1 of 7 patients (14%) with non-small cell lung cancer, and in 1 of 11 patients (9%) with squamous cell cancer of head and neck (Table 1). PIK3CA mutation status was not significantly associated with age, gender, or race (Fisher's exact test).
Table 2. Characteristics of 25 patients with PIK3CA mutations.
| Case number | Tumor type | Histology | PIK3CA mutations | Other mutations | Response (RECIST %) | TTP (weeks) |
|---|---|---|---|---|---|---|
| 1 | Ovarian | Clear cell carcinoma | c1047 | BRAF c600 | PR (-34) | 17.9+ |
| 2 | Ovarian | High-grade endometrioid carcinoma | c1047 | None | - | - |
| 3 | Ovarian | Clear cell carcinoma | c1049 | None | SD (-4) | 23.3 |
| 4 | Ovarian | High-grade carcinoma | c542 | None | SD (-6) | 17.9+ |
| 5 | Ovarian | High-grade carcinoma | c546 | KRAS c61 | PR (-34) | 30.6+ |
| 6 | Colorectal | Moderately differentiated adenocarcinoma | c1047 | KRAS c12 | PD (+87) | 7.9 |
| 7 | Colorectal | Moderately differentiated adenocarcinoma | c545 | KRAS c12 | - | - |
| 8 | Colorectal | Moderately differentiated adenocarcinoma | c1049 | KRAS c12, c13 | SD (-2) | 6.4+ |
| 9 | Colorectal | Moderately differentiated adenocarcinoma | c1047 | KRAS c12 | - | - |
| 10 | Colorectal | Moderately differentiated adenocarcinoma | c545 | KRAS c12 | SD (-3) | 5.3 |
| 11 | Colorectal | Moderately differentiated adenocarcinoma | c545 | KRAS c12 | SD (+14) | 16.1+ |
| 12 | Colorectal | Moderately differentiated adenocarcinoma | c545 | None | SD (-5) | 11+ |
| 13 | Colorectal | Moderately differentiated adenocarcinoma | c1047 | None | PDa | 2.9 |
| 14 | Colorectal | Moderately differentiated adenocarcinoma | c545 | KRAS c61 | - | - |
| 15 | Endometrial | High-grade endometrial | c1047 | None | PR (-37) | 35.3 |
| 16 | Endometrial | Endometrial adenocarcinoma, grade 2 | c1047 | None | PR (-60) | 59+ |
| 17 | Endometrial | Endometrial adenocarcinoma, grade 2 | c1049 | None | PD (+46) | 5.6 |
| 18 | Breast | Lobular carcinoma, ER+, PR+, HER2/neu- | c1047 | None | PR (-37) | 25+ |
| 19 | Breast | Ductal carcinoma, grade 2, ER+, PR+, HER2/neu- | c1047 | None | - | - |
| 20 | Cervix | Moderately differentiated squamous cell carcinoma | c545 | None | SD (-4) | 19.1 |
| 21 | Cervix | Moderately/poorly differentiated squamous cell carcinoma | c545,c549 | None | PR (-100) | 8.9 |
| 22 | Head & Neck | Poorly differentiated squamous cell carcinoma | c1043 | None | - | - |
| 23 | Lungs | Adenocarcinoma | c545 | None | - | - |
| 24 | Pancreas | Poorly differentiated adenocarcinoma | c545 | KRAS c12 | - | - |
| 25 | Small Intestine | Poorly differentiated adenocarcinoma | c1047 | None | PDa | 8.1 |
TTP, time to progression; c, codon; PR, partial response; SD, stable disease; PD, progressive disease; ER+, estrogen receptor positive; PR+, progesteron receptor positive; HER2/neu-, HER2/neu receptor negative.
clinical progression
not progressing at the time of analysis
Simultaneous RAS and PIK3CA mutations
Preclinical data suggest that activation of RAS mediates resistance to PI3K inhibitors (8). Therefore, RAS mutation status was investigated whenever possible.
KRAS mutations in exon 2 were assessed in 130 patients and identified in 33 individuals (25%) (Table 3). The presence of KRAS mutations was significantly associated with PIK3CA mutations (p=0.03; Fisher's exact test). Indeed, forty-five percent (9 of 20) of patients with a PIK3CA mutation (who were also tested for a KRAS mutation) also had a KRAS mutation, whereas only 22% of patients (24 of 110) without a PIK3CA mutation (who were also tested for KRAS) harbored a KRAS mutation. Of the 33 patients with KRAS mutations, 9 (27%) had simultaneous PIK3CA mutations. In contrast, of the 97 patients without a KRAS mutation, only 11 (11%) had a PIK3CA mutation (p = 0.03). Of the nine patients with simultaneous PIK3CA and KRAS mutations, disease distribution was as follows: colorectal cancer, n=7; pancreatic cancer, n=1; ovarian cancer, n=1 (Table 2).
Table 3. PIK3CA, RAS (K- or N-), and BRAF mutations.
| Oncogene | Mutated (%) | Total tested |
|---|---|---|
| PIK3CA mutations | 25 (11.5) | 217 |
| KRAS mutations | 33 (25) | 130 |
| NRAS mutations | 2 (3) | 62 |
| BRAF mutations | 11 (9) | 122 |
| RAS or BRAF mutations | 46 (31) | 145 |
| KRAS mutations in mutated PIK3CA | 9 (45) | 20 |
| RAS or BRAF mutations in mutated PIK3CA | 10 (50) | 20 |
NRAS c61 mutations were detected in 2 patients (3%) of 62 tested. Both patients had melanoma with wild-type PIK3CA.
Simultaneous BRAF and PIK3CA mutations
BRAF exon 15 mutations were assessed in 122 patients, of whom 11 (9%) had a c600 mutation (Table 3). BRAF mutations were found in patients with melanomas, n=7; colorectal cancer, n=3; and ovarian cancer, n=1. Only one patient had a simultaneous PIK3CA and BRAF mutation detected (ovarian cancer) (Table 2).
Response in patients with PIK3CA mutations treated with PI3K/AKT/mTOR inhibitors
Seventeen of 25 patients with an underlying PIK3CA mutation were enrolled in clinical trials that included a PI3K/AKT/mTOR inhibitor. These patients were refractory to a median of four prior therapies (range,1 to 12). Of these patients, 6 had colorectal cancer, 4 had ovarian cancer, 3 had endometrial cancer, 2 had squamous cell cervical cancer, 1 had small intestine cancer, and 1 had breast cancer (Table 2). Sixteen patients received anti-mTORC1-based regimens and 1 patient was treated with an anti-PI3K-based regimen (Table 4) (11). A PR was observed in 6 (35%) patients. Duration of response was 8.9, 17.9+, 25+, 30.6+, 35.3, and 59+ weeks (with the plus sign indicating ongoing response at the time period noted). (Figures 1 and 2). Seven (41%) patients had stable disease (SD), including 4 (23.5%) patients with prolonged stable disease lasting for more than 16 weeks. In total, 10 patients (59%) achieved either stable disease for over 16 weeks or a PR. Four (23.5%) patients had progressive disease (PD) (two with radiological and two with clinical progression). In comparison, patients without documented PIK3CA mutations (meaning they had no mutation or tumor tissue was unavailable for testing) treated on the same protocols demonstrated a significantly lower response rate of 6% (15 of 241) (p=0.001). Fisher's exact test was used to assess the associations among response (PR, SD or PD) and other patient characteristics, such as age, gender, race, number of prior therapies (> 3 prior therapies vs. ≤ 3 therapies), type of PIK3CA mutation (exon 9 vs. exon 20), and KRAS mutation. None of these variables was significantly associated with response.
Table 4. Therapeutic regimens used to treat patients with PIK3CA mutations.
| Regimen | Mechanism of action | Patients (Case number*) |
% | Reference |
|---|---|---|---|---|
| Temsirolimus | mTORC1 inhibitor | 5 (4, 10, 11, 15, 17) |
29 | NCT00877773 |
| Temsirolimus, bevacizumab | mTORC1 inhibitor, anti-VEGF monoclonal antibody | 2 (8, 13) |
12 | NCT00610493 |
| Temsirolimus, liposomal doxorubicin, bevacizumab | mTORC1 inhibitor, anti-VEGF monoclonal antibody, topo II alpha inhibitor | 8 (1, 3, 5, 12, 16, 18, 20, 21) |
47 | NCT00761644 |
| Temsirolimus, topotecan, bortezomib | mTORC1 inhibitor, topoisomerase I inhibitor, proteasome inhibitor | 1 (25) |
6 | NCT00770731 |
| XL147, carboplatin, paclitaxel | PI3K inhibtor, alkylating agent, microtubule stabilizing agent | 1 (6) |
6 | Wheler et al.11 |
Case numbers are depicted in Table 2
NCT: clinicaltrials.gov identifier
Figure 1. Waterfall plot of patients with PIK3CA mutations treated with PI3K/AKT/mTOR inhibitors.
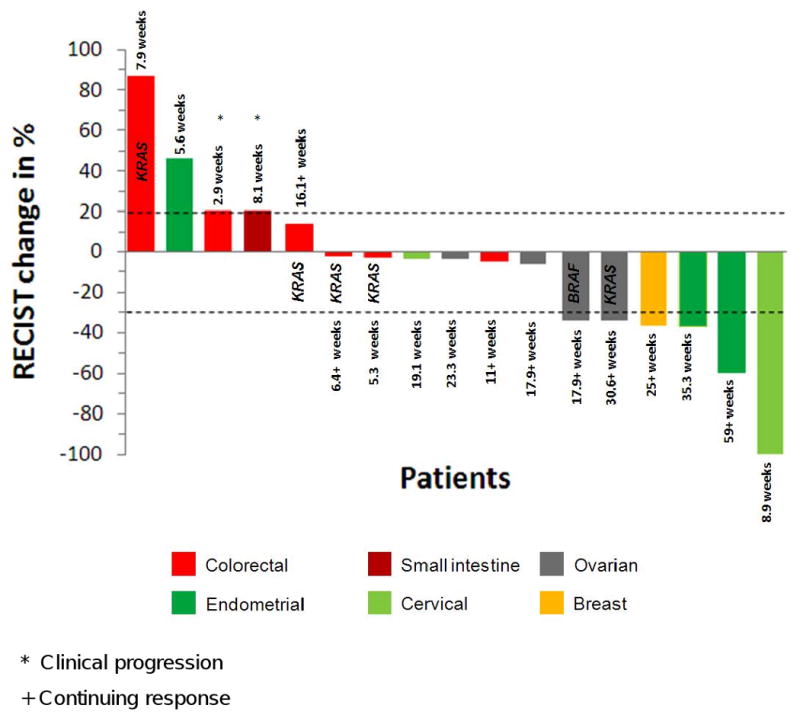
Six PRs (5 confirmed) and 6 minor responses less than PR were observed. The overall response rate was 35%. The best response was complete resolution of all measurable disease with persistence of non-measurable disease in a patient with squamous cell cervical carcinoma treated with an mTOR-based regimen. Four patients with colorectal cancer and 1 patient with ovarian cancer had simultaneous KRAS mutations. One patient with ovarian cancer had a simultaneous BRAF mutation.
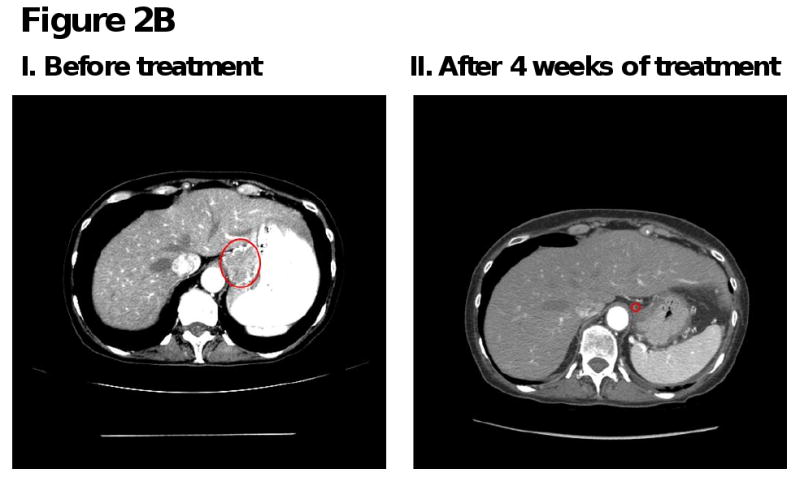
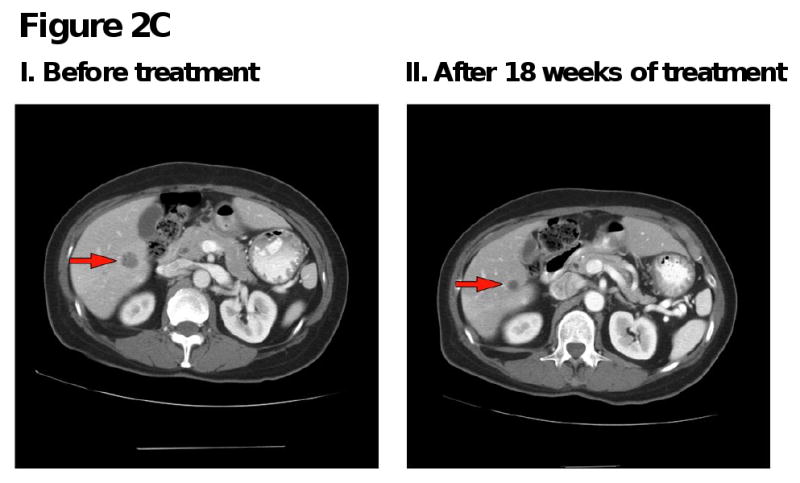
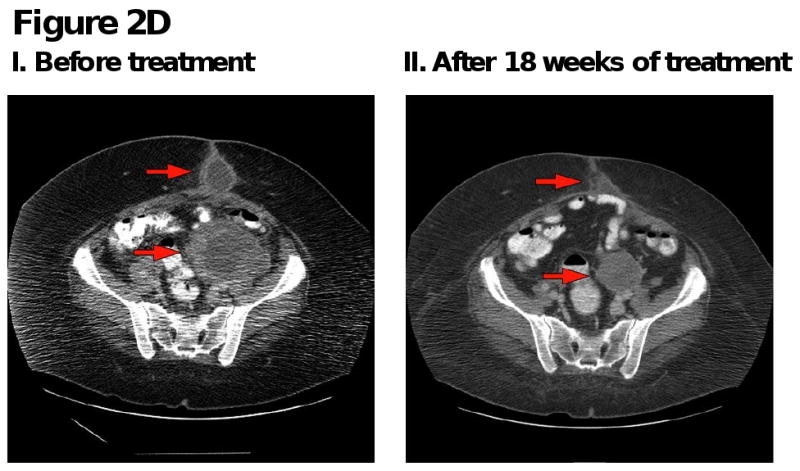
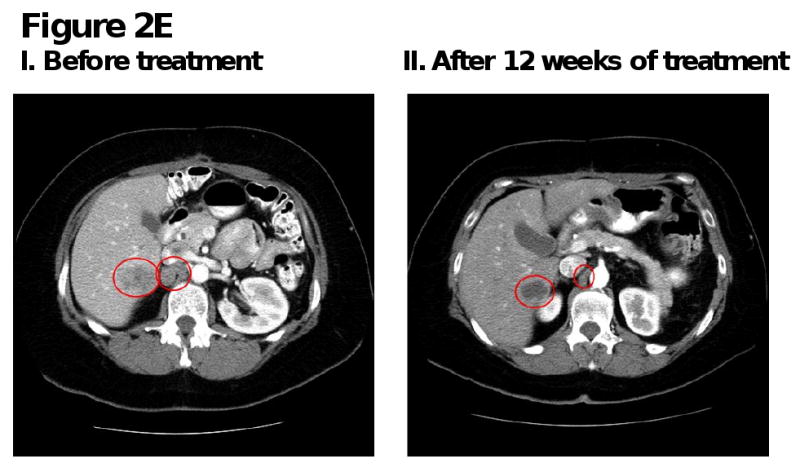
Figure 2. Computed tomography (CT) scans of responding patients. Red arrows or red circles indicate locations of metastases.
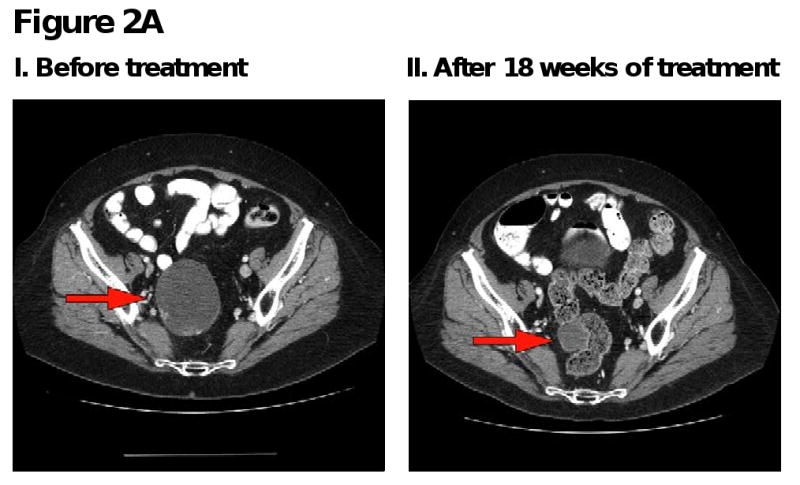
A. Patient with endometrial cancer demonstrating partial response in pelvic mass.
B. Patient with squamous cell cervical cancer demonstrating partial response in gastrohepatic metastasis.
C. Patient with endometrial cancer demonstrating partial response in liver metastases.
D. Patient with ovarian cancer demonstrating partial response in pelvic and subcutaneous metastases.
E. Patient with ovarian cancer demonstrating partial response in liver and aortocaval metastases.
Discussion
We determined that mutations in exon 9 or exon 20 of the PIK3CA proto-oncogene were present in 25 of 217 patients (11.5%) with diverse tumor types, with the incidence being highest (9 – 21%) in patients with endometrial, ovarian, colorectal, breast, cervical cancer, non-small cell lung cancer, and squamous cell cancer of head and neck. Although the number of patients in each tumor type is limited in our study, previous reports have also documented PIK3CA mutations in these tumor types with an incidence as follows: 23%-36% of endometrial cancers (12, 13), 14%-32% of colon cancers (12, 14, 15), 4%-12% of ovarian cancers (16-18), 18-40% of breast cancers usually associated with expression of hormone receptors or HER2/neu (12, 16, 17, 19), 8%-14% of cervical squamous cell cancers (12, 20), and in 11%-33% of squamous cell cancers of the head and neck (12, 21).
Previous preclinical observations have demonstrated that activation of the RAS/RAF/MEK pathway mediates resistance to PI3K inhibitors in PIK3CA-mutant tumors (8). Therefore, we examined our patients for co-existence of PIK3CA mutations with RAS (K- or N-) or BRAF mutations. Forty-five percent of patients (9 of 20) with a PIK3CA mutation (who were also tested for a KRAS mutation) also had a KRAS mutation, whereas only 22% of patients (24 of 110) without a PIK3CA mutation (who were also tested for KRAS) harbored a KRAS mutation. Of the 33 patients with KRAS mutations, 9 (27%) had simultaneous PIK3CA mutations. In contrast, of the 97 patients without KRAS mutation, only 11 (11%) had a PIK3CA mutation (p = 0.03). These results suggest that these mutations (PIK3CA and RAS) commonly co-exist. We also observed that 7 of 9 patients (78%) with colorectal cancer who harbored a PIK3CA mutation also had a KRAS mutation. This rate of dual mutations is similar to that in a previously reported study, which showed mutant KRAS in 56% of patients with colorectal cancer and PIK3CA mutations (14). In one of five patients with PIK3CA-mutant ovarian cancer, we detected a simultaneous KRAS mutation and, in another, a simultaneous BRAF mutation. In contrast, a study from the Middle East showed no coexistence of mutated KRAS or BRAF mutations with PIK3CA mutations in ovarian cancer, though the incidence of PIK3CA mutations in the population studied was quite low (4%) (18).
Whenever possible, our patients with PIK3CA mutations were entered on trials utilizing targeted inhibitors of the PI3K/AKT/mTOR pathway. Their overall response rate on these trials was 35%. Responses were seen in patients with cervical, endometrial, ovarian cancer, and breast cancer (Figure 1). In contrast, of the 241 patients without documented PIK3CA mutations treated on the same protocols, only 15 (6%) responded (p=0.001). The latter response rate is similar to the 4% to 11% response rate reported by our group and others when patients are treated on Phase I trials without molecular selection (22-24). It should be noted that it is conceivable that some of the small group of patients without PIK3CA mutations who responded to PI3K/AKT/mTOR axis inhibitors had other aberrations in PIK3CA not detected by our assay or had other abnormalities such as PTEN loss, that are known to activate PIK3CA (6). Indeed, we have previously shown that PTEN loss can be detected in about 20% of patients in the phase I setting (25).
Consistent with our data, clinical trials with therapies directed against well-defined targets have shown improved results when patients are selected for the presence of those targets, even in the phase I setting (where patients tend to be heavily pretreated and refractory/resistant to multiple conventional drugs), though mostly these trials have reported results in a disease-specific setting. Examples include imatinib mesylate (a KIT and BCR-ABL kinase inhibitor), which demonstrated response rates of over 50% in patients with gastrointestinal stromal tumors (a disorder characterized by KIT kinase mutations) or BCR-ABL-positive chronic myelogenous leukemia (1, 26). More recently, patients with NSCLC and an underlying EML4-ALK fusion also demonstrated a response rate over 50% after treatment with the ALK inhibitor crizotinib as did patients with metastatic malignant melanoma who had an underlying BRAF mutation responded to the BRAF inhibitor PLX4032 (4, 27). In contrast, epidermal growth factor (EGFR) receptor tyrosine kinase inhibitors were initially tested in an unselected patient population and had only modest activity (28). Subsequent “bench to the bedside” forays demonstrated that anti-EGFR tyrosine kinase inhibitors are far more effective in patients with lung cancer and an underlying EGFR mutation (29).
One question that arises is whether or not the detection of additional mutations that might confer resistance would provide more predictive information. In this regard, our patients with colorectal cancer and a simultaneous KRAS mutation did not respond to PI3K/AKT/mTOR axis therapy, which is in agreement with preclinical data suggesting that KRAS activation mediates resistance to PI3K inhibitors (8). In contrast, two patients with ovarian cancer and simultaneously occurring KRAS or BRAF mutations achieved a PR with PI3K/AKT/mTOR axis inhibitors, thus suggesting that such resistance is not absolute or that RAS- or RAF-mutant colorectal cancers behave differently than RAS- or RAF-mutant ovarian cancers.
In conclusion, mutations in PIK3CA occur in a subset of patients with several common cancers. In the current study, the response rate for patients with heavily-pretreated, diverse, advanced cancers and PIK3CA mutations who were given PI3K/AKT/mTOR axis inhibitors was significantly higher than that for patients without documented PIK3CA mutations treated on the same trials. The latter observation is consistent with data that demonstrate low response rates on traditional phase I trials, where molecular testing is not used. One hypothesis that could be generated from this data is that selecting PIK3CA-mutant patients for treatment with PI3K/AKT/mTOR axis inhibitors may predict response independent of underlying histology. Patients with colorectal cancer and the concomitant presence of KRAS and PIK3CA mutations did not respond, consistent with previous experiments indicating that the RAS/RAF/MEK pathway serves as a driver of resistance to PI3K inhibitors. Since the number of patients in our series was small and no randomization occurred, these data must be interpreted cautiously. However, it appears that screening for PIK3CA (and RAS or RAF) mutations warrants further investigation in the application of targeted PI3K/AKT/mTOR inhibitors to the clinic.
Supplementary Material
Acknowledgments
We thank Ms. Joann Aaron for scientific review and editing of this article. This study was supported in part by Grant Number RR024148 from the National Center for Research Resources, a component of the NIH Roadmap for Medical Research (http://nihroadmap.nih.gov/clinicalresearch/overview-translational.asp).
Footnotes
Conflict of Interest: None of the authors have any conflict of interest relevant to the subject of this manuscript.
References
- 1.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001 Apr 5;344(14):1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 2.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002 Aug 15;347(7):472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 3.Braiteh F, Kurzrock R. Uncommon tumors and exceptional therapies: paradox or paradigm? Mol Cancer Ther. 2007 Apr;6(4):1175–9. doi: 10.1158/1535-7163.MCT-06-0674. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart DJ, Kurzrock R. Cancer: the road to Amiens. J Clin Oncol. 2009 Jan 20;27(3):328–33. doi: 10.1200/JCO.2008.18.9621. [DOI] [PubMed] [Google Scholar]
- 6.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009 Aug;9(8):550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 7.Ligresti G, Militello L, Steelman LS, et al. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle. 2009 May 1;8(9):1352–8. doi: 10.4161/cc.8.9.8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ihle NT, Lemos R, Jr, Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009 Jan 1;69(1):143–50. doi: 10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuo Z, Chen SS, Chandra PK, et al. Application of COLD-PCR for improved detection of KRAS mutations in clinical samples. Mod Pathol. 2009;22(8):1023–31. doi: 10.1038/modpathol.2009.59. [DOI] [PubMed] [Google Scholar]
- 10.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000 Feb 2;92(3):205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 11.Wheler JJ, T AM, Bailey HH, Attia S, Falchook GS, Laird AD, Nguyen L, Scheffold C, Kurzrock R. A phase I safety and pharmacokinetics (PK) study of the PI3K inhibitor XL147 (SAR245408) in combination with paclitaxel (P) and carboplatin (C) in patients with advanced solid tumors. AACR-NCI-EORTC: Molecular targets and cancer therapeutics; 2009; 11/17/2009; Boston. 2009. p. 59. [Google Scholar]
- 12.Forbes SA, Tang G, Bindal N, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010 Jan;38(Database issue):D652–7. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005 Dec 1;65(23):10669–73. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- 14.Ogino S, Nosho K, Kirkner GJ, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol. 2009 Mar 20;27(9):1477–84. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004 Apr 23;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 16.Campbell IG, Russell SE, Choong DY, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004 Nov 1;64(21):7678–81. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- 17.Levine DA, Bogomolniy F, Yee CJ, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005 Apr 15;11(8):2875–8. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- 18.Abubaker J, Bavi P, Al-Haqawi W, et al. PIK3CA alterations in Middle Eastern ovarian cancers. Mol Cancer. 2009;8:51. doi: 10.1186/1476-4598-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008 Aug 1;68(15):6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyake T, Yoshino K, Enomoto T, et al. PIK3CA gene mutations and amplifications in uterine cancers, identified by methods that avoid confounding by PIK3CA pseudogene sequences. Cancer Lett. 2008 Mar 8;261(1):120–6. doi: 10.1016/j.canlet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Qiu W, Schonleben F, Li X, et al. PIK3CA mutations in head and neck squamous cell carcinoma. Clin Cancer Res. 2006 Mar 1;12(5):1441–6. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horstmann E, McCabe MS, Grochow L, et al. Risks and benefits of phase 1 oncology trials, 1991 through 2002. N Engl J Med. 2005 Mar 3;352(9):895–904. doi: 10.1056/NEJMsa042220. [DOI] [PubMed] [Google Scholar]
- 23.Jain RK, Lee JJ, Hong D, et al. Phase I oncology studies: evidence that in the era of targeted therapies patients on lower doses do not fare worse. Clin Cancer Res. Feb 15;16(4):1289–97. doi: 10.1158/1078-0432.CCR-09-2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurzrock R, Benjamin RS. Risks and benefits of phase 1 oncology trials, revisited. N Engl J Med. 2005 Mar 3;352(9):930–2. doi: 10.1056/NEJMe058007. [DOI] [PubMed] [Google Scholar]
- 25.Garrido-Laguna I, Janku F, Tsimberidou A, et al. Phosphatase and tensin homologue (PTEN) loss and response to phase I trials targeting PI3K/AKT/mTOR pathway in patients with advanced cancer. J Clin Oncol. 2010;28:e13018. [Google Scholar]
- 26.van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001 Oct 27;358(9291):1421–3. doi: 10.1016/s0140-6736(01)06535-7. [DOI] [PubMed] [Google Scholar]
- 27.Kwak E, C DR, Clark J, Shapiro GI, Maki RG, Ratain MJ, Solomon B, Bang Y, Ou S, Salgia R. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066. J Clin Oncol. 2009;27(15s) abstr 3509. [Google Scholar]
- 28.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005 Jul 14;353(2):123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 29.Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer--is it becoming a reality? Nat Rev Clin Oncol. 2010 Jul;7(7):401–14. doi: 10.1038/nrclinonc.2010.64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.