

Special dendrictic cells invade target tissue at late autoimmune inflammation stage and may terminate inflammation by induction of T cell apoptosis.
Keywords: autoimmunity, apoptosis, tolerance, kidney
Abstract
DCs play critical roles in promotion of autoimmunity or immune tolerance as potent APCs. In our anti-GBM GN model, WKY rats develop severe T cell-mediated glomerular inflammation followed by fibrosis. A DC-like cell population (CD8αα+CD11c+MHC-II+ED1–) was identified in the inflamed glomeruli. Chimera experiments demonstrated that the CD8αα+ cells were derived from BM. The CD8αα+ cells infiltrated glomeruli at a late stage (Days 28–35), coincident with a rapid decline in glomerular inflammation before fibrosis. The CD8αα+ cells isolated from inflamed glomeruli were able to migrate rapidly from the bloodstream into inflamed glomeruli but not into normal glomeruli, suggesting that the migration was triggered by local inflammation. Despite high-level expression of surface and cellular MHC class II molecules, in vitro experiments showed that this CD8αα+ DC-like cell induced apoptosis but not proliferation in antigen-specific CD4+ T cells from T cell lines or freshly isolated from lymph nodes; they were not able to do so in the absence of antigens, suggesting induction of apoptosis was antigen-specific. Furthermore, apoptotic T cells were detected in a large number in the glomeruli at Day 32, coincident with the infiltration of the cells into glomeruli, suggesting that the cells may also induce T cell apoptosis in vivo. A potential role of this CD8αα+ DC-like population in peripheral immune tolerance and/or termination of autoimmune inflammation was discussed.
Introduction
As APCs, DCs play a key role in the immunological reactions throughout the body. With their potent antigen-presentation capacities, DCs can activate naïve T cells and thus, function as a pivotal connection between innate and acquired immunity [1]. DCs are also crucial for autoimmune pathogenesis as a promoter of autoimmunity [2, 3]. On the other hand, DCs may also participate in the maintenance of self-tolerance. A subset of DC is able to induce anergy in T cells, and regulatory DCs can stimulate the differentiation of Tregs [4, 5]. More recently, the CD103+ DC has been identified in a mucosal/dermal lymph node to induce development of Tregs [6, 7]. A CD8+ DC in the spleen is able to induce T cell apoptosis through the FasL pathway [8]. A recent study has shown that a DC of myeloid origin may be responsible for CD8+ T cell tolerance to tumor antigens [9]. Thymic DCs participate in negative selection by inducing apoptosis in autoreactive pre-T cells. The common feature among the above DCs, which play or may play a role in self-tolerance, is that regulation or apoptosis induction of T cells is located in lymphoid organs prior to activation of autoreactive T cells.
Anti-GBM GN is one of the earliest recognized human autoimmune diseases. DCs have long been suspected to play critical roles in various types of GN [10]. Previous studies in humans suggest a role of DCs in promotion of GN pathogenesis [11]. More recent studies were based on animal models for various types of GN [12–17]. One study characterized DCs derived from BM in GN or in healthy mice and found CD11+ cells in the renal interstitium, which may activate T cells [12]. A potential DC with the phenotype of OX6 (MHC class II)+OX42 (CD11b/c)+ from renal mesangium of healthy rats has been described [13]. The authors suggested that those DCs may play roles in GN pathogenesis or acute allograft rejection. Another study identified a potential DC population with the MHC II+ED2– phenotype among glomerular-infiltrating leukocytes in the Thy1.1 GN model [14]. Those previous studies in humans and animals have suggested a possible role of DCs in the initiation of self-reactive T cell activation and promotion of GN pathogenesis.
We have developed a rat model for anti-GBM GN. In our model, severe GN is mediated by autoreactive CD4+ T cells through the immunization of the T cell epitope pCol(28–40) from the autoantigen collagen 4α3 chain or by the transfer of activated T cells into WKY rats [18–20]. Influx of CD4+ T cells into the glomeruli is detectable as early as Day 14 postimmunization [21]. In the present study, we investigated inflammatory cells that had infiltrated target tissue glomeruli. A CD8αα+ DC-like population among those cells was identified. This BM-derived CD8αα+ cell population infiltrated only inflamed tissue and more importantly, induced apoptosis in antigen-specific T cells in vitro, suggesting their unique role in autoimmunity.
MATERIALS AND METHODS
Antibodies
Biotin-labeled mAb to rat CD3 (G.4.18), CD11b/c (OX42) and integrin αE2 (rat DC marker, OX62), CD178 (FasL; MFL4), and CD11c (8A2); PE-labeled mAb to rat CD4 (OX35), CD8α (OX8), and CD8β (clone 341); and FITC-labeled mAb to rat CD8α (OX8), RT.1B (OX6), CD11b (OX42), and various rat/mouse IgG isotype controls were obtained from PharMingen (San Diego, CA, USA). Antibody to CD68 (ED1) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). SR-13, a mAb reactive with rat GBM and TBM, was kindly provided by Dr. Yoshikau Sado (Okayama University, Japan). Anti-rat CD32 mAb (D34-485, PharMingen) was used for Fc block. Anti-rat IgG antibody, HRP-labeled anti-rat IgG antibody, and purified rat IgG were from Southern Biotechnology (Birmingham, AL, USA).
Induction of anti-GBM GN
All procedures involving animals in this study were approved by the Institutional Animal Welfare Committee. Female Wistar Kyoto (WKY) rats (4–6 weeks of age) were purchased from Harlan (Indianapolis, IN, USA) and allowed to acclimate for 3 days in the animal facility at the University of Texas Houston Health Science Center (Houston, TX, USA). Rats were immunized with peptide pCol(28–40) (0.15 μmol), emulsified in CFA. Rats immunized with CFA alone served as controls. GN was evaluated by albuminuria and renal histopathology. Urine albumin concentration was semiquantitated on the SDS-PAGE. Renal tissues were fixed in 4% PFA for immunochemical staining or immunofluorescence.
Isolation of renal tissue-infiltrating inflammatory cells
Renal cortex was sliced, placed in a cold DMEM medium containing 10% FCS, ground against a #100 mesh, and rinsed repeatedly. The single cells were first separated by a cell constrainer (80 μm) and designated as “interstitial cells.” The glomeruli were then isolated to a purity of ∼95% following an established method [21, 22]. The purified glomeruli were incubated with collagenase IV and Accutase® at 37°C for 30 min with periodic stirring. Digested glomerular fragments were eliminated by centrifugations (300 rpm, 5 min), and the single cells were collected by centrifugation (1000 rpm, 10 min). Those cells were designated as “glomerular cells.” Isolated cells were used for further culture or for flow cytometry analyses. As a result of high expression levels of FcγRII on DCs and other APCs, the cells were first incubated with anti-rat CD32 mAb to block nonspecific binding of Fc. The cells were then stained with antibodies or isotype control Ig and analyzed with a flow cytometer (FACSCalibur, Becton Dickinson, San Jose, CA, USA).
Purification of glomerular CD8αα+CD11+ cells
The isolated, bulk glomerular-infiltrating inflammatory cells were adjusted to 107 cells/ml and plated to Φ75 mm culture dishes (4 ml/dish). The cells were allowed to adhere to the dish for 2 h at 37°C. Nonadhesive cells, mostly T cells, were removed by gentle washing with 37°C medium. This procedure was repeated a 2nd time. The plates were incubated further at 37°C overnight and were washed with 37°C medium, and the suspended cells were collected into a conical tube. After 2 washings with complete medium using centrifugation, the cells were counted and used for other experiments. To obtain a highly purified DC population (>95%), the positive selection method was applied using anti-rat CD8α antibody (OX8)-conjugated magnetic microbeads (107 cells/3 μl beads, Miltenyi Biotec, Germany). Before positive selection, CD8+CD3+ T cells were removed using magnetic bead-conjugated anti-CD6 mAb (OX52).
CD8αα+CD11+ DC labeling and transfer
DCs, freshly purified by the magnetic bead method, were washed with HBSS, adjusted to 107cells/ml, and labeled by CFSE (Molecular Probes, Eugene, OR, USA), following a published method [17]. The labeled cells (2×106 cells/rat) were injected into untreated or immunized rats. The recipients were killed 24 h after the cell transfer. Different organs, including lung, liver, pancreas, spleen, lymph nodes, and kidney, were removed and fixed in 3% of PFA or snap-frozen. Serial frozen sections (3 μm), which cut through 10 glomeruli, were counterstained with SR-13 to reveal GBM or TBM. The labeled cells were counted under a fluorescent microscope (Eclipse 80i, Nikon, Japan) and expressed as total cells/glomerulus.
Generation of T cell lines and T cell stimulation
An established method was followed to generate T cell lines [19]. Briefly, lymphocytes were prepared from the immunized rats, and T cells were isolated with a T cell enrichment column (R&D Systems, Minneapolis, MN, USA). The T cells were stimulated by pCol(28–40) peptides in the presence of irradiated thymocytes for 4 days and harvested by a sequence of two Histopaque (Sigma Chemical Co., St. Louis, MO, USA) gradient centrifugations. The cells were allowed to rest for 7 days in the presence of 2.5% of supernatant of Con A (Sigma Chemical Co.)-stimulated normal rat splenocytes. The stimulation was repeated 6 times. T cells (2×105 cells) from a cell line were incubated with isolated CD8αα+CD11+ cells (2×105 cells) in 96-well plates in 200 μl complete T cell medium. As a control, thymocytes were used as APCs. Peptide pCol(28–40) (1–30 μM) was added to each well in triplicate. The cells were incubated at 37°C in a humidified, 5% CO2 atmosphere for 72 h, pulsed with 3[H]-thymidine, 0.5 μCi/well, for 18 h (ICN, Costa Mesa, CA, USA), and harvested onto glass fiber filters using a semiautomatic cell harvester. The incorporated radioactivity was measured by a liquid scintillation counter (Beckman Coulter, Fullerton, CA, USA). The results were expressed as Δcpm (mean triplicate cpm with antigen–mean triplicate cpm without antigen).
Detection of dying or apoptotic T cells
To detect dying or dead cells, T cells were labeled with CFSE and incubated in a 48-well plate with the DCs (see Generation of T cell lines and T cell stimulation above) [20]. Cells were sampled daily from 1 well, and total labeled cells were counted. At the same time, the harvested cells were incubated with PI and analyzed with a flow cytometer. Dead or dying cells showed reduced CFSE intensity and were PI-positive. Proliferating cells were displayed as a series of peaks of reduced CFSE intensities. To detect apoptosis or apoptotic cells, multiple methods were used. A DNA fragmentation assay was used for detection of apoptosis. Briefly, the T cells were harvested 3 days after incubation, and the cellular DNA was isolated, which was separated on a 1% agarose gel. For in vitro experiments, an ApopTag peroxidase in situ apoptosis detection kit was used to detect apoptotic T cells after they were incubated under various conditions (Chemicon, Billerica, MA, USA). Two-color flow cytometry was also used for identifying apoptotic CD4+ T cells, which were isolated from draining lymph nodes, were stained by a PE-labeled anti-CD4 antibody and FITC-labeled Annexin V (BioVision, Mountain View, CA, USA) after in vitro incubation with CD8αα+ DCs.
BM chimera construction
WKY rats at the age of 6 weeks were irradiated with a γ-ray at a lethal dose (80 Gy) in the animal facility of M.D. Anderson Cancer Center (Houston, TX, USA) and transferred with 15 × 106 BM cells from LEW rats, which have identical MHC molecules (RT.1l type). The BM recipients were fed with neomycin containing water (2 mg/ml) for 21 days. The chimerism was confirmed by genotyping on PBL DNA (see below). The chimeras were immunized further with pCol(28–40) 28 days after irradiation and killed at Day 30 postimmunization. Inflammatory cells were isolated from the glomeruli of immunized chimeras and fractionated into CD8+ and CD8– populations using anti-CD8α antibody magnetic beads. Genomic DNA was isolated from the cells. As controls, genomic DNAs were also isolated from the tails, ears, BM, and PBL. The lengths and sequences of three pairs of primers for 3 polymorphic DNA microsatellites (D2Rat199, D3Rat201, and D10Rat11) were obtained through the website www.broad.mit.edu/rat/public and used for PCR-based determination of the genotypes of a microsatellite of cells or tissues.
RESULTS
Identification of CD8+CD11+ cells in inflamed glomeruli
After immunization with nephritogenic T cell epitope pCol(28–40), CD4+ T cells first targeted glomeruli through the recognition of RT.1B/D+ (MHC class II molecules) cells in Bowman's capsule or mesangial cells in WKY rats (Fig. 1A). Glomerular inflammation was histologically detectable at Day 20 postimmunization (Fig. 1B). After Day 30, the glomerular inflammation was replaced gradually by fibrosis (Fig. 1C) [18, 20]. Infiltrating leukocytes were isolated from the target tissue glomeruli and analyzed. To ensure accuracy, the kidneys were perfused with culture medium before isolation, and the glomeruli were isolated to nearly 100% purity (Fig. 2A). Infiltrating leukocytes, together with glomerular cells, were then released from glomeruli after a brief digestion (Fig. 2B). Cells, morphologically similar to DCs, were present among the infiltrating cells (Fig. 2B, right panel). Glomerular cells were analyzed at different time-points postimmunization. Flow cytometry did not detect a significant number of leukocytes in the normal glomeruli (Day 0, Fig. 2C). Glomerular-infiltrating leukocytes, especially CD4+ T cells, were detected by flow cytometry in significant numbers as early as Day 14 and plateaued at Days 30–35 (Fig. 2C). Following a sharp drop in their numbers, the leukocytes vanished completely by approximately Day 40, accompanied by progressive glomerular fibrosis (Fig. 2C). The most abundant cells were CD4+ cells at all time-points examined (Fig. 2C); CD11+ cells were the second-most plentiful, especially at the peak inflammation (Day 30; Fig. 2D). A CD8+ population was also detected (Fig. 2C). However, the CD8 expression level of this population was lower than that of PBL CD8+ T cells (Fig. 2C). The CD8+ cells did not express CD68, as they did not react with antibody ED1 (Fig. 2E). Flow cytometry showed further that a small percentage (5–11%) of CD8+ cells was also CD3+, suggesting that they were CD8+ T cells (Fig. 2F). However, the majority (>90%) of CD8+ cells did not express CD3 (Fig. 2F). A detailed analysis showed that those CD8+CD3– cells were also CD11+ (Fig. 2D). Thus, there were 2 distinct CD11+ populations in the glomeruli: CD8–CD11+ and CD8+CD11+ cells (Fig. 2D, left panel). The CD8+CD11+ cells were present only in the glomeruli and not in the interstitium (Fig. 2D, middle panel). This population was not a result of contamination from PBL, as PBL contained a trace amount of CD8+CD11+ cells (Fig. 2D, right panel). CD8–CD11+ cells will not be discussed in the present study. We focused on the CD8+CD11+ population. A significant number of the CD8+CD11+ population was detected only after Day 25 when T cells were already present [21]; a rapid increase in their number was seen at or after Day 30, and they accounted for 22.3% of all CD11+ cells (Fig. 2D). Indirect immunofluorescence of the kidney sections confirmed the presence of a significant number of CD8+CD11+ cells in glomeruli of immunized rats (Fig. 3). The CD8+ cells in the glomeruli did not express CD68 (ED1 antibody), a marker for rat macrophages (Fig. 3A–D). Time-course observation with immunofluorescence showed that at Day 20, only a few, if any, of the CD8+ cells were present in the inflamed glomeruli, which were characterized by the presence of numerous ED1+ macrophages (Fig. 3A). At Day 28, more CD8+ cells were detected in the glomeruli, coexiting with ED1+ macrophages (Fig. 3B). However, the 2 cell populations distributed at different locations and patterns. Fewer CD8+ cells were seen at Day 35, when fibrosis had advanced (Fig. 3C). The CD8+ cells infiltrated the inflamed glomeruli through Bowman's space (Fig. 3E). High magnification showed that CD8+ cells in the glomeruli had an uneven surface with projections, which was morphologically different from macrophages or T cells (Fig. 3E and F). Expression of CD11 in CD8+ cells was confirmed (Fig. 3G). The CD8+ cells in the inflamed glomeruli were quantitated by counting the cells on serial sections, which cut through at least 10 glomeruli (Fig. 3H and I). A tendency similar to that in flow cytometry analysis was observed: the CD8+ cells invaded glomeruli at a much later stage than ED1+ macrophages (Fig. 3J).
Figure 1. Anti-GBM GN in WKY rats induced by T cell epitope pCol(28–40).

(A) Immunofluorescence shows polarized invasion of CD4+ T cells (green) into a glomerulus through Bowman's capsule at an early stage of disease (Day 17; ×400 original magnification). Note a cluster of RT.1B+ cells (red) in the proximity of CD4 cells. (B) A glomerulus with inflammation at Day 20 (H&E staining; ×300 original magnification). (C) A glomerulus at Day 45 with severe fibrosis (Periodic acid-Schiff staining; ×300 original magnification). (D–F) Kidney sections from a CFA-immunized rat with the identical staining methods as A–C Arrowheads in D outline a glomerulus.
Figure 2. Presence of a CD8+CD11+ population in inflamed glomeruli of immunized WKY rats.
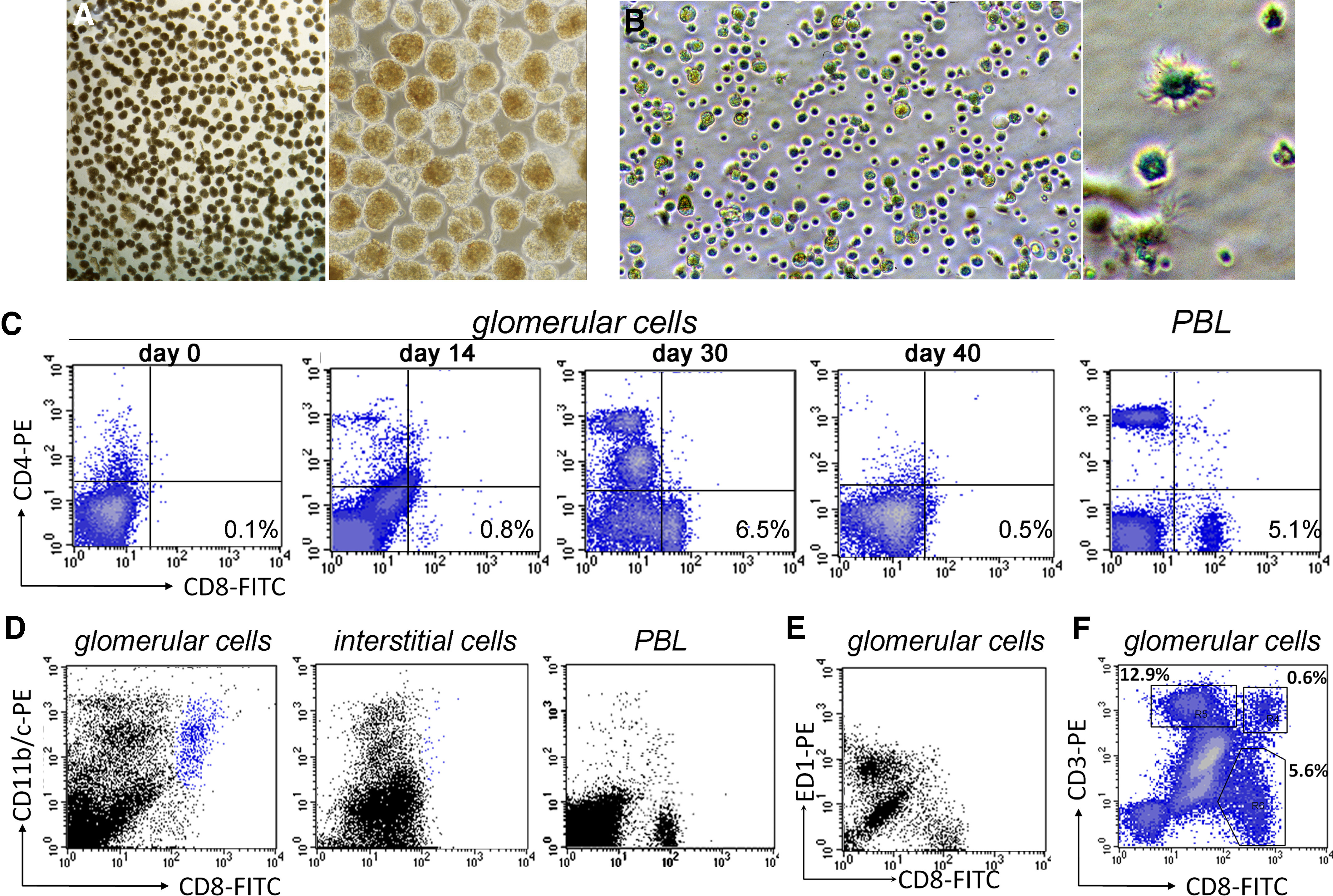
(A) Left panel, micrograph at a low magnification shows purified glomeruli for isolation of glomerular cells (×20 original magnification). Right panel shows purified glomeruli at a higher magnification (×100 original magnification). (B) Left panel is a phase-contrast micrograph showing cells released from the glomeruli after digestion (×200 original magnification). A DC-like cell among infiltrating cells in the right panel (×600 original magnification). (C) Flow cytometries show a CD8+ cell population, detectable at Day 30, among cells isolated from glomeruli. PBLs were used as controls. (D) Flow cytometries on the cells isolated from glomeruli or interstitium of a WKY rat 30 days after immunization show the presence of a CD8+CD11+ cell population (blue) in glomeruli but not in the interstitium. PBLs were used as a control. (E) Flow cytometry shows two distinct cell populations that expressed CD8 and CD68 (ED1), respectively. (F) Flow cytometry shows two distinct CD8+ cell populations among GICs: a minor CD8+CD3+ T cell population and a major CD8+CD3– population.
Figure 3. Invasion of CD8αα+ cells into glumeuli at a later inflammatory stage than ED1+ macrophages.

(A–C) Two-color immunofluorescence detection of CD8+ cells (green) and ED1+ macrophages (red) in the glomeruli at the times postimmunization, as indicated. Glomeruli are outlined by arrowheads. The sections are counter-stained by DAPI; ×300 original magnification. (D) Immunofluorescence shows coexistence of a CD8+ cell (green) with ED1+ macrophages (red; Day 28; ×400 original magnification). (E) Immunofluorescence shows a CD8+ cell, which displays an irregular shape, in Bowman's space (Day 28; ×350 original magnification). (F) Images at a high magnification show 2 CD8+ cells in the glomeruli (×600 original magnification). Note that projections from the cells are visible. (G) Coexpression of CD8 and CD11b/c molecules in CD8+ cells (Day 28; ×400 original magnification). Arrowhead in the synthesized picture indicates epithelial cells of Bowman's capsule. (H and I) Confocal micrographs show CD8+ cells (red) in a normal glomerulus (H) or in a glomerulus 28 days after immunization (I). GBM (green) was counter-stained with mAb SR13. (J) Average numbers of CD8αα+CD11+ cells/glomerulus at different times postimmunization. The cells are quantitated by counting the cells on serial sections that cut through 10 glomeruli.
CD8+CD11+ cell is a DC-like cell
We next cultured the inflammatory cells isolated from inflamed glomeruli of immunized WKY rats at Day 30. The cells that adhered to a plastic plate were mainly CD8–CD11+ and CD8+CD11+ cells. Nonadhesive cells included CD8+CD3+ T cells and were removed by washing at the beginning. After overnight culture, the adhesive CD8+CD11+ cells were collected by gentle washing. Upon culturing, the CD8+CD11+ cells, which reattached to the plastic plate, showed the shape of a DC (Fig. 4A and B). Morphologically, these cells resembled lymphoid DCs. Then, glomerular CD8+CD3– cells were purified using magnetic bead sorting. To ensure purity of CD8+CD3– cells, glomerular CD8+ T cells were pre-removed by mAb OX52 to pan-T cell marker CD6 before positive selection using anti-rat CD8α antibody-conjugated magnetic beads. The majority of the purified CD8+CD11+ cells (72%) expressed higher levels of CD11c and middle levels of CD11b (Fig. 4C). A minor portion (23%) of the cells expressed low levels of CD11c and CD11b. Thus, the purified CD8+ cells formed 2 distinct populations, which were designated as CD11chigh and CD11clow populations (Fig. 4C).
Figure 4. Characterization of CD8αα+ cells that have infiltrated glomeruli after immunization with pCol(28–40) in WKY rats.
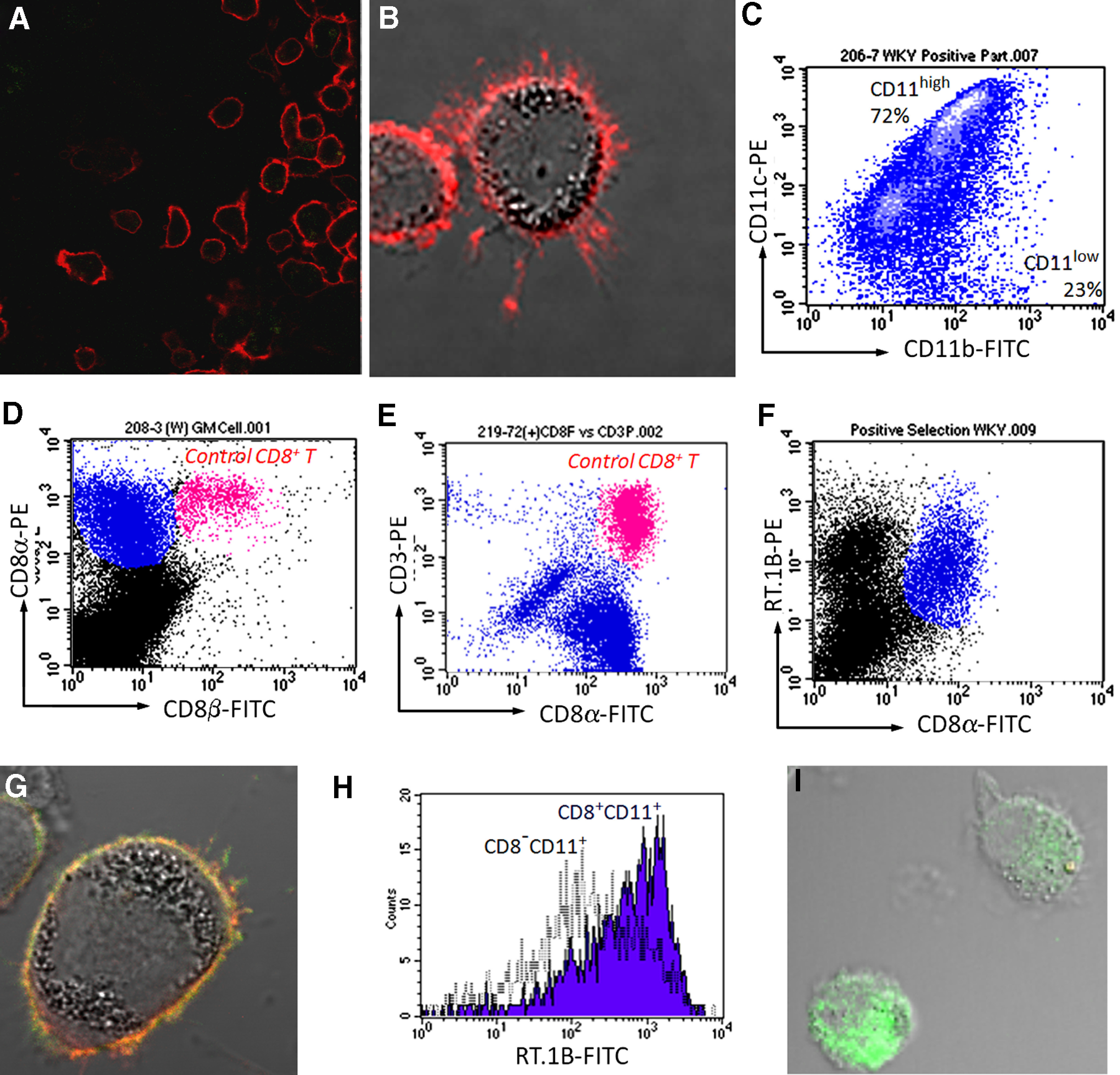
(A) A confocal image of isolated CD8+CD11+ cells after they were stained for CD8α (red; ×200 original magnification). (B) Overlapped fluorescent and DIC image of a CD8+CD11+ cell reveals numerous dendrite-like projections extending from the cell (×600 original magnification). (C) Purified CD8α+CD11+ cells (blue) show two distinct subpopulations (CD11high and CD11low) with different expression levels of CD11c and CD11b. (D) Glomerular (GM)-infiltrating CD8α+CD11+ cells (blue) did not express CD8β, as revealed by flow cytometry. Note that a population of CD8+ T cells (red), added as an internal control, expressed CD8α and CD8β. (E) Flow cytometry shows that purified glomerular CD8α+ cells (blue) did not express CD3. Note that CD8+ T cells (red), added as an internal control, expressed CD8α and CD3. (F) Flow cytometry on GICs shows expression of MHC class II (RT.1B) on CD8αα+ cells (blue). (G) Confocal image demonstrates expression of MHC class II (RT.1B, green) on a CD8αα+ cell (red; ×600 original magnification). (H) A representative histogram shows a higher level of MHC class II RT.1B in CD8α+CD11+ cells (blue) than CD8α–CD11+ cells after an overnight culture. (I) Overlapped fluorescent and DIC image reveals intracellular MHC class II molecule RT.1B (green) in a CD8α+CD11+ cell. Surface RT.1B was blocked before permeablization (×600 original magnification).
Glomerular-infiltrating CD8+ T cells have been described in several types of GN models [23–25]. However, CD8+CD11c+CD3– cells were not reported. We confirmed further that the CD8+CD11c+CD3– cells, which represented more than 90% of CD8+ cells identified in our model, were not of the CD8+ T cells. First, flow cytometry showed that the CD8+CD11+ cells in our model did not react with the antibody to CD8β, suggesting an αα homodimer of the CD8 molecule in contrast to an αβ dimer of the CD8 molecule in CD8+ T cells (Fig. 4D). Thus, the cells were a CD8αα+CD11+ phenotype. Next, the CD8αα+CD11+ cells were purified as described above; as we expected, none of the purified CD8αα+CD11+ cells reacted to anti-CD3 antibody (Fig. 4E, blue population). As an internal control, CD8+ T cells from lymph nodes, which were added to purified CD8αα+CD11+ cells, proved positive to CD3 and CD8 (Fig. 4E, red population). Thus, the glomerular CD8αα+CD11+ cells in our model were not CD8+ T cells. More importantly, flow cytometry and immunofluorescence detected expression of MHC class II (RT.1B) on the surface of the CD8αα+CD11+ cells (Fig. 4F and G). After overnight culture, the CD8αα+CD11+ cells showed a higher level of surface MHC class II (RT.1B) than CD8α–CD11+ cells (probably macrophages; Fig. 4H), suggesting an active synthesis of MHC class II molecules by those cells. To confirm this result further, the cell surface RT.1B was blocked by nonlabeled antibody followed by permeablization with PFA; intracellular RT.1B was readily detectable (Fig. 4I). A subset of NK cells expresses CD8α but not CD8β, and they may be positive for CD11b as well. However, NK cells do not express MHC class II. In addition, the CD8αα+CD11+ cells were morphologically similar to DC but not NK cells (Figs. 2B, 3E and F, and 4A and B). Flow cytometry showed further that CD8αα+CD11c+ cells did not express CD94, a marker for NK cells (data not shown). Thus, the CD8αα+CD11+ cells were not a subset of NK cells. In summary, all of the above experiments suggested that the glomerular CD8αα+CD11c+ cells may be a type of DCs. However, these cells did not express OX62, a common marker for rat DCs (data not shown) [26]. Thus, this population was not a typical rat DC.
The CD8αα+CD11c+ MHC+ cells invade glomeruli at a late inflammatory stage
CD8αα+CD11c+ cells were purified from WKY rats at Day 30 or 35 postimmunization with a positive-selection method using anti-CD8, antibody-conjugated magnetic beads to a purity of over 90%. The purified cells were then labeled with CFSE and transferred immediately into normal or immunized WKY rats. The kidneys of recipients were collected 24 h after the transfer. The WKY rats, which had been immunized 10, 20, or 35 days ago (2 rats/group), were designated as Groups –10d, –20d, and –35d. The labeled cells, which had migrated into the glomeruli, were quantitated by counting the CFSE cells on serial sections as described previously (Fig. 5). In the normal kidneys (0d group), only very few, if any, labeled cells (0.09±0.02 cells/glomerulus) were found in the glomeruli (Fig. 5A). In the rats from the –10d group, which showed no sign of glomerular damage, the number of the labeled cells (0.15±0.05 cells/glomerulus) was similar to that seen in the normal rats. However, the number of transferred CD8αα+CD11c+ cells in glomeruli increased significantly in the rats from the –20d group, which had developed proteinuria as well as histologically recognizable glomerular inflammation (Fig. 5B). Rats from the –35d group showed a >6-fold increase over the –20d group in the number of labeled cells that had migrated into the glomeruli (6.8±0.72 cells/glomerulus; Fig. 5C–E). The labeled cells were not found in a significant number in other tissues, such as the liver and lungs, in any rats. The labeled CD8αα+CD11c+ cells, which had migrated into the glomeruli, displayed dendrite-like extensions (Fig. 5C and D). We have reported previously that labeled antigen-specific T cells are able to migrate into normal glomeruli [20]. Thus, unlike the antigen-specific T cells in our model, the CD8αα+CD11c+ cells may invade glomeruli only at mid- to late-inflammatory stages. Glomerular inflammation peaked at Day 30, followed by a rapid decline; the inflammation was replaced gradually by glomerular fibrosis (Figs. 1 and 2). Thus, invasion of the CD8+ cells was coincident with the decline of inflammation.
Figure 5. CD8αα+CD11+ cells migrate into glomeruli at a late inflammatory stage but not normal glomeruli.
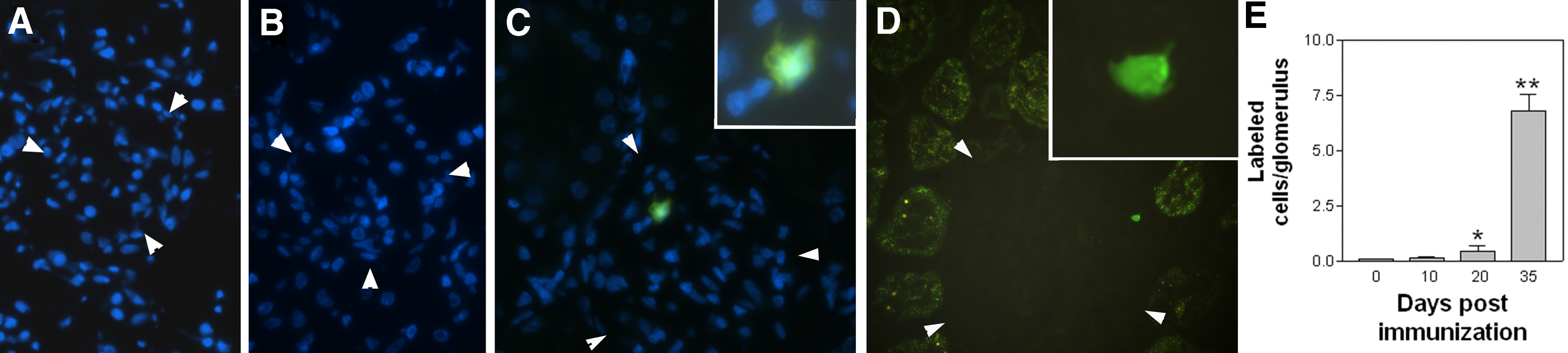
(A–D) Fluorescent micrographs show transferred, CFSE-labeled CD8αα+CD11+ cells (green) in glomeruli of WKY rats, which had been immunized 0 (A), 20 (B), and 35 days (C and D) previously (×300 original magnification). Nuclei were counterstained by DAPI, and glomeruli are outlined by arrowheads. (C and D) Insets display transferred CD8αα+CD11+ cells at a high magnification, showing visible dendrite-like projections (×600 original magnification). (E) Summary of numbers of transferred CD8αα+CD11+ cells present in glomeruli at different time-points after immunization. *P < 0.05, and **P < 0.005 in t test between Day 0 and other groups.
The CD8αα+CD11c+ MHC+ cells are derived from BM
The origin of the glomerular CD8αα+CD11c+ cells was investigated. WKY and LEW rats belong to the MHC RT.1l type, and thus, they are immune-compatible [21]. A BM chimera was constructed using WKY as host and LEW as a BM donor. The chimera, designated as WL, was immunized with nephritogenic T cell epitope pCol(28–40) 28 days post-BM transfer. The cells were isolated from glomeruli 30 days after immunization. These cells, designated as GICs, were sorted further into CD8+ and CD8– populations using magnetic beads. Flow cytometry analysis showed the CD8+ population to be over 90% purity as a CD8+CD11c+ population (Fig. 6A). Both cell populations were subject for genotyping using 3 polymorphic DNA microsatellites (D2Rat199, D3Rat201, and D10Rat11). Several other tissues or cells, such as BM, PBL, the tail, and the ear, were used as controls (Fig. 6B). PCR, on the genomic DNA, demonstrated that CD8+ cells were the LEW genotype, and CD8– cells had a mixed genotype of LEW and WKY (Fig. 6B). As we expected, BM of chimera rats had a LEW genotype, and other tissues were mainly the WKY type (Fig. 6B). Thus, the glomerular-infiltrating CD8αα+CD11c+ cells were derived from BM.
Figure 6. Glomerular-infiltrating CD8αα+CD11+ cells are derived from BM.
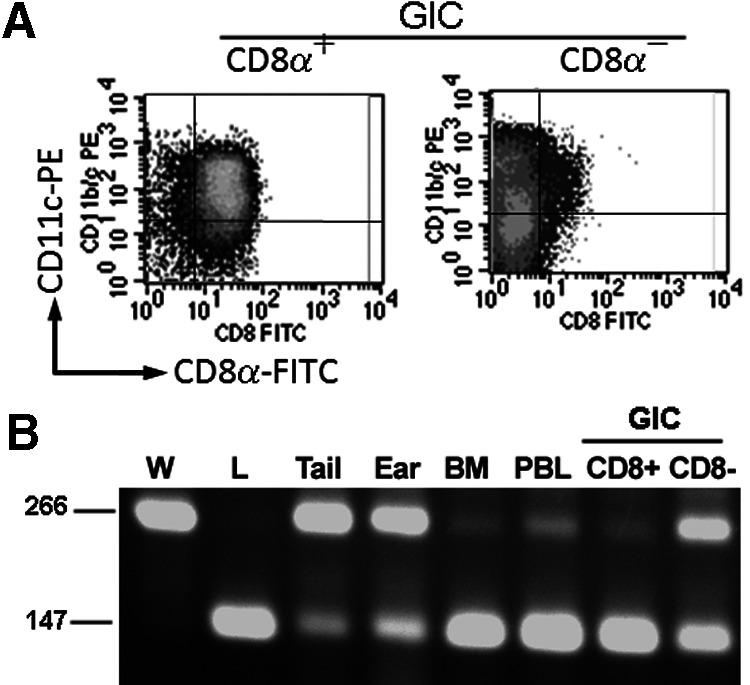
(A) Flow cytometry shows a CD8αα+ cell population positively selected from GIC of a LEW to WKY (WL) BM chimera 30 days after immunization (left panel). The right panel shows the CD8α– population after positive selection. (B) PCR genotyping of various tissues or cells from a WL chimera using polymorphic microsatellites. The result of a representative microsatellite D2Rat199 is shown. W, WKY; L, LEW. Sizes of D2Rat199 for WKY and LEW are indicated at left.
The CD8αα+CD11c+ DC induces death of antigen-specific T cells
Active expression of MHC class II on the glomerular-infiltrating CD8αα+ cells prompted us to hypothesize that by presenting GBM autoantigen to T cells, CD8αα+CD11c+ cells might be an excellent APC in establishing and promoting glomerular inflammation, especially at the initiation stage of the disease. If it were true, the CD8αα+CD11c+ cells, especially those isolated from an early stage of GN, would show potent APC function in activation of T cells. As only a trace number of CD8αα+CD11c+ GICs was available in WKY at an early stage, the glomerular CD8αα+CD11c+ cells were isolated 30 days after immunization, when the CD8αα+CD11c+ cells were most abundant. As we have shown earlier, the cells isolated at this stage were composed of a major CD8αα+CD11chigh population and a minor CD8αα+CD11clow population (refer to Fig. 4C). A multiple panning method was used for isolation to avoid potential disturbance in their function. The purified cells showed 66% purity as the CD8αα+CD11chighRT.1B (MHC II)+ population. To eliminate contamination of other APCs, ED1+ macrophages were removed by magnetic beads. The purified CD8αα+CD11chighRT.1B+ population contained only a trace number of other RT.1B+ cells (1.7%; Fig. 7A) and was used for experiments. As a positive control for T cell activation, the pCol(28–40)-specific T cells were incubated with irradiated, thymic APCs (standard APCs for rats) in the presence of antigen; a vigorous T cell proliferation, as revealed by a high cpm as a result of incorporation of 3H-thymdine, was observed (Fig. 7B). Incubation of the pCol(28–40)-specific T cells with CD8αα+CD11+ cells in the presence of pCol(28–40) led to an unusually lower cpm than T cells alone (Fig. 7B), suggesting the death of pCol(28–40)-specific T cells. Death of T cells was morphologically confirmed at Day 3 postincubation under phase-contrast microscope (Fig. 7E). However, T cells that were incubated with CD8αα+CD11+ cells in the absence of pCol(28–40) survived, suggesting that antigen pCol(28–40) triggered cell death. This was confirmed by 6 experiments using 3 independent pCol(28–40)-specific T cell lines. Thus, the death of T cells induced by CD8αα+CD11+ cells was antigen-dependent. To determine when the death of T cells occurred, the CSFE-labeled T cells were incubated with the CD8αα+CD11+ cells in the presence or absence of the antigen. A rapid decrease in the CSFE-labeled T cells occurred at Day 3 after incubation (Fig. 7C). On the other hand, only a slight reduction in the number of the labeled T cells was observed in the incubation without the antigen (Fig. 7C). After the cells were stained by PI, flow cytometry demonstrated a large dead/dying T cell population as a PI+CSFE+ population at Day 3 (Fig. 7D, left panel). As a control, a vigorous T cell proliferation, evidenced by multiple CSFE peaks, was observed when they were incubated with thymocytes and the peptide (Fig. 7D, 2nd panel from right). Glomerular CD8αα+CD11+ cells were positively selected to a high purity of 91% with magnetic beads (Fig. 7F). The glomerular cells left from positive selection were designated as non-CD8+ cells. Incubation of the purified CD8+ cells and pCol(28–40) with T cells (98%) leads to a >90% reduction in the number of labeled T cells at Day 5 (Fig. 7G). On the other hand, incubation with non-CD8+ glomerular cells resulted in an ∼20% reduction in labeled T cells (Fig. 7G).
Figure 7. CD8αα+CD11+ cells induce death of antigen-specific T cells through antigen presentation.

(A) Glomerular-infiltrating CD8αα+CD11+ DCs were isolated for experiments using a panning method and shown as a CD8α+RT.1B+ population (upper right quadrant). Only a small population of CD8α–RT.1B (MHC II)+ was present (upper left quadrant). (B) Proliferative responses of pCol(28–40)-specific T cells after incubation with the CD8αα+CD11+ DC (CD8) or thymocytes (ThyC) in the presence or absence of pCol(28–40). Cell proliferation is expressed as Δcpm. (C) Changes in numbers of CFSE-labeled pCol(28–40)-specific T cells after incubation with the CD8αα+CD11+ cells and pCol(28–40) (●) or with the CD8αα+CD11+ cells alone (○). Note a sharp drop between Days 2 and 3 in the T cell number only after incubation with the CD8αα+CD11+ cells and pCol(28–40). (D) Death of CFSE-labeled T cells after incubation with CD8αα+CD11+ cells (DC) in the presence of pCol(28–40) (left panel). Dead cells are shown as PI+CFSE+. In contrast, vigorous proliferation of T cells, shown as serial peaks of CFSE+ cells, was observed after incubation with thymocytes and pCol(28–40) (2nd panel from right). Cell death did not occur in other controls as indicated. (E) Phase-contrast micrographs show cells when pCol(28–40)-specific T cells were incubated with CD8αα+CD11+ cells and pCol(28–40). CD8αα+CD11+ cells (arrows) and T cells (arrowheads) were present at Day 0, but T cells were absent at Day 3. (F) Flow cytometries show purified CD8+ cells (upper panel) and pCol(28–40)-specific T cells (lower panel). (G) T cell death, expressed as reductions in the number of labeled cells, after incubation with purified glomerular CD8+ cells or non-CD8+ cells in the presence of pCol(28–40).
The CD8αα+CD11+ MHC+ cells induce death of T cells through apoptosis
We next investigated the mechanism of T cell death. DNA fragmentation was detected only in T cells after a 3-day incubation with CD8αα+CD11c+ DCs in the presence of pCol(28–40), suggesting the occurrence of apoptosis in T cells (Fig. 8A). In contrast, DNA fragmentation was not detected in the T cells from the incubation without pCol(28–40) and other controls (Fig. 8A). TUNEL staining of T cells further demonstrated apoptosis in the T cells after they were incubated with CD8αα+CD11c+ cells and pCol(28–40) (Fig. 8B). In contrast, apoptosis did not occur in T cells in the controls (Fig. 8C). Apoptosis occurred 2–3 days after incubation with CD8αα+CD11c+ DCs in the presence of pCol(28–40), coinciding with the peak death of the T cells. Apoptosis in CD4+ T cells in the incubation was confirmed further by using FITC-labeled Annexin V, which binds to apoptotic cells at an early stage. A large CD4+Annexin-V+ population was detectable at Days 2 and 3 after incubation (data not shown). However, all T cells used for tests were from T cell lines, and their phenotype might have been altered artificially. We next tested whether CD8αα+CD11c+ cells could induce apoptosis in T cells, which were freshly isolated from draining lymph nodes of immunization sites. We have shown previously that the cells from the draining lymph nodes contain an expanding pCol(28–40)-specific T cell population. The T cells were enriched by a negative-selection column to avoid possible disturbance and were incubated with the CD8αα+CD11c+ cells at a ratio of 2:1 in the presence of pCol(28–40). The incubation without CD8αα+CD11c+ cells was used as a control. The cells were analyzed by flow cytometry after a 2-day incubation. The lymphocytes, which had been incubated with CD8αα+CD11c+ cells, showed a significant increase in the size of the CD4+Annexin V+ population (21%; Fig. 8D and E). In contrast, the lymphocytes without CD8αα+CD11c+ DCs showed a much smaller population of CD4+Annexin V+ (4% of total CD4+ cells; Fig. 8E). Those results suggest that the CD8αα+CD11c+ DCs induce T cell apoptosis, not because of artificial modification of T cell phenotype as a result of long-term culture.
Figure 8. CD8αα+CD11+ cells induce T cell death through apoptosis.

(A) A representative DNA fragmentation test in pCol(28–40)-specific T cells 3 days after incubation with CD8αα+CD11+ cells (CD8) in the presence or absence of pCol(28–40). Other controls are also shown. T1 and T2 are two independent pCol(28–40)-specific T cell lines. (B) TUNEL staining for apoptotic T cells after 3-day incubation with CD8αα+CD11+ cells and pCol(28–40). Inset, a high magnification showing nuclear staining of apoptotic T cells. (C) TUNEL staining for T cells incubated with thymic APC and pCol(28–40). Note a positive cell. (D) Flow cytometry reveals a population of CD4+Annexin V+ cells among unfractionated lymph node cells (LNC) after a 2-day incubation with CD8αα+ cells and pCol(28–40). (E) Histogram based on the gate (R4 in C) demonstrates a higher percentage (21%) of Annexin V+ cells in the CD4+ population in the incubation with CD8αα+ cells than that without CD8αα+ cells (5%) at Day 2.
Occurrence of T cell apoptosis in glomeruli is coincident with infiltration of CD8αα+CD11c+ cells
As we have shown above, invasion of the CD8+ cells was coincident with a decline of inflammation. We next investigated if glomeruli-infiltrating T cells would undergo apoptosis when the CD8+ cells infiltrate glomeruli. Using caspase 3 as a marker for preapoptotic cells, we observed apoptotic T cells at Days 22, 32, and 42 postimmunization. At Day 22 postimmunization, many CD3+ T cells were present in the glomeruli. However, none of these glomeruli-infitrating CD3+ T cells showed positive for caspase 3 (Fig. 9A). As we described before, there were only a few, if any, CD8αα+ DCs presented at that time-point. In contrast, 15–25% of CD3+ T cells were also caspase 3+ at Day 32 when the number of CD8αα+ DCs increased greatly (refer to Figs. 1 and 3). A few non-CD3+ cells also showed caspase 3. At Day 42, a fibrotic crescent had formed in the glomeruli, and CD3+ T cells were no longer present. Caspase 3 was detected in some other types of cells located in the fibrotic crescent (Fig. 9C). The glomerular cells were isolated at Days 22, 32, or 42 and analyzed with flow cytometry after double-staining by anti-CD4 antibody and Annexin V (Fig. 10). Glomerular cells isolated at Day 22 postimmunization had a small CD4+Annexin+ population (∼5%). In contrast, cells isolated at Day 32 contained a CD4+Annexin V+ population, which accounted for 25–37% of the whole CD4+ cell population (Fig. 10A and C). The presence of the CD4+Annexin V+ population was not caused by an isolation procedure, as lymphocytes isolated from lymph nodes under an identical isolation procedure from the same individual showed a small CD4+Annexin+ population (<5%; Fig. 10B). We attempted to isolate infiltrating leukocytes at Day 42 when fibrosis had developed. A few, if any, leukocytes were presented. In summary, the occurrence of T cell apoptosis in glomeruli was coincident with infiltration of CD8+ cells.
Figure 9. Immunofluorescent detection of apoptotic T cells in glomeruli at different time-points postimmunization.

CD3+ T cells were stained red and caspase 3, an apoptotic marker, green. (A) A glomerulus at Day 22. Note that many CD3+ T cells were present. (B) A glomerulus at Day 32 when number of the infiltrating CD8αα+CD11+ cells reached peak. (C) A glomerulus at Day 42. Note that fibrosis replaced inflammation, and the CD3+ cell was absent. Arrows indicate a fibrotic crescent. Glomeruli are outlined by arrowheads.
Figure 10. Flow cytometry detection of apoptotic T cells in glomeruli postimmunization.
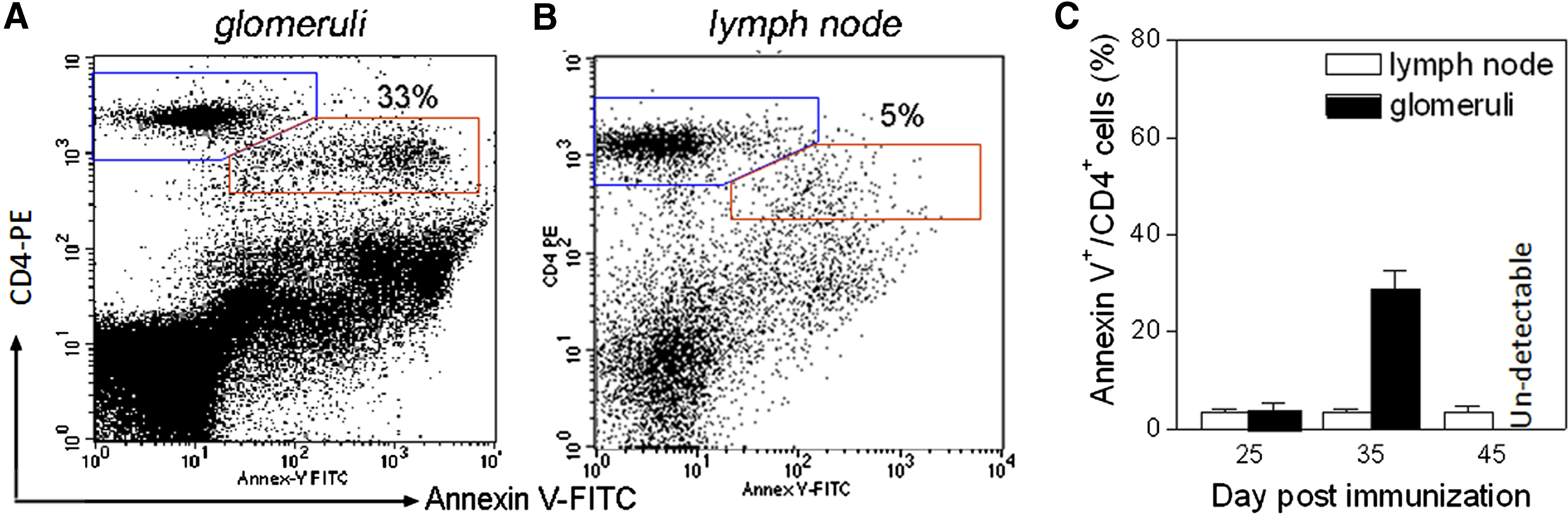
(A) A representative flow cytometry chart shows a normal (blue gate, CD4+Annexin V–) and an apoptotic T cell population (red gate, CD4+Annexin V+) among glomerular cells at Day 32. (B) Flow cytometry shows a lack of apoptotic CD4+ T cells among cells isolated from lymph nodes of the same individual. (C) Summary of apoptotic CD4+ T cell population as indicated. The apoptotic populations (CD4+Annexin V+) are expressed as percentage of total CD4+ cells.
DISCUSSION
In our anti-GBM GN model, we have identified a subset of GICs with a phenotype of CD8αα+CD11b/c+MHC+OX62–, which is derived from BM and actively infiltrates the inflamed glomeruli. Many inflammatory cells, which included CD4+/CD8+ T cells and various types of macrophages, have been described previously in several anti-GBM GN models. The identified CD8αα+ cell population in the present study does not belong to those cells described previously. A macrophage subset, which expresses CD4 and CD8 as well as a high level of FasL, has been reported in rats [27]. However, the CD8αα+ cells described in this study do not express CD4 (Fig. 2C). In several rat models for spinal cord injury or EAE, a CD8+ phagocyte population has been reported [28]. However, this subset of macrophages was identified by the ED1 antibody. In contrast, the CD8αα+ cells in our model do not express ED1 (Figs. 2E and 3A). A subset of γδ T cells is CD8αα+. However, in addition to their expression of many markers or molecules specific to DCs, the CD8αα+ cells do not express CD3 and thus, are clearly not γδ T cells. In addition, the CD8αα+ cells were morphologically different from γδ T cells (Figs. 3–5). The CD8αα+CD11+ cells also do not belong to CD8+ NK cells, as the CD8+ NK cells are morphologically different and more importantly, do not express MHC class II. Furthermore, the CD8αα+CD11+ cells lack expression of CD94, a marker for NK cells. Based on these results, we conclude that the CD8αα+ cell population described in the present study may be a subset of myeloid CD8αα+ DCs.
It remains unclear to which subset this CD8αα+ DC belongs. First, it shares many markers with rat cDCs. However, there are several differences between them. cDCs usually migrate into secondary lymphoid organs and reside there for the rest of their lifespan [29]. On the other hand, the CD8αα+ DC, described here, invades inflamed target tissue. cDC is a potent APC for T cell activation. In contrast, the CD8αα+ DC in this paper induced T cell apoptosis. In addition, the CD8αα+ DC lacks OX62, which is a common marker for cDC [26]. Second, a study reported that transfer of a splenic CD8+ DC reduced severity of EAE in a rat model [30]. However, the splenic DC was able to stimulate T cell proliferation in vitro, although it was less potent than other DCs. More importantly, it was not determined whether those transferred DCs migrate into the target tissue CNS, and the DC was not characterized further [30]. Thus, it is difficult to compare the two subsets of DCs. There are several subsets of DCs that may be involved in immune tolerance through induction of T cell apoptosis or other mechanisms. However, those DCs usually reside in the lymphoid organs [2–8]. For example, the CD103+ DC, which resides in mucosal/dermal lymph nodes, induces development of Tregs [6, 7]. Third, the splenic killer DC has been reported in a rat cancer model [31]. However, this killer DC induces apoptosis in tumor cells but not T cells. Furthermore, this DC lacks CD8 expression. Obviously, this killer DC is different from the CD8αα+ DCs in our study. Finally, DC subsets isolated from renal interstitial tissue or local lymph nodes have been analyzed in a murine model for glomerular injury [32]. Those DCs, including CD8+ DCs, were able to simulate T cells and promote glomerular injury. In summary, our results suggest that the CD8αα+ DCs may not belong to DC subsets described above. More characterization of the CD8αα+ DC is required.
This new population of DC-like cells was able to induce apoptosis in T cells through its APC functions in vitro. Although induction of T cell apoptosis by some type of APCs, including DCs, has been reported, this DC is unique as a result of the following characteristics. First, many other types of DCs or APCs, which induce apoptosis or anergy of autoreactive T cells, are usually located in lymphoid organs such as spleen and lymph nodes [8]. Thus, the potential autoreactive T cells are eliminated before they have a chance to migrate into the target tissue to cause any damage [4–9]. In contrast, this new type of DC invades the inflamed glomeruli specifically. Second, numerous previous studies have described different types of DCs in autoimmune diseases, including several types of GNs [12–17, 32]. DCs reported in those studies generally promote or initiate T cell-mediated pathogenesis through their APC function. For example, the isolated DCs from a renal tissue showed potent APC function and induced proliferation of antigen-specific T cells in vitro and in vivo [12, 13, 32]. However, the CD8αα+ DC from inflamed glomeruli in our study induces apoptosis in antigen-specific T cells in vitro in the presence of the antigen. We also showed the occurrence of apoptosis in glomeruli-infiltrating T cells in vivo during peak time for DC infiltration, suggesting that the CD8αα+ DC may induce T cell apoptosis in vivo as well.
Our finding raises an interesting question about the function or role of this subset of BM-derived DCs in autoimmune anti-GBM GN. It seems unlikely that this CD8αα+ DC-like population may participate in initiating and promoting T cell-mediated inflammation in the glomeruli because of the following reasons. First, in vitro experiments showed that the CD8αα+ cells were able to induce apoptosis in unmanipulated, freshly isolated T cells (Fig. 8). Thus, it is unlikely that they activate T cells in vivo. Second, this CD8αα+ cell population only infiltrated glomeruli at a much later stage than ED1+ macrophages. Thus, they may not be required for initiating glomerular inflammation. If this CD8αα+ cell population does not participate in establishing glomerular inflammation, what other roles may it play? A critical question about its role in autoimmune GN is whether this CD8αα+ cell population induces apoptosis in T cells in vivo. We have demonstrated that apoptosis in a large number of glomerular T cells was coincident with invasion of this CD8αα+ cell population. Glomerular inflammation peaked at Day 30, followed by a rapid decline; the inflammation was replaced gradually by glomerular fibrosis (Figs. 1 and 2). Thus, invasion of this CD8αα+ cell population was also coincident with a decline in inflammation. All of those results suggest that this CD8αα+ cell population may terminate T cell-mediated inflammation by inducing antigen-specific T cell apoptosis. Interestingly, several previous studies have associated apoptosis and CD8+ population with relief of autoimmune diseases. Inhibition of TRAIL-mediated apoptosis enhanced the severity of autoimmune diseases, such as thyroiditis, Type I diabetes, and arthritis in animal models [33–36]. Transfer of splenic CD8 DCs reduced severity of EAE in a rat model [30]. Induced experimental autoimmune uveoretinitis in rats will recover spontaneously; the recovery is associated with an influx of a CD8+ population, and depletion of CD8+ cells exaggerates the disease [37]. Similarly, another study found that the depletion of CD8+ cells leads to a severe late airway response, a model for asthma [38]. Although the CD8+ population has not been well-characterized in both studies, those results suggest that the CD8+ may terminate the T cell-mediated disease.
ED1+ macrophages have been reported to play critical roles in several GN models. Thus, it is worthwhile to emphasize differences between this CD8αα+ DC-like population and ED1+ macrophages in our model. First, this CD8αα+ cell population infiltrated gomeruli at a much later stage than ED1+ macrophages. This was well demonstrated by a time-course study using 2-color immunofluorescence (Fig. 3) and cell transfer experiments (Fig. 5). Second, although the CD8αα+ cells were scattered in an entire glomerulus, ED1+ cells usually clustered in an inflammatory focus (Fig. 3). Third, the CD8αα+ cells were bigger and had a more irregular surface with projections (Figs. 3E and F and 5A and B). More importantly, the CD8αα+ cells expressed a higher level of MHC class II than macrophages (Fig. 4H). Furthermore, the CD8αα+ cells were able to induce apoptosis in T cells. Thus, these two cell populations may function differently in the pathogenesis of anti-GBM GN. An important role of macrophages in the promotion of autoimmune tissue inflammation has been demonstrated repeatedly. It would be interesting to ask what role this new type of CD8αα+ DC-like cells may play.
In summary, we have identified a subset of BM-derived CD8αα+CD11c+ DC-like cells, which was able to migrate into inflamed autoimmune target tissues at a late inflammatory stage. Our finding suggests that this DC-like population may not participate in establishing or enhancing autoimmune inflammation. It would be interesting to ask what role this cell population may play in anti-GBM GN. We have demonstrated that these DC-like cells induce T cell apoptosis in vitro in an antigen-specific manner. We further showed a peak of apoptosis in glomeruli-infiltraitng T cells, which was coincident with infiltration of this DC-like population. Those results lead to a hypothetic mechanism for immune tolerance, by which this special BM-derived DC terminates autoimmune inflammation by the induction of apoptosis in autoreactive T cells in the target glomeruli tissues.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health R01-DK60029 (Y-H.L.), R01-DK077857 (Y-H.L.), and partially by R01-HD049613 (Y-H.L.). J.R. was supported by National Institutes of Health T32-DE015355. We thank Dr. John McMahon of University of Texas Houston for the critical reading of our manuscript and Dr. Yoshikazu Sado of Okayama University, Japan, for providing mAb SR13. We also thank the animal facility at the University of Texas M.D. Anderson Cancer Center for its help in the irradiation procedure.
Footnotes
- BM
- bone marrow
- cDC
- conventional DC
- EAE
- experimental allergic encephalomyelitis
- GBM
- glomerular basement membrane
- GIC
- glomeruli-infiltrating cell
- GN
- glomerulonephritis
- LEW rat
- Lewis rat
- TBM
- tubular basement membrane
- Treg
- regulatory T cell
- WKY
- Wistar Kyoto
AUTHORSHIP
J.W. planned and conducted experiments and summarized data; C.Z. conducted experiments and summarized data; J.R. conducted experiments; C.C.Y.W. conducted experiments; M.L.M. collaborated and consulted; R.C.T. consulted and provided the facility; Y-H.L. planned and directed the project and prepared manuscripts.
REFERENCES
- 1.Banchereau J., Steinman R. M. (1998) Dendritic cells and the control of immunity. Nature 392, 245–252. [DOI] [PubMed] [Google Scholar]
- 2.Cravens P. D., Lipsky P. E. (2002) Dendritic cells, chemokine receptors and autoimmune inflammatory diseases. Immunol. Cell Biol. 80, 497–505. [DOI] [PubMed] [Google Scholar]
- 3.Eriksson U., Ricci R., Hunziker L., Kurrer M. O., Oudit G. Y., Watts T. H., Sonderegger I., Bachmaier K., Kopf M., Penninger J. M. (2003) Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 9, 1484–1490. [DOI] [PubMed] [Google Scholar]
- 4.Chorny A., Gonzalez-Rey E., Fernandez-Martin A., Pozo D., Ganea D., Delgado M. (2005) Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc. Natl. Acad. Sci. USA 102, 13562–13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coombes J.L., Siddiqui K. R. R., Arancibia-Cárcamo C. V., Hall J., Sun C. M., Belkaid Y., Powrie F. (2007) A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β- and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guilliams M., Crozat K., Henri S., Tamoutounour S., Grenot P., Devilard E., de Bovis B., Alexopoulou L., Dalod M., Malissen B. (2010) Skin-draining lymph nodes contain dermis-derived CD103– dendritic cells that constitutively produce retinoic acid and induce Foxp3+ regulatory T cells. Blood 115, 1958–1968. [DOI] [PubMed] [Google Scholar]
- 7.Heath W. R., Carbone F. R. (2001) Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19, 47–64. [DOI] [PubMed] [Google Scholar]
- 8.Suss G., Shortman K. (1996) A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 183, 1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kusmartsev S., Nagaraj S., Garbrilovich D. I. (2005) Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J. Immunol. 175, 4583–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuzic S., Ritz E., Waldherr R. (1992) Dendritic cells in glomerulonephritis. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 62, 357–363. [DOI] [PubMed] [Google Scholar]
- 11.Peterson K. S., Huang J. F., Zhu J., D′Agati V., Liu X., Miller N., Erlander M. G., Jackson M. R., Winchester R. J. (2004) Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J. Clin. Invest. 113, 1722–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kruger T., Benke D., Eitner F., Lang A., Wirtz M., Hamilton-Williams E. E., Engel D., Giese B., Muller-Newen G., Floege J., Kurts C. (2004) Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J. Am. Soc. Nephrol. 15, 613–621. [DOI] [PubMed] [Google Scholar]
- 13.Gieseler R., Hoffmann P. R., Kuhn R., Fayyazi A., Stojanovic T., Schlemminger R., Peters J. H. (1997) Enrichment and characterization of dendritic cells from rat renal mesangium. Scand. J. Immunol. 46, 587–596. [DOI] [PubMed] [Google Scholar]
- 14.Roy-Chaudhury P., Wu B., McDonald S., Haites N. E., Simpson J. G., Power D. A. (1995) Phenotypic analysis of the glomerular and periglomerular mononuclear cell infiltrates in the Thy 1.1 model of glomerulonephritis. Lab. Invest. 72, 524–531. [PubMed] [Google Scholar]
- 15.Ghebrehiwet B., Peerschke E. I. (2004) Role of C1q and C1q receptors in the pathogenesis of systemic lupus erythematosus. Curr. Dir. Autoimmun. 7, 87–97. [DOI] [PubMed] [Google Scholar]
- 16.Wang J., Lo J. C., Foster A., Yu P., Chen H. M., Wang Y., Tamada K., Chen L., Fu Y. X. (2001) The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J. Clin. Invest. 108, 1771–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lan H. Y., Nikolic-Paterson D. J., Atkins R. C. (1993) Immune events in lymphoid tissues during experimental glomerulonephritis. Pathology 25, 159–166. [DOI] [PubMed] [Google Scholar]
- 18.Wu J., Hicks J., Ou C., Singleton D., Borillo J., Lou Y. H. (2001) Glomerulonephritis induced by recombinant Col4α3NC1 is not associated with antibody to GBM: a potential T cell mediated mechanism. J. Immunol. 167, 2388–2395. [DOI] [PubMed] [Google Scholar]
- 19.Wu J., Hicks J., Borillo J., Glass W. F., II, Lou Y. H. (2002) CD4+ T cells specific to glomerular basement membrane antigen induce glomerulonephritis. J. Clin. Invest. 109, 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J., Borillo J., Glass W. F., II, Hicks J., Ou C., Lou Y. H. (2003) T cell epitope of glomerular basement membrane antigen induces severe glomerulonephritis. Kidney Int. 64, 1292–1301. [DOI] [PubMed] [Google Scholar]
- 21.Robertson J., Wu J., Arends J., Zhou C., Adrogue H. E., Chan J. T., Lou Y. H. (2008) Spontaneous recovery from early glomerular inflammation is associated with resistance to anti-GBM glomerulonephritis: tolerance and autoimmune tissue injury. J. Autoimmun. 30, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu J., Arends J., Borillo J., Zhou C., Merszei J., McMahon J., Lou Y. H. (2004) A self T cell epitope induces autoantibody response: mechanism for production of antibodies to diverse glomerular basement membrane antigens. J. Immunol. 172, 4567–4574. [DOI] [PubMed] [Google Scholar]
- 23.Tipping P. G., Huang X. R., Qi M., Van G. Y., Tang W. W. (1998) Crescentic glomerulonephritis in CD4- and CD8-deficient mice: requirement for CD4 but not CD8 cells. Am. J. Pathol. 152, 1541–1548. [PMC free article] [PubMed] [Google Scholar]
- 24.Khan S. B., Allen A. R., Bhangal G., Smith J., Lobb R. R., Cook H. T., Pusey C. D. (2003) Blocking VLA-4 prevents progression of experimental crescentic glomerulonephritis. Nephron Exp. Nephrol. 95, e100–e110. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds J., Norgan V. A., Bhambra U., Smith J., Cook H. T., Pusey C. D. (2002) Anti-CD8 monoclonal antibody therapy is effective in the prevention and treatment of experimental autoimmune glomerulonephritis. J. Am. Soc. Nephrol. 13, 359–369. [DOI] [PubMed] [Google Scholar]
- 26.Matsuno K., Ezaki T., Kudo S., Uehara Y. A. (1996) Life stage of particle-laden rat dendritic cells in vivo: their terminal division, active phagocytosis, and translocation from the liver to the draining lymph. J. Exp. Med. 183, 1865–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baba T., Iwasaki S., Maruoka T., Suzuki A., Tomaru U., Ikeda H., Yoshiki T., Kasahara M., Ishizu A. (2008) Rat CD4+CD8+ macrophages kill tumor cells through an NKG2D- and granzyme/perforin-dependent mechanism. J. Immunol. 180, 2999–3006. [DOI] [PubMed] [Google Scholar]
- 28.Schroeter M., Stoll G., Weissert R., Hartung H. P., Lassmann H., Jander S. (2003) CD8+ phagocyte recruitment in rat experimental autoimmune encephalomyelitis. Am. J. Pathol. 163, 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yrlid U., Macpherson G. (2003) Phenotype and function of rat dendritic cell subsets. APMIS 111, 756–765. [DOI] [PubMed] [Google Scholar]
- 30.Pettersson A., Wu X. C., Ciumas C., Lian H., Chirsky V., Huang Y. M., Bjelke B., Link H., Xiao B. G. (2004) CD8α+ dendritic cells and immune protection from experimental allergic encephalomyelitis. Clin. Exp. Immunol. 137, 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chauvin C., Philippeau J. M., Hémont C., Hubert F. X., Wittrant Y., Lamoureux F., Trinité B., Heymann D., Rédini F., Josien R. (2008) Killer dendritic cells link innate and adaptive immunity against established osteosarcoma in rats. Cancer Res. 68, 9433–9440. [DOI] [PubMed] [Google Scholar]
- 32.Heymann F., Meyer-Schwesinger C., Hamilton-Williams E. E., Hammerich L., Panzer U., Kaden S., Quaggin S. E., Floege J., Gröne H. J., Kurts C. (2009) Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J. Clin. Invest. 119, 1286–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song K., Chen Y., Göke R., Wilmen A., Seidel C., Göke A., Hilliard B., Chen Y. (2000) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J. Exp. Med. 191, 1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S. H., Cao Z., Wolf J. M., van Antwerp M., Baker J. R., Jr. (2005) Death ligand tumor necrosis factor-related apoptosis-inducing ligand inhibits experimental autoimmune thyroiditis. Endocrinology 146, 4721–4726. [DOI] [PubMed] [Google Scholar]
- 35.Mi Q. S., Ly D., Lamhamedi-Cherradi S. L., Salojin K. V., Zhou L., Grattan M., Meagher C., Zucker P., Chen Y. H., Nagle J., Taub D., Delovitch T. L. (2003) Blockade of tumor necrosis factor-related apoptosis-inducing ligand exacerbates type 1 diabetes in NOD mice. Diabetes 52, 1967–1975. [DOI] [PubMed] [Google Scholar]
- 36.Kiener P. A., Davis P. M., Rankin B. M., Klebanoff S. J., Ledbetter J. A., Starling G. C., Liles W. C. (1997) Human monocytic cells contain high levels of intracellular Fas ligand: rapid release following cellular activation. J. Immunol. 159, 1594–1598. [PubMed] [Google Scholar]
- 37.Calder V. L., Zhao Z. S., Wang Y., Barton K., Lightman S. L. (1993) Effects of CD8 depletion on retinal soluble antigen induced experimental autoimmune uveoretinitis. Immunology 79, 255–262. [PMC free article] [PubMed] [Google Scholar]
- 38.Allakhverdi Z., Lamkhioued B., Olivenstein R., Hamid Q., Renzi P. M. (2000) CD8 depletion-induced late airway response is characterized by eosinophilia, increased eotaxin, and decreased IFN-γ expression in rats. Am. J. Respir. Crit. Care Med. 162, 1123–1131. [DOI] [PubMed] [Google Scholar]