

Fyn-dependent expression of the cationic transient receptor potential channel 1 contributes to mast cell Ca2+ influx, cortical F-actin depolymerization, and degranulation.
Keywords: calcium, IgE, transient receptor potential cation channel 1
Abstract
MC degranulation requires the influx of calcium from the extracellular environment. Orai1/STIM1 is essential to MC SOCE, as shown in rat peritoneal MCs, the rat MC lines (RBL-2H3), or in Orai1 null embryo liver-derived, cultured MCs. However, minimal information exists about the role of other calcium channels expressed on these cells. Here, we demonstrate that the nonselective TRPC1 participates in FcεRI-mediated calcium entry in mouse BMMCs. We found that Fyn null MCs, which have an impaired degranulation response, expressed reduced levels of TRPC1, had normal depletion of intracellular calcium stores but an impaired calcium influx, and failed to depolymerize cortical F-actin (a key step for granule-plasma membrane fusion). Partial RNAi silencing of TRPC1 expression in WT MCs (to the level of Fyn null MCs) mimicked the Fyn null defect in calcium influx, cortical F-actin depolymerization, and MC degranulation. Ectopic expression of Fyn or TRPC1 in Fyn null MCs restored calcium responses and cortical F-actin depolymerization and increased MC degranulation. Together with our findings that expression of Orai1 is not altered in Fyn null MCs, our findings suggest that TRPC1 participates in calcium influx and other key events required for MC degranulation. This demonstrates that in addition to a role described previously for Orai1 in promoting MC degranulation, nonselective cation channels participate in promoting the exocytotic response.
Introduction
Ag-induced aggregation of the IgE antibody-occupied FcεRI on MCs causes the release of preformed and newly synthesized allergic mediators from these cells. The first-recognized intracellular step in this process is the Lyn kinase-dependent phosphorylation of FcεRI [1–3], which initiates events that lead to amplification of the signaling required for cellular responses, such as exocytosis (degranulation), eicosanoid, and cyokine production and release (reviewed in refs. [4–6]). Recent evidence has shown that in the absence of Lyn kinase, FcεRI phosphorylation is severely impaired [7–10], yet many MC effector responses appear to be normal or enhanced. Several studies have implicated the Src family kinase Fyn as an important player in facilitating MC effector responses [9–11]. A deficiency of Fyn kinase in MCs caused impaired degranulation [12] and defective cytokine and leukotriene production [13].
It has been suggested [14] that a defect in the Ca2+-independent reorganization of microtubules (a step required for granule movement [15–17]) may underlie the defective degranulation in the absence of Fyn. However, the ability to restore normal degranulation in Fyn null MCs with calcium ionophores and phorbol esters [12] and the failure of phorbol esters alone to elicit MC degranulation suggest that a defect in a Ca2+-dependent step may also contribute to this impairment. This view has received support from a recent report [18], published during the course of this work, demonstrating a defective Ca2+ influx in Fyn null MCs. However, the identity of the Ca2+ channel affected by Fyn deficiency was not determined, and how the defect in Ca2+ influx might affect MC degranulation was unexplored.
The rapid dissolution of cortical F-actin upon FcεRI stimulation of MCs has been shown to be an important step in facilitating the granule-plasma membrane fusion that is required for release of granule contents to the extracellular environment [19, 20]. Multiple studies have demonstrated that signals generated by small GTPases such as Rac and Rho, which regulate the F-actin cytoskeleton rearrangement, promote MC degranulation [21–23]. In addition, agents that destabilize F-actin, such as cytochalasins or latrunculins, enhance the extent of MC degranulation, suggesting that the presence of cortical F-actin is inhibitory to this event [24–28]. Nonetheless, the most convincing evidence that the cortical F-actin restricts MC degranulation comes from recent work [20], using atomic force and laser-scanning confocal microscopy, which demonstrates that granule fusion occurs only at plasma membrane sites that are devoid of cortical F-actin.
In the current work, we set out to explore the mechanisms that contribute to the defective degranulation of Fyn null MCs. Here, we report that Fyn null MCs are unable to depolymerize cortical F-actin normally, which limits MC degranulation. This was linked to a defect in Ca2+ influx, a step that is required for depolymerization of cortical F-actin and MC degranulation [4–6]. We identified the abnormal expression of TRPC1 (a nonselective calcium channel expressed in MCs) as a contributory factor and established its role by RNAi silencing in WT MCs. These findings provide novel evidence of a role for this nonselective calcium channel in FcεRI-mediated MC degranulation.
MATERIALS AND METHODS
General reagents
Mouse anti-DNP-IgE was produced as described [29]. DNP-HSA (DNP36-HSA, Ag), ionomycin, 2-APB, and FITC- and rhodamine-conjugated phalloidin (FITC- or rhodamine-phalloidin) were from Sigma-Aldrich (St. Louis, MO, USA). Fura-2 AM, Fluo-4 AM, and Fura Red AM were from Molecular Probes (Eugene, OR, USA). 45CaCl2 was from MP Biomedicals (Solon, OH, USA). Murine IL-3 and SCF were from PeproTech (Rocky Hill, NJ, USA). FBS and RPMI 1640 were from Invitrogen (Carlsbad, CA, USA).
Mice, BMMC cultures, IgE sensitization, and cell activation
BM was isolated from femurs of sex- and age-matched (8- to 12-week-old) WT and fyn–/– mice [129Sv×C57/BL6, (N5)], which were maintained and used in accordance with NIH guidelines, and the animal study proposal was approved by the Institutional Animal Care and Use Committee. BMMCs were cultured in RPMI 1640, supplemented with 10% FBS and 20 ng/ml each SCF and IL-3, essentially as described previously [12, 30]. Cultures were monitored by FACScan (Becton Dickinson, San Diego, CA, USA) for surface expression of FcεRI and used for experiments when >95% of the cells were FcεRI+. Cultures of 4–6 weeks were used in these experiments. Conditions for IgE sensitization and for cell activation were described previously [9, 31, 32].
Degranulation assay, cell lysates, and immunoblotting
Degranulation was determined by the release of the granule enzyme, β-hexosaminidase, as described previously [33, 34]. Degranulation is expressed as the percent of total cellular β-hexosaminidase in the supernatant.
Ag-stimulated cells [2×107 cells in 500 μl Tyrode's BSA buffer with 10 ng/ml Ag (unless otherwise indicated)] were placed on ice and lysed [9, 13]. Lysates were centrifuged for 10 min at 12,000 g at 4°C, and proteins were resolved by 8% SDS-PAGE (Invitrogen). The procedure for immunoblotting was performed as described previously [9, 12]. In brief, proteins were electrophoretically transferred onto a nitrocellulose membrane (Invitrogen). After the membrane was soaked in Odyssey blocking buffer (Li-Cor Biosciences, Lincoln, NE, USA), proteins were probed with antibodies to TRPC1 (Abcam, Cambridge, MA, USA) or TRPC6 (Alomone Labs, Israel). The immunoreactive proteins were visualized with IRDye800-conjugated goat anti-mouse IgG (Rockland Immunochemicals, Boyertown, PA, USA) or Alexa Fluor 680-conjugated goat anti-rabbit IgG (Invitrogen) and detected by an Odyssey infrared imaging system (Li-Cor Biosciences).
Gene modification of BMMCs
For ectopic expressions of genes, lentivirus gene transduction was used. Viral supernatants were produced by transfecting the packaging cell 293LTV with Fyn kinase in the pLenti6 vector using Lipofectamine 2000 (Invitrogen). Cells were also mock-transfected with LacZ/pLenti6 vector as a negative control. The procedure for infection was described previously [35]. After infection, cells were washed and allowed to grow in IL-3- and SCF-containing medium for 2 days (as above) before initiating selection of transduced cells with 8 μg/ml blasticidin S (Invitrogen). Following 2 weeks of selection, cells were analyzed for FcεRI expression and used when >95% of the cells expressed this receptor.
For transient gene transfection and gene silencing, nucleofection (Amaxa, Switzerland) of BMMCs was performed according to the manufacturer's instructions with minor modification. After BMMCs were washed twice in PBS, 5 × 106 cells were resuspended in 100 μl mouse macrophage nucleofection solution (Amaxa). pEYFP-actin (Clontech, Palo Alto, CA, USA) and/or siRNA against mouse TRPC (2 μM; Sigma-Aldrich) were added/5 × 106 cells, and the samples were transferred into certified cuvettes (Amaxa) and transfected by using a mouse macrophage nucleofection program. WT cells transfected with pEYFP and scramble sequence (nontargeting) siRNA (Sigma-Aldrich) were used as a control for pEYFP-actin and TRPC1 siRNA, respectively.
Measurement of changes in intracellular Ca2+ and Ca2+ influx
The procedures for Ca2+ fluorimetry on cell populations were described previously [32]. For single cell analysis, two Ca2+ indicator dyes, Fluo-4 AM/Fura Red AM (Molecular Probes), were used. BMMCs were loaded with Fluo-4 AM (2 μM) and Fura Red AM (10 μM) for 30 min at 37°C. The cells were rinsed 3 times with Tyrodes buffer. A Zeiss LSM-510 Meta confocal microscope with a 488-nm wavelength light from an argon laser was used for ratio-metric measurements. The fluorescence of Fluo-4 and Fura Red was detected through a band-pass filter (505–530 nm) and a long-pass filter (>560 nm), respectively. Calcium responses are reported as the ratio of Fluo-4 to Fura Red normalized as a fold increase relative to cells in the absence of Ag. Fluorescence images were collected every 5 s, and the intensity of fluorescence was quantified using Zeiss LSM-510 Meta software.
45Ca2+ uptake was initiated by addition of 45Ca2+ (5 μCi/sample), with or without 10 ng/ml Ag. For some experiments, this was done in the presence or absence of 10 or 20 μM 2-APB. At an indicated time, the reactions were terminated by washing the cells 3 times with ice-cold PBS containing 1% BSA. The cells were lysed with 400 μl water, and the intracellular radioactivity was detected by liquid scintillation counting (LS1801, Beckman, Fullerton, CA, USA). The amount of Ca2+ taken up was calculated using the specific activity of Ca2+ in the extracellular buffer. Data were normalized to equal amounts of cellular protein. To determine the amount of protein, cells were lysed with 0.5% Triton X-100 and centrifuged at 12,000 g for 10 min. The amount of proteins in cell lysate was determined by DC protein assay (Bio-Rad, Hercules, CA, USA).
Measurement of F-actin content in BMMCs
For single cell measurements of F-actin content, Ag-stimulated and -unstimulated cells were washed with ice-cold PBS and fixed with 4% paraformaldehyde for 30 min at room temperature. Fixed cells were permeabilized and stained with 0.1% Triton X-100 and 2.5 μM FITC-phalloidin in PBS for 30 min at room temperature. After washing the cells, FITC-phalloidin was excited with an argon ion laser at 488 nm, and fluorescence emission of >505 nm was measured by confocal laser fluorescence microscopy, LSM-510 Meta (Carl Zeiss MicroImaging, Inc., Thornwood, NY, USA). For detection of fluoresence in cells with actin-YFP and rhodamine-phalloidin, the fluorescence of actin-YFP and rhodamine-phalloidin was detected through a band-pass filter (505–530 nm) and a long-pass filter (>560 nm), respectively.
For real-time imaging of actin-YFP in a cortical membrane region, culture dishes plated with cells were mounted on an inverted confocal microscope (LSM 510; Carl Zeiss MicroImaging, Inc.), equipped with a 63 × 1.40 oil objective (Plan-Apochromat; Carl Zeiss MicroImaging, Inc.). Excitation of YFP was with a 488-nm argon ion laserline, and YFP fluorescence from the actin-YFP fusion protein was recorded through an emission filter (LP505; Carl Zeiss MicroImaging, Inc.). For all experiments, the same pinhole size was used. To quantitate the fluorescence of the cortical membrane region, we used ImageJ software (NIH). Fluoresence was measured on the whole cell, cytosol, and noncell background. To quantitate the fluorescence of the cortical membrane region, the data were obtained as follows: Fcortical region = (Fwhole cell–Fbackgrownd) – (Fcytosol–Fbackgrownd), where Fwhole cell is the intensity of the actin-YFP at each time-point, Fbackgrownd is the intensity of background fluorescence, and Fcytosol is the intensity at the cytosol. The data at each time-point [Fcortical actin(t)] were normalized to the fluoresence intensity at time 0 [F(0)].
Quantitative real-time PCR analysis
RNA isolation and generation of cDNA have been described previously [32]. Quantitative gene expression analysis was performed by real-time PCR on an ABI 7700 Fast real-time PCR system (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed in triplicate according to the Taqman Universal 2× master mix and run on the ABI/PRISM 7700 sequence detector system (Applied Biosystems). The amount of mRNA for TRPC and Orai isoforms was normalized to the amount of mouse GAPDH RNA and calculated, according to the comparative threshold method as described by Applied Biosystems.
Electrophysiological measurements
BMMCs grown on poly-D-lysine and laminin-coated coverslips were transferred to the recording chamber and perfused with a standard external solution [145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, 10 mM glucose, pH 7.4 (NaOH)]. The patch pipette had resistances between 3 and 5 mOsm after filling with the standard intracellular solution that contained the following: 145 mM cesium methane sulfonate, 8 mM NaCl, 10 mM MgCl2, 10 mM Hepes, 10 mM EGTA, pH 7.2 (caesium hydroxide). Osmolarity for all of the solutions was adjusted with mannose to 300 ± 5 mOsm using a vapor pressure Osmometer (Wescor, Logan, UT, USA). Whole cell patch clamp experiments were performed in the standard whole cell configuration at room temperature (22–25°C) using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA). Generation of the current was assessed by the amplitude at –80 mV, taken from the currents recorded during voltage ramps ranging from –90 to 90 mV over a period of 1 s imposed every 4 s (holding potential was 0 mV) and digitized at a rate of 1 kHz. Liquid-junction potentials were <8 mV and were not corrected. Capacitative currents and series resistance were determined and minimized. For analysis, current recorded during the first ramp was used for leak subtraction of the subsequent current records.
Statistical analysis
Results were analyzed using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA). The statistical significance of differences between groups was determined by unpaired, two-tailed Student's t tests or by one-way ANOVA as appropriate. Two-way ANOVA was used to examine the overall effects of genotype and time on the change in cortical F-actin levels. Differences were considered significant if the P value were ≤0.05 with the confidence intervals of 95%. All results are shown as mean ± sem of at least four independent experiments performed with distinct samples.
Online Supplemental materials
Five supplemental figures are available online. Supplemental Fig. 1 shows that cortical F-actin depolymerization in MCs is dependent on calcium. Supplemental Fig. 2 demonstrates that drug-mediated inhibition of SOCE inhibits cortical F-actin depolymerization, calcium uptake, and degranulation. Supplemental Fig. 3 shows that LAT deficiency also causes an impairment of calcium uptake and cortical F-actin depolymerization that mimics that seen in Fyn null MCs. Supplemental Fig. 4 shows the real-time quantitation of mRNA expression for TRPC and Orai isoforms in WT and Fyn null MCs.
RESULTS
F-actin depolymerization is defective in the absence of Fyn
We initially explored whether Fyn might play a role in cortical F-actin depolymerization. Using FITC-labeled phalloidin in fixed and permeabilized, nonstimulated and Ag-stimulated WT and Fyn null MCs, measurement of the fluorescence intensity (Fig. 1A, upper panels and fluorescence intensity maps) revealed significant differences in cortical F-actin depolymerization (P<0.007) between WT and Fyn null MCs after Ag stimulation. In WT cells, Ag stimulation (1 min) caused a marked reduction in the numbers of cells retaining cortical F-actin as well as in the fluorescence intensity of those cells retaining some cortical F-actin. Ag stimulation of Fyn null MCs also caused a decrease in the numbers of cells retaining a cortical F-actin ring, but this decrease was less marked relative to WT cells, and more cells retained strong cortical fluorescences (Fig. 1A and fluorescence intensity maps). To investigate the dynamics of the rearrangement of cortical F-actin, we first determined if ectopic expression of actin-YFP would mimic the loss of cortical F-actin fluorescence seen when cells were stained with rhodamine-labeled phalloidin. As shown in Fig. 1B, a similar loss of actin-YFP signal was observed, and the peak loss of the cortical F-actin signal was seen 1–2 min poststimulation. Recovery of cortical F-actin fluorescence occurred between 4 and 5 min poststimulation. Thus, actin-YFP showed similar dynamics to rhodamine-labeled phalloidin (Fig. 1B). Analysis of actin dynamics (using actin-YFP) revealed that Fyn null MCs were impaired in their ability to depolymerize but not polymerize F-actin, achieving a higher percentage of cortical F-actin than seen in WT cells (Fig. 1C). Collectively, these experiments demonstrated that a deficiency in Fyn expression impairs the depolymerization of the cortical F-actin ring, which is known to limit MC degranulation [19, 20, 26].
Figure 1. FcεRI stimulation of Fyn null MCs reveals altered actin dynamics.
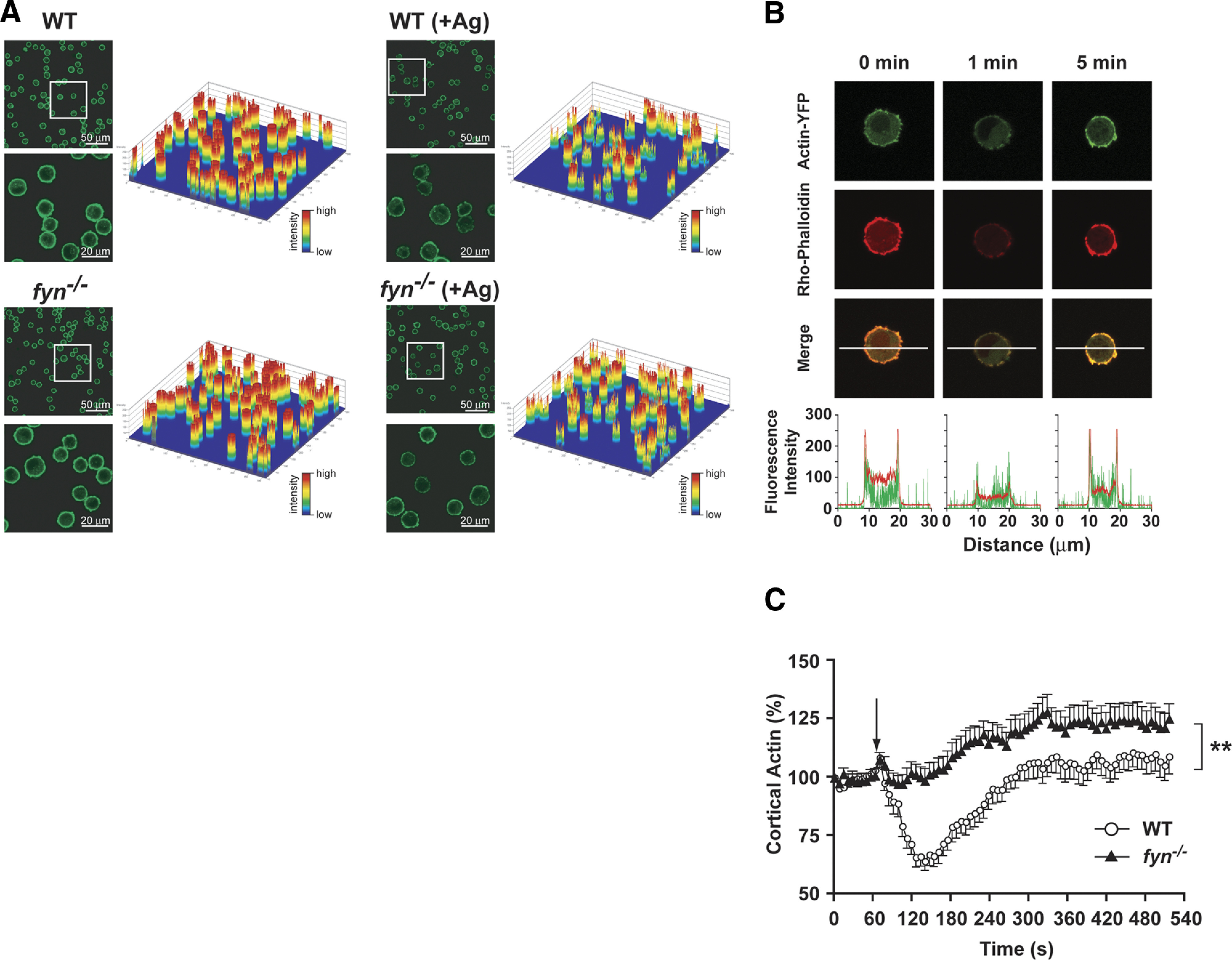
(A) Single cell analysis of actin deploymerization in WT and Fyn null MCs. FcεRI-dependent changes in F-actin content were measured by the amount of fluorescent phalloidin bound to F-actin at the indicated time. The fluoresence intensity of individual cells is shown as an intensity plot. Insets in low-magnification field show the regions selected for higher magnification in the lower panels. One representative of three individual experiments is shown. (B) Distribution of actin-YFP and rhodamine-labeled phalloidin. WT MCs were transiently transfected with actin-YFP (green, top panel) and counterstained with rhodamine-labeled phalloidin (red, middle panel) prior to or after activation with Ag for the indicated time. The merged images (yellow, bottom panel) indicate that actin-YFP and rhodamine-labeled phalloidin were localized predominantly in the cortical region and showed a strong colocalization. Recovery of actin-YFP and rhodamine-labeled phalloidin signals was observed at 5 min poststimulation (intensity profile). (C) Real-time actin-YFP dynamics in WT or Fyn null MCs. Actin dynamics were determined by measuring the amount of fluorescent actin-YFP in the cortical region with time (see Materials and Methods). Arrow indicates the time of Ag addition. Data are mean ± se of 4 individual experiments (23–26 cells were monitored in total). Statistical significance relative to WT cells was **P < 0.01.
In other cell types, it is well known that actin depolymerization requires an increase in intracellular calcium (reviewed in ref. [36]). As shown in Supplemental Fig. 1A, Ag-dependent stimulation of WT and Fyn null MCs, in the absence of extracellular calcium, resulted in cortical F-actin polymerization but not depolymerization, demonstrating the requirement of Ca2+ influx for the latter. Ionomycin, a calcium ionophore produced by Streptomyces conglobatus, caused a rapid depolymerization of cortical F-actin, which persisted for more than 8 min post-treatment (Supplemental Fig. 1B). The rate and extent of F-actin depolymerization were identical between WT and Fyn null MCs, demonstrating that the actin depolymerization defect in Fyn null MCs was not a result of abnormalities in the cytoskeletal machinery. Thus, the findings suggested a possible defect in Ca2+ mobilization as an underlying mechanism for the defect observed in Fyn null MCs.
To explore this possibility further, we used the pharmacological agent, 2-APB, a known inhibitor of SOCE through inhibition of calcium influx and its release from endoplasmic stores [37, 38], to block Ca2+ influx in WT MCs. FcεRI stimulation of 2-APB (20 μM)-treated MCs caused a marked inhibition of cortical F-actin depolymerization, whereas polymerization was unaffected (Supplemental Fig. 2A). 2-APB was also highly effective in inhibiting 45Ca2+ uptake (Supplemental Fig. 2B). Some inhibition of 45Ca2+ uptake was also seen at a 10-μM concentration of 2-APB. Analysis of MC degranulation demonstrated that 10 and 20 μM 2-APB had an inhibitory effect on the release of MC granule contents (Supplemental Fig. 2C), whereas poststimulation treatment with APB had no effect, suggesting that once Ca2+ entry occurs, exocytosis is unimpaired. These findings showed that the inhibition of Ca2+ entry in WT cells mimics the impaired cortical F-actin depolymerization and defective degranulation of Fyn null MCs.
Ag-stimulated Ca2+ entry is defective in Fyn null MCs
To assess the Ca2+ response of Fyn null MCs accurately, we used Fura-2-based fluorimetry and single cell calcium imaging analysis. As shown in Fig. 2A, the Ca2+ mobilization of WT and Fyn null MCs differed in the extent of the response, whereas the initial rise was similar. This suggested normal intracellular store depletion but defective Ca2+ influx. To evaluate this possibility, experiments were conducted in the absence of extracellular Ca2+, allowing the monitoring of intracellular Ca2+ store depletion. As shown in Fig. 2B, Ag stimulation of WT and Fyn null MCs revealed no significant difference in the intracelluar depletion of Ca2+ stores. However, replenishment of extracellular Ca2+ at ∼6 min post-Ag stimulation revealed a diminshed influx of Ca2+ in the Fyn null MC.
Figure 2. Calcium mobilization is defective in Fyn null MCs but is unrelated to the depletion of intracellular calcium stores.
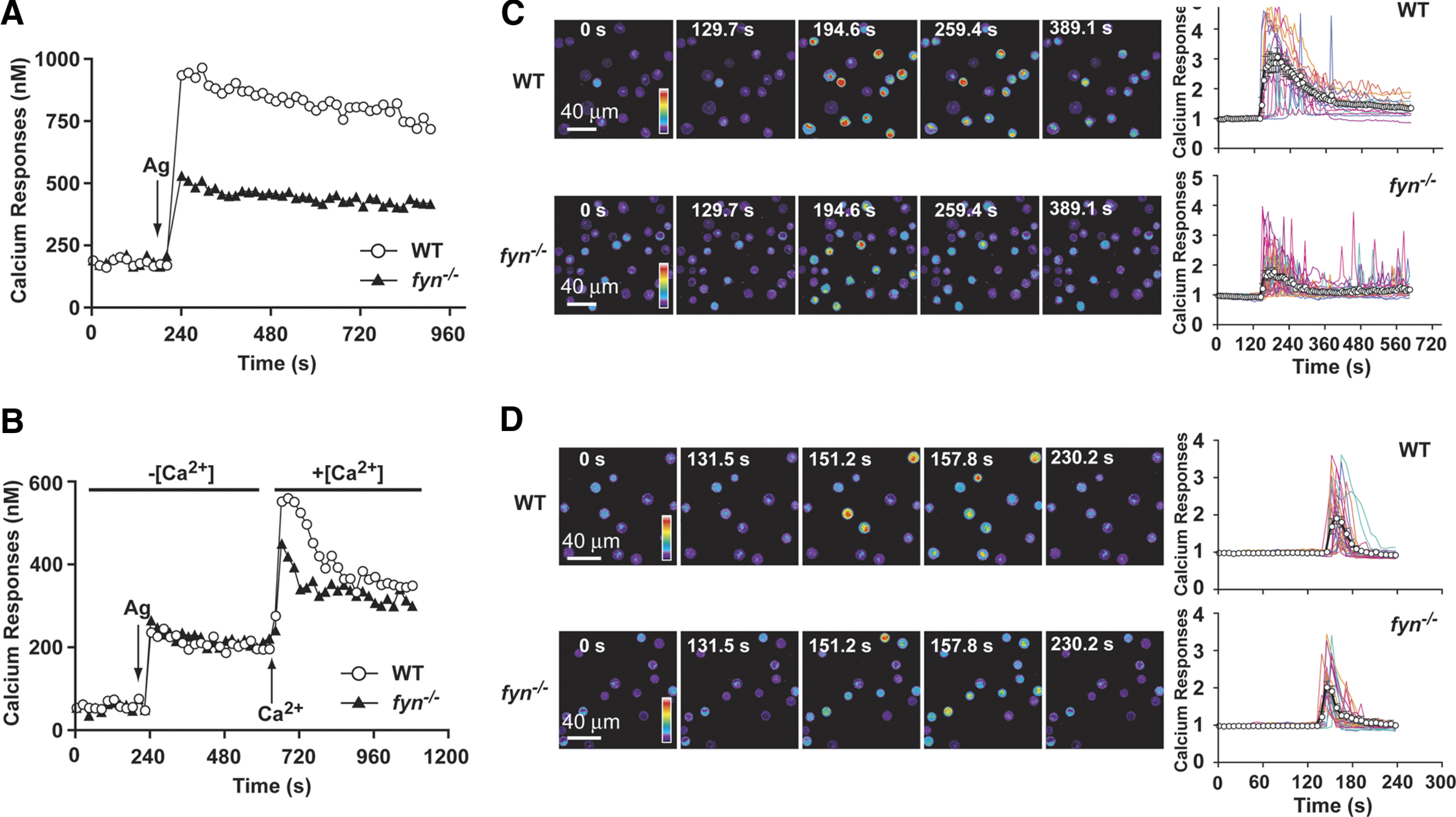
(A) IgE-sensitized WT or Fyn null MCs were loaded with Fura-2. MCs were stimulated with Ag (as indicated by arrow), and changes in the intracellular calcium concentration were monitored by fluorimetry for the indicated time. One representative of 4 experiments is shown. (B) WT and Fyn null MCs were treated in the same manner as above but were stimulated in the absence of extracellular Ca (–[Ca2+]), which was subsequently replenished to the extracellular medium (+[Ca2+]) as indicated. One representative of 3 experiments is shown. (C) Single cell analysis of calcium responses in WT and Fyn null MCs in calcium containing medium (+[Ca2+]). WT and Fyn null MCs were loaded with the calcium-binding Fluo-3 fluorescent dye and stimulated with Ag. Change in fluorescence intensity was monitored with time (as indicated). Reverse psuedo-colored images are used to distinguish the intensity of the individual cell response to the stimulus. (D) Single cell analysis of calcium responses in WT and Fyn null MCs in the absence of extracellular calcium. (C and D) Graphs were generated by capturing the fluorecense intensity of a single cell with time (128–326 cells were monitored in total). An averaged response is shown by the line with open circles.
To distinguish whether the impaired Ca2+ influx of Fyn null MCs was distributed uniformly among cells in the population or resulted from the abberant response of a subset of nonresponding cells, we performed single cell Ca2+ imaging analysis. As shown in Fig. 2C, the extent of the Ca2+ response of individual Fyn null MCs in the presence of extracellular Ca2+ was generally decreased relative to WT cells. Although the number of MCs responding did not differ markedly between the two genotypes, a large proportion of Fyn null MCs showed reduced intensity or oscillating Ca2+ signals when compared with the more sustained responses seen in WT cells (see graphs in Fig. 2C). Averaging the Ag-dependent Ca2+ response of individual Fyn null versus WT MCs (∼298 vs. 326 cells were analyzed for the respective genotype) demonstrated that the large proportion of transiently responding cells was a major factor in the reduced population responses (Fig. 2C). Similar experiments were conducted in the absence of extracellular Ca2+ (Fig. 2D). Here, the number of Fyn null MCs (186 cells were analyzed) showing a reduced or oscillating response was not significantly different than WT cells (128 cells were analyzed). Thus, the mean response of Ag-dependent depletion of intracellular Ca2+ stores for individual Fyn null MCs was quite similar to that of WT cells (Fig. 2D). To determine further whether FcεRI-dependent, early signals leading to depletion of intracellular Ca2+ stores were affected by Fyn deficiency, we stimulated Ca2+ entry by use of the sarco/endoplasmic reticulum Ca2+ ATPase inhibitor, thapsigargin. As shown in Fig. 3A, thapsigargin induced a robust Ca2+ response from WT but not from Fyn null MCs in the presence of extracellular Ca2+. Thapsigargin stimulation of Fyn null MCs also showed defective uptake of 45Ca2+ (Fig. 3B). In contrast, the intracellular Ca2+ response to thapsigargin was identical for WT and Fyn null MCs (Fig. 3C). Collectively, these findings isolate the major defect of Fyn null MCs to an impaired ability to maintain the normal influx of Ca2+.
Figure 3. Thapsigargin stimulation of Fyn null MCs reveals normal depletion of intracellular calcium stores but impaired calcium influx.

(A) Thapsigargin (0.5 μM) induced a robust Ca2+ response from WT but not from Fyn null MCs in the presence of extracellular Ca2+. The arrow indicates the time of thapsigargin addition. Data are the mean ± se of all cells analyzed, and the statistical significance relative to WT (without treatment) was ***P < 0.001. One representative of 3 experiments is shown. (B) Thapsigargin stimulation of Fyn null MCs revealed a marked defect in 45Ca2+ uptake when compared with WT MCs. Data are the mean ± se of quadruplicate samples, and the statistical significance relative to WT (without treatment) was ***P < 0.001. One representative of 3 independent experiments is shown. (C) Thapsigargin-induced mobilization of intracellular Ca2+ in the absence of extracellular Ca2+ showed no difference in WT and Fyn null MCs. The arrow indicates the time of thapsigargin addition. Data are the mean ± se of all cells analyzed (42–51 cells were monitored in total). One representative of 3 experiments is shown. NS, Not significant.
In subsequent experiments, we explored the relationship between impaired calcium influx (45Ca2+) and actin depolymerization. As shown in Supplemental Fig. 3A, Fyn null MCs were defective in Ag-dependent 45Ca2+ influx, with an approximate 3-fold reduction in calcium uptake. To verify that a defect in Ca2+ uptake translates to impaired actin depolymerization, we studied these responses in LAT null MCs. These cells were described previously to have an impairment in SOCE, but unlike for Fyn null MCs, this defect is a result of the inability to activate PLCγ and thus, to initiate SOCE effectively through depletion of intracellular Ca2+ stores [33, 39]. Τherefore, LAT null MCs would be expected to be impaired in the influx of 45Ca2+ (Supplemental Fig. 3B). Moreover, like Fyn null MCs, LAT null MCs also failed to depolymerize cortical F-actin (Supplemental Fig. 3C). These findings demonstrated that the defective Ca2+ influx in Fyn and LAT null MCs is strongly linked to an impairment in cortical F-actin depolymerization. Importantly, like Fyn null MCs [12], LAT null MCs were also shown previously to be impaired in degranulation [33].
We next explored if replenishment of Ca2+ could restore the exocytotic response of Fyn null MCs. In these experiments, ionomycin was used at a concentration (150 nM) that elicits minimal MC degranulation on its own (Fig. 4A). However, in conjunction with Ag stimulation, this concentration of ionomycin augmented the degranulation response of WT MCs (Fig. 4A). Fyn null MCs showed impaired degranulation (50–>90% inhibition, depending on the Ag concentration; Fig. 4A). Treatment of these cells with ionomycin alone caused some modest degranulation relative to WT cells, but this was increased markedly upon Ag stimulation. At a concentration of 10 ng/ml Ag, the degranulation response of Fyn null MCs treated with ionomycin was similar to that of untreated or treated WT MCs (Fig. 4A), whereas Fyn null MCs stimulated with Ag alone showed a profound impairment. Importantly, the intracellular Ca2+ concentration of ionomycin-treated and Ag-stimulated Fyn null MCs mirrored that of Ag-stimulated WT MCs and was slightly less than that of ionomycin-treated and Ag-stimulated WT MCs (Fig. 4B), showing the association between increased Ca2+ responses and the extent of exocytosis. Moreover, cortical F-actin depolymerization in Ag-stimulated, ionomycin-treated WT and Fyn null MCs was identical (Fig. 4C). These findings demonstrated that Ca2+ replenishment can restore the degranulation response of Fyn null MCs, which is associated with increased intracellular Ca2+ and cortical F-actin depolymerization.
Figure 4. Calcium replenishment in Fyn null MCs restores normal F-actin depolymerization and degranulation.

(A) Degranulation of WT or Fyn null MCs was measured by the release of hexosaminidase after Ag and/or ionomycin stimulation (at the indicated concentrations). Data are mean ± se from 3 individual experiments. Statistical significance relative to appropriate control was ***P < 0.001. (B) WT and Fyn null MCs were loaded with calcium indicator dye (Fluo-4 AM and Fura Red AM) and activated by exposure to Ag (10 ng/ml) and/or ionomycin (150 nM). Calcium signals were determined by measuring the fluorescent ratio (Fluo-4/Fura Red) with time and reported as fold-increase relative to response in the absence of Ag. One representative of 4 experiments is shown. Data shown are a mean ± se. (C) Actin dynamics was measured in WT or Fyn null MCs, which were activated as in B. An arrow indicates the time of Ag and ionomycin addition. An averaged response is shown in the graph (12–16 cells were monitored in total). Data shown are a mean ± se.
Expression of Ca2+ channel components in Fyn null MCs
The finding of a defective Ca2+ influx in Fyn null MCs and the demonstration that Ca2+ replenishment could restore normal actin depolymerization and degranulation led us to explore the Ca2+ currents activated by Ag stimulation of these cells. Patch-clamp analysis revealed a defective inward current in Fyn null MCs relative to WT MCs (Fig. 5A and C). This defect was marked with a 5-fold reduction in peak current. Analysis of the I–V relationship (Fig. 5B and D) demonstrated a nonselective Ca2+ current in WT and Fyn null MCs with a marked impairment of inward and outward currents in the absence of Fyn. Thapsigargin stimulation of WT MCs (Fig. 5E and F) versus Fyn null MCs (Fig. 5G and H) also revealed this defect, consistent with the previous finding (Fig. 3) that thapsigargin stimulation of Fyn null MCs did not overcome the defect in Ca2+ influx. These findings suggested that the defect in Fyn null MCs was a result of the functional loss of a nonselective Ca2+ current, rather than the loss of the selective Ca2+ current (calcium ion release-activated calcium ion channels modulator or Orai1 [40, 41]) described previously, shown to be expressed in MCs. The apparent difference in the characteristics of the Ca2+ current could result from the source or culture of MCs used, as Orai1 null MCs were derived from embryonic liver [40, 41], whereas the cells used herein are cultured BMMCs.
Figure 5. Patch-clamp analysis reveals a defective, nonselective Ca2+ current in Fyn null MCs.

Whole-cell, patch-clamp measurements (see Materials and Methods) of inward current in Ag-stimulated WT (A) and Fyn null MCs (C) or thapsigargin (Thps)-stimulated WT (E) or Fyn null MCs (G). I–V relationship for WT or Fyn null MCs stimulated with Ag (B and D) or thapsigargin (F and H), respectively. Note the difference of the picoamperes/picofarad (pA/pF) scale. One representative of at least 3 independent experiments is shown.
We next explored the mRNA expression of some of the Ca2+ channels (TRPC family members and Orai) whose mRNA is known to be expressed in MCs [42, 43]. Although the presence of mRNA for TRPC1, -4, and -5, as well as Orai1, -2, and -3, was verified, we were unable to confirm the presence of mRNA for TRPC3 or TRPC4 in MCs by conventional or real-time PCR (Supplemental Fig. 4A and B). Strikingly, real-time PCR revealed a loss of TRPC1 mRNA expression in Fyn null MCs, whereas TRPC5 and TRPC6 expression showed no significant change (Supplemental Fig. 4A). In addition, no significant differences were detected in the mRNA expression of the Orai family members between WT and Fyn null MCs (Supplemental Fig. 4B). The finding of reduced TRPC1 expression in Fyn null MCs was confirmed subsequently by Western blot (Fig. 6A and B).
Figure 6. RNAi silencing of TRPC1 expression in WT MCs mimics the functional defects observed in Fyn null MCs.

(A) mRNA levels of TRPC1 in WT, WT MCs treated with TRPC1-specific siRNA [indicated as WT (+siRNA)], and Fyn null MCs were determined by real-time PCR. Data are mean ± se from 3 individual experiments. Statistical significance was **P < 0.01. (B) Expression of TRPC1 protein was determined by Western blots. Data are mean ± se from 4 individual experiments. Statistical significance relative to WT was *P < 0.05. (C) Scrambled sequence siRNA (nontargeting siRNA) was used as a control for WT and Fyn null MCs and showed no effect on TRPC1 or TRPC6 protein expression nor on cellular responses. (D) Calcium responses observed in WT, TRPC1 siRNA WT [WT (+siRNA)], and Fyn null MCs. Calcium was measured as described in Materials and Methods. Graph is representative of 3 independent experiments. Data are mean ± se. (E) Whole cell patch-clamp measurements of WT, TRPC1 siRNA WT, and Fyn null MCs. (F) I–V relationship of the current in WT, siRNA WT, and Fyn null MCs. (G) Mean current densities observed in WT, TRPC1 siRNA WT, and Fyn null MCs. Statistical significance relative to WT was ***P < 0.001. (H) TRPC1 siRNA WT MCs show defective degranulation. Statistical significance relative to appropriate control was ***P < 0.001. (I) Silencing of TRPC1 in WT cells inhibits cortical F-actin depolymerization. An arrow indicates the time of Ag addition. Mean ± se is shown in the graph (15–31 cells were monitored in total). *P < 0.05.
To test whether TRPC1 regulates Ca2+ influx in MCs, we used siRNA in WT MCs to obtain cells that expressed similar levels of TRPC1 mRNA and protein, as seen in Fyn null MCs (Fig. 6A and B). As shown in Fig. 6C and D, silencing of TRPC1 in WT cells to similar levels as seen in Fyn null MCs resulted in a cell with a Ca2+ response that was almost identical to that of Fyn null MCs. The specific TRPC1 siRNA and the nontargeting siRNA did not alter the levels of TRPC6 protein expression. Patch-clamp studies were consistent, and the aforementioned result was the current of the WT MCs, where TRPC1 was partially silenced and mirrored that of Fyn null MCs (Fig. 6E–G). Assessment of the exocytotic response and of cortical F-actin deploymerization in response to Ag stimulation showed that loss of TRPC1 from WT MCs also led to decreased MC degranulation and a failure to depolymerize cortical F-actin (Fig. 6H and I). However, normal degranulation was observed when TRPC1-silenced or Fyn null MCs were stimulated by ATP, substance P, or thrombin (Supplemental Fig. 5), demonstrating that TRPC1 is dispensable for degranulation via a purinergic or several G protein-coupled receptors. Collectively, these findings demonstrated that the loss of TRPC1 expression in WT MCs mirrors the functional defects observed in Fyn null MCs upon Ag challenge.
Ectopic expression of Fyn or of TRPC1 in Fyn null MCs induces recovery of FcεRI-stimulated Ca2+ entry, cortical F-actin depolymerization, and degranulation
To determine if Fyn kinase was required for the normal expression of TRPC1 and its function, we stably transduced Fyn null MCs with Fyn kinase. As shown in Fig. 7A, ectopic expression of Fyn restored the expression of TRPC1 to levels that were similar to those in WT cells. We then evaluated the Ca2+ response of Fyn-reconstituted cells and found that this response was also restored (Fig. 7B). Moreover, MC degranulation and the ability of these cells to depolymerize cortical F-actin were also restored (Fig. 7C and D), demonstrating that TRPC1 expression, activation of Ca2+ fluxes, cortical F-actin depolymerization, and MC degranulation required the presence of Fyn kinase.
Figure 7. Ectopic expression of Fyn kinase in Fyn null MCs restores TRPC1 expression, calcium responses, cortical F-actin depolymerization, and degranulation.

(A) Fyn null MCs stably transduced with Fyn kinase show normal expression of TRPC1 protein. Statistical significance relative to WT MCs is **P < 0.01. (B) Ectopic expression of Fyn kinase in Fyn null MCs restores enhanced calcium responses, which were measured as described in Materials and Methods. Data shown are mean ± se of 3 independent experiments. (C) Ectopic expression of Fyn kinase in Fyn null MCs restores degranulation to the level of WT MCs. One representative of 4 experiments is shown. Data are mean ± se. Statistical significance relative to Fyn null MCs was **P < 0.01. (D) Actin dynamics were measured in WT and Fyn kinase-transduced Fyn null MCs. The arrow indicates the time of Ag addition. Mean response (±se) is shown (19–26 cells were monitored in total).
To confirm directly that a deficiency in TRPC1 was a contributory factor in the defective reponse of Fyn null MCs, we expressed TRPC1 in Fyn null MCs (Fig. 8A). Expression of TRPC1 in viable Fyn null MCs led to a significant increase in Ag-dependent Ca2+ responses (Fig. 8B), increased MC degranulation (Fig. 8C), and normal cortical F-actin depolymerization (Fig. 8D) in viable cells. Thus, collectively, these findings show that TRPC1 participates in the FcεRI signal transduction cascade required for normal Ca2+ responses and MC degranulation.
Figure 8. Ectopic expression of TRPC1 in Fyn null MCs restores calcium responses, degranulation, and cortical F-actin depolymerization.

(A) Fyn null MCs stably transduced with TRPC1 express normal levels of TRPC1. One representative of 3 experiments is shown. (B) Fyn null MCs transduced with TRPC1 show enhanced calcium responses, which were measured as described in Materials and Methods. The arrow indicates the time of Ag addition. Data are the mean ± se of all cells analyzed from 3 independent experiments. (C) Degranulation is enhanced by expression of TRPC1 in Fyn null MCs. Data are mean ± se from 3 individual experiments. Statistical significance relative to Fyn null MCs was **P < 0.01. (D) Cortical F-actin depolymerization is restored by expression of TRPC1 in Fyn null MCs. The arrow indicates the time of Ag addition. Mean response (±se) is shown (16–24 cells were monitored in total).
DISCUSSION
Studies about MCs have shown that Fyn kinase is required for normal phosphatidylinositol 3-OH kinase (PI3K) activity [12, 13], phospholipase D activation [44], MC degranulation [9, 12], cytokine and leukotrine production [13], Kit and integrin-dependent Rac activation, cytoskeletal reorganization, chemotaxis [45, 46], and microtubule reorganization [14]. The loss of Fyn expression in MCs unmasked the contribution of TRPC1 in cortical F-actin deploymerization and in the degranulation of these cells.
As the early recognition of the crucial role for Ca2+ in MC degranulation [47] and the discovery of a selective Ca2+ current in rat peritoneal MCs and the tumor-derived MC line RBL-2H3 [48] are important for Ca2+ influx, much of the effort to understand MC activation and to develop therapeutic strategies to intervene in allergic disease has been focused on identifying this Ca2+ channel (reviewed in ref. [49]). Recently, the identity of this selective Ca2+ channel was revealed with the identification and cloning of the Orai1 family of calcium channels [40, 41]. In embryo liver-derived MCs, the absence of Orai1 was shown to cause a marked defect in exocytosis [50]. However, MCs also express other ion channels that can flux Ca2+ [43]. The presence of purinergic P2X channels, voltage-dependent Ca2+ channels, the TRP channels, and others has been described [42, 43]. Nonetheless, their role in MC biology and function is not well understood. TRPC channels, including TRPC1, have been reported to contribute to SOCE [51–56], and their activity is coupled to intracellular endoplasmic stores via the sensor STIM1 [57, 58]. RNA silencing of TRPC1 and TRPC5 in various cell types (including RBL-2H3) caused a marked reduction in Ca2+ entry [42, 59–61], demonstrating that the expression of Orai1 and STIM1 in the same cell was insufficient to initiate full Ca2+ entry. This suggests that other Ca2+ channels could contribute to Ca2+ influx in MCs. In support of this postulate, two recent studies [62, 63] demonstrated that Ca2+ influx causes TRPC5 activation, which then contributes to sustained influx of Ca2+. It may be possible that other TRPC channels may function in a similar manner. Importantly, although embryonic liver-derived MCs from Orai1 null mice showed a defective Ca2+ entry, some Ca2+ entry was still apparent [50], thus suggesting other potential contributors. We find that in BMMCs grown in IL-3 and SCF, a nonselective Ca2+ current is observed. This view of heterogeneity in Ca2+ currents is supported further by an analysis of ion channel expression in human lung, skin, and cord blood-derived MCs, which showed the presence of TRPC1 mRNA only in skin MCs [43], suggesting that the microenvironment is a likely determinant of the expression of TRP channels in MCs in vivo. We emphazise that our findings do not exclude a role for Orai1 or for TRPC5 and -6 in regulating Ca2+ influx in Fyn null or WT MCs. Relative to the effects of Orai1 deletion in MCs [50], reducing the levels of TRPC1 in MCs caused a less-marked, inhibitory effect on Ca2+ entry, yet it was sufficient to reduce cortical F-actin depolymerization and degranulation. Although it has been reported that TRPC1 mRNA levels are normal in Fyn null MCs [18], the reported experiments did not use quantitative PCR approaches (as used herein) nor was there verification of protein expression. Moreover, this study [18] reported that thapsigargin-induced Ca2+ fluxes were unimpaired in Fyn null MCs. However, modest differences in the extent of the thapsigargin-induced Ca2+ response were observed yet not explained, and the direct measurement of the uptake of radiolabeled Ca2+ in response to thapsigargin was not performed. The apparent discrepancy in our findings with the prior work [18] could result from the different concentrations of thapsigargin used for stimulation. We found that a concentration of 0.5 μM (used in this study) revealed considerable differences, whereas the use of 1.0 μM (used in the cited study [18]) masked these differences. Regardless, our findings of a role for TRPC1 in MC degranulation are also consistent with preliminary studies in MCs derived from TRPC1 null mice, which show a defect in degranulation (Nevenka Medic, Dean D. Metcalfe, and Alasdair M. Gilfillan, personal communication, NIAID, NIH, January 19, 2010).
Multiple studies [19, 20, 25, 26, 64] have shown that the cortical F-actin supresses MC degranulation. Deng and colleagues [20] demonstrated recently that F-actin-devoid, crater-like regions of the plasma membrane are the sites where granule membrane-plasma membrane fusion occurs. Nonetheless, a dual role for F-actin is suggested by a requirement for F-actin polymerization in MC degranulation after profound depletion of actin filaments [65]. Deficiencies in proteins, such as the Wiskott-Aldrich syndrome protein (which interact with the actin-polymerizing complex actin-related protein 2/3 [66]) and the Wiskott-Aldrich syndrome protein-interaction protein (which stabilizes actin filaments), also showed the requirement for actin microfilaments in normal MC degranulation [67, 68]. Interestingly, however, we did not observe a marked defect in cortical F-actin polymerization in Fyn null MCs. Therefore, our findings suggest that Fyn kinase is not essential to actin polymerization in MCs but instead, to the expression of TRPC1, which accounts for a significant level of Ca2+ influx and thus, cortical F-actin depolymerization.
Microtubules are also an important cytoskeletal component that is central to MC secretory granule transport [15, 16]. Fyn has been shown to be a component of microtubule nucleation in MCs [17]. This calcium-independent movement of granules was shown to be defective in Fyn null MCs [14]. Although our findings are not inconsistent with a defect in microtubule-dependent granule movement in Fyn null MCs, we find that restoration of Ca2+ influx can overcome this apparent defect and restore normal MC degranulation. This argues that the major defect in Fyn null MCs is the inability to influx normally the Ca2+ required for key steps, such as cortical F-actin depolymerization and granule-membrane fusion, which regulate MC degranulation.
Fyn kinase and other Src family tyrosine kinases have been implicated in the regulation of the activity of TRPC family members [69–71]. In particular, TRPC3 and -6 were shown to be phosphorylated and to complex with Src kinases, a potential mechanism by which receptor activation can control TRPC activity. The restoration of Fyn null MC responses by expression of TRPC1 suggests that Fyn phosphorylation of TRPC1 is not vital to its activity, although a compensatory effect by other Src kinases cannot be excluded. On the other hand, the previous observation that Fyn kinase is necessary for normal PI3K responses, which contribute to MC degranulation [12, 13], is the most likely explanation for the partial reconstitution of degranulation by expression of TRPC1 alone (Fig. 8). The observation that silencing of TRPC1 in WT BMMCs closely mimics the Ca2+ influx defect, the loss of actin depolymerization, and the impaired MC degranulation seen in Fyn null MCs provides a strong argument that TRPC1 contributes to these responses. The contribution of TRPC1 to degranulation appears to be FcεRI-selective, as stimulation of TRPC1- or Fyn-deficient MCs via a purinergic receptor or GPCRs did not manifest a defect (Supplemental Fig. 5). Regardless, these findings demonstrate that the nonselective cation channel TRPC1 is involved in the Ca2+ response of MCs and show that such channels contribute to MC degranulation. Given that TRPC1 mRNA was found to be expressed in human skin MCs [43], the findings may be of possible therapeutic importance to skin diseases, where MCs play a role.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Programs of the NIAMS and the National Institute of Dental and Craniofacial Research, NIH. We are grateful for the support of the Flow Cytometry Section, the Laboratory Animal Care and Use Section, and the Light Imaging Section of the Office of Science and Technology, NIAMS. We thank Drs. Nevenka Medic, Dean D. Metcalfe, and Alasdair Gilfillan (NIAID, NIH) for personal communication.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- 2-APB
- 2-aminoethoxydiphenyl borate
- Ca2+
- calcium ion
- CaCl2
- calcium chloride
- FcεRI
- high-affinity receptor for IgE
- I–V
- current-to-voltage
- LAT
- linker for activation of T cells
- MC
- mast cell
- mOsm
- milliohms
- NIAID
- National Institute of Allergy and Infectious Diseases (Bethesda, MD, USA)
- NIAMS
- National Institute of Arthritis and Musculoskeletal and Skin Diseases (Bethesda, MD, USA)
- NIH
- National Institutes of Health (Bethesda, MD, USA)
- pEYFP
- plasmid encoding enhanced yellow fluorescent protein
- RBL-2H3
- rat basophilic leukemia 2H3 cloned cell
- RNAi
- RNA interference
- SCF
- stem cell factor
- siRNA
- silencing RNA
- SOCE
- store-operated calcium entry
- STIM1
- stromal-interacting molecule 1
- TRPC
- transient receptor potential cation channel subfamily C member 1
AUTHORSHIP
R.S., X.L., A.O., L.A., Y.Y,. U.B., and I.A. contributed by conducting experiments, summarizing data, and/or generating necessary reagents or cells. R.S. and J.R. conceived and directed this study. R.S., U.B., I.A., and J.R. participated in the writing of the manuscript.
DISCLOSURE
The authors have no conflicting financial interests.
REFERENCES
- 1.Eiseman E., Bolen J. B. (1992) Engagement of the high-affinity IgE receptor activates src protein-related tyrosine kinases. Nature 355, 78–80. [DOI] [PubMed] [Google Scholar]
- 2.Paolini R., Jouvin M. H., Kinet J. P. (1991) Phosphorylation and dephosphorylation of the high-affinity receptor for immunoglobulin E immediately after receptor engagement and disengagement. Nature 353, 855–858. [DOI] [PubMed] [Google Scholar]
- 3.Pribluda V. S., Pribluda C., Metzger H. (1994) Transphosphorylation as the mechanism by which the high-affinity receptor for IgE is phosphorylated upon aggregation. Proc. Natl. Acad. Sci. USA 91, 11246–11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner H., Kinet J. P. (1999) Signaling through the high-affinity IgE receptor FcεRI. Nature 402 (Suppl.), B24–B30. [DOI] [PubMed] [Google Scholar]
- 5.Blank U., Rivera J. (2004) The ins and outs of IgE-dependent mast-cell exocytosis. Trends Immunol. 25, 266–273. [DOI] [PubMed] [Google Scholar]
- 6.Rivera J., Gilfillan A. M. (2006) Molecular regulation of mast cell activation. J. Allergy Clin. Immunol. 117, 1214–1225, quiz 1226. [DOI] [PubMed] [Google Scholar]
- 7.Nishizumi H., Yamamoto T. (1997) Impaired tyrosine phosphorylation and Ca2+ mobilization, but not degranulation, in Lyn-deficient bone marrow-derived mast cells. J. Immunol. 158, 2350–2355. [PubMed] [Google Scholar]
- 8.Kawakami Y., Kitaura J., Satterthwaite A. B., Kato R. M., Asai K., Hartman S. E., Maeda-Yamamoto M., Lowell C. A., Rawlings D. J., Witte O. N., Kawakami T. (2000) Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cell activation. J. Immunol. 165, 1210–1219. [DOI] [PubMed] [Google Scholar]
- 9.Odom S., Gomez G., Kovarova M., Furumoto Y., Ryan J. J., Wright H. V., Gonzalez-Espinosa C., Hibbs M. L., Harder K. W., Rivera J. (2004) Negative regulation of immunoglobulin E-dependent allergic responses by Lyn kinase. J. Exp. Med. 199, 1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernandez-Hansen V., Smith A. J., Surviladze Z., Chigaev A., Mazel T., Kalesnikoff J., Lowell C. A., Krystal G., Sklar L. A., Wilson B. S., Oliver J. M. (2004) Dysregulated FcεRI signaling and altered Fyn and SHIP activities in Lyn-deficient mast cells. J. Immunol. 173, 100–112. [DOI] [PubMed] [Google Scholar]
- 11.Kitaura J., Kawakami Y., Maeda-Yamamoto M., Horejsi V., Kawakami T. (2007) Dysregulation of Src family kinases in mast cells from epilepsy-resistant ASK versus epilepsy-prone EL mice. J. Immunol. 178, 455–462. [DOI] [PubMed] [Google Scholar]
- 12.Parravicini V., Gadina M., Kovarova M., Odom S., Gonzalez-Espinosa C., Furumoto Y., Saitoh S., Samelson L. E., O′Shea J. J., Rivera J. (2002) Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat. Immunol. 3, 741–748. [DOI] [PubMed] [Google Scholar]
- 13.Gomez G., Gonzalez-Espinosa C., Odom S., Baez G., Cid M. E., Ryan J. J., Rivera J. (2005) Impaired FcεRI-dependent gene expression and defective eicosanoid and cytokine production as a consequence of Fyn deficiency in mast cells. J. Immunol. 175, 7602–7610. [DOI] [PubMed] [Google Scholar]
- 14.Nishida K., Yamasaki S., Ito Y., Kabu K., Hattori K., Tezuka T., Nishizumi H., Kitamura D., Goitsuka R., Geha R. S., Yamamoto T., Yagi T., Hirano T. (2005) FcεRI-mediated mast cell degranulation requires calcium-independent microtubule-dependent translocation of granules to the plasma membrane. J. Cell Biol. 170, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin-Verdeaux S., Pombo I., Iannascoli B., Roa M., Varin-Blank N., Rivera J., Blank U. (2003) Analysis of Munc18–2 compartmentation in mast cells reveals a role for microtubules in granule exocytosis. J. Cell Sci. 116, 325–334. [DOI] [PubMed] [Google Scholar]
- 16.Smith A. J., Pfeiffer J. R., Zhang J., Martinez A. M., Griffiths G. M., Wilson B. S. (2003) Microtubule-dependent transport of secretory vesicles in RBL-2H3 cells. Traffic 4, 302–312. [DOI] [PubMed] [Google Scholar]
- 17.Macurek L., Draberova E., Richterova V., Sulimenko V., Sulimenko T., Draberova L., Markova V., Draber P. (2008) Regulation of microtubule nucleation from membranes by complexes of membrane-bound γ-tubulin with Fyn kinase and phosphoinositide 3-kinase. Biochem. J. 416, 421–430. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez-Miranda E., Ibarra-Sanchez A., Gonzalez-Espinosa C. (2010) Fyn kinase controls FcεRI receptor-operated calcium entry necessary for full degranulation in mast cells. Biochem. Biophys. Res. Commun. 391, 1714–1720. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen E. H., Braun K., Johansen T. (1989) Reorganization of the subplasmalemmal cytoskeleton in association with exocytosis in rat mast cells. Histol. Histopathol. 4, 473–477. [PubMed] [Google Scholar]
- 20.Deng Z., Zink T., Chen H. Y., Walters D., Liu F. T., Liu G. Y. (2009) Impact of actin rearrangement and degranulation on the membrane structure of primary mast cells: a combined atomic force and laser scanning confocal microscopy investigation. Biophys. J. 96, 1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price L. S., Norman J. C., Ridley A. J., Koffer A. (1995) The small GTPases Rac and Rho as regulators of secretion in mast cells. Curr. Biol. 5, 68–73. [DOI] [PubMed] [Google Scholar]
- 22.Norman J. C., Price L. S., Ridley A. J., Koffer A. (1996) The small GTP-binding proteins, Rac and Rho, regulate cytoskeletal organization and exocytosis in mast cells by parallel pathways. Mol. Biol. Cell 7, 1429–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen J. D., Jaffer Z. M., Park S. J., Burgin S., Hofmann C., Sells M. A., Chen S., Derr-Yellin E., Michels E. G., McDaniel A., Bessler W. K., Ingram D. A., Atkinson S. J., Travers J. B., Chernoff J., Clapp D. W. (2009) p21-Activated kinase regulates mast cell degranulation via effects on calcium mobilization and cytoskeletal dynamics. Blood 113, 2695–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koffer A., Tatham P. E., Gomperts B. D. (1990) Changes in the state of actin during the exocytotic reaction of permeabilized rat mast cells. J. Cell Biol. 111, 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narasimhan V., Holowka D., Baird B. (1990) Microfilaments regulate the rate of exocytosis in rat basophilic leukemia cells. Biochem. Biophys. Res. Commun. 171, 222–229. [DOI] [PubMed] [Google Scholar]
- 26.Frigeri L., Apgar J. R. (1999) The role of actin microfilaments in the down-regulation of the degranulation response in RBL-2H3 mast cells. J. Immunol. 162, 2243–2250. [PubMed] [Google Scholar]
- 27.Torigoe C., Song J., Barisas B. G., Metzger H. (2004) The influence of actin microfilaments on signaling by the receptor with high-affinity for IgE. Mol. Immunol. 41, 817–829. [DOI] [PubMed] [Google Scholar]
- 28.Tolarova H., Draberova L., Heneberg P., Draber P. (2004) Involvement of filamentous actin in setting the threshold for degranulation in mast cells. Eur. J. Immunol. 34, 1627–1636. [DOI] [PubMed] [Google Scholar]
- 29.Liu F. T., Bohn J. W., Ferry E. L., Yamamoto H., Molinaro C. A., Sherman L. A., Klinman N. R., Katz D. H. (1980) Monoclonal dinitrophenyl-specific murine IgE antibody: preparation, isolation, and characterization. J. Immunol. 124, 2728–2737. [PubMed] [Google Scholar]
- 30.Razin E., Ihle J. N., Seldin D., Mencia-Huerta J. M., Katz H. R., LeBlanc P. A., Hein A., Caulfield J. P., Austen K. F., Stevens R. L. (1984) Interleukin 3: a differentiation and growth factor for the mouse mast cell that contains chondroitin sulfate E proteoglycan. J. Immunol. 132, 1479–1486. [PubMed] [Google Scholar]
- 31.Olivera A., Urtz N., Mizugishi K., Yamashita Y., Gilfillan A. M., Furumoto Y., Gu H., Proia R. L., Baumruker T., Rivera J. (2006) IgE-dependent activation of sphingosine kinases 1 and 2 and secretion of sphingosine 1-phosphate requires Fyn kinase and contributes to mast cell responses. J. Biol. Chem. 281, 2515–2525. [DOI] [PubMed] [Google Scholar]
- 32.Olivera A., Mizugishi K., Tikhonova A., Ciaccia L., Odom S., Proia R. L., Rivera J. (2007) The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity 26, 287–297. [DOI] [PubMed] [Google Scholar]
- 33.Saitoh S., Arudchandran R., Manetz T. S., Zhang W., Sommers C. L., Love P. E., Rivera J., Samelson L. E. (2000) LAT is essential for FcεRI-mediated mast cell activation. Immunity 12, 525–535. [DOI] [PubMed] [Google Scholar]
- 34.Blank U., Rivera J. (2006) Assays for regulated exocytosis of mast cell granules. Curr. Protoc. Cell Biol. Chapter 15, Unit 15.11. [DOI] [PubMed] [Google Scholar]
- 35.Furumoto Y., Brooks S., Olivera A., Takagi Y., Miyagishi M., Taira K., Casellas R., Beaven M. A., Gilfillan A. M., Rivera J. (2006) Cutting edge: lentiviral shRNA silencing of PTEN in human mast cells reveals constitutive signals that promote cytokine secretion and cell survival. J. Immunol. 176, 5167–5171. [DOI] [PubMed] [Google Scholar]
- 36.Malacombe M., Bader M. F., Gasman S. (2006) Exocytosis in neuroendocrine cells: new tasks for actin. Biochim. Biophys. Acta 1763, 1175–1183. [DOI] [PubMed] [Google Scholar]
- 37.Park M. K., Lee K. K., Uhm D. Y. (2002) Slow depletion of endoplasmic reticulum Ca(2+) stores and block of store-operated Ca(2+) channels by 2-aminoethoxydiphenyl borate in mouse pancreatic acinar cells. Naunyn Schmiedebergs Arch. Pharmacol. 365, 399–405. [DOI] [PubMed] [Google Scholar]
- 38.DeHaven W. I., Smyth J. T., Boyles R. R., Bird G. S., Putney J. W., Jr. (2008) Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 283, 19265–19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saitoh S., Odom S., Gomez G., Sommers C. L., Young H. A., Rivera J., Samelson L. E. (2003) The four distal tyrosines are required for LAT-dependent signaling in FcεRI-mediated mast cell activation. J. Exp. Med. 198, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., Kinet J. P. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P. G. (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. [DOI] [PubMed] [Google Scholar]
- 42.Ma H. T., Peng Z., Hiragun T., Iwaki S., Gilfillan A. M., Beaven M. A. (2008) Canonical transient receptor potential 5 channel in conjunction with Orai1 and STIM1 allows Sr2+ entry, optimal influx of Ca2+, and degranulation in a rat mast cell line. J. Immunol. 180, 2233–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bradding P., Okayama Y., Kambe N., Saito H. (2003) Ion channel gene expression in human lung, skin, and cord blood-derived mast cells. J. Leukoc. Biol. 73, 614–620. [DOI] [PubMed] [Google Scholar]
- 44.Choi W. S., Hiragun T., Lee J. H., Kim Y. M., Kim H. P., Chahdi A., Her E., Han J. W., Beaven M. A. (2004) Activation of RBL-2H3 mast cells is dependent on tyrosine phosphorylation of phospholipase D2 by Fyn and Fgr. Mol. Cell. Biol. 24, 6980–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samayawardhena L. A., Hu J., Stein P. L., Craig A. W. (2006) Fyn kinase acts upstream of Shp2 and p38 mitogen-activated protein kinase to promote chemotaxis of mast cells towards stem cell factor. Cell. Signal. 18, 1447–1454. [DOI] [PubMed] [Google Scholar]
- 46.Samayawardhena L. A., Kapur R., Craig A. W. (2007) Involvement of Fyn kinase in Kit and integrin-mediated Rac activation, cytoskeletal reorganization, and chemotaxis of mast cells. Blood 109, 3679–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foreman J. C., Mongar J. L., Gomperts B. D. (1973) Calcium ionophores and movement of calcium ions following the physiological stimulus to a secretory process. Nature 245, 249–251. [DOI] [PubMed] [Google Scholar]
- 48.Hoth M., Penner R. (1992) Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356. [DOI] [PubMed] [Google Scholar]
- 49.Vig M., Kinet J. P. (2007) The long and arduous road to CRAC. Cell Calcium 42, 157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vig M., DeHaven W. I., Bird G. S., Billingsley J. M., Wang H., Rao P. E., Hutchings A. B., Jouvin M. H., Putney J. W., Kinet J. P. (2008) Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 9, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X., Cheng K. T., Bandyopadhyay B. C., Pani B., Dietrich A., Paria B. C., Swaim W. D., Beech D., Yildrim E., Singh B. B., Birnbaumer L., Ambudkar I. S. (2007) Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(−/−) mice. Proc. Natl. Acad. Sci. USA 104, 17542–17547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu X., Singh B. B., Ambudkar I. S. (2003) TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the S5–S6 region. J. Biol. Chem. 278, 11337–11343. [DOI] [PubMed] [Google Scholar]
- 53.Ambudkar I. S., Ong H. L., Liu X., Bandyopadhyay B. C., Cheng K. T. (2007) TRPC1: the link between functionally distinct store-operated calcium channels. Cell Calcium 42, 213–223. [DOI] [PubMed] [Google Scholar]
- 54.Parekh A. B., Putney J. W., Jr. (2005) Store-operated calcium channels. Physiol. Rev. 85, 757–810. [DOI] [PubMed] [Google Scholar]
- 55.Venkatachalam K., van Rossum D. B., Patterson R. L., Ma H. T., Gill D. L. (2002) The cellular and molecular basis of store-operated calcium entry. Nat. Cell Biol. 4, E263–E272. [DOI] [PubMed] [Google Scholar]
- 56.Montell C. (2005) The TRP superfamily of cation channels. Sci. STKE 2005, re3. [DOI] [PubMed] [Google Scholar]
- 57.Huang G. N., Zeng W., Kim J. Y., Yuan J. P., Han L., Muallem S., Worley P. F. (2006) STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 8, 1003–1010. [DOI] [PubMed] [Google Scholar]
- 58.Lopez J. J., Salido G. M., Pariente J. A., Rosado J. A. (2006) Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J. Biol. Chem. 281, 28254–28264. [DOI] [PubMed] [Google Scholar]
- 59.Ong H. L., Cheng K. T., Liu X., Bandyopadhyay B. C., Paria B. C., Soboloff J., Pani B., Gwack Y., Srikanth S., Singh B. B., Gill D. L., Ambudkar I. S. (2007) Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 282, 9105–9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng K. T., Liu X., Ong H. L., Ambudkar I. S. (2008) Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 283, 12935–12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cohen R., Torres A., Ma H. T., Holowka D., Baird B. (2009) Ca2+ waves initiate antigen-stimulated Ca2+ responses in mast cells. J. Immunol. 183, 6478–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gross S. A., Guzman G. A., Wissenbach U., Philipp S. E., Zhu M. X., Bruns D., Cavalie A. (2009) TRPC5 is a Ca2+-activated channel functionally coupled to Ca2+-selective ion channels. J. Biol. Chem. 284, 34423–34432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blair N. T., Kaczmarek J. S., Clapham D. E. (2009) Intracellular calcium strongly potentiates agonist-activated TRPC5 channels. J. Gen. Physiol. 133, 525–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oka T., Hori M., Tanaka A., Matsuda H., Karaki H., Ozaki H. (2004) IgE alone-induced actin assembly modifies calcium signaling and degranulation in RBL-2H3 mast cells. Am. J. Physiol. Cell Physiol. 286, C256–C263. [DOI] [PubMed] [Google Scholar]
- 65.Pendleton A., Koffer A. (2001) Effects of latrunculin reveal requirements for the actin cytoskeleton during secretion from mast cells. Cell Motil. Cytoskeleton 48, 37–51. [DOI] [PubMed] [Google Scholar]
- 66.Machesky L. M., Insall R. H. (1998) Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 8, 1347–1356. [DOI] [PubMed] [Google Scholar]
- 67.Kettner A., Kumar L., Anton I. M., Sasahara Y., de la Fuente M., Pivniouk V. I., Falet H., Hartwig J. H., Geha R. S. (2004) WIP regulates signaling via the high affinity receptor for immunoglobulin E in mast cells. J. Exp. Med. 199, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pivniouk V. I., Snapper S. B., Kettner A., Alenius H., Laouini D., Falet H., Hartwig J., Alt F. W., Geha R. S. (2003) Impaired signaling via the high-affinity IgE receptor in Wiskott-Aldrich syndrome protein-deficient mast cells. Int. Immunol. 15, 1431–1440. [DOI] [PubMed] [Google Scholar]
- 69.Hisatsune C., Kuroda Y., Nakamura K., Inoue T., Nakamura T., Michikawa T., Mizutani A., Mikoshiba K. (2004) Regulation of TRPC6 channel activity by tyrosine phosphorylation. J. Biol. Chem. 279, 18887–18894. [DOI] [PubMed] [Google Scholar]
- 70.Vazquez G., Wedel B. J., Kawasaki B. T., Bird G. S., Putney J. W., Jr. (2004) Obligatory role of Src kinase in the signaling mechanism for TRPC3 cation channels. J. Biol. Chem. 279, 40521–40528. [DOI] [PubMed] [Google Scholar]
- 71.Kawasaki B. T., Liao Y., Birnbaumer L. (2006) Role of Src in C3 transient receptor potential channel function and evidence for a heterogeneous makeup of receptor- and store-operated Ca2+ entry channels. Proc. Natl. Acad. Sci. USA 103, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.