

Abstract
High glucose (HG) has been shown to induce insulin resistance in both type 1 and type 2 diabetes. However, the molecular mechanism behind this phenomenon is unknown. Insulin receptor substrate (IRS) proteins are the key signaling molecules that mediate insulin’s intracellular actions. Genetic and biological studies have shown that reductions in IRS1 and/or IRS2 protein levels are associated with insulin resistance. In this study we have shown that proteasome degradation of IRS1, but not of IRS2, is involved in HG-induced insulin resistance in Chinese hamster ovary (CHO) cells as well as in primary hepatocytes. To further investigate the molecular mechanism by which HG induces insulin resistance, we examined various molecular candidates with respect to their involvement in the reduction in IRS1 protein levels. In contrast to the insulin-induced degradation of IRS1, HG-induced degradation of IRS1 did not require IR signaling or phosphatidylinositol 3-kinase/Akt activity. We have identified glycogen synthase kinase 3β (GSK3β or GSK3B as listed in the MGI Database) as a kinase required for HG-induced serine332 phosphorylation, ubiquitination, and degradation of IRS1. Overexpression of IRS1 with mutation of serine332 to alanine partially prevents HG-induced IRS1 degradation. Furthermore, overexpression of constitutively active GSK3β was sufficient to induce IRS1 degradation. Our data reveal the molecular mechanism of HG-induced insulin resistance, and support the notion that activation of GSK3β contributes to the induction of insulin resistance via phosphorylation of IRS1, triggering the ubiquitination and degradation of IRS1.
Introduction
Insulin resistance is a major pathophysiological problem in both type 1 and type 2 diabetes mellitus, and is a key component of the metabolic syndrome (DeFronzo et al. 1982, Fourlanos et al. 2004). Insulin resistance is characterized by a progressive reduction in the response of insulin-sensitive tissues to a normal level of insulin (Kahn 1994). The molecular events that mediate insulin’s actions in its target tissues have been well detailed in recent years due to extensive genetic and molecular studies (Virkamaki et al. 1999, Lee & White 2004, Taniguchi et al. 2006). In comparison, the molecular basis of insulin resistance is considerably less clear.
Insulin receptor substrate (IRS) proteins are major docking proteins that mediate insulin/insulin-like growth factor 1 (IGF1) signaling in various insulin-sensitive tissues (White 2006). IRS1, like other IRS proteins, contains many tyrosyl phosphorylation sites for IR tyrosine kinase (Sun et al. 1993). Tyrosine phosphorylation of IRS1 creates binding sites for proteins containing the Src homology-2 domain including phosphatidylinositol 3-kinase (PI3K), the GRB2/SOS complex, SHP-2 (or PTPN11 as listed in the MGI Database), c-fyn, and NCK (Sun et al. 1993, White 2006). These interactions lead to the activation of multiple signaling pathways required for insulin’s pleiotropic actions (White 2006). In addition, IRS1 also contains multiple serine/threonine phosphorylation sites (Sun et al. 1991). It is subject to phosphorylation by a wide array of protein kinases that are involved in diverse signaling pathways including those stimulated by insulin, inflammation, nutrients, and cellular stress (Sun & Liu 2009). Many serine/threonine kinases that phosphorylate IRS1 have been identified, including glycogen synthase kinase 3β (GSK3β or GSK3B), p70S6K, protein kinase Cθ (PKCθ), ERK, JNK, MTOR, and AMPK among others (Sun & Liu 2009). Most IRS1 serine/threonine phosphorylation events attenuate insulin signaling, and are thought to induce insulin resistance.
Studies in humans, animals (Saad et al. 1992, Rondinone et al.1997), and cultured cells(Lee et al. 2000, Zhande et al.2002) have reported an inverse correlation between IRS1 protein levels and insulin resistance. Mice deficient in IRS1 are insulin resistant and growth retarded (Araki et al. 1994, Lee et al. 2000); diabetes develops when the loss of IRS1 is combined with a heterozygous null IR mutation (Bruning et al. 1997). Chronic insulin treatment, a well-established method for inducing insulin resistance (Del Prato et al. 1994, Koopmans et al. 1997), induces ubiquitin-mediated proteasome degradation of IRS1, leading to cellular insulin resistance (Sun et al. 1999, Lee et al. 2000, Rui et al. 2002, Zhande et al. 2002). Recently, an in vivo siRNA-based study demonstrated the essential role of IRS1 in the regulation of gluconeogenic gene expression in the liver, such that knockdown of Irs1 resulted in the upregulation of gluconeogenic genes (Taniguchi et al. 2005). These studies emphasize the importance of cellular levels of IRS1 in insulin sensitivity.
It has been reported that chronic hyperglycemia can induce insulin resistance in both humans and animals (Vuorinen-Markkola et al. 1992, Buren et al. 2003). In contrast to that induced by hyperinsulinemia, hyperglycemia-catalyzed resistance is less straightforward. It is unclear whether high glucose (HG) inhibits insulin signaling directly through metabolite effects, secondarily by stimulation of excessive insulin secretion, via a combination of the two aforementioned mechanisms, or through some yet undiscovered mechanism. From cell-based studies and research in type 1 diabetes, it can be concluded that hyperglycemia certainly does not require secondary hyperinsulinemia to antagonize insulin function. However, even in insulin-free systems, the mechanism of hyperglycemia-induced insulin resistance is unclear. Activation of PKC, degradation of IRS2, and generation of reactive oxygen species have all been proposed, with no conclusive model to date (Berti et al. 1994, Ide et al. 1994, Pillay et al. 1996, Nakajima et al. 2000).
GSK3β is a serine kinase negatively regulated by insulin that was originally identified as a key regulator of glycogen synthesis. Insulin activates AKT/protein kinase B through a well-defined mechanism mediated by the IRS1/PI3K pathway. This leads to the phosphorylation of GSK3β at serine9, resulting in its inactivation (Cross et al. 1995, Shaw et al. 1997, Frame & Cohen 2001). In addition to glycogen synthesis, GSK3β plays roles in many other biological functions including Wnt signaling (crucial in development) and NFκB (or NFKB) signaling (a key mediator of the inflammatory response; Frame & Cohen 2001, Lee & Kim 2007). GSK3β is also thought to be involved in insulin resistance. In type 2 diabetes, GSK3β activity is elevated twofold in both basal and insulin-stimulated muscle (Nikoulina et al. 2000). Additionally, inhibition of GSK3β improves insulin’s action and glucose metabolism in human smooth muscle (Nikoulina et al. 2002). GSK3β has been shown to directly phosphorylate IRS1 in vitro and in vivo at serine332 and impair insulin signaling (Eldar-Finkelman & Krebs 1997, Liberman & Eldar-Finkelman 2005). Thus, GSK3β may play a causative role in the development of insulin resistance.
In this study, we have investigated the molecular mechanism of HG-induced insulin resistance in both Chinese hamster ovary (CH O) cells and primary mouse hepatocytes. We report that HG promotes proteasome-mediated degradation of IRS1. Unlike insulin, HG-induced degradation is independent of IR signaling and PI3K activity. We have identified GSK3β as a key kinase that mediates HG-induced degradation of IRS1.
Materials and Methods
Cells and transfection
CHO cells and CHO/IR cells (CHO cells overexpressing the IR) were grown in F12 medium supplemented with 10% fetal bovine serum (FBS). They were cultured at 37 °C in a humidified incubator maintained at 5% CO2.
CHO/IR cells overexpressing His-tagged IRS1 (IRS1His6), wild-type IRS1, IRS1 with mutation of serine332 to alanine, or IRS1 with mutation of serine332/336 to alanine were obtained by transfection of CHO/IR cells with pCMV/His/IRS1His6, pCMV/His/IRS1, pCMV/His/IRS1S332A, or pCMV/His/IRS1S332/336A constructs respectively using lipofectamine (Invitrogen; Zhande et al. 2002, Liberman & Eldar-Finkelman 2005). Transfected cells were selected for resistance to 10 mM histidinol for 3 weeks before being used in the experiments.
Adenoviral infection
For the expression of GSK3β variants, recombinant adenoviruses were generated and used as previously described (Finlay et al. 2004). In total, 40% confluent CHO/IR cells were incubated with 200 MOI of the following adenoviruses: AdV-Fluc (virus control), AdV-GSK3β-KD (kinase-deficient mutant), or AdV-GSK3βS9A (constitutively active mutant). Infections were performed in F12 medium containing 2% FBS for 2 h. The medium was then removed, and the cells were washed twice with PBS. F12 medium containing 10% FBS was added again, and the cells were allowed to recover for at least 20 h. The adenovirus-infected CHO/IR cells were then used in the experiments as described.
Primary hepatocyte isolation and culture
Primary mouse hepatocytes (C57/BJ6) were isolated by a modified noncirculating collagenase perfusion method as described (Berry & Friend 1969, Zhande et al. 2006).
Prolonged HG exposure
For glucose exposure in CHO cells, the cells were cultured to 80–85% confluency, and were then incubated with low glucose (5·5 mM or 1 g/l) or HG (25 mM or 4·5 g/l) DMEM containing 0·25% BSA for various time points. The medium was refreshed every 24 h if incubation was done for longer than 24 h. For glucose exposure in primary mouse hepatocytes, the cells were cultured in low glucose (5·5 mM) or HG (25 mM) DMEM with 10% FBS. In some cases, insulin (5 or 10 nM) or inhibitors, including cycloheximide (0·5 mM), MG132 (25 μM), LY294002 (50 μM), LiCl (20 mM), rapamycin (200 nM), and PD98059 (0·1 mM), were also included in the medium. At the end of the incubation, cells were left unstimulated or stimulated with 10 nM insulin for 10 min before being lysed in an appropriate buffer.
His-tagged protein purification
CHO/IR cells overexpressing His6-tagged IRS1 (IRS1His6; Zhande et al. 2002) were washed once with ice-cold PBS containing 100 μM Na3VO4 and 1 mM phenylmethyl-sulfonyl fluoride (PMSF), and lysed in 1 ml lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole, 1% Tween 20, 1 mM PMSF, 100 μM Na3PO4, 50 μg/ml aprotinin, 50 μg/ml leupeptin, and 50 μg/ml microcystin) for 10 min. Lysates were centrifuged at 13 000 g for 15 min, and the supernatants were mixed with Ni-NTA beads (Qiagen; 200 μl per 10 cm plate of cells). The beads with IRS1His6 were washed with a wash buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 20 mM imidazole, 1% Tween 20, 1 mM PMSF, 100 μM Na3PO4, 50 μg/ml aprotinin, and 50 μg/ml leupeptin) three times, and were then denatured in Laemmli sample buffer containing 0·1 M dithiothreitol (DTT) and boiled for 5 min.
Whole cell lysate preparation
CHO cells, CHO/IR cells, or primary mouse hepatocytes were lysed directly in Laemmli sample buffer containing 0·1 M DTT, sonicated, and boiled for 5 min.
Immunoblotting analysis
Samples from Ni-NTA purification or whole cell lysates were separated by SDS-7·5% or 10% PAGE, and transferred onto nitrocellulose membranes. Membranes were briefly washed with tris buffered saline (TBS; 20 mM Tris–HCl, pH 8·0, and 0·15 M NaCl) and blocked (5% skim milk in TBST (TBS containing 0·05% Tween 20)) for 2 h at room temperature or overnight at 4 °C, followed by incubation with the corresponding antibodies in the appropriate blocking solution (5% skim milk–TBST or 5% BSA–TBST): αPY (PY20), αIRβ (C-19) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), αPS9-GSK3βα-P44/42 ERK1/2, αERK1/2, αPT24 FoxO1, αGSK3β (Cell Signaling Technology, Inc.), αubiquitin (FK2, Biomol International, LP, Plymouth Meeting, PA, USA), αIRS1, αIRS2, and αPS332IRS1. Membranes were then washed three times with TBST and probed with HRP-conjugated protein A (Santa Cruz) for 30 min, or HRP-conjugated rat anti-mouse (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) or HRP-conjugated anti-rabbit antibody (Jackson ImmunoResearch Laboratories, Inc.) for 1 h. After incubation with secondary antibodies, membranes were washed three times with TBST and once with TBS. Specific proteins were visualized using ECL (GE Healthcare, Pittsburg, PA, USA; Zhande et al. 2002).
Results
HG induces degradation of IRS1, but not of IRS2
We have shown previously that prolonged insulin treatment can induce degradation of IRS1, but not of IRS2, in CHO cells overexpressing the human IR (CHO/IR; Sun et al. 1999, Zhande et al. 2002). To investigate if HG could lead to the same effect, CHO/IR cells were exposed to normal glucose (5·5 mM) or HG (25 mM) for various lengths of time. Reductions in IRS1 protein levels were observed after 72 h of exposure (Fig. 1A, lanes a–d and f versus lane e). This was not due to changes in cell number since nonspecific bands (NS) and GSK3β bands were constant at all time points. To ensure that this effect was not limited to CHO/IR cells, we performed similar experiments using primary mouse hepatocytes. We obtained comparable data, although the reduction (50%) in IRS1 protein levels was observed after 22 h of exposure to HG (Fig. 1B, panel 1). Interestingly, this effect was specific to the IRS1 molecule, since HG exposure had no effect on IRS2 protein levels under the same conditions (Fig. 1B, panel 2).
Figure 1.

Effect of high glucose on IRS1 protein levels. (A) CHO/IR cells were incubated in DMEM–0·25% BSA containing 5·5 mM (solid lines) or 25 mM glucose (dashed lines) for the indicated time periods. The medium was refreshed every 24 h. (B) Primary mouse hepatocytes were incubated in DMEM–10% FBS containing 5·5 or 25 mM glucose for 22 h. (C) CHO/IR cells were incubated in DMEM–0·25% BSA containing 5·5 or 25 mM glucose in the presence or absence of cycloheximide (0·5 mM) and MG132 (25 μM) for 14 h. After treatment(s), all cells were lysed in Laemmli sample buffer containing 0·1 M DTT. IRS1 and IRS2 proteins in the cell lysates were detected by immunoblotting analysis using anti-IRS1 and anti-IRS2 antibodies respectively as described in Materials and methods. Results are representative of at least three separate experiments. The densities for western blot bands were quantified by Image J software. The ratio of density for IRS1 and GSK3β proteins was calculated for each lane, and the value for normal (5·5 mM) was considered as 1 and compared with other treatments for each individual experiment. Data are expressed as means±S.E.M., n=3.
Intracellular protein levels are determined by the balance between protein synthesis and degradation. To evaluate the contribution of impaired synthesis versus enhanced degradation to HG-induced reduction of IRS1 protein levels, CHO/IR cells were coincubated with HG and cycloheximide, a protein translation inhibitor, for 14 h. Without cycloheximide, the reduction in IRS1 protein levels by HG was not detectable after 14 h of exposure (Fig. 1C, lane b versus lane a). When cycloheximide was present, a significant reduction in IRS1 protein levels by HG was observed (Fig. 1C, lane d versus lane c). This suggests that the effect of HG on IRS1 is not mediated by suppression of IRS1 gene transcription or mRNA translation, but rather that HG acts by augmenting IRS1 protein degradation. Indeed, MG132, a relatively specific proteasome inhibitor, significantly blocked HG-induced degradation of IRS1 (Fig. 1C, lanes e–f versus lanes c–d), implying the involvement of the ubiquitin-mediated proteasome degradation pathway.
To test if the reduction in IRS1 protein levels could impact insulin signaling, we examined insulin-stimulated tyrosyl phosphorylation of IRS proteins and downstream signaling events in primary hepatocytes. HG impaired insulin signaling, including insulin-induced tyrosine phosphorylation of IRS proteins (30%), threonine24 phosphorylation of FoxO1 (20%), serine9 phosphorylation of GSK3β threonine202/tyrosine204 phosphorylation of ERK (35%), and serine473 phosphorylation of AKT (65%; Fig. 2, lane c versus lane b).
Figure 2.

Effect of high glucose on insulin signaling. Primary mouse hepatocytes were incubated in DMEM–10% FBS containing 5·5 or 25 mM glucose for 22 h. Cells were challenged with insulin (10 nM) for 10 min before being lysed in Laemmli sample buffer containing 0·1 M DTT. Signal proteins in the cell lysates were detected by immunoblotting analysis using the corresponding antibodies. Quantification of western blot was done in the same way as described in Fig. 1. Data are expressed as means± S.E.M., n = 3.
GSK3β is required for HG-induced IRS1 degradation
To investigate the signaling pathway that mediates HG-induced degradation of IRS1, we used several specific inhibitors for various serine/threonine kinases that are involved in the modulation of insulin sensitivity. These include LiCl (GSK3β), rapamycin (MTOR), PD98059 (MEK), and LY294002 (PI3K). Among these tested inhibitors, LiCl was the only inhibitor that was capable of completely blocking HG-induced degradation of IRS1 (Fig. 3A, panel 1, lane d versus lanes b, f, and h; Fig. 4C, panels 1 and 3, lane b versus lane d), suggesting the involvement of GSK3β in HG-induced degradation of IRS1. Since phosphorylation of GSK3β at serine9 inactivates the kinase, we next checked the phosphorylation level of GSK3β. Indeed, HG exposure led to a significant reduction in GSK3β serine9 phosphorylation (activation; Fig. 3A, panel 2, lane b versus lane a), and LiCl enhanced the phosphorylation of GSK3β (inactivation; Fig. 3A, panel 2, lanes c–d versus lanes a–b). The effect of LiCl on GSK3β phosphorylation and HG-induced degradation of IRS1, but not of IRS2, was also observed in primary hepatocytes (Fig. 3B, panels 1–3, lanes c–d versus lanes a–b). PD98059, rapamycin, and LY294002 were unable to block the reduction of GSK3β phosphorylation or degradation of IRS1 induced by HG (Fig. 3A, panels 1–2, lanes e–h versus lanes a–b; Fig. 4C, panels 1–4, lanes a–b versus lanes c–d). These data strongly suggest a specific role for GSK3β in the mediation HG-induced degradation of IRS1.
Figure 3.

Involvement of GSK3β in HG-induced IRS1 degradation. (A) CHO/IR cells were incubated in DMEM–0·25% BSA with cycloheximide (0·5 mM) and 5·5 or 25 mM glucose containing various inhibitors: LiCl (20 mM), rapamycin (200 nM), or PD98059 (0·1 mM) for 16 h. (B) Primary mouse hepatocytes were incubated in DMEM–10% FBS with 5·5 or 25 mM glucose for 22 h. In lanes c–d, LiCl (20 mM) was added to the medium and incubated for the last 8 h. (C) CHO/IR cells were infected with blank adenovirus or adenovirus containing the kinase-deficient GSK3β mutant or the constitutively active GSK3β mutant (200 MOI) for 20 h. After treatment(s), all cells were lysed in Laemmli sample buffer containing 0·1 M DTT. Lysates were processed for immunoblotting analysis as described in Materials and Methods. Results are representative of at least two separate experiments. Quantification of western blots (B) was done in the same way as described in Fig. 1. Data are expressed as means ± S.E.M., n = 3.
Figure 4.

Independence of insulin signaling and PI3K activity for HG-induced degradation of IRS1. (A) CHO and CHO/IR cells were incubated in DMEM–0·25% BSA containing 5·5 or 25 mM glucose for 72 h. (B) CHO and CHO/IR cells were incubated in DMEM–0·25% BSA containing 5·5 or 25 mM glucose in the presence or absence of cycloheximide (0·5 mM) for 16 h. (C) CHO and CHO/IR cells were incubated in DMEM–0·25% BSA containing cycloheximide (0·5 mM) and 5·5 or 25 mM glucose in the presence or absence of LY294002 (50 μM) and/or insulin (10 nM) for 16 h. (D) CHO/IR cells were incubated in DMEM–0·25% BSA containing cycloheximide (0·5 mM) and 5·5 or 25 mM glucose in the presence or absence of insulin (5 nM) for 6, 12, or 24 h. After treatment(s), all cells were lysed in Laemmli sample buffer containing 0·1 M DTT. Lysates were processed for immunoblotting analysis as described in Materials and methods. Results are representative of at least two separate experiments. (E) Densities of IRS1 and GSK3β in (D) were quantified by Image J software. The ratio of the IRS1:GSK3β density was plotted against time. NG, normal glucose (5·5 mM) and HG, high glucose (25 mM).
It is well known that many kinase inhibitors are not as specific as commercially advertised (Bain et al. 2007). To verify if activation of GSK3β is sufficient to induce IRS1 degradation, we overexpressed a GSK3β kinase-deficient mutant as well as a constitutively active GSK3β (serine9 mutated to alanine) by adenoviral infection of CHO/IR cells. Overexpression of the constitutively active GSK3β mutant significantly reduced IRS1 protein levels (Fig. 3C, lane c versus lane a), whereas overexpression of the kinase-deficient mutant slightly increased the level of IRS1 protein (Fig. 3C, lane b versus lane a). Since high levels of endogenous GSK3β are present in CHO/IR cells, the kinase-deficient mutant presumably served as a dominant negative mutant to block endogenous GSK3β activity towards IRS1. This result demonstrates that activation of GSK3β is sufficient to prompt the degradation of IRS1.
HG and insulin induce degradation of IRS1 through distinct signaling pathways
Insulin-induced IRS1 proteasome degradation depends on the activity of the IR tyrosine kinase and PI3K (Sun et al. 1999, Zhang et al. 2000). We first tested whether the IR was also required for HG-induced degradation by comparing CHO/IR cells to parental CHO cells. Although CHO/IR cells expressed significant quantities of IR when compared with CHO cells, a comparable degree of degradation of IRS1 was observed in both cell lines after 72 h of HG exposure (Fig. 4A, lanes d and b versus lanes a and c). This pattern was maintained even when protein synthesis was blocked with cycloheximide (Fig. 4B, lanes d and h versus lanes c and g). Thus, IR is not required for HG-induced degradation of IRS1.
Next, we tested whether PI3K activity was required for HG-induced degradation of IRS1 by stimulating cells with insulin, or blocking PI3K activity with LY294002, a potent PI3K inhibitor. CHO and CHO/IR cells were incubated with or without insulin (10 nM) in the presence of normal glucose (5·5 mM) or HG (25 mM) for 16 h in the presence of cycloheximide (Fig. 4C). As expected, IR was required for insulin-induced degradation under normal glucose conditions, which was evidenced by the observation that insulin induced IRS1 degradation only in CHO/IR cells, but not in CHO cells (Fig. 4C, panels 1 and 3, lane e versus lane a). LY294002 completely blocked insulin-induced degradation of IRS1 (Fig. 4C, panel 3, lane g versus lanes e and a), suggesting that PI3K mediates the insulin-induced degradation of IRS1. In contrast, HG alone reduced IRS1 in both CHO and CHO/IR cell lines even when PI3K activity was blocked (Fig. 4C, panels 1 and 3, lanes b and d versus lanes a and c), indicating that IR and PI3K are not required for HG-induced degradation of IRS1.
The fact that HG and insulin imposed opposite effects on GSK3β activity (HG activates GSK3β while insulin inhibits GSK3β; Fig. 4C, panels 2 and 4) prompted us to examine whether insulin signaling had any effect on HG-induced degradation of IRS1. In CHO/IR cells, although 10 nM insulin induced the inhibition of GSK3β (increased phosphorylation; Fig. 4C, panel 4, lanes e–f), it induced the degradation of IRS1 under both normal and HG conditions (Fig. 4C, panel 3, lanes e–f), suggesting a mechanism independent of GSK3β activity. However, blocking PI3K activity by LY294002 only prevented insulin-induced degradation under normal glucose conditions, but not under HG conditions (Fig. 4C, panel 3, lanes g–h versus lanes e–f), which is consistent with the fact that HG-induced degradation of IRS1 is independent of PI3K activity. Thus, HG and insulin induce degradation of IRS1 via distinct pathways.
Although 10 nM insulin failed to prevent the HG-induced degradation of IRS1 in CHO/IR cells (sensitive to insulin), it did prevent HG-induced IRS1 degradation to a modest extent in CHO cells (less sensitive to insulin; Fig. 4C, panels 1 and 3, lanes e–f versus lanes a–d). There is a possibility for the effect of insulin on HG-induced degradation of IRS1 being minimized by the insulin-induced degradation of IRS1 during overnight exposure of CHO/IR cells to high-dose insulin (10 nM). To investigate if short-term exposure to a low dose of insulin could reveal any effect of insulin on HG-induced activation of GSK3β and degradation of IRS1, CHO/IR cells were treated with normal and HG in conjunction with cycloheximide in the presence or absence of a low dose of insulin (5 nM) for various time periods. Five-nanomolar insulin in normal glucose induced degradation of IRS1 at a slow rate. In comparison, HG induced IRS1 degradation at a much faster rate (Fig 4D, panel 1, lanes b–d versus lanes e–g, and Fig 4E). Interestingly, HG-induced degradation of IRS1 was greatly reduced in the presence of 5 nM insulin (Fig. 4D, lanes h–j versus lanes b–d and e–g; Fig. 4E). The dynamic changes in HG-induced degradation of IRS1 by insulin were consistent with changes in GSK3β activity (Fig. 4D, panel 2), further supporting the involvement of GSK3β in HG-induced degradation of IRS1.
HG promotes the phosphorylation of serine332 in IRS1
GSK3β has been shown to negatively regulate insulin signaling by phosphorylating IRS1 at serine332 after phosphorylation of the priming site serine336 by PKCβII (Eldar-Finkelman & Krebs 1997, Liberman & Eldar-Finkelman 2005, Liberman et al. 2008). To investigate if IRS1 is phosphorylated at this specific site during HG exposure, we used a phospho-specific antibody against the site to monitor the specific phosphorylation of IRS1. Indeed, serine332 phosphorylation was increased in HG-treated cells, and was completely blocked by LiCl (Fig. 5A, lanes b and c versus lane a), suggesting that GSK3β is the potential kinase that mediates the phosphorylation of IRS1 by HG. To further investigate the role of serine332 in the HG-induced degradation of IRS1, two nonphosphorylatable serine→alanine mutants (Ser→ Ala332 and Ser→Ala332/336) were overexpressed in CHO/IR cells (Liberman & Eldar-Finkelman 2005), as well as in wild-type IRS1 control. Transfected cells were subjected to HG treatment and examined for IRS1 degradation. Both ser→ala mutants prevented HG-induced degradation of IRS1 to a significant extent (Fig. 5B, lanes d and f versus lane b). Considering the high ratio of endogenous versus over-expressed IRS1 proteins, the degree to which IRS1 was protected from degradation by the mutants is potentially greater than that suggested by the immunoblots. Thus, serine332 phosphorylation in IRS1 by GSK3β is required for and appears to be the principle site with respect to HG-induced degradation of IRS1.
Figure 5.

Serine332 phosphorylation of IRS1 in HG-induced IRS1 degradation. (A) CHO/IR/IRS1 cells were incubated in DMEM–0·25% BSA containing cycloheximide (0·5 mM) and MG132 (25 μM) and 5·5 or 25 mM glucose in the presence or absence of LiCl (20 mM) for 24 h. (B) CHO/IR cells overexpressing wild-type IRS1, IRS1S332A, or IRS1S332/336A mutants were incubated in DMEM–0·25% BSA containing cycloheximide (0·5 mM) and 5·5 or 25 mM glucose for 16 h. After treatment(s), all cells were lysed in Laemmli sample buffer containing 0·1 M DTT. S332 phosphorylation of IRS1, IRS1 fprotein, and the β-subunit of insulin receptor in the lysates were detected by immunoblotting analysis using the corresponding antibodies as described in Materials and Methods. Results are representative of at least three separate experiments. Quantification of western blot (B) was done in the same way as described in Fig. 1. Data are expressed as means± S.E.M., n = 3.
GSK3β activity is required for HG-induced ubiquitination of IRS1
As shown in Fig. 1C, HG-induced IRS1 degradation was blocked by MG132, a 26S protease inhibitor, indicating the involvement of ubiquitin-mediated proteasome degradation. To confirm the ubiquitination of IRS1 during HG exposure, ubiquitination of IRS1 was measured in the presence of MG132 (to preserve the ubiquitinated IRS1). Indeed, HG increased IRS1 ubiquitination (Fig. 6, lane b versus lane a). Consistent with our data, LiCl inhibited GSK3β activity (Fig. 3A), reduced phosphorylation of IRS1 at serine332 (Fig. 5A), and reduced IRS1 ubiquitination (Fig. 6, lane b versus lanes a and b). These data support the conclusion that HG-induced ubiquitination and degradation of IRS1 are mediated by GSK3β.
Figure 6.
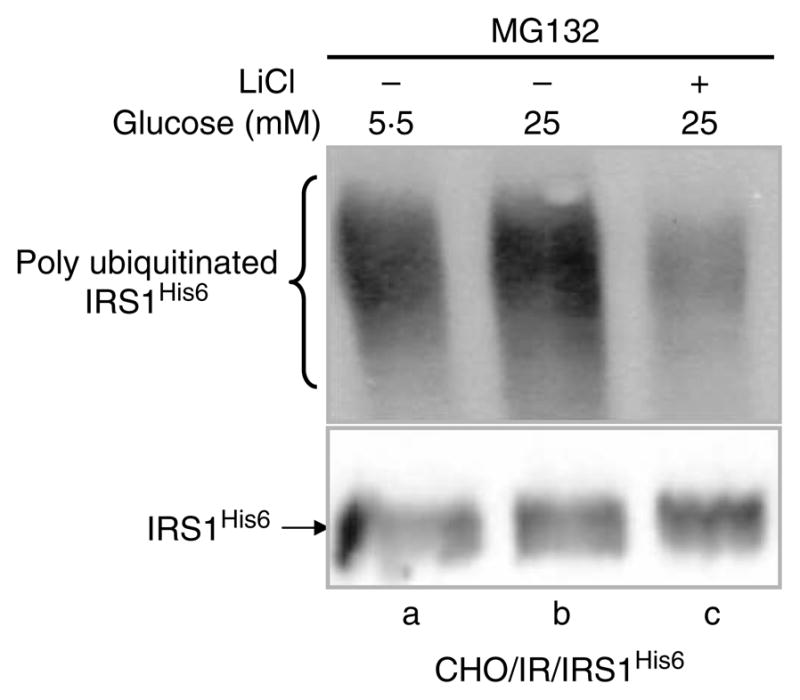
Ubiquitination of IRS1 induced by HG. CHO/IR/IRS1His6 cells were incubated in DMEM–0·25% BSA containing MG132 (25 μM) and 5·5 or 25 mM glucose in the presence or absence of LiCl (20 mM) for 24 h. After treatments, His6-tagged IRS1 (IRS1His6) was purified using Ni-NTA beads and detected by immunoblotting with ubiquitin antibody (FK2) and IRS1 antibody respectively. Results are representative of two separate experiments.
Discussion
IRS1 is one of the major IRSs in the insulin signaling pathway, and is required for normal insulin signaling (Lee & White 2004, Taniguchi et al. 2006). IRS1 protein levels are controlled by many factors including insulin/IGF1, glucocorticoids, oxidative stress, and certain nutrients (free fatty acids, amino acids, and glucose). The ubiquitin-mediated proteasome degradation of IRS1 is an important cellular control mechanism for insulin sensitivity (Sun et al. 1999, Rui et al. 2002, Zhande et al. 2002). Here, we have shown that long-term HG exposure (mimicking chronic hyperglycemia) also induces ubiquitin-mediated proteasome degradation of IRS1. Interestingly, HG-induced degradation of IRS1 is mediated by a mechanism distinct from that of insulin, in that it is IR independent and does not require PI3K activity. More importantly, we have shown that the activation of GSK3β is required for HG-induced degradation of IRS1. HG activates GSK3β, which phosphorylates IRS1 at serine332, leading to the ubiquitination and proteasome-mediated degradation of IRS1. Thus, our study, for the first time, provides direct evidence that phosphorylation of IRS1 is linked to its ubiquitin-mediated proteasome degradation, and reveals a molecular mechanism for HG-induced degradation of IRS1.
We and others have reported previously that insulin/IGF1 can induce IRS1 degradation through a ubiquitin-mediated proteasome degradation pathway (Sun et al. 1999, Lee et al. 2000, Rui et al. 2002, Zhande et al. 2002). In this study, we have clearly shown that although both insulin and HG activate the ubiquitin-mediated proteasome pathway to degrade IRS1, there exist at least two distinct upstream signaling chains that ultimately lead to ubiquitination of IRS1 (Fig. 7). Contrary to the insulin-induced degradation of IRS1, which requires both the IR tyrosine kinase and PI3K activities (Sun et al. 1999, Lee et al. 2000, Zhang et al. 2000, Rui et al. 2002), HG-induced proteasome degradation is an IR- and PI3K-independent process. This conclusion is supported by the fact that overexpression of the IR and/or inhibition of PI3K did not significantly affect HG-incited IRS1 degradation (Fig. 4). Although high concentrations of insulin and HG individually lead to the degradation of IRS1, insulin at low concentrations, for short periods of time, seems to inhibit HG-induced degradation of IRS1. This can be clearly explained by our finding that insulin and HG have opposing effects on GSK3β phosphorylation. Insulin, through activation of PI3K/Akt, phosphorylates serine9 on GSK3β, inhibiting kinase activity. In contrast, HG activates GSK3β through unknown mechanism by reducing serine9 phosphorylation. Since insulin-induced degradation of IRS1 is a GSK3β-independent process (LiCl cannot block insulin-induced IRS1 degradation, data not shown), long-term insulin exposure – even at lower concentrations that initially counter HG’s effects on GSK3β – would eventually trigger insulin-induced proteasome degradation (Fig. 4D, 6- and 12-h versus 24-h insulin co-treatment with HG).
Figure 7.

Model of HG- and insulin-induced proteasome degradation of IRS1. Insulin-induced degradation of IRS1 is mediated by a PI3K-dependent pathway; whereas high glucose-induced degradation of IRS1 depends on GSK3β activity. Although both high glucose and insulin induce the degradation of IRS1 under certain conditions (low insulin concentration and short treatment times), insulin can block the high glucose-induced degradation of IRS1 through the inhibition of GSK3β activity.
The central finding in this study is the identification of GSK3β as the key component in HG-induced degradation of IRS1. There are a few conflicting reports that describe the effect of HG on GSK3β activity. In cancer cells, increasing glucose uptake and high rates of glucose utilization induced the phosphorylation of GSK3α/β (inhibition) in a PKC-mediated process, leading to cell survival (Zhao et al. 2007). In glomerular mesangial cells, HG-stimulated GSK3β activity augmented apoptosis by antagonizing Wnt signaling, an effect preventable by LiCl treatment (Lin et al. 2006). GSK3β activity was found to be higher in STZ-treated hyperglycemic rats due to decreased AKT activity (Gurusamy et al. 2006). HG has also been shown to downregulate both basal and insulin-stimulated glycogen synthase in rat-1 fibroblasts secondary to the stimulation of GSK3β (Singh & Crook 2000). Our current study clearly shows that HG reduces GSK3β serine9 phosphorylation without changing GSK3β protein levels. More importantly, we have demonstrated in several experiments that GSK3β is necessary and sufficient for the mediation of HG-induced degradation of IRS1. First, an inhibitor of GSK3β blocks HG-induced proteasome degradation of IRS1; secondly, HG reduces GSK3β phosphorylation at serine9, which is indicative of activation; and finally, overexpression of constitutively active GSK3β increases IRS1 degradation, whereas overexpression of kinase-deficient GSK3β slightly blocks basal IRS1 degradation.
The mechanism by which GSK3β mediates IRS1 degradation is unknown. Our results provide a possible mechanism explaining how GSK3β mediates the ubiquitination of IRS1. Serine/threonine phosphorylation is known to be an important signal for the ubiquitination and proteasome degradation of cellular proteins (Nash et al. 2001). GSK3β phosphorylates IRS1 at serine332 and attenuates insulin’s action (Liberman & Eldar-Finkelman 2005). Our results clearly show that inhibition of GSK3β not only blocks the proteasome degradation of IRS1, but also prevents IRS1 serine332 phosphorylation and its subsequent ubiquitination. Furthermore, mutation of serine332 to alanine prevents IRS1 degradation induced by HG. Taken together, the data support a model where HG activates GSK3β promoting serine332 phosphorylation of IRS1. This phosphorylation tags IRS1 for ubiquitination, leading to proteasome-mediated destruction.
Glucosamine, a glucose metabolite, has also been shown to induce insulin resistance in different cells and animals (DeFronzo et al. 1982, Rossetti et al. 1995, Patti 1999, Heart et al. 2000). We were unable to detect the effect of glucosamine on IRS1 ubiquitination and proteasome degradation in our cell systems (data not shown), indicating that glucose and glucosamine might induce insulin resistance via different mechanisms as suggested in the literature (DeFronzo et al. 1982, Nelson et al. 2000, 2002, Han et al. 2003, Duan et al. 2005). Exactly how glucose activates GSK3β remains to be revealed in a future study.
Much evidence in the literature suggests that GSK3β is involved in insulin resistance. GSK3 is constitutively active in unstimulated cells, and is inactivated by a variety of extracellular stimuli, notably insulin (Woodgett 1990). Elevated GSK3 activity and expression have been observed in obese and diabetic humans and rodents (Eldar-Finkelman et al. 1999, Nikoulina et al. 2000, Ciaraldi et al. 2002) and in the adipose tissue of high-fat-fed mice (Eldar-Finkelman et al. 1999). Overexpression of human GSK3β (fivefold) in the skeletal muscle of mice results in hyperglycemia, hyperinsulinemia, hyperlipidemia, a reduction in glycogen levels, and a significant decrease in IRS1 protein levels (Pearce et al. 2004). Homozygous GSK3β knockout is embryonically lethal, suggesting that it plays an important role in early development (Hoeflich et al. 2000). However, GSK3α knockout animals are viable, and demonstrate enhanced glucose and insulin sensitivity (MacAulay et al. 2007). Insulin-stimulated Akt and GSK3β phosphorylation is higher in GSK3α KO livers than in the wild-type littermates. Interestingly, hepatic IRS1 expression is markedly increased in these animals (MacAulay et al. 2007). Although our experiments did not test GSK3α, it is very likely that GSK3α is also involved in HG-induced IRS1 degradation. Thus, our studies are consistent with these reports, and further provide the molecular mechanism through which GSK3 is involved in insulin resistance.
Acknowledgments
Funding
This work was supported by a grant from NIH Grant R01 DK060128 to XJS.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Araki E, Lipes MA, Patti ME, Bruning JC, Haag B, III, Johnson RS, Kahn CR. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochemical Journal. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MN, Friend DS. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. Journal of Cell Biology. 1969;43:506–520. doi: 10.1083/jcb.43.3.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti L, Mosthaf L, Kroder G, Kellerer M, Tippmer S, Mushack J, Seffer E, Seedorf K, Haring H. Glucose-induced translocation of protein kinase C isoforms in rat-1 fibroblasts is paralleled by inhibition of the insulin receptor tyrosine kinase. Journal of Biological Chemistry. 1994;269:3381–3386. [PubMed] [Google Scholar]
- Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1997;88:561–572. doi: 10.1016/s0092-8674(00)81896-6. [DOI] [PubMed] [Google Scholar]
- Buren J, Liu HX, Lauritz J, Eriksson JW. High glucose and insulin in combination cause insulin receptor substrate-1 and -2 depletion and protein kinase B desensitisation in primary cultured rat adipocytes: possible implications for insulin resistance in type 2 diabetes. European Journal of Endocrinology. 2003;148:157–167. doi: 10.1530/eje.0.1480157. [DOI] [PubMed] [Google Scholar]
- Ciaraldi TP, Nikoulina SE, Henry RR. Role of glycogen synthase kinase-3 in skeletal muscle insulin resistance in type 2 diabetes. Journal of Diabetes and its Complications. 2002;16:69–71. doi: 10.1016/s1056-8727(01)00193-3. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Hendler R, Simonson D. Insulin resistance is a prominent feature of insulin-dependent diabetes. Diabetes. 1982;31:795–801. doi: 10.2337/diab.31.9.795. [DOI] [PubMed] [Google Scholar]
- Del Prato S, Leonetti F, Simonson DC, Sheehan P, Matsuda M, DeFronzo RA. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia. 1994;37:1025–1035. doi: 10.1007/BF00400466. [DOI] [PubMed] [Google Scholar]
- Duan W, Paka L, Pillarisetti S. Distinct effects of glucose and glucosamine on vascular endothelial and smooth muscle cells: evidence for a protective role for glucosamine in atherosclerosis. Cardiovascular Diabetology. 2005;4:16. doi: 10.1186/1475-2840-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. PNAS. 1997;94:9660–9664. doi: 10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Schreyer SA, Shinohara MM, LeBoeuf RC, Krebs EG. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes. 1999;48:1662–1666. doi: 10.2337/diabetes.48.8.1662. [DOI] [PubMed] [Google Scholar]
- Finlay D, Patel S, Dickson LM, Shpiro N, Marquez R, Rhodes CJ, Sutherland C. Glycogen synthase kinase-3 regulates IGFBP-1 gene transcription through the thymine-rich insulin response element. BMC Molecular Biology. 2004;5:15. doi: 10.1186/1471-2199-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC. Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia. 2004;47:1661–1667. doi: 10.1007/s00125-004-1507-3. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochemical Journal. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurusamy N, Watanabe K, Ma M, Prakash P, Hirabayashi K, Zhang S, Muslin AJ, Kodama M, Aizawa Y. Glycogen synthase kinase 3β together with 14-3-3 protein regulates diabetic cardiomyopathy: effect of losartan and tempol. FEBS Letters. 2006;580:1932–1940. doi: 10.1016/j.febslet.2006.02.056. [DOI] [PubMed] [Google Scholar]
- Han DH, Chen MM, Holloszy JO. Glucosamine and glucose induce insulin resistance by different mechanisms in rat skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism. 2003;285:E1267–E1272. doi: 10.1152/ajpendo.00255.2003. [DOI] [PubMed] [Google Scholar]
- Heart E, Choi WS, Sung CK. Glucosamine-induced insulin resistance in 3T3-L1 adipocytes. American Journal of Physiology. Endocrinology and Metabolism. 2000;278:E103–E112. doi: 10.1152/ajpendo.2000.278.1.E103. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Ide R, Maegawa H, Kikkawa R, Shigeta Y, Kashiwagi A. High glucose condition activates protein tyrosine phosphatases and deactivates insulin receptor function in insulin-sensitive rat 1 fibroblasts. Biochemical and Biophysical Research Communications. 1994;201:71–77. doi: 10.1006/bbrc.1994.1670. [DOI] [PubMed] [Google Scholar]
- Kahn CR. Insulin action, diabetogenes, and the cause of type II diabetes (Banting Lecture) Diabetes. 1994;43:1066–1084. doi: 10.2337/diab.43.8.1066. [DOI] [PubMed] [Google Scholar]
- Koopmans SJ, Ohman L, Haywood JR, Mandarino LJ, DeFronzo RA. Seven days of euglycemic hyperinsulinemia induces insulin resistance for glucose metabolism but not hypertension, elevated catecholamine levels, or increased sodium retention in conscious normal rats. Diabetes. 1997;46:1572–1578. doi: 10.2337/diacare.46.10.1572. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Research and Clinical Practice. 2007;77:S49–S57. doi: 10.1016/j.diabres.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Lee YH, White MF. Insulin receptor substrate proteins and diabetes. Archives of Pharmacological Research. 2004;27:361–370. doi: 10.1007/BF02980074. [DOI] [PubMed] [Google Scholar]
- Lee AV, Gooch JL, Oesterreich S, Guler RL, Yee D. Insulin-like growth factor I-induced degradation of insulin receptor substrate 1 is mediated by the 26S proteasome and blocked by phosphatidylinositol 3′-kinase inhibition. Molecular and Cellular Biology. 2000;20:1489–1496. doi: 10.1128/mcb.20.5.1489-1496.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman Z, Eldar-Finkelman H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. Journal of Biological Chemistry. 2005;280:4422–4428. doi: 10.1074/jbc.M410610200. [DOI] [PubMed] [Google Scholar]
- Liberman Z, Plotkin B, Tennenbaum T, Eldar-Finkelman H. Coordinated phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 and protein kinase CβII in the diabetic fat tissue. American Journal of Physiology. Endocrinology and Metabolism. 2008;294:E1169–E1177. doi: 10.1152/ajpendo.00050.2008. [DOI] [PubMed] [Google Scholar]
- Lin CL, Wang JY, Huang YT, Kuo YH, Surendran K, Wang FS. Wnt/beta-catenin signaling modulates survival of high glucose-stressed mesangial cells. Journal of the American Society of Nephrology. 2006;17:2812–2820. doi: 10.1681/ASN.2005121355. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3α-specific regulation of murine hepatic glycogen metabolism. Cell Metabolism. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Yamauchi K, Shigematsu S, Ikeo S, Komatsu M, Aizawa T, Hashizume K. Selective attenuation of metabolic branch of insulin receptor down-signaling by high glucose in a hepatoma cell line, HepG2 cells. Journal of Biological Chemistry. 2000;275:20880–20886. doi: 10.1074/jbc.M905410199. [DOI] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- Nelson BA, Robinson KA, Buse MG. High glucose and glucosamine induce insulin resistance via different mechanisms in 3T3-L1 adipocytes. Diabetes. 2000;49:981–991. doi: 10.2337/diabetes.49.6.981. [DOI] [PubMed] [Google Scholar]
- Nelson BA, Robinson KA, Buse MG. Defective Akt activation is associated with glucose- but not glucosamine-induced insulin resistance. American Journal of Physiology. Endocrinology and Metabolism. 2002;282:E497–E506. doi: 10.1152/ajpendo.00438.2001. [DOI] [PubMed] [Google Scholar]
- Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49:263–271. doi: 10.2337/diabetes.49.2.263. [DOI] [PubMed] [Google Scholar]
- Nikoulina SE, Ciaraldi TP, Mudaliar S, Carter L, Johnson K, Henry RR. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes. 2002;51:2190–2198. doi: 10.2337/diabetes.51.7.2190. [DOI] [PubMed] [Google Scholar]
- Patti ME. Nutrient modulation of cellular insulin action. Annals of the New York Academy of Sciences. 1999;892:187–203. doi: 10.1111/j.1749-6632.1999.tb07796.x. [DOI] [PubMed] [Google Scholar]
- Pearce NJ, Arch JR, Clapham JC, Coghlan MP, Corcoran SL, Lister CA, Llano A, Moore GB, Murphy GJ, Smith SA, et al. Development of glucose intolerance in male transgenic mice overexpressing human glycogen synthase kinase-3β on a muscle-specific promoter. Metabolism. 2004;53:1322–1330. doi: 10.1016/j.metabol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Pillay TS, Xiao S, Olefsky JM. Glucose-induced phosphorylation of the insulin receptor. Functional effects and characterization of phosphorylation sites. Journal of Clinical Investigation. 1996;97:613–620. doi: 10.1172/JCI118457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondinone CM, Wang LM, Lonnroth P, Wesslau C, Pierce JH, Smith U. Insulin receptor substrate (IRS) 1 is reduced and IRS-2 is the main docking protein for phosphatidylinositol 3-kinase in adipocytes from subjects with non-insulin-dependent diabetes mellitus. PNAS. 1997;94:4171–4175. doi: 10.1073/pnas.94.8.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti L, Hawkins M, Chen W, Gindi J, Barzilai N. In vivo glucosamine infusion induces insulin resistance in normoglycemic but not in hyperglycemic conscious rats. Journal of Clinical Investigation. 1995;96:132–140. doi: 10.1172/JCI118013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. Journal of Biological Chemistry. 2002;277:42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- Saad MJ, Araki E, Miralpeix M, Rothenberg PL, White MF, Kahn CR. Regulation of insulin receptor substrate-1 in liver and muscle of animal models of insulin resistance. Journal of Clinical Investigation. 1992;90:1839–1849. doi: 10.1172/JCI116060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M, Cohen P, Alessi DR. Further evidence that the inhibition of glycogen synthase kinase-3β by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Letters. 1997;416:307–311. doi: 10.1016/s0014-5793(97)01235-0. [DOI] [PubMed] [Google Scholar]
- Singh LP, Crook ED. The effects of glucose and the hexosamine biosynthesis pathway on glycogen synthase kinase-3 and other protein kinases that regulate glycogen synthase activity. Journal of Investigative Medicine. 2000;48:251–258. [PubMed] [Google Scholar]
- Sun XJ, Liu F. Phosphorylation of IRS proteins Yin-Yang regulation of insulin signaling. Vitamins and Hormones. 2009;80:351–387. doi: 10.1016/S0083-6729(08)00613-4. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Rothenberg PL, Kahn CR, Backer JM, Araki E, Wilden PA, Cahill DA, Goldstein BJ, White MF. The structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352:73–77. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Crimmins DL, Myers MG, Jr, Miralpeix M, White MF. Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Molecular and Cellular Biology. 1993;13:7418–7428. doi: 10.1128/mcb.13.12.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XJ, Goldberg JL, Qiao LY, Mitchell JJ. Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes. 1999;48:1359–1364. doi: 10.2337/diabetes.48.7.1359. [DOI] [PubMed] [Google Scholar]
- Taniguchi CM, Ueki K, Kahn R. Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. Journal of Clinical Investigation. 2005;115:718–727. doi: 10.1172/JCI23187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nature Reviews. Molecular Cell Biology. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Virkamaki A, Ueki K, Kahn CR. Protein–protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. Journal of Clinical Investigation. 1999;103:931–943. doi: 10.1172/JCI6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorinen-Markkola H, Koivisto VA, Yki-Jarvinen H. Mechanisms of hyperglycemia-induced insulin resistance in whole body and skeletal muscle of type I diabetic patients. Diabetes. 1992;41:571–580. doi: 10.2337/diab.41.5.571. [DOI] [PubMed] [Google Scholar]
- White MF. Regulating insulin signaling and β-cell function through IRS proteins. Canadian Journal of Physiology and Pharmacology. 2006;84:725–737. doi: 10.1139/y06-008. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO Journal. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhande R, Mitchell JJ, Wu J, Sun XJ. Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Molecular and Cellular Biology. 2002;22:1016–1026. doi: 10.1128/MCB.22.4.1016-1026.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhande R, Zhang W, Zheng Y, Pendleton E, Li Y, Polakiewicz RD, Sun XJ. Dephosphorylation by default, a potential mechanism for regulation of insulin receptor substrate-1/2, Akt, and ERK1/2. Journal of Biological Chemistry. 2006;281:39071–39080. doi: 10.1074/jbc.M605251200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Hoff H, Sell C. Insulin-like growth factor I-mediated degradation of insulin receptor substrate-1 is inhibited by epidermal growth factor in prostate epithelial cells. Journal of Biological Chemistry. 2000;275:22558–22562. doi: 10.1074/jbc.M000412200. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, Wofford JA, Dimascio LN, Ilkayeva O, Kelekar A, et al. Glycogen synthase kinase 3α and 3β mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Molecular and Cellular Biology. 2007;27:4328–4339. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]