

Abstract
Complement component C3, the central player in the complement cascade and the pro-inflammatory cytokine IL-1β is expressed by activated glial cells and may contribute to neurodegeneration. This study examines the regulation of the expression of C3 by IL-1β in astroglial cells focusing on the role of the upstream kinase MKK6, p38-α MAPK and C/EBP-β isoforms (LAP1, LAP2 or LIP) in astroglial cells. Activation of human astroglial cell line, U373 with IL-1β, led to the induction of C3 mRNA and protein expression as determined by real-time RT-PCR and western blot analysis, respectively. This induction was suppressed by the pharmacological inhibitor of p38 MAPK (i.e., SB202190-HCl), suggesting the involvement of p38 MAPK in C3 gene expression. IL-1β also induced C3 promoter activity in U373 cells in a MAP kinase- and C/EBP-β-dependent manner. Cotransfection of C3 luciferase reporter construct with constitutively active form of the upstream kinase in the MAP kinase cascade, i.e., MKK6 (the immediate upstream activator of p38 kinase) resulted in marked stimulation of the promoter activity, whereas, overexpression of a dominant negative forms of MKK6 and p38α MAPK inhibited C3 promoter activity. Furthermore, a mutant form of C/EBP-β, LAPT235A showed reduction in IL-1β mediated C3 promoter activation. These results suggest that the p38α MAPK and MKK6 play prominent roles in IL-1β and C/EBP-β mediated C3 gene expression in astrocytes.
Keywords: Complement C3, IL-1β, C/EBP-β, MKK6, p38 MAPK, astrocytes
INTRODUCTION
The pro-inflammatory cytokine, interleukin-1β (IL-1β) is one of the most potent and best characterized signals that trigger astrocyte activation in most neurodegenerative diseases [Simi et al., 2007]. Astrocytes, the predominant glial cells in the brain [Aldskogius and Kozlova, 1998] are known to play an important role in modulating neuroimmune processes and likely play a protective role in limiting injury that are triggered in most neurodegenerative disease [Escartin and Bonvento, 2008]. In the context of HIV-1 infection of the CNS, studies demonstrate that IL-1β expression is increased in infiltrating macrophages, microglia and astrocytes [Zhao et al., 2001; Xing et al., 2009].
The complement component C3 that plays a central role in the activation of complement system is also induced in most neurodegenerative diseases [see reviews, Bonifati and Kishore, 2007]; Yanamadala and Friedlander, 2010]. Its activation is required for both classical and alternative complement activation pathways [see reviews, Datta and Rappaport, 2006; Lambris et al., 2008]. Inflammatory cytokines such as IL-1β, interferon-γ, and TNF-α are known to induce expression of several complement components including C3 in astrocytes [Barnum et al., 1992, 1993; Gasque et al., 1992; Rus et al., 1992; Maranto et al., 2008]. HIV-1 and HIV-1 proteins gp41 and Nef up-regulate the synthesis of complement factor C3 in astrocytes and neurons [Bruder et al., 2004; Speth et al., 2001, 2002] SIV infection of the CNS in rhesus macaques also induces synthesis of C3 in infiltrating macrophages, astrocytes and neurons [Speth et al., 2004]. Complement expression and activation in the brain is postulated to play both neuroprotective and neurodegenerative roles [Bonifati and Kishore, 2007; Yanamadala and Friedlander, 2010].
We have demonstrated by electrophoretic mobility shift assay and transient transfection assay that IL-1β regulates C3 promoter expression through the activation of C/EBP in a p38-MAPK dependent manner in astrocytic cells [Maranto et al., 2008]. However, the role of different isoforms of C/EBP-β, the kinase upstream of p38-MAPK i.e., MKK6 and role of C/EBP-β phosphorylation in regulation of C3 gene remains to be elucidated. C/EBP-β, a member of the bZIP family of transcription factors is expressed in mammalian cells as three alternate translation products [Nerlov, 2007], 49- and 45-kDa proteins in human cells termed as LAP1 and LAP2 (liver enriched activating protein) respectively, and a 20-kDa protein known as LIP (liver-enriched inhibitory protein) [Eaton et al., 2001]. The N-terminal region of LAP corresponds to the transactivation domain, whereas LIP lacks this transactivation domain and acts as an inhibitor of transcription [Eaton et al., 2001]. Studies have demonstrated that phosphorylation of C/EBP-β can modulate its DNA binding activity or alter its transactivation potential [Nakajima et al., 1993; Aouadi et al., 2007; Wegner et al., 1992, Piwien-Pilipuk et al., 2001; Trautwein et al., 1994; Chinery et al., 1997; Buck et al., 1999].
In this study, we have investigated the role of p38-α MAPK in IL-1β mediated regulation of the endogenous C3 gene, and further elucidated the role of MKK6 and the different isoforms of C/EBP-β (LAP1, LAP2 or LIP), and C/EBP-β phosphorylation in IL-1β mediated induction of C3 promoter activity in astrocytic cells.
MATERIALS AND METHODS
Materials
p38 MAPK inhibitor, 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole-HCl (SB202190.HCl) and recombinant human IL-1β were purchased from EMD BioSciences (San Diego, CA) and R&D Systems Inc. (Minneapolis, MN), respectively. Precast 4–20% gradient SDS-polyacrylamide gels were obtained from Lonza (Walkersville, MD). Phospho-specific and total p38MAPK antibodies were purchased from Cell Signaling Technology (Danver, MA) and C3 antibody (H-300) from Santa Cruz Biotechnology, Santa Cruz, CA).
Cell culture
Human astrocytic cell line U373-MG were maintained as monolayer cultures in a humidified 5% CO2 atmosphere at 37°C in Dulbecco’s minimal essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 20 units/ml penicillin and 20 μg/ml of streptomycin.
Expression vectors and reporter constructs
Expression vectors for C/EBP β isoforms (LAP1), (LAP2) and LIP (amino acids 199–345) [Kukimoto et al., 2006] were kindly provided by Dr. I. Kukimoto, National Institute of Infectious Disease, Tokyo, Japan. The expression vectors for MKK6b (wild type), MKK6b(E) (a constitutively active mutant of MKK6b in which serine207 and threonine211 are replaced with glutamic acid), MKK6b(A) (a dominant negative mutant of MKK6b in which lysine82 is replaced with alanine) [Huang et al., 1997] were kindly provided by Dr. J. Han, The Scripps Research Institute, LaJolla, CA. The expression vector for p38α MAPK (AF) (a double mutant in which threonine188 and tyrosine190 are substituted with phenylalanine) [Enslen et al., 1998] were kindly provided by Dr. Roger Davies, U Mass, Worcester, MA. Plasmids encoding hLAP (also known as NF-IL-6) and a mutant hLAP where Thr235 was mutated to Ala (hLAP-T235A) [Nakajima et al., 1993], were kindly provided by Dr. S. Akira, Osaka University, Japan courtesy of J. Schwartz, University of Michigan, Ann Arbor, MI.
The C3 reporter plasmid contains the −1030 to +58 region of the C3 promoter cloned upstream of the luciferase reporter gene in pGL3 vector (Promega, Madison, WI) as described previously [Maranto et al., 2008].
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from U373 cells using Trizol reagent as per manufacturer’s instruction (Invitrogen, CA). cDNA was generated from 1 μg total RNA using the Qiagen RT-PCR kit (Qiagen, Valencia, CA, USA), in a 25 μl final reaction volume. Real-time PCR reactions were performed in triplicate using 1/10 of each cDNA, 1 μM of forward and reverse primer, and 1×QuantiFast SYBR Green Mix for real-time PCR (Qiagen, Valencia, CA) in 25 μl volume in Roche Optical 96-well plates (Roche Applied Science, Indianapolis, IN). PCR reactions were run in a Roche Light Cycler 480 machine and SDS v2.2 software was used to analyse results by the comparative Ct Method (ΔΔCt). Relative levels of C3 mRNA were quantified after normalization with corresponding levels of β-actin mRNA.
Protein lysate preparation and Western blot analysis
Cell lysates were prepared using M-PER mammalian protein extraction reagent in presence of HALT phosphatase and protease inhibitor as per manufacturer’s instruction (Thermo Scientific, Rockland, IL). Protein concentration in lysates was determined using BCA reagent (Thermo Scientific, Rockland, IL) and stored at −70°C until use. Cell lysates containing equal amounts of protein were electrophoresed on 4–20% SDS-polyacrylamide gels and transferred to PVDF membranes. The membranes were then processed using the Fast Western Blot, SuperSignal West Femto kit (Thermo Scientific Rockland, IL). Blots were incubated with primary antibodies against phospho-specific and total p38MAPK antibodies, and C3 antibody diluted in antibody dilution buffer at appropriate dilutions and then incubated with HRP conjugated secondary antibodies. Membranes were washed 3 times in fast blot wash buffer for 10 min each before the detection of the bands using Super Signal West Femto chemiluminescent substrate (Thermo Scientific, Rockford, IL).
Transient transfection, protein estimation and Luciferase assay
For transient transfection experiments U373-MG cells were seeded at a density of 2×105 cells in six-well plate a day before transfection. Transfections were performed with 2 μg of reporter plasmid and indicated amounts of the relevant expression vector or corresponding empty vector using Superfect transfection reagent as per manufacturer’s protocol (Qiagen, Valencia, CA). After 3 h, the cells were washed with phosphate-buffered saline and replaced with DMEM containing 10% FBS. In experiments assessing effects of IL-1β, cells were treated with IL-1β (10ng/ml) 24h post-transfection. After 48h, cells were lysed in 1X reporter lysis buffer (Promega, Madison, WI) and extracts were prepared for luciferase reporter assay [Maranto et al., 2008]. Protein concentration in lysates was determined using BCA reagent (Pierce, Rockland, IL). Luciferase activity was assessed using luciferase assay system (Promega, Madison, WI) and relative light unit was measured using a FB12 Luminometer (Zylux, Oak Ridge, TN). Data are representative for at least three independent experiments and are expressed as fold increase in RLU/μg of protein, which was calculated relative to basal level of C3 promoter activity (set to 1) and corrected for empty vector effects for each expression vector.
Statistical analyses
Results are mean+/−SEM of three independent experiments. Student’s t-test was used for statistical analysis and p< 0.05 was considered statistically significant.
RESULTS
IL-1β induces C3 gene expression in U373 cells in a p38 MAPK dependent manner
We first determined whether the inflammatory cytokine IL-1β stimulated C3 gene expression in the U373 astrocytoma cell line. With the availability of specific inhibitors such as SB202190.HCl which selectively inhibits p38MAPK α- and β-isoforms [Lee et al., 1994] it is possible to investigate the isoform of p38 MAPK that plays a role in IL-1β mediated C3 gene regulation.
Using quantitative real-time reverse transcription-PCR, we demonstrate that IL-1β treatment induces C3 expression by ~15-fold in comparison to untreated cells; and inhibition of p38-α MAPK attenuated the expression of IL-1β mediated C3 expression by 50 percent in comparison to IL-1β alone treated cells (Fig. 1A). The relative amounts of C3 mRNA in SB202190-HCl alone treated cells remained unaltered in comparison to the untreated cells (Fig. 1A).
Figure 1. The effect of p38 MAPK inhibitor on IL-1β mediated C3 gene induction in U373 cells.
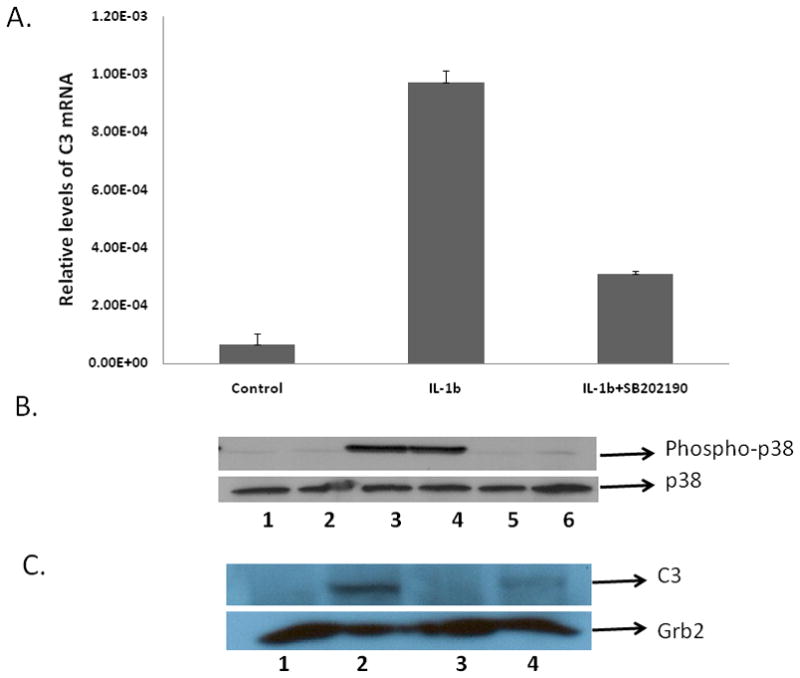
A. Role of p38 MAPK in C3 mRNA regulation in IL-1β treated cells U373 cells were either cultured without addition (control) or were pretreated with 10 μM of SB202190-HCl for 30 min alone or were pretreated with 10 μM of SB202190-HCl for 30 min and then treated with IL-1β (10ng/ml) or treated with IL-1β (10ng/ml) alone for 4h. Total RNA was prepared, followed by RT-PCR and real-time PCR for C3 mRNA. Each sample was normalized for the corresponding values of β-actin mRNA as control. Relative quantification of the C3 mRNAs was calculated using the Roche analysis software (based on the 2nd derivative maximum; Roche, Indianapolis, IN, USA).
B. Role of IL-1β on p38 MAPK phosphorylation. U373 cells were either cultured without addition (control, lane 1) or were pretreated with 10 μM of SB202190-HCl for 30 min alone (lane 2), or treated with IL-1β (10ng/ml) alone for 30 min (lanes 3–4), or were pretreated with 10μM of SB202190-HCl for 30 min and then treated with IL-1β (10ng/ml) (lanes 5–6). Protein lysates were prepared and levels of phosphorylated p38 and total p38 were determined by western blot analysis using phospho-p38 and total p38 antibodies (Cell Signaling Technology, MA).
C. Role of p38 MAPK in C3 protein expression in IL-1β treated cells. U373 cells were either cultured without addition (control, lane 1); treated with IL-1β (10ng/ml) alone (lane 2); pretreated with 10 μM of SB202190-HCl for 30 min alone (lane 3); pretreated with 10 μM of SB202190-HCl for 30 min and then treated with IL-1β (10ng/ml) (lane 4). Protein lysates were prepared 24 h post treatment with IL-1β, followed by western blot analysis using C3 antibody. The membranes were probed with Grb2 antibody as loading control. The experiments were repeated twice.
In order to validate the role of p38 MAPK in IL-1β-mediated alterations in C3 expression we also determined the activation of p38 by western blot analysis in untreated astrocytic cells, in cells treated with IL-1β for 30 min and in cells pretreated with SB202190-HCl (10μM) and then treated with IL-1β. p38 MAPK activation (phosphorylation) was detected using a phospho-specific p38 MAPK antibody that recognizes p38 dually phosphorylated at threonine 180 and tyrosine 182. In order to control for cytokine induced changes in p38 kinase, total p38 MAPK expression was also examined in parallel blot. Treatment with IL-1β, or the p38 inhibitor SB202190-HCl (alone or in combination) had no effect on total p38 MAPK expression (Fig. 1B). IL-1β alone, however, caused significant increase (~6.5 fold) in p38 MAPK phosphorylation over basal level (lanes 3–4), which was attenuated by pretreatment with SB202190-HCl (Fig. 1B, lanes 5–6).
Western blot analysis of C3 expression in astrocytic cells demonstrate that IL-1β treatment induces C3 protein expression significantly in comparison to untreated cells wherein C3 expression is undetectable; and inhibition of p38-α MAPK attenuated the expression of IL-1β mediated C3 expression by 50 percent in comparison to IL-1β alone treated cells (Fig. 1C).
Taken together, these experiments provide strong evidence for a role of IL-1β and p38 MAPK in C3 expression and supports our earlier observations that transfection of astrocytic cells with dominant negative p38-α MAPK inhibits IL-1β mediated C3 promoter activation (Maranto et al., 2008).
IL-1β mediated C3 induction is attenuated by dominant negative MKK6b(A) mutant and induced by constitutively active MKK6b (E) mutant
To elucidate the role of the upstream kinase of p38 MAPK in IL-1β mediated C3 promoter activation, we examined the effects of previously characterized MKK6b expression constructs to modulate, either positively or negatively, the endogenous p38 MAPK activity. In several cell systems, p38MAPK activity can be stimulated by co-expression of p38 MAPK with MKK6b or by expression of the constitutively active mutant MKK6b(E) (Ser207 and Thr211 replaced with Glu) [Han et al., 1996; Huang et al., 1997]. The results illustrated in Fig. 2 demonstrate that IL-1β induced the C3 promoter by 15-fold while the constitutively active MKK6b(E) isoform almost doubled the fold stimulation by IL-1β. Conversely, the dominant negative mutant MKK6b(A) inhibited IL-1β mediated C3 promoter induction. These observations taken together demonstrate that MKK6 is an upstream kinase that regulates IL-1β mediated C3 gene induction.
Figure 2. Role of MKK6b in IL-1β mediated C3 promoter activation.
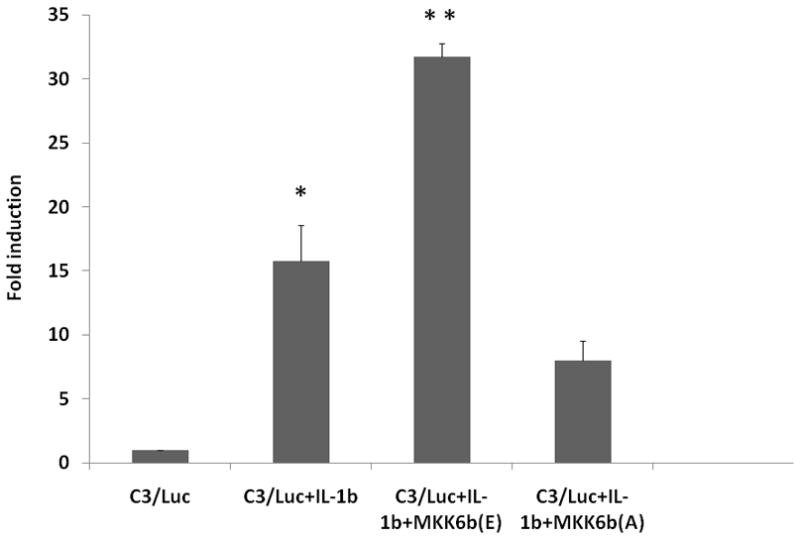
Fold induction of C3 promoter activity in U373 cells transiently co-transfected with C3/Luc reporter vector and 1 μg of empty expression vector or expression vector for MKK6b(A) or MKK6b(E) and then treated 24h post transfection with IL-1β (10ng/ml). Cells were harvested 48h post transfection to assess luciferase activity. RLU/s was normalized to protein concentration and expressed as fold induction wherein the activity in C3/Luc alone transfected cells is set at 1. Statistical significance (*, p<0.05 between control and IL-1β treated cells; **, p<0.05 between IL-1β alone treated cells and IL-1β treated MKK6(E) transfected cells) was analyzed by Student’s t-test.
C/EBP-β isoforms LAP1 and LAP2 transactivate C3 promoter in astrocytes
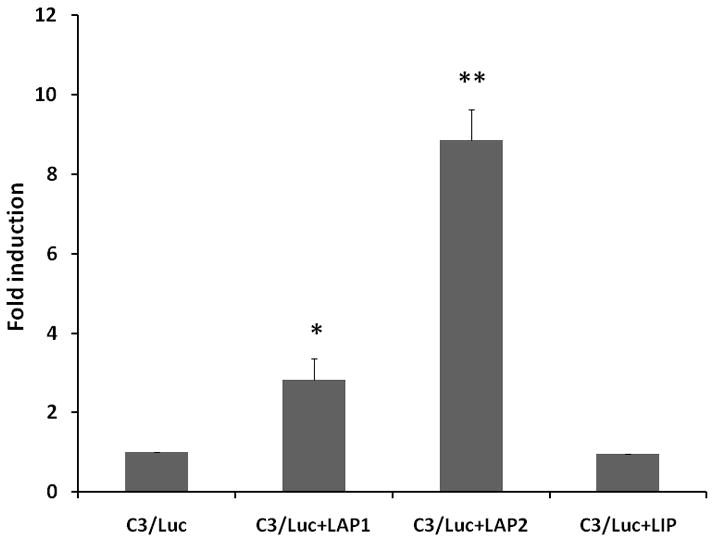
Our earlier studies [Maranto et al., 2008] also demonstrated that C/EBP-β was a component of the DNA-protein interaction that occurred at the bZIP1/bZIP2 site [Juan et al., 1993], and C/EBP-β isoform LAP induced both versions of the C3 promoters, the full length (−1030 to +58) and minimal promoter (−199 to +1) constructs harboring the C/EBP-binding site. C/EBP-β, a member of the bZIP family of transcription factors, is expressed in mammalian cells as three alternate translation products [Nerlov, 2007], 49- and 45-kDa proteins in human cells known as LAP1 and LAP2 (liver enriched activating protein) respectively, and a 20-kDa protein known as LIP (liver-enriched inhibitory protein) [Eaton et al., 2007]. We assessed the effects of all three isoforms on C3 gene induction in these studies. To assess the role of different C/EBP-β isoforms in C3 promoter activation, U373 cells were co-transfected with C3/Luc (C3 promoter region −1030 to +58) promoter construct and expression vectors coding for C/EBP-β isoforms (Fig. 3A) or empty vector. Overexpression of C/EBP-β isoforms, LAP1 and LAP2 had a profound effect on the induction of C3 promoter activity in comparison to vector alone control (Fig. 3B). On the contrary, LIP had no inducible effect on C3 promoter activation (Fig 3B).
Figure 3. Role of C/EBP-β isoforms in C3 promoter activation.
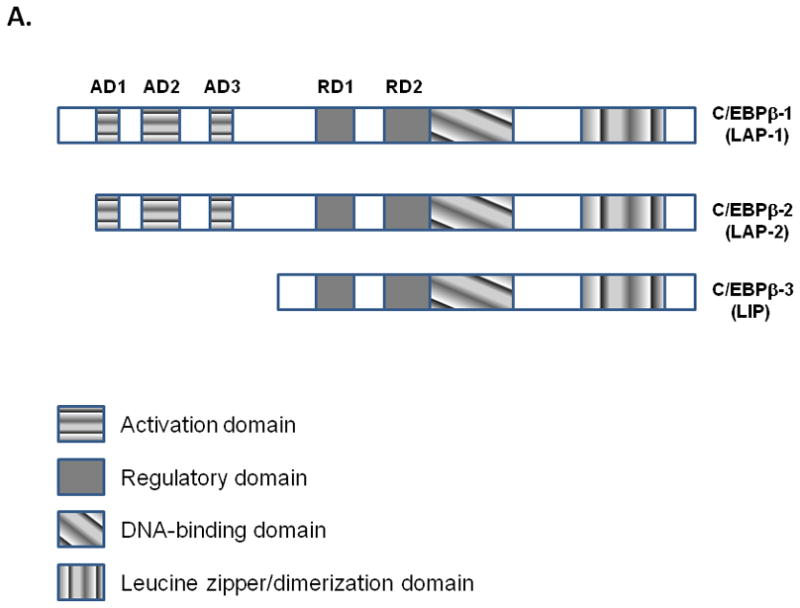
A. Schematic representation of the three isoforms of C/EBP-β generated as a result of alternate translation.
B. Fold induction of C3 promoter activity in U373 cells transiently co-transfected with C3/Luc reporter vector and 1 μg of empty expression vector or expression vector for C/EBP-β1 (LAP1) or C/EBP-β2 (LAP2) or LIP. Cells were harvested 48h post transfection to assess luciferase activity. RLU/s was normalized to protein concentration and expressed as fold induction wherein the activity in C3/Luc alone transfected cells is set at 1. Statistical significance (*, p<0.05 between C3/Luc alone and C3/Luc plus LAP1 transfected cells; **, p<0.05 between between C3/Luc alone and C3/Luc plus LAP2 transfected cells) was analyzed by Student’s t-test.
Role of p38-α MAPK in C/EBP-β mediated C3 promoter activation
Using the strategy of ectopic expression of a dominant negative kinase to investigate the role of MAP kinases in IL-1β signaling, we demonstrated that transfection of astrocytic cells with dominant negative p38-α MAPK inhibited IL-1β mediated C3 promoter activation [Maranto et al., 2008]. To test whether p38 MAPK is involved in LAP2 mediated C3 promoter activation in astrocytes, we examined the effects of inhibition of this kinase by using a specific inhibitor SB202190-HCl [Lee et al., 1994] and dominant negative p38-α MAPK mutant (Thr180 and Tyr182 replaced with Ala and Phe, AF). As shown in Figure 4, both SB202190-HCl and dominant negative p38-α MAPK inhibited LAP2 mediated C3 promoter activity. Taken together, these experiments provide strong evidence for a role of p38-α MAPK in LAP2 dependent C3 promoter transactivation.
Figure 4. Role of p38-α MAPK in C/EBP-β mediated C3 promoter activation.
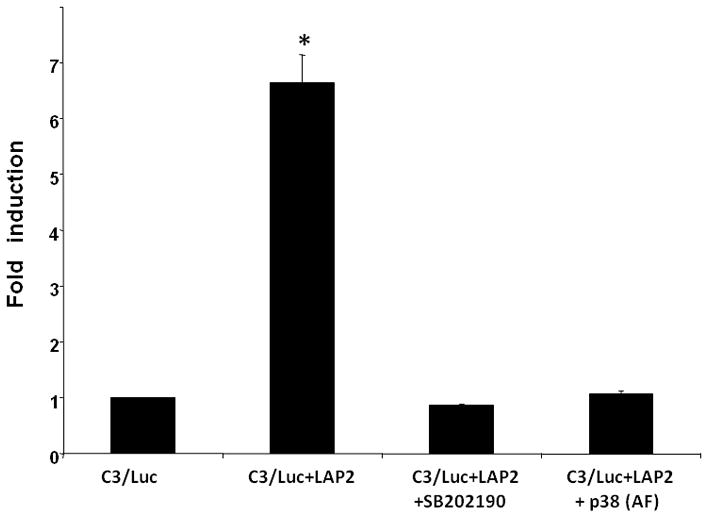
Fold induction of C3 promoter activity in U373 cells transiently co-transfected with C3/Luc reporter vector and 1 μg of empty expression vector or expression vector for (LAP2) and p38-α mutant (AF) or treated with p-38 MAPK inhibitor, SB202190-HCl (10 μM) 24hr after transfection. Cells were harvested 48h post transfection to assess luciferase activity. RLU/s was normalized to protein concentration and expressed as fold induction wherein the activity in C3/Luc alone transfected cells is set at 1. Statistical significance (*, p<0.05 between C3/Luc alone and C3/Luc plus LAP2 transfected cells) was analyzed by Student’s t-test.
Role of MKK6 in C3 promoter activation by LAP2
To investigate the role of p38 MAPK signaling pathway in the regulation of C3 promoter activation by LAP2 more directly, we used expression constructs of MKK6b to modulate either positively or negatively the endogenous p38 MAPK activity. The results in Fig. 5 demonstrate that the constitutively active MKK6b(E) isoform further induced LAP2 mediated induction. Conversely, the dominant negative mutant MKK6b(A) inhibited the induction. These observations taken together demonstrate that MKK6 is an upstream kinase that regulates C/EBP-β mediated C3 gene induction.
Figure 5. Role of MKK6b in LAP2 mediated C3 promoter activation.
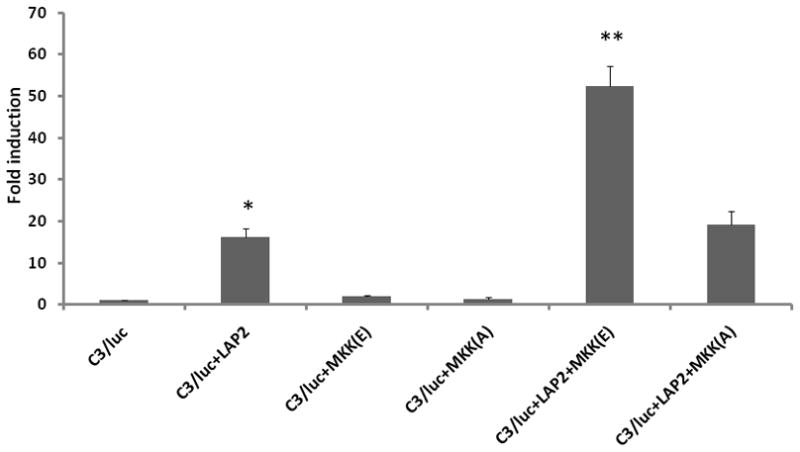
Fold induction of C3 promoter activity in U373 cells transiently co-transfected with C3/Luc reporter vector and 1 μg of empty expression vector or expression vector for MKK6b(A) or MKK6b(E) and LAP2. Cells were harvested 48h post transfection to measure luciferase activity. RLU/s was normalized to protein concentration and expressed as fold induction wherein the activity in C3/Luc alone transfected cells is set at 1. Statistical significance (*, p<0.05 between C3/Luc alone and C3/Luc plus LAP2 transfected cells; **, p<0.05 between between C3/Luc plus MKK6b(E) alone and C3/Luc plus LAP2 and MKK6b(E) transfected cells) was analyzed by Student’s t-test.
Role of C/EBP-β phosphorylation in C3 promoter activation
It has been postulated that C/EBP-β contains negative regulatory regions, and that phosphorylation(s) within these domains may regulate C/EBP-β function. Thus, C/EBP-β may be present in cells as a repressed transcription factor that becomes activated upon phosphorylation. Phosphorylation of Thr235 of human LAP has been implicated to be a key determinant of its transactivation capacity since the MAPK consensus site is highly conserved throughout species, encompassing Thr188 in murine LAP and the homologous Thr235 in human LAP [Nakimura et al., 1992]. We therefore assessed whether IL-1β mediated phosphorylation of LAP at its MAPK consensus site is required for LAP to be transcriptionally active. LAP or mutant LAPT235A (Thr235 mutated to Ala) [Nakimura et al., 1992] were coexpressed with a luciferase reporter gene driven by the C3 promoter in U373 cells. Overexpression of mutant LAPT235A decreased C3 promoter activity by 13-fold in comparison to wild type LAP expression (Fig. 6). Stimulation of cells transfected with wild type LAP by IL-1β treatment further activated C3 promoter activity by another 10-fold (Fig. 5). In contrast, stimulation of C3 promoter activity in the presence of mutant LAPT235A by IL-1β was similar to that of the wild type LAP (Fig. 6). These findings suggest that phosphorylation of LAP at Thr235 in the MAPK consensus site is required for LAP to be transcriptionally active in the context of the C3 promoter in response to IL-1β.
Figure 6. Phosphorylation of C-EBP-β is essential for C3 promoter activation by IL-1β.
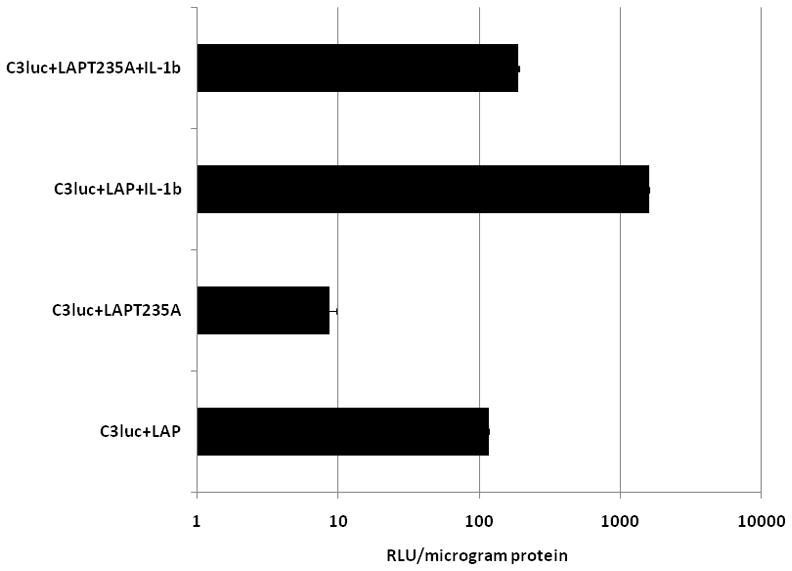
Relative luciferase activity in U373 cells transiently co-transfected with C3/Luc reporter vector and 1μg of empty expression vector or expression vector for wild type LAP or mutant LAPT235A. 24h post transfection cells were treated with IL-1β (10ng/ml). Cells were harvested 48h post transfection to assess for luciferase activity.
Discussion
The precise signaling mechanisms whereby IL-1β activates expression of C3 are not fully understood. The major and novel findings of this study are: 1) IL-1β induces p38 MAPK phosphorylation and C3 gene expression; 2) the proximal C3 promoter is differentially regulated by the C/EBP-β family of transcription factors, C/EBP-β isoform LAP1 and LAP2 are positive regulators while LIP is a negative regulator; 3) p38-α MAPK plays a significant role in both IL-1β and LAP2 mediated C3 promoter activation; and 4) MKK6b is the upstream kinase that regulates both IL-1β and LAP2 mediated C3 promoter activation.
The C/EBP family of transcription factors includes at least six members namely C/EBP-α, -β, -γ, -δ, -ε and -ζ [Ramji and Foka, 2002]. Our observations on the differential transactivation role of the C/EBP-β isoforms in C3 promoter activation in astrocytic cells are in agreement with earlier studies in hepatoma cell line in which it was demonstrated that C/EBP-β isoform LAP2 transactivates C3 expression, while the full length isoform of C/EBP-β, LAP1 did not transactivate the C3 promoter [Juan et al., 1993]. Similar observation was also reported in the context of cyclin D1 promoter activation by C/EBPβ-1 and -2 [Eaton et al., 2001]. The N-terminal transactivation domain of C/EBP-β common to both C/EBPβ-1 and -2 is known to interact with factors such as TBP, TFIIB, and CBP/p300 [Nerlov and Ziff, 1995; Mink et al., 1997]. Although we do not know which factor(s) C/EBPβ-2 is interacting with to transactivate the C3 promoter, it is possible that the additional amino acids of C/EBPβ-1 block this interaction, either sterically, or by interacting with another distinct set of factors such as the SWI-SNF chromatin remodeling complex.
p38 MAPK belongs to a family of MAPKs and consists of four isoforms in mammalian cells [Turjanski et al., 2007] and is primarily activated by proinflammatory cytokines, and cellular stress [Raingeaud et al., 1995; Han et al., 1994; Minet et al., 2001]. The dual specificity kinases that activate p38 MAPK are MKK3 and MKK6 [Enslen at al., 1998; Han et al., 1996; Han et al., 1997]. MKK6 functions as an upstream kinase for all p38 MAPK isoforms [Han et al., 1997; Enslen et al., 1998]. With the availability of specific inhibitors such as SB202190.HCl which selectively inhibits p38MAPK α- and β-isoforms [Lee et al., 1994], it is possible to investigate the isoform of p38 MAPK that plays a role in IL-1β and C/EBP-β mediated C3 gene regulation. Our studies, utilizing this strategy demonstrate that indeed p38-α MAPK plays a significant role in LAP and IL-1β mediated C3 gene induction. Both SB202190-HCl and dominant negative p38-α MAPK inhibited C/EBP-β mediated C3 promoter activity, while overexpression of dominant negative p38-α MAPK inhibited IL-1β mediated C3 promoter activation. The reduced activation of C3 promoter by IL-1β in presence of SB202190-HCl and dominant negative p38-α MAPK suggest that regions outside of the C/EBP-binding site also play a role in C3 gene regulation. Interestingly, the C3 promoter has also been shown to be regulated by NFκB in other cell types [Moon et al., [1999]; Andoh et al., [2000]].
Our studies demonstrating that dominant negative MKK6 (MKK6b(A) inhibits both IL-1β and LAP2 mediated C3 promoter activation are in agreement with studies in fibroblast like-synoviocytes wherein IL-1β induced IL-8, IL-6, and matrix metalloproteinase-3 protein production was significantly inhibited in DN MKK6-transfected cells [Inoue et al., 2005]. The observation that the constitutively active isoform of MKK6b induces both IL-1β- and LAP2- mediated C3 promoter activity are consistent with similar observations in BV2 microglial cells wherein IP10 promoter is constitutively activated by overexpression of the constitutively active isoform of MKK6b [Shen et al, 2006].
C/EBP-β contains phosphorylation sites for multiple protein kinases, including Ras-MAPK, p38 MAPK, calcium/calmodulin-dependent protein kinase, glycogen-synthase kinase 3 (GSK-3), protein kinase A (PKA), protein kinase C (PKC) and p90 ribosomal S kinase (p90rsk) [Nakajima et al., 1993; Aouadi et al., 2007; Wegner et al., 1992, Piwien-Pilipuk et al., 2001; Trautwein et al., 1994; Chinery et al., 1997; Buck et al., 1999]. Previous studies have demonstrated that phosphorylation of C/EBP-β can modulate its DNA binding activity or alter its transactivation potential [Nakajima et al., 1993; Aouadi et al., 2007; Wegner et al., 1992, Piwien-Pilipuk et al., 2001; Trautwein et al., 1994; Chinery et al., 1997; Buck et al., 1999]. Our observation that wild type LAP2 activates C3 promoter and IL-1β further induces this activation while the mutant isoform LAPT235A does not activate C3 promoter activity suggests that phosphorylation of Thr235 is a critical determinant for LAP2 interaction with its cognate recognition sequence on C3 promoter.
Based on these observations, we propose a model where the activation of MKK6b and p38α MAPK activity by IL-1β can further activate C/EBP-β to result in sustained induction of C3 (Fig. 7).
Figure 7. Model illustrating the mechanism of IL-1β mediated C3 gene regulation in astrocytes.
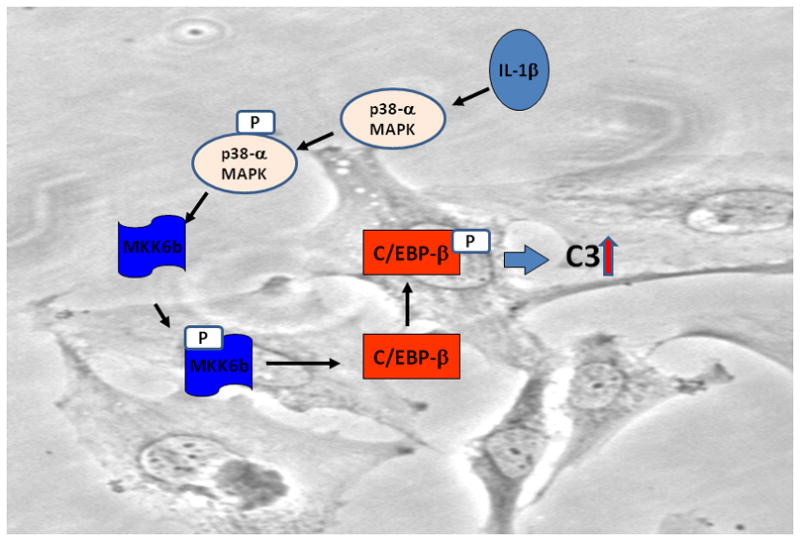
Binding of IL-1β to its cognate receptor induces phosphorylation of MKK6b which in turn activates p38-α MAPK. Phosphorylation of p38-α MAPK activates C/EBP-β to induce transcription of C3 gene.
Acknowledgments
The study has been supported by NIH/NIDA Career development Award 5K01DA022147 to PD, additional support was provided by an NIH/NINDS grant R01 NS047031 to JR.
References
- Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interactions following axon injury. Prog Neurobiol. 1998;55:1–26. doi: 10.1016/s0301-0082(97)00093-2. [DOI] [PubMed] [Google Scholar]
- Andoh A, Fujiyama Y, Shimada M, Bamba T. Modulation of complement component (C3 and factor B) biosynthesis by a histone deacetylase inhibitor in human intestinal epithelial cells. Int J Mol Med. 2000;6:51–54. [PubMed] [Google Scholar]
- Aouadi M, Jager J, Laurent K, Gonzalez T, Cormont M, Binétruy B, Le Marchand-Brustel Y, Tanti JF, Bost F. p38MAP Kinase activity is required for human primary adipocyte differentiation. FEBS Lett. 2007;581:5591–5596. doi: 10.1016/j.febslet.2007.10.064. [DOI] [PubMed] [Google Scholar]
- Barnum SR, Jones JL, Benveniste EN. Interferon-gamma regulation of C3 gene expression in human astroglioma cells. J Neuroimmunol. 1992;38:275–282. doi: 10.1016/0165-5728(92)90020-l. [DOI] [PubMed] [Google Scholar]
- Barnum SR, Jones JL, Benveniste EN. Interleukin-1 and tumor necrosis factor-mediated regulation of C3 gene expression in human astroglioma cells. Glia. 1993;7:225–236. doi: 10.1002/glia.440070306. [DOI] [PubMed] [Google Scholar]
- Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44:999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Bruder C, Hagleitner M, Darlington G, Mohsenipour I, Würzner R, Höllmüller I, Stoiber H, Lass-Flörl C, Dierich MP, Speth C. HIV-1 induces complement factor C3 synthesis in astrocytes and neurons by modulation of promoter activity. Mol Immunol. 2004;40:949–961. doi: 10.1016/j.molimm.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Buck M, Poli V, van der Geer P, Chojkier M, Hunter T. Phosphorylation of rat serine 105 or mouse threonine 217 in C/EBP beta is required for hepatocyte proliferation induced by TGF alpha. Mol Cell. 1999;4:1087–1092. doi: 10.1016/s1097-2765(00)80237-3. [DOI] [PubMed] [Google Scholar]
- Chinery R, Brockman JA, Dransfield DT, Coffey RJ. Antioxidant-induced nuclear translocation of CCAAT/enhancer-binding protein beta. A critical role for protein kinase A-mediated phosphorylation of Ser299. J Biol Chem. 1997;272:30356–30361. doi: 10.1074/jbc.272.48.30356. [DOI] [PubMed] [Google Scholar]
- Datta PK, Rappaport J. HIV and complement: hijacking an immune defense. Biomed Pharmacother. 2006;60:561–568. doi: 10.1016/j.biopha.2006.07.087. [DOI] [PubMed] [Google Scholar]
- Eaton EM, Hanlon M, Bundy L, Sealy L. Characterization of C/EBPbeta isoforms in normal versus neoplastic mammary epithelial cells. J Cell Physiol. 2001;189:91–105. doi: 10.1002/jcp.1139. [DOI] [PubMed] [Google Scholar]
- Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- Escartin C, Bonvento G. Targeted activation of astrocytes: a potential neuroprotective strategy. Mol Neurobiol. 2008;38:231–241. doi: 10.1007/s12035-008-8043-y. [DOI] [PubMed] [Google Scholar]
- Gasque P, Julen N, Ischenko AM, Picot C, Mauger C, Chauzy C, Ripoche J, Fontaine M. Expression of complement components of the alternative pathway by glioma cell lines. J Immunol. 1992;149:1381–1387. [PubMed] [Google Scholar]
- Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996;271:2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- Han J, Wang X, Jiang Y, Ulevitch RJ, Lin S. Identification and characterization of a predominant isoform of human MKK3. FEBS Lett. 1997;403:19–22. doi: 10.1016/s0014-5793(97)00021-5. [DOI] [PubMed] [Google Scholar]
- Huang S, Jiang Y, Li Z, Nishida E, Mathias P, Lin S, Ulevitch RJ, Nemerow GR, Han J. Apoptosis signaling pathway in T cells is composed of ICE/Ced-3 family proteases and MAP kinase kinase 6b. Immunity. 1997;6:739–49. doi: 10.1016/s1074-7613(00)80449-5. [DOI] [PubMed] [Google Scholar]
- Inoue T, Hammaker D, Boyle DL, Firestein GS. Regulation of p38 MAPK by MAPK kinases 3 and 6 in fibroblast-like synoviocytes. J Immunol. 2005;174:4301–4306. doi: 10.4049/jimmunol.174.7.4301. [DOI] [PubMed] [Google Scholar]
- Juan TS, Wilson DR, Wilde MD, Darlington GJ. Participation of the transcription factor C/EBP delta in the acute-phase regulation of the human gene for complement component C3. Proc Natl Acad Sci USA. 1993;90:2584–2588. doi: 10.1073/pnas.90.7.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukimoto I, Takeuchi T, Kanda T. CCAAT/enhancer binding protein beta binds to and activates the P670 promoter of human papillomavirus type 16. Virology. 2006;346:98–107. doi: 10.1016/j.virol.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Maranto J, Rappaport J, Datta PK. Regulation of complement component C3 in astrocytes by IL-1beta and morphine. J Neuroimmune Pharmacol. 2008;3:43–51. doi: 10.1007/s11481-007-9096-9. [DOI] [PubMed] [Google Scholar]
- Minet E, Michel G, Mottet D, Raes M, Michiels C. Transduction pathways involved in Hypoxia-Inducible Factor-1 phosphorylation and activation. Free Radic Biol Med. 2001;31:847–855. doi: 10.1016/s0891-5849(01)00657-8. [DOI] [PubMed] [Google Scholar]
- Moon MR, Parikh AA, Pritts TA, Fischer JE, Cottongim S, Szabo C, Salzman AL, Hasselgren PO. Complement component C3 production in IL-1beta-stimulated human intestinal epithelial cells is blocked by NF-kappaB inhibitors and by transfection with ser 32/36 mutant IkappaBalpha. J Surg Res. 1999;82:48–55. doi: 10.1006/jsre.1998.5503. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Kinoshita S, Sasagawa T, Sasaki K, Naruto M, Kishimoto T, Akira S. Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc Natl Acad Sci USA. 1993;90:2207–2211. doi: 10.1073/pnas.90.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov C. The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007;17:318–324. doi: 10.1016/j.tcb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Piwien-Pilipuk G, Van Mater D, Ross SE, MacDougald OA, Schwartz J. Growth hormone regulates phosphorylation and function of CCAAT/enhancer-binding protein beta by modulating Akt and glycogen synthase kinase-3. J Biol Chem. 2001;276:19664–19671. doi: 10.1074/jbc.M010193200. [DOI] [PubMed] [Google Scholar]
- Ramji DP, Foka P. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J. 2002;365:561–575. doi: 10.1042/BJ20020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Rus HG, Kim LM, Niculescu FI, Shin ML. Induction of C3 expression in astrocytes is regulated by cytokines and Newcastle disease virus. J Immunol. 1992;148:928–933. [PubMed] [Google Scholar]
- Shen Q, Zhang R, Bhat NR. MAP kinase regulation of IP10/CXCL10 chemokine gene expression in microglial cells. Brain Res. 2006;1086:9–16. doi: 10.1016/j.brainres.2006.02.116. [DOI] [PubMed] [Google Scholar]
- Simi A, Tsakiri N, Wang P, Rothwell NJ. Interleukin-1 and inflammatory neurodegeneration. Biochem Soc Trans. 2007;35:1122–1126. doi: 10.1042/BST0351122. [DOI] [PubMed] [Google Scholar]
- Speth C, Stockl G, Mohsenipour I, Wurzner R, Stoiber H, Lass-Florl C, Dierich MP. Human immunodeficiency virus type 1 induces expression of complement factors in human astrocytes. J Virol. 2001;75:2604–2615. doi: 10.1128/JVI.75.6.2604-2615.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speth C, Schabetsberger T, Mohsenipour I, Stockl G, Wurzner R, Stoiber H, Lass-Florl C, Dierich MP. Mechanism of human immunodeficiency virus-induced complement expression in astrocytes and neurons. J Virol. 2002;76:3179–3188. doi: 10.1128/JVI.76.7.3179-3188.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speth C, Williams K, Hagleitner M, Westmoreland S, Rambach G, Mohsenipour I, Schmitz J, Würzner R, Lass-Flörl C, Stoiber H, Dierich MP, Maier H. Complement synthesis and activation in the brain of SIV-infected monkeys. J Neuroimmunol. 2004;151:45–54. doi: 10.1016/j.jneuroim.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Trautwein C, van der Geer P, Karin M, Hunter T, Chojkier M. Protein kinase A and C site-specific phosphorylations of LAP (NF-IL6) modulate its binding affinity to DNA recognition elements. J Clin Invest. 1994;93:2554–2561. doi: 10.1172/JCI117266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turjanski AG, Vaqué JP, Gutkind JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- Wegner M, Cao Z, Rosenfeld MG. Calcium-regulated phosphorylation within the leucine zipper of C/EBP beta. Science. 1992;256:370–373. doi: 10.1126/science.256.5055.370. [DOI] [PubMed] [Google Scholar]
- Xing HQ, Hayakawa H, Izumo K, Kubota R, Gelpi E, Budka H, Izumo S. In vivo expression of proinflammatory cytokines in HIV encephalitis: an analysis of 11 autopsy cases. Neuropathology. 2009;29:433–442. doi: 10.1111/j.1440-1789.2008.00996.x. [DOI] [PubMed] [Google Scholar]
- Yanamadala V, Friedlander RM. Complement in neuroprotection and neurodegeneration. Trends Mol Med. 2010;16:69–76. doi: 10.1016/j.molmed.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- Zhao ML, Kim MO, Morgello S, Lee SC. Expression of inducible nitric oxide synthase, interleukin-1 and caspase-1 in HIV-1 encephalitis. J Neuroimmunol. 2001;115:182–191. doi: 10.1016/s0165-5728(00)00463-x. [DOI] [PubMed] [Google Scholar]