

Abstract
There has been an explosion in our knowledge of the pathways and mechanisms by which the immune system can influence the brain and behavior. In the context of inflammation, pro-inflammatory cytokines can access the central nervous system and interact with a cytokine network in the brain to influence virtually every aspect of brain function relevant to behavior including neurotransmitter metabolism, neuroendocrine function, synaptic plasticity, and neurocircuits that regulate mood, motor activity, motivation, anxiety and alarm. Behavioral consequences of these effects of the immune system on the brain include depression, anxiety, fatigue, psychomotor slowing, anorexia, cognitive dysfunction and sleep impairment; symptoms that overlap with those which characterize neuropsychiatric disorders, especially depression. Pathways that appear to be especially important in immune system effects on the brain include the cytokine signaling molecules, p38 mitogen activated protein kinase and nuclear factor kappa B; indoleamine 2,3 dioxygenase and its down stream metabolites, kynurenine, quinolinic acid and kynurenic acid; the neurotransmitters, serotonin, dopamine and glutamate; and neurocircuits involving the basal ganglia and anterior cingulate cortex. A series of vulnerability factors including aging and obesity as well as chronic stress also appear to interact with immune to brain signaling to exacerbate immunologic contributions to neuropsychiatric disease. The elucidation of the mechanisms by which the immune system influences behavior yields a host of targets for potential therapeutic development as well as informing strategies for the prevention of neuropsychiatric disease in at risk populations.
Keywords: Immune system, Cytokines, Inflammation, Brain, Behavior, Neuropsychiatric Disorders
1. Introduction
Reciprocal interactions between the immune system and the brain have attracted considerable attention regarding the role of the immune system in neuropsychiatric diseases especially major depression. During the last several decades, research in the field referred to as “Psychoneuroimmunology” has demonstrated an intricate network of bi-directional relationships between the immune system and the brain. Alterations in immune function have been found in depressed patients with major depression and include early reports of immune suppression (e.g., reduced natural killer cell activity and reduced lymphocyte proliferation) followed by evidence of increased inflammatory activity (e.g., increased circulating levels of inflammatory markers) (Kronfol et al., 1983; Irwin and Gillin, 1987; Maes et al., 1993; Anisman et al., 1999; Zorilla et al., 2001). Much of the recent interest in the role of the immune system in depression has focused on increased inflammation associated with depression. Pro-inflammatory cytokines, including interleukin (IL)-1, IL-6 and tumor necrosis factor (TNF)-alpha, are released by activated immune cells during the host response to pathogen invasion as well as in the context of tissue injury and psychosocial stress. These soluble factors appear to represent primary mediators of the communication between the immune system and the brain, and not only help orchestrate cellular responses to immune challenge, but also coordinate the behavioral changes that are necessary for recovery. During the course of immune challenge, the release of pro-inflammatory cytokines is usually transient and regulated by anti-inflammatory mechanisms. Consequently, the behavioral effects triggered by the activation of the inflammatory response develop as an adaptative, temporary and controlled reactionof the central nervous system (CNS) to immune signals. Nevertheless, when immune challenge becomes chronic and/or unregulated, as is observed in patients receiving chronic cytokine treatments or those exposed to chronic medical illness and/or stress, the behavioral effects of cytokines and the resultant inflammatory response may contribute to the development of clinically relevant behavioral symptoms and neuropsychiatric diseases, including major depression. These immune-based behavioral disorders, as well as the pathophysiological mechanisms involved will be discussed in this review along with relevant treatment implications.
2. Behavioral Effects of Cytokines
a. Sickness Behavior
A rich database has been developed that substantiates the capacity of pro-inflammatory cytokines to induce, in addition to fever and activation of the hypothalamic-pituitary-adrenal (HPA) axis, a constellation of behavioral symptoms referred to as sickness behavior (Dantzer, 2001). Sickness behavior is typically associated with the behavioral changes seen in humans and laboratory animals suffering from microbial infections and includes depressive-like behavior, anhedonia, fatigue, psychomotor slowing, decreased appetite, sleep alterations and increased sensitivity to pain (Hart, 1988; Kent et al., 1992). Relevant to its mediation by pro-inflammatory cytokines, sickness behavior can be reliably reproduced by administration of pro-inflammatory cytokines separately or by treatment with cytokine-inducers, including endotoxin or lipopolysaccharide (LPS) or infectious agents such as Salmonella typhi and Bacille Calmette-Guerin (BCG), an attenuated form of Mycobacterium bovis(Yirmiya et al., 1999; Brydon et al., 2008; Harrison et al., 2009a; Harrison et al., 2009b; O’Connor et al., 2009b). Supporting further the notion that pro-inflammatory cytokines are the key mediators of sickness behavior, administration of cytokine antagonists, such as IL-1 receptor antagonist (IL-1ra), or anti-inflammatory cytokines such as IL-10, can block the behavioral effects of treatment with IL-1 and/or LPS in laboratory rodents (Kent et al., 1992; Bluthe et al., 1995; Avitsur et al., 1997; Dantzer et al., 2008).
In terms of symptom expression, sickness behavior shares many overlapping features with symptoms of depression. In animals, depressive-like behavior, reflected by decreased sucrose consumption and increased immobility (or behavioral despair) in the forced-swimming test or tail-suspension test, can be induced by acute or chronic treatment with inflammatory challenges (Yirmiya, 1996; Anisman et al., 2005; Dantzer et al., 2008; Moreau et al., 2008). Interestingly, chronic (but not acute) treatment with antidepressants, including imipramine and tianeptine, can reduce, or even abolish, the depressive-like effects of LPS treatment in rats (Yirmiya, 1996; Castanon et al., 2001). With respect to the temporal aspects of symptom expression, sickness behavior develops rapidly after administration of LPS in animals and usually peaks 2–6 hours post-treatment. Sickness behavior gradually resolves after 6 hours and evolves into depressive-like behavior 24 hours post-LPS (Frenois et al., 2007; Dantzer et al., 2008).
b. Cytokine-induced Neuropsychiatric Symptoms in Clinical Settings
The close resemblance of sickness behavior with symptoms of depression has led to the hypothesis that cytokines and inflammatory factors may be involved in the pathophysiology of neuropsychiatric disorders such as depression (Raison et al., 2006; Dantzer et al., 2008; Miller et al., 2009). Supporting this hypothesis is the high prevalence of psychiatric co-morbidity, including depression, in patients afflicted with chronic medical conditions associated with inflammation such as rheumatoid arthritis, cancer, infectious diseases, autoimmune disorders, and cardiovascular disease (Evans et al., 2005). In addition, there is a rich literature demonstrating elevated concentrations of inflammatory markers including pro-inflammatory cytokines, acute phase reactant, chemokines and adhesion molecules in the blood and cerebrospinal fluid of both medically ill and medically healthy patients with major depression (Zorrilla et al., 2001; Howren et al., 2009; Miller et al., 2009; Dowlati et al., 2010). Significant associations between markers of inflammation and individual neuropsychiatric symptoms including fatigue and cognitive dysfunction have also been described (Musselman et al., 2001; Meyers et al., 2005; Jehn et al., 2006; Lutgendorf et al., 2008; Bower et al., 2009). In depressed patients, TNF-alpha, IL-6, and C reactive protein (CRP) have been found to be the most reliably elevated across studies as demonstrated in three meta-analyses of the literature in this area (Zorrilla et al., 2001; Howren et al., 2009; Dowlati et al., 2010). Of note, increased inflammatory markers in depressed patients have also been associated with non-response to treatment with conventional antidepressant medications as well as early life stress, which is likewise associated with an antidepressant treatment non response (Sluzewska et al., 1997; Lanquillon et al., 2000; Nemeroff et al., 2003; Danese et al., 2007; Danese et al., 2008).
Strong support for the role of inflammatory processes in the pathophysiology of depression also comes from findings obtained in patients undergoing cytokine therapy. Because of their immunomodulatory effects, cytokines such as interferon (IFN)-alpha are currently used for the treatment of cancers and viral infections (e.g., chronic hepatitis C). Despite its clinical efficacy, IFN-alpha is frequently associated with significant psychiatric morbidity, notably in the form of the development of major depression in up to 45 percent of patients (Musselman et al., 2001; Capuron and Miller, 2004; Raison et al., 2005). To further assess the role of cytokines in the pathophysiology of depression, our group and others have used IFN-alpha administration in patients with malignant melanoma or chronic hepatitis C as a way to study cytokine effects on the brain and behavior in humans. IFN-alpha is an innate immune cytokine released early in viral infection that has both antiviral and antiproliferative activities (Roitt et al., 1998). In addition, IFN-alpha is a potent stimulator of pro-inflammatory cytokines, not only at the periphery but also within the CNS (Taylor and Grossberg, 1998; Raison et al., 2009). Relevant to this notion, recent data indicate that IFN-alpha treatment in patients with hepatitis C is associated with activation of CNS inflammatory pathways as reflected by increased cerebrospinal fluid (CSF) concentrations of IFN-alpha along with elevated CSF concentrations of IL-6 and monocyte chemoattractant protein (MCP)-1 (Raison et al., 2009).
Dimensional analyses of symptom expression in IFN-alpha treated patients have shown that administration of the cytokine is responsible for the development of two behavioral syndromes (i.e., a neurovegetative syndrome versus a mood and cognitive syndrome) with distinct phenomenology and responsiveness to antidepressants (Capuron et al., 2002a; Capuron and Miller, 2004). The neurovegetative syndrome, characterized by symptoms of fatigue, psychomotor slowing, anorexia and altered sleep patterns, develops rapidly in almost every individual exposed to cytokines and persists for the duration of cytokine exposure. In contrast, the mood and cognitive syndrome, characterized by symptoms of depressed mood, anxiety, memory and attentional disturbances, usually appears at later stages of cytokine treatment (between the first and third month of IFN-alpha therapy), especially in patients with intrinsic vulnerability factors (Figure 1). In addition, whereas IFN-alpha induced mood and cognitive symptoms appear to be responsive to antidepressant treatment, the neurovegetative symptoms (fatigue in particular) are antidepressant non-responsive (Capuron et al., 2002a; Capuron and Miller, 2004). With respect to the notion of vulnerability factors, the hyper-responsiveness of the stress response system, characterized by HPA axis hyper-reactivity, together with preexisting subclinical depressive traits were found to represent potent risk factors for the development of cytokine-induced depression (Capuron et al., 2003b; Capuron et al., 2004; Raison et al., 2005). Recent advances have been made regarding the pathophysiological mechanisms underlying the mood/cognitive versus neurovegetative effects of cytokine therapy. As discussed in more detail below, impaired tryptophan/serotonin metabolism was found to be involved in the development of IFN-alpha-induced mood and cognitive symptoms (Capuron et al., 2002b; Capuron et al., 2003a; Raison et al., 2010b). In contrast, neurovegetative symptoms, including fatigue and anergia, were found to correlate with changes in basal ganglia activity, possibly related to alterations in dopamine (DA) metabolism (Capuron et al., 2007b).
Figure 1. Temporal Evolution of the Neuropsychiatric Symptoms Induced by Chronic Interferon-alpha Therapy.

Interferon (IFN)-alpha therapy induces two types of behavioral symptoms with differential time course and responsiveness to antidepressants. The neurovegetative symptoms (e.g., fatigue, anergia and psychomotor slowing) develop rapidly (as soon as week 1 [W1]) in almost every individuals exposed to cytokines and persist during the duration of IFN-alpha therapy. These symptoms are minimally responsive to antidepressant treatment. In contrast, the mood and cognitive symptoms (e.g., depressed mood, anxiety, irritability, memory and attentional disturbance) develop in vulnerable patients at later stages of IFN-alpha therapy (between weeks 8–12) and are highly responsive to antidepressant medication (Capuron et al., 2002a).
3. Immune to Brain Signaling
a. How do Cytokine Signals Access the Brain?
Cytokines are relatively large molecules that do not freely pass through the blood brain barrier. Nevertheless, data indicate that cytokine signals are able to reach the brain through humoral, neural and cellular pathways (Figure 2). These pathways are comprised of at least five non-exclusive mechanisms, including 1) passage of cytokines through leaky regions of the blood-brain barrier, including the choroid plexus and circumventricular organs, 2) active transport via saturable cytokine-specific transport molecules on brain endothelium, 3) activation of endothelial cells, responsible for the subsequent release of second messengers (e.g., prostaglandins [PGE2] and nitric oxide) within the brain parenchyma, 4) transmission of cytokine signals via afferent nerve fibers, such as the vagus nerve, and 5) entry into the brain parenchyma of peripherally activated monocytes (Watkins et al., 1995; Plotkin et al., 1996; Rivest et al., 2000; Konsman et al., 2002; Quan and Banks, 2007; D’Mello et al., 2009). Data have shown that activation of these specific mechanisms differentially mediates cytokine effects on the CNS. For instance, whereas subdiaphragmatic vagotomy was found to block the LPS-induced sickness behavior in rats, it did not affect the LPS-induced synthesis and release of pro-inflammatory cytokines at the periphery nor the pyrogenic effects of systemic injection of IL-1beta (Bluthe et al., 1994; Dantzer et al., 1998; Luheshi et al., 2000).
Figure 2. Communication Pathways from the Periphery to the Brain.

Different pathways by which cytokine signals access the brain have been identified.
- Humoral pathway: Pro-inflammatory cytokines released by activated monocytes and macrophages access the brain through leaky regions of the blood-brain barrier such as the choroid plexus and circumventricular organs (CVOs). Within the brain parenchyma, the activation of endothelial cells is responsible for the subsequent release of second messengers (e.g., prostaglandins [PGE2] and nitric oxide [NO]) that act on specific brain targets.
- Neural pathway: Pro-inflammatory cytokines released by activated monocytes and macrophages stimulate primary afferent nerve fibers in the vagus nerve. Sensory afferents of the vagus nerve relay information to brain areas through activation of the nucleus of the tractus solitarius (NTS) and area postrema.
- Cellular Pathway: A cellular pathway has been recently described by which pro-inflammatory cytokines, notably TNF-alpha, are able to stimulate microglia to produce monocyte chemoattractant protein-1 (MCP-1), which in turn is responsible for the recruitment of monocytes into the brain (D’ Mello et al., 2009).
Abbreviations: Interleukin-6: IL-6; interleukin-1β: IL-1β; tumor-necrosis factor: TNF
b. Cytokine Network in the Brain
Cytokines function as part of an integrated network, each of them inducing their own synthesis while also inducing the synthesis of other pro-inflammatory cytokines, including TNF-alpha, IL-1 and IL-6, in a “cascade” fashion (Taylor and Grossberg, 1998). Within the brain, there is a cytokine network consisting of cells, e.g., neurons, microglia and astrocytes, which are able to produce cytokines, express cytokine receptors and amplify cytokine signals (Rothwell et al., 1996; Haas and Schauenstein, 1997). This neural substrate of immune signals underlies the potent effects of peripheral pro-inflammatory cytokines on pathways involved in the pathophysiology of neuropsychiatric disorders, including the activation of the HPA axis and corticotropin-releasing hormone (CRH) and the alteration of the metabolism of key monoamines (e.g., serotonin, DA, norepinephrine) (Dantzer et al., 1999; Capuron and Miller, 2004; Raison et al., 2006; Capuron et al., 2007a; Miller et al., 2009).
4. Pathophysiological Mechanisms by which Cytokines Affect the Brain
A number of pathways by which cytokines may influence behavior have been identified. These pathways include effects on neurotransmitter function, neuroendocrine activity, neural plasticity and alterations of brain circuitry (Figure 3).
Figure 3. Pathways to Cytokine-Induced Pathology.
Once cytokine signals reach the brain, they can interact with virtually every pathophysiologic domain relevant to mood regulation. These include effects on neurocircuits that regulate motor activity and motivation (basal ganglia) and mood, anxiety, arousal and alarm (anterior cingulate cortex); effects on growth factors such as brain derived neurotrophic factor, neurogenesis and ultimately synaptic plasticity; effects on the metabolism of monoamine neurotransmitters such as serotonin and dopamine as well as excitatory amino acid neurotransmitters such as glutamate; and effects on neuroendocrine function leading to glucocorticoid resistance and altered glucocorticoid secretion.
Abbreviations: Interleukin-6: IL-6; interleukin-1β: IL-1β; tumor-necrosis factor: TNF
a. Effects on Neurotransmitters
i. Serotonin
Probably the best studied neurotransmitter system as it relates to the effects of cytokines on the brain and behavior is serotonin. A number of studies in laboratory animals have indicated that acute exposure to a variety of cytokines and cytokine-inducers such as LPS can alter the turnover of serotonin in multiple brain regions (Dunn and Wang, 1995; Dunn et al., 1999; Anisman et al., 2005). Regarding the clinical literature, a compelling case can also be made that in the context of chronic exposure to cytokines, serotonin is involved. Indeed, data have demonstrated that the serotonin reuptake inhibitor, paroxetine, can significantly reduce depressive symptoms in the context of chronic administration of IFN-alpha (Musselman et al., 2001; Raison et al., 2007), and at least two studies have demonstrated that polymorphisms in the serotonin transporter are associated with the development of depression during IFN-alpha therapy (Bull et al., 2008; Lotrich et al., 2009). In addition, chronic IFN-alpha treatment leads to a downregulation of serotonin receptor 1A (Cai et al., 2005). Further relevant to serotonin, special attention has been paid to the role of the enzyme, indoleamine 2,3 dioxygenase (IDO) and the signaling molecule p38 mitogen activated protein kinase (MAPK).
1. IDO
IDO can be stimulated by a number of cytokines alone or in combination including IFN-alpha, IFN-gamma and TNF-alpha through activation of a number of inflammatory signaling pathways such as signal transducer and activator of transcription 1a (STAT1a), interferon regulatory factor (IRF)-1, NF-kB and p38 MAPK (Fujigaki et al., 2006). When activated, IDO breaks down tryptophan (TRP), the primary amino acid precursor of serotonin, into kynurenine (KYN) (Schwarcz and Pellicciari, 2002). The breakdown of TRP is believed to contribute to reduced serotonin availability which is not only relevant for the regulation of T cell function but also for CNS serotonin synthesis (Schwarcz and Pellicciari, 2002; Dantzer et al., 2008). Indeed, acute depletion of dietary tryptophan has been shown to precipitate depressive symptoms in vulnerable populations of depressed subjects (Delgado et al., 1994). Consistent with the role of IDO in cytokine-induced depression, decreased TRP and increased KYN in the peripheral blood have been associated with the development of depression in patients administered IFN-alpha (Bonaccorso et al., 2002; Capuron et al., 2002b; Capuron et al., 2003a). Moreover, blockade of IDO has been shown to inhibit the development of depressive-like behavior in mice treated with LPS or BCG (O’Connor et al., 2009a; O’Connor et al., 2009b).
It should be noted that in addition to depletion of TRP, the generation of KYN also appears to have important effects on neurotransmitter function and behavior (Wichers et al., 2005). For example, administration of KYN alone has been shown to induce depressive-like behavior in mice (O’Connor et al., 2009a). In addition, based on the differential expression of relevant metabolic enzymes, KYN is preferentially converted to kynurenic acid (KA) in astrocytes and quinolinic acid (QUIN) in microglia (Schwarcz and Pellicciari, 2002). Kynurenic acid has been shown to inhibit the release of glutatmate, which, by extension, may inhibit the release of DA, whose release is regulated in part by glutamatergic activity (Borland and Michael, 2004). Indeed, intrastriatal administration of KA has been shown to dramatically reduce extracellular DA in the rat striatum (Wu et al., 2007). In contrast, QUIN, promotes glutamate release through activation of N-methyl-D-aspartate (NMDA) receptors (Schwarcz and Pellicciari, 2002). QUIN also induces oxidative stress, which in combination with glutamate release may contribute to CNS excitotoxicity (Rios and Santamaria, 1991; Schwarcz and Pellicciari, 2002; Muller and Schwarz, 2007; McNally et al., 2008).
Data from patients treated with IFN-alpha indicate that the generation of KYN in the peripheral blood is associated with significant increases in KYN, QUIN and KA in the CSF (Raison et al., 2010b). Increases in CSF KYN and QUIN were in turn correlated with depression. Increased CSF QUIN and KYN were also correlated with CSF MCP-1, consistent with the notion that peripheral blood macrophages, which are ~20 times more capable of converting KYN to QUIN than microglia, may be attracted to the brain during IFN-alpha administration and contribute to QUIN production (Guillemin and Brew, 2004; D’Mello et al., 2009; Raison et al., 2010b). Given the capacity of cytokines like IFN-alpha and cytokine inducers like LPS and BCG to activate IDO pathways, the generation of KYN, QUIN and KA may represent an especially unique aspect of the effects of cytokines and inflammation on behavior.
2. p38 MAPK
There has been increasing interest in the role of MAPK pathways including p38 in the impact of cytokines on neurotransmitter metabolism. For example, several studies using pharmacologic agonists and cytokines (IL-1 and TNF-alpha) to stimulate p38 have shown that activation of p38 signaling pathways in vitro can up-regulate both the expression and activity of the membrane transporters for serotonin as well as norepinephrine in cell lines and rat brain synaptasome preparations (Zhu et al., 2005; Zhu et al., 2006). Recently, this work has been extended in vivo with the demonstration that LPS-induced activation of p38 MAPK can upregulate serotonin transporter expression and activity in the hippocampus in association with increased extracellular clearance of serotonin (Zhu et al., 2010). Consistent with these findings is that activated p38 in peripheral blood mononuclear cells has been associated with decreased CSF concentrations of the serotonin metabolite, 5-hydroxyindoleacetic acid (5-HIAA), in juvenile rhesus monkeys that were maternally abused as infants (Sanchez et al., 2007). Taken together with the influence of cytokines on serotonin synthesis (via effects on TRP), these data suggest that cytokines may exert a “double hit” on both monoamine synthesis and reuptake, thus contributing to reduced serotonin availability.
ii. Dopamine
Like with serotonin, cytokines have been shown to influence both the synthesis and reuptake of dopamine (as well as its release, as discussed above in relation to KA). For example, administration of species specific IFN-alpha to rats has been shown to decrease CNS concentrations of tetrahydrobiopterin (BH4) and DA in association with the stimulation of nitric oxide (NO) (Kitagami et al., 2003). Tetrahydrobiopterin is an important enzyme co-factor for tyrosine hydroxlylase, which converts tyrosine to L-DOPA and is the rate limiting enzyme in DA synthesis. Tetrahydrobiopterin is also required for NO synthesis, and therefore increased NO generation is associated with increased BH4 utilization (and thus decreased BH4 to support tyrosine hydroxylase activity). Treatment with an inhibitor of NO synthase was found to reverse IFN-alpha’s inhibitory effects on brain concentrations of both BH4 and DA (Kitagami et al., 2003). Activation of microglia is associated with increased NO production (Zielasek and Hartung, 1996), suggesting that cytokine influences on BH4 via NO may be a common mechanism by which cytokines reduce DA availability in relevant brain regions. Like with the serotonin transporter, cytokine-induced activation of MAPK pathways has also been shown to upregulate the activity of the dopamine transporter (Moron et al., 2003).
iii. Glutamate/Excitotoxicity
Another neurotransmitter pathway that is receiving increasing interest in the development and treatment of depression as well as the impact of cytokines on the brain and behavior is glutamate. When activated in the brain, cytokines and associated inflammatory mediators have been shown to stimulate the release of glutamate from glial elements and decrease glutamate reuptake, in part through downregulation of glutamate transporters(Pitt et al., 2003; Volterra and Meldolesi, 2005; Tilleux and Hermans, 2007; Ida et al., 2008). Of particular concern, excessive release of glutamate by astrocytes can access extrasynaptic NMDA receptors, which mediate excitotoxicity and decreased production of trophic factors including brain derived neurotrophic factor (BDNF) (Hardingham et al., 2002; Haydon and Carmignoto, 2006). In addition to excessive glutamate, cytokines can also stimulate astrocytes and microglia to release reactive oxygen and nitrogen species that in combination with QUIN (see above) can amplify oxidative stress and further endanger relevant cell types including neurons and oligodendrocytes, which are especially vulnerable to oxidative damage (Rios and Santamaria, 1991; Schwarcz and Pellicciari, 2002; Buntinx et al., 2004; Matute et al., 2006; Thornton et al., 2006; Ida et al., 2008; Li et al., 2008; McTigue and Tripathi, 2008). These potential effects of cytokines and central inflammatory processes on glia may in turn contribute to the loss of glial elements including oligodendrocytes and astrocytes in multiple mood-relevant brain regions including the subgenual prefrontal cortex and amygdala. Loss of glial elements in these brain regions has emerged as a fundamental morphologic abnormality in major depression (Ongur et al., 1998; Hamidi et al., 2004; Rajkowska and Miguel-Hidalgo, 2007).
b. Effects on Neuroendocrine Function
Alteration in the function of the HPA axis is a hallmark of neuropsychiatric disorders especially major depression (Pariante and Miller, 2001). Therefore, considerable attention has been paid to the impact of cytokines on HPA axis function. A rich literature has demonstrated that acute administration of cytokines and cytokine inducers can potently activate the HPA axis, consistent with the HPA axis hyperactivity that characterizes many depressed patients. For example, cytokines, especially when administered acutely, have been shown to stimulate the expression and release of CRH, adrenocorticotropic hormone (ACTH) as well as cortisol, all of which have been found to be elevated in patients with major depression (Besedovsky and del Rey, 1996; Pariante and Miller, 2001; Pace et al., 2007). As noted above, acute activation of the HPA axis by the cytokine, IFN-alpha, has also been shown to predict the development of depressive symptoms in IFN-alpha-treated humans, likely related to the role of sensitized CRH pathways in the vulnerability to cytokine-induced depression (Capuron et al., 2003b). Nevertheless, in both laboratory animals and humans, chronic administration of cytokines or inflammatory stimuli is not associated with ongoing activation of the HPA axis response (Capuron et al., 2003b; Harbuz et al., 2003; Wichers et al., 2007). Instead, studies have shown that chronic cytokine exposure or chronic medical illness is associated with flattening of the diurnal cortisol curve and increased evening cortisol concentrations which have been associated with both adverse behavioral effects (depression and fatigue) as well as poor disease outcome in several medical disorders including cardiovascular disease and cancer (Sephton et al., 2000; Matthews et al., 2006; Raison et al., 2010a).
One mechanism by which chronic cytokine exposure may influence HPA axis function is through inhibitory effects on the receptor for the glucocorticoid, cortisol, the end product of HPA axis activation (Pace and Miller, 2009). By stimulating a number of relevant inflammatory signaling molecules including NF-kB, p38 MAPK and STAT5, cytokines have been shown to reduce the function of glucocortioid receptors (GR) through disruption of GR translocation from the cytoplasm to nucleus as well as through nuclear protein-protein interactions which inhibit GR-DNA binding (Pace et al., 2007). Cytokines can also influence GR expression, leading to decreased GR alpha, the active form of the receptor, and increased GR beta, a relatively inert GR isoform (Pace et al., 2007). Decreased responsiveness to glucocorticoids (or glucocorticoid resistance) as manifested by increased cortisol concentrations following dexamethasone (DEX) administration in the DEX suppression test (DST) and the DEX-CRH test and decreased glucocorticoid-mediated inhibition of in vitro immune responses are well described in patients with major depression (Pariante and Miller, 2001). Of note, flattening of the cortisol slope has also been associated with nonsuppression on the DEX-CRH test in patients with breast cancer (Spiegel et al., 2006). Thus, cytokines have exhibited the capacity to replicate many of the typical HPA axis features associated with the development of major depression including altered glucocorticoid secretion and impaired GR function. Given the fundamental role of endogenous glucocorticoids in inhibiting inflammatory responses, cytokine-induced alterations in HPA axis function and decreased GR function might serve to further exacerbate unrestrained inflammation.
c. Effects on Neural Plasticity (Neurogenesis)
Although under physiological conditions cytokines provide trophic support to neurons and enhance neuronal integrity, in the context of chronic stress, excessive cytokine production has been shown to lead to depressive-like behavior in part through their disruption of growth factor production and neurogenesis. Neuogenesis is believed to play a fundamental role in the maintenance of neural integrity, and reduced neurogenesis especially in the hippocampus is a hallmark of chronic exposure to stress in laboratory animals (Duman and Monteggia, 2006). Not only does chronic stress activate cytokine expression, in particular IL-1, in the hippocampus, but several studies have shown that blockade of IL-1 through the administration of IL-1ra or transplantation of IL-1ra secreting neural precursor cells or the use of IL-1 knockout mice reverses the reduced growth factor production (i.e. BDNF) and decreased neurogenesis that is associated with chronic stress (while also reversing stress-induced behavioral changes) (Ben Menachem-Zidon et al., 2008; Goshen et al., 2008; Koo and Duman, 2008). In vitro studies have suggested that the effects of IL-1 on neurogenesis appear to occur through activation of NF-kB signaling pathways (Koo and Duman, 2008), while in vivo studies indicate that endogenous glucocorticoids are also involved (Goshen et al., 2008).
d. Effects on Neurocircuitry
Neuroimaging studies have begun to reveal the neurocircuits that appear to be targeted by cytokines. Neurocircuits in both cortical and subcortical brain regions are involved including most notably the basal ganglia and the subgenual and dorsal aspects of the anterior cingulate cortex (ACC). The basal ganglia play an important role in motor activity and motivation, whereas the subgenual and dorsal ACC are implicated in depression (subgenual ACC) and anxiety, arousal and alarm (dorsal ACC), respectively (Alexander et al., 1990; Eisenberger and Lieberman, 2004; Lozano et al., 2008). Consistent with the notion that cytokines may preferentially target DA neurotransmission and ultimately the basal ganglia are data that IFN-alpha as well as Salmonella typhi vaccination induce motor slowing which in IFN-alpha-treated patients is correlated with the development of both depression and fatigue (Brydon et al., 2008; Majer et al., 2008). In addition, neuroimaging studies have found that both IFN-alpha and Salmonella typhi vaccination induce altered metabolic activity (as revealed by positron emission tomography) and blood flow (as measured by functional magnetic resonance imaging) in relevant basal ganglia nuclei including the nucleus accumbens, putamen, and substantia nigra (Capuron et al., 2007b; Brydon et al., 2008). In patients administered Salmonella typhi, altered activity in the substantia nigra was correlated with peripheral blood IL-6 responses to vaccination (Brydon et al., 2008).
Administration of Salmonella typhi has also been associated with increased activation of the subgenual ACC, which correlated with depressed mood (Harrison et al., 2009a). Of note, the subgenual ACC [Broadman Area (BA) 25] is the target of successful attempts to restore normal mood in highly treatment resistant depressed patients (Lozano et al., 2008). Activation of the dorsal ACC (BA 24) has also been described in patients receiving IFN-alpha and in patients administered Salmonella typhi (Capuron et al., 2005; Harrison et al., 2009a). Increased activity in the dorsal ACC has been associated with high-trait anxiety, neuroticism, obsessive compulsive disorder and bipolar disorder (Eisenberger and Lieberman, 2004), all of which are associated with increased anxiety and arousal. Interestingly, in a recent study, stress-induced inflammatory responses (peripheral blood soluble TNF receptor 2) were associated with increased activity in the dorsal ACC, consistent with the notion that cytokines may preferentially target dorsal ACC circuits to increase arousal and alarm (Slavich et al., 2010). It has been suggested that by targeting the basal ganglia (which mediate motor activity and motivation) as well as the dorsal ACC, cytokines may be addressing competing survival priorities in a vulnerable, wounded or infected animal, which are to reduce activity to allow fighting of infection and wound healing while fostering hypervigilance to protect against future attack (Miller, 2008).
5. Sources of Chronic Inflammation
Chronic or non-resolving inflammation may originate from either pathophysiological (e.g., inflammatory diseases, immune-based disorders, T cell dysfunction) or non-pathological conditions including aging and obesity.
a. T Cell Dysfunction
Recent advances in immunology have increasingly recognized the importance of T cell subsets in the regulation of inflammatory responses including the relative role of T regulatory cells (T reg) and T effector cells that produce IL-17 (T Helper (Th)17 cells).
Interestingly, one of the first observations regarding the immune system in depression was that peripheral blood mononuclear cells from depressed patients exhibited impaired proliferation in response to the T cell mitogens, phytohemagglutinin and concanavalin A(Kronfol et al., 1983; Schleifer et al., 1984; Stein et al., 1991; Irwin and Miller, 2007; Irwin, 2008). Although the early findings were not always replicated, meta-analytic approaches to the literature in this area have reached the consensus that statistically reliable decreases in lymphocyte responses to T cell mitogens are apparent in depressed individuals (Zorrilla et al., 2001; Irwin and Miller, 2007). In vivo measures of cell-mediated immune function have also indicated decreased T cell activity in depressed patients including reduced skin responses to commonly encountered T cell-relevant antigens (Hickie et al., 1993; Sephton et al., 2009) as well as reduced T cell responses to varicella-zoster virus antigen ex vivo (Irwin et al., 1998).
The mechanisms of impaired T cell proliferation in depression in humans have yet to be established, however, a number of possibilities have been considered. Flow cytometric assessments have revealed that CD4+ T cells from depressed patients exhibit evidence of accelerated spontaneous apoptosis as well as increased expression of the receptor for Fas (CD95), which mediates apoptotic signaling by Fas-ligand (Eilat et al., 1999; Ivanova et al., 2007; Szuster-Ciesielska et al., 2008). T cell proliferation in patients with depression may also be disrupted by inflammatory cytokines, such as TNF-alpha, which has been shown to be elevated in depressed patients (Miller et al., 2009). Both in vitro and in vivo studies have demonstrated that chronic exposure of T cells to TNF-alpha decreases T cell proliferation and cytokine production (Cope et al., 1994; Cope et al., 1997; Lee et al., 2008). The effects of TNF-alpha on T cell function can be reversed by injections of monoclonal antibodies to TNF-alpha (Cope et al., 1994; Bayry et al., 2007; Lee et al., 2008), and are related in part to disruption of signaling through the T cell receptor (Cope et al., 1994; Cope et al., 1997; Lee et al., 2008). It should be noted that although glucocorticoids are well known to suppress T cell function, no relationship has been found between increased cortisol secretion and decreased in vitro proliferative responses to T cell mitogens in depressed patients (Kronfol et al., 1986; Raison and Miller, 2003). Indeed, if anything, peripheral blood lymphocytes from patients with depression exhibit decreased responsiveness to the in vitro inhibitory effects of glucocorticoids on T cell proliferation and the in vivo effects of glucocorticoids on T cell redistribution (Pariante and Miller, 2001; Bauer et al., 2002; Bauer et al., 2003; Raison and Miller, 2003). These findings of glucocorticoid resistance in T cells are consistent with reduced GR function as described above.
Despite both functional and genetic data indicating that alterations in T cells may be involved in depression (Wong et al., 2008), few studies have directly examined the relative expression and function of relevant T cell subsets beyond the characterization of CD4+ and CD8+ T cell phenotypes and mitogen-induced T cell proliferation (Zorrilla et al., 2001; Irwin and Miller, 2007). Given recent developments in immunology regarding the role of specific T cell subsets in the regulation of inflammation, the relative expression and function of T cell subsets including T reg cells and certain T effector cells including Th17 cells as well as Th1 and Th2 cells are of special relevance.
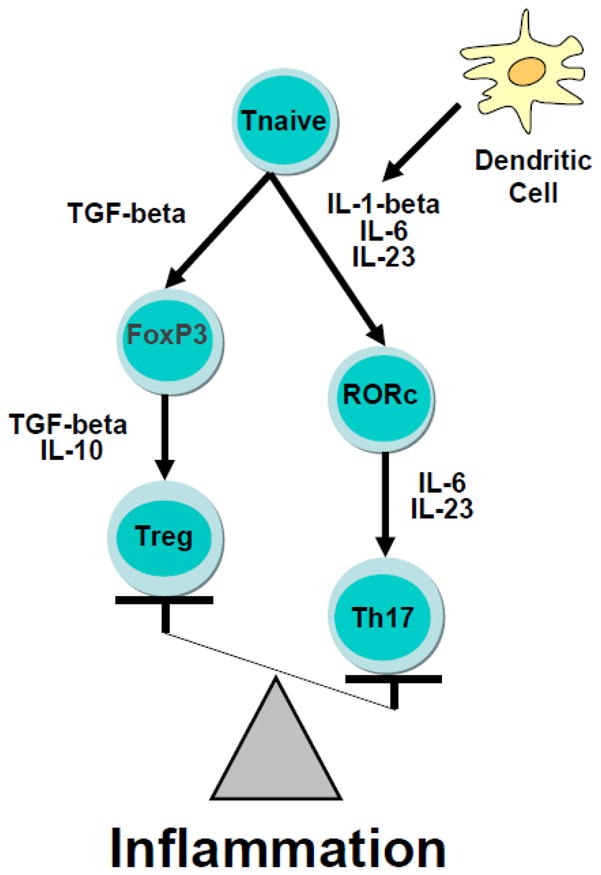
Most studies to date have imputed the function of relevant T cell subsets through the measurement of peripheral blood concentrations of cytokines that are regulated by these cell subsets (although it is recognized that many of these cytokines can be produced by multiple cell types other than T cells). Based on the available data, the findings in this area have been somewhat contradictory with some studies supporting evidence of increased Th1 cell activity (e.g. increased IFN-gamma) and others supporting increased Th2 cell activity (e.g. increased IL-4) (Schwarz et al., 2001; Myint et al., 2005; Pavon et al., 2006; Kim et al., 2007; Song et al., 2009). Of relevance to T reg, reduced IL-10 and transforming growth factor (TGF)-beta as well as an increased IL-6/IL-10 ratio have been found in depressed patients and are believed to be consistent with reduced T reg expression and/or function (Sutcigil et al., 2007; Dhabhar et al., 2009). Of note, 2 studies have directly examined CD4+CD25+ T reg expression in depression. One study found decreased T reg percentage in association with reduced IL-10 and TGF-beta in depressed patients versus controls (Li et al., 2010b), and a second study reported that antidepressant treatment led to increased CD4+CD25+ T reg percentage, which was associated with decreased IL-1-beta (Himmerich et al., 2010). These results support the hypothesis that CD4+CD25+ regulatory T cells are decreased in depressed patients, and given the role of T reg in reducing inflammatory responses, may contribute to increased inflammation in certain depressed individuals. Relevant to increased inflammation in depression, Th1 cells have long been identified as producers of pro-inflammatory mediators, such as TNF-alpha, and other contributors to both autoimmune and inflammatory disorders (Damsker et al., 2010). More recently however, T cells that produce IL-17 (Th17 cells) have been recognized as a distinct, highly pro-inflammatory T cell subset that may play an even more important role in inflammatory disease (Acosta-Rodriguez et al., 2007; Annunziato et al., 2007; Wilson et al., 2007; Tesmer et al., 2008; Damsker et al., 2010). In conjunction with IL-6 and IL-23, IL-1-beta is the most effective inducer of IL-17 expression in naive T cells in humans (Boissier et al., 2008). Based on these developments regarding the regulation of inflammation by T reg and Th17 cell subsets, examination of the hypothesis that high inflammation in patients with major depression is a result of increased Th17 activity in conjunction with reduced T reg function warrants further consideration (Figure 4).
Figure 4. An Inflammatory Imbalance of T cell Subsets.

Chronic inflammation may be maintained as the result of a preferential differentiation of T cell subsets toward a T helper (Th17) phenotype at the cost of T regulatory cell (T reg) development. Exposure to cytokines derived from dendritic cells and other immune cell types including the proinflammatory cytokines, IL-1β and IL-6, in conjunction with IL-23, increases the expression of the transcription factor, retinoid-related orphan receptor (ROR)c, which ultimately drives differentiation of the naïve T cell to the highly proinflammatory Th17 cell subset. In contrast, the anti-inflammatory cytokines TGF-beta and IL-10 promote the development of the counter-regulatory, anti-inflammatory T regulatory cell subset.
Abbreviations: FoxP3: Forkhead box P3; Interleukin-6: IL-6; interleukin-1β: IL-1β; Interleukin-10: IL-10; transforming growth factor: TGF
b. Non-pathological Conditions: Normal Aging and Obesity
Normal aging is a situation characterized by a chronic low-grade inflammatory state, with an over-expression of circulating inflammatory factors, including pro-inflammatory cytokines, to the detriment of anti-inflammatory factors (Fagiolo et al., 1992; Fagiolo et al., 1993). While the clear origin of chronic inflammation in aging remains to be elucidated, several possibilities including immunosenescence-related dysregulation, deficient antioxidant defenses, and impaired pro- versus anti-inflammatory regulatory mechanisms may be involved. Within the brain, this inflammatory characteristic of aging, referred to as inflammaging (Franceschi et al., 2000; Franceschi et al., 2007), manifests primarily by the chronic activation of perivascular and parenchymal macrophages/microglia expressing pro-inflammatory cytokines, while the number of astrocytes increases (Floyd, 1999; Ye and Johnson, 1999; Akiyama et al., 2000). This condition is accompanied by an increase in the brain production of reactive oxygen species (ROS), leading to greater susceptibility to neuronal damage and death (Coyle and Puttfarcken, 1993). Moreover, recent data suggest that aging represents a typical ‘priming’ condition for microglia cells, which become more reactive to noxious stimuli and immune challenge (Dilger and Johnson, 2008). More specifically, it was found in mice that aging was associated with increased markers of activated or primed microglia together with increased inflammation, and that, upon subsequent immune stimulation with LPS administered peripherally, primed microglial cells were responsible for the overproduction of pro-inflammatory cytokines within the CNS (Godbout et al., 2005; Dilger and Johnson, 2008). In terms of behavioral outcomes, this state was associated with an exacerbation of sickness behavior and cognitive alterations following peripheral or central administration of LPS to aged mice (Godbout et al., 2005; Chen et al., 2008; Henry et al., 2009).
Similar to aging, chronic inflammation is a fundamental characteristic of obesity. Obesity is characterized by an excessive accumulation of fat and its diagnosis involves the measurement of the body mass index (BMI), which is calculated by the ratio of the person’s weight (kg) divided by the height (in m2). The BMI is considered as one of the best clinical indices of adiposity, with differential severity ranging from overweight to severe and morbid obesity. Obesity is a chronic condition which is associated with low-grade inflammatory processes, with increased circulating levels of acute phase proteins (CRP in particular) and pro-inflammatory cytokines being observed in both adult and younger obese subjects (Bastard et al., 1999; Wellen and Hotamisligil, 2003). Part of systemic inflammation in obesity originates from adipose tissue in which macrophages accumulate and potently secrete inflammatory factors (Bastard et al., 1999; Wellen and Hotamisligil, 2003; Clement et al., 2004) (Figure 5). Interestingly, weight loss significantly improves the inflammatory profile of obese subjects (e.g., decreased pro-inflammatory factors and increased anti-inflammatory molecules) and regulate inflammation-related genes in white adipose tissue (Bastard et al., 2000; Clement et al., 2004; Cancello et al., 2005; Forsythe et al., 2008). Recent experimental data show evidence of inflammatory processes (ROS generation, elevated PGE2 levels, increased cyclooxygenase (COX)-2 expression, increased pro-inflammatory cytokines) in several brain areas, including the cortex and hippocampus, of animals fed a high-fat diet known to induce obesity (De Souza et al., 2005; Zhang et al., 2005). In addition, recent findings suggest that high-fat diet-related chronic inflammation may participate in the development of obesity through alteration in leptin and insulin signalling in the hypothalamus (Thaler and Schwartz, 2010). Moreover, consistent with the notion of priming, obesity was found to be associated with an increased reactivity of microglial cells under basal conditions and in response to immune challenge (Bilbo and Tsang, 2010). More specifically, it was shown that microglia were activated in the hippocampus of pups born from diet-induced obese female rats at birth, and that, at weaning and in adulthood, these same rats exhibited higher LPS-induced pro-inflammatory cytokine expression in the periphery and hippocampus in comparison to controls (Bilbo and Tsang, 2010). Finally, in nonhuman-primates, increased pro-inflammatory cytokines and markers of activated microglia were measured in the hypothalamus of third-trimester fetuses of high-fat diet fed mothers (Grayson et al., 2010).
Figure 5. Obesity-related Chronic Inflammation.

In obesity, fat accumulation leads to the development of chronic low grade inflammation. This systemic inflammation originates primarily from the adipose tissue in which macrophages accumulate and secrete pro-inflammatory cytokines and monocyte chemoattractant protein-1 (MCP-1), which in turn increases macrophage infiltration. Adipocytes in obesity are also able to secrete inflammatory factors, including tumor necrosis factor (TNF)-alpha and adipokines and related hormones (e.g., increased secretion of leptin, resistin and visfatin and decreased secretion of adiponectin) which also contribute to non-resolving inflammation.
c. Relevance for Vulnerability to Neuropsychiatric Symptoms
The prevalence of behavioral symptoms, including depression and cognitive dysfunction, is particularly elevated in the elderly and obese subjects. Given the potential role of inflammation in psychopathology, it is thus highly possible that chronically activated inflammatory signals in aging and/or obesity, although more subtle and expressed at a lower grade, may contribute to increased vulnerability to neuropsychiatric disorders. This hypothesis is supported by recent clinical findings obtained in our group indicating that activated inflammatory processes, in conjunction with poor antioxidant status and altered tryptophan metabolism, were associated with reduced quality of life in the elderly (Capuron et al., 2009). Consistent with these findings, data obtained in patients with the metabolic syndrome indicate that inflammation is one major determinant of the higher prevalence of depressive symptoms in this patient population (Capuron et al., 2008). With respect to obesity, a recent study by our group has shown that adiposity in a sample of obese women is associated with increased concentrations of inflammatory markers (IL-6, CRP and adipokines) that correlate with scores on the depression and anxiety aspects of neuroticism, with higher inflammation predicting higher anxiety and depression (Capuron et al., 2010). Interestingly, and as documented by other groups, surgery-induced weight loss was associated with reduced inflammation (Cancello et al., 2005; Emery et al., 2007), which was associated with significant improvement in emotional status (Capuron et al., 2010). Regarding cognitive dysfunction, similar associations have been documented between inflammatory status and cognitive performance/decline among overweight and obese women (Sweat et al., 2008) and aged individuals with the metabolic syndrome (Yaffe et al., 2004). These findings are consistent with experimental data indicating concomitant increases in brain inflammation and behavioral alterations in aged laboratory animals (Godbout and Johnson, 2009; Laye, 2010) or in animals with diet-induced obesity (Pistell et al., 2010). Interestingly, in this regard, it was found that lifelong caloric restriction in aged rats was associated with reduced secretion of inflammatory markers from adipose tissue together with higher physical performance scores in contrast to lifelong ad-libitum feeding, which was associated with increased inflammation and significant decline in physical performance (You et al., 2007). Altogether, these data suggest that chronic low-grade inflammation may play a role in the increased vulnerability to neuropsychiatric disorders in the elderly or obese subjects. Given the growing elderly population and increasing prevalence of overweight and obesity in modern societies, strategies to regulate chronic inflammatory processes in these conditions and to prevent inflammation-related neuropsychiatric disturbances may appear particularly relevant and urgently required.
6. Translational Implications
Given the vast knowledge base that has been elaborated regarding the capacity of inflammatory cytokines to influence behavior as well as the data detailing the potential mechanisms involved, there are a wide array of potential pharmacological and behavioral targets that may be relevant for the development of new treatments or prevention strategies for neuropsychiatric disorders including depression.
a. Pharmacological Targets
Probably the most obvious targets for pharmacologic intervention are the cytokines themselves and their signaling pathways. Biologic therapies, which are approved for use in the United States and Europe, that inhibit TNF-alpha and IL-1 have already been extensively studied in autoimmune and inflammatory disorders, and drugs which target a limited number of inflammatory signaling pathways [e.g., COX 1 and 2] are readily available or are in development. Data from the use of anti-cytokine therapies have already indicated an impact on depressed mood. For example in a large double blind placebo-controlled trial in patients with psoriasis, patients who received the TNF-alpha antagonist, etanercept, showed significant improvement in depressive symptoms that was independent of changes in symptoms associated with disease activity (e.g. skin lesions and joint pain) (Tyring et al., 2006). In addition, in medically healthy patients with major depression, the COX-2 inhibitor, celecoxib, was shown to significantly enhance antidepressant efficacy of the norepinephrine reuptake inhibitor, reboxetine, in a small double blind placebo-controlled trial (Muller et al., 2006). Albeit limited, these data suggest that targeting cytokines or their signaling pathways may have antidepressant activity. However, no study has specifically examined depressed patients with increased inflammatory markers and/or patients who may be at special risk for depression with increased inflammation including patients who are treatment resistant, obese and/or have a history of childhood maltreatment. Moreover, there is no data on the potential efficacy of antagonists of other relevant cytokine signaling pathways that appear to be especially germane to the mechanisms by which cytokines influence neurotransmitter metabolism, neuroendocrine function and synaptic plasticity including p38 MAPK and NF-kB. Finally, chemokines such as MCP-1, which can attract monocytes to multiple tissue sites including the brain where they can perpetuate inflammatory responses (D’Mello et al., 2009), are another class of target that may have unique applicability to behavioral disorders associated with increased inflammation.
Studies in both laboratory animals and humans also provide strong evidence for antagonizing IDO and the kynurenine pathway. Indeed, the IDO antagonist, 1-methyl tryptophan, has shown efficacy in animal models of sickness behavior as a consequence of LPS or BCG administration (O’Connor et al., 2009a; O’Connor et al., 2009b). Given the additional role of IDO and TRP depletion in inhibiting T cell function, there has been interest in IDO inhibitors to improve T cell activity in inflammatory states and cancer, thus indicating broad interest in the development of pharmacologic agents that target IDO (Johnson et al., 2009). Efforts in immunology that also may be relevant to immune-based behavioral disorders are pharmacologic strategies that target IL-17 or Th17 cells through the inhibition of cytokines or transcription factors which promote Th17 differentiation including IL-23 and retinoic acid receptor (RAR)-related orphan receptor C (RORc) (Boissier et al., 2008; Tesmer et al., 2008). Such mutual interests in the development of pharmacologic agents relevant to immunobiology and neurobiology underline the opportunities for cross-fertilization among disciplines that are presented by brain-immune system interactions.
Finally, there is considerable interest in drugs that antagonize glutamate for the treatment of depression. For example, administration of the glutamate (NMDA) receptor antagonist, ketamine, has led to rapid and dramatic improvements in mood in patients with treatment resistant depression (aan het Rot et al., 2010). Studies in animals suggest that these effects may result from the development of new synapses in the brain as result of activation of the enzyme mTOR (Li et al., 2010a). Interestingly, mTOR has been shown to inhibit NF-kB (Weichhart et al., 2008). Thus glutamate antagonists may not only reduce glutamate-mediated excitotoxicity as a result of cytokine-induced effects on glial glutamate reuptake and release but also may serve to inhibit inflammatory responses through effects on NF-kB.
b. Non-pharmacological Interventions
Given the epidemic of obesity in modern society, it is apparent that the relationship among depression, obesity and inflammation warrants serious attention. Treatments focused on weight loss including dietary modifications and exercise are clearly indicated both as part of the treatment of depression as well as its prevention. In addition, in the context of the immunomodulatory and anti-inflammatory effects of specific nutritional factors, including antioxidants and polyunsaturated fatty acids (omega-3 fatty acids in particular) (Laye, 2010; Su et al., 2010), the possibility to prevent or to modulate neuropsychiatric symptomatology in a context of chronic low-grade inflammation using nutritional interventions is an important consideration. Finally, given data in laboratory animals and humans that stress can activate the inflammatory response through stimulation of sympathetic pathways (in conjunction with parasympathetic withdrawal) (Miller et al., 2009), behavioral interventions focused on stress management and coping as well as therapies that modulate sympathetic/parasympathetic tone (e.g. meditation and yoga) should also be considered.
7. Conclusions
Brain-immune system interactions have opened up new vistas regarding the treatment and prevention of behavioral disorders. Given the interest in inflammation as a common mechanism of multiple diseases including cardiovascular disease, diabetes and cancer, the role of inflammation in neuropsychiatric diseases such as depression puts the neurosciences and psychiatry in lock step with other medical disciplines in identifying and targeting immune relevant molecules and pathways for the treatment of disease. Depression has received the greatest amount of attention to date. Nevertheless, in light of the impact of cytokines on neurotransmitter systems and neurocircuitry relevant to a broader spectrum of neuropsychiatric diseases (e.g., anxiety disorders, personality disorders, neurodegenerative diseases such as Parkinson’s and Alzheimer’s disease), it is likely that the importance of brain-immune system interactions may have wide applicability for the development of novel immune-based therapeutics for many neuropsychiatric illnesses.
Abbreviations
- ACC
Anterior Cingulate Cortex
- ACTH
Adrenocorticotropic Hormone
- BCG
Bacille Calmette-Guerin
- BDNF
Brain Derived Neurotrophic Factor
- BH4
Tetrahydrobiopterin
- BMI
Body Mass Index
- CNS
Central Nervous System
- COX
Cyclooxygenase
- CRH
Corticotropin-Releasing Hormone
- CRP
C Reactive Protein
- CSF
Cerebrospinal Fluid
- DA
Dopamine
- DEX
Dexamethasone
- DST
Dexamethasone Suppression Test
- GR
Glucocorticoid Receptor
- 5-HIAA
5-hydroxyindoleacetic acid
- HPA
Hypothalamic-Pituitary-Adrenal Axis
- IDO
Indoleamine 2,3 Dioxygenase
- IFN
Interferon
- IL
Interleukin
- IL-1ra
Interleukin-1 Receptor Antagonist
- IRF
Interferon Regulatory Factor
- KA
Kynurenic Acid
- KYN
Kynurenine
- LPS
Lipopolysaccharide
- MAPK
Mitogen Activated Protein Kinase
- MCP-1
Monocyte Chemotactic Protein-1
- NF-kB
Nuclear factor-kB
- PGE
Prostaglandin
- QUIN
Quinolinic Acid
- NMDA
N-Methyl-D-Aspartate
- NO
Nitric Oxide
- RAR
Retinoic Acid Receptor
- RORc
Receptor-related Orphan Receptor C
- ROS
Reactive Oxygen Species
- STAT
Signal Transducer Activator of Transcription
- TGF
Transforming Growth Factor
- Th17
T Helper 17 cells
- TNF
Tumor Necrosis Factor
- T reg
T regulatory cells
- TRP
Tryptophan
Footnotes
Conflict of Interest: The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, Mathew SJ. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67:139–145. doi: 10.1016/j.biopsych.2009.08.038. [DOI] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord. 2000;14(Suppl 1):S47–53. doi: 10.1097/00002093-200000001-00008. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD, DeLong MR. Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res. 1990;85:119–146. [PubMed] [Google Scholar]
- Anisman H, Ravindran AV, Griffiths J, Merali Z. Endocrine and cytokine correlates of major depression and dysthymia with typical or atypical features. Molecular Psychiatry. 1999;4:182–188. doi: 10.1038/sj.mp.4000436. [DOI] [PubMed] [Google Scholar]
- Anisman H, Merali Z, Poulter MO, Hayley S. Cytokines as a precipitant of depressive illness: animal and human studies. Curr Pharm Des. 2005;11:963–972. doi: 10.2174/1381612053381701. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avitsur R, Pollak Y, Yirmiya R. Different receptor mechanisms mediate the effects of endotoxin and interleukin-1 on female sexual behavior. Brain Res. 1997;773:149–161. doi: 10.1016/s0006-8993(97)00927-x. [DOI] [PubMed] [Google Scholar]
- Bartrop RW, Luckhurst E, Lazarus L, Kiloh LG, Penny R. Depressed lymphocyte function after bereavement. Lancet. 1977;1:834–836. doi: 10.1016/s0140-6736(77)92780-5. [DOI] [PubMed] [Google Scholar]
- Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, Vidal H, Hainque B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab. 2000;85:3338–3342. doi: 10.1210/jcem.85.9.6839. [DOI] [PubMed] [Google Scholar]
- Bastard JP, Jardel C, Delattre J, Hainque B, Bruckert E, Oberlin F. Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation. 1999;99:2221–2222. [PubMed] [Google Scholar]
- Bauer ME, Papadopoulos A, Poon L, Perks P, Lightman SL, Checkley S, Shanks N. Dexamethasone-induced effects on lymphocyte distribution and expression of adhesion molecules in treatment-resistant depression. Psychiatry Res. 2002;113:1–15. doi: 10.1016/s0165-1781(02)00243-3. [DOI] [PubMed] [Google Scholar]
- Bauer ME, Papadopoulos A, Poon L, Perks P, Lightman SL, Checkley S, Shanks N. Altered glucocorticoid immunoregulation in treatment resistant depression. Psychoneuroendocrinology. 2003;28:49–65. doi: 10.1016/s0306-4530(02)00009-4. [DOI] [PubMed] [Google Scholar]
- Bayry J, Siberil S, Triebel F, Tough DF, Kaveri SV. Rescuing CD4+CD25+ regulatory T-cell functions in rheumatoid arthritis by cytokine-targeted monoclonal antibody therapy. Drug Discov Today. 2007;12:548–552. doi: 10.1016/j.drudis.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Ben Menachem-Zidon O, Goshen I, Kreisel T, Ben Menahem Y, Reinhartz E, Ben Hur T, Yirmiya R. Intrahippocampal transplantation of transgenic neural precursor cells overexpressing interleukin-1 receptor antagonist blocks chronic isolation-induced impairment in memory and neurogenesis. Neuropsychopharmacology. 2008;33:2251–2262. doi: 10.1038/sj.npp.1301606. [DOI] [PubMed] [Google Scholar]
- Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev. 1996;17:64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Tsang V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. Faseb J. 2010;24:2104–2115. doi: 10.1096/fj.09-144014. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Beaudu C, Kelley KW, Dantzer R. Differential effects of IL-1ra on sickness behavior and weight loss induced by IL-1 in rats. Brain Res. 1995;677:171–176. doi: 10.1016/0006-8993(95)00194-u. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Walter V, Parnet P, Laye S, Lestage J, Verrier D, Poole S, Stenning BE, Kelley KW, Dantzer R. Lipopolysaccharide induces sickness behaviour in rats by a vagal mediated mechanism. C R Acad Sci III. 1994;317:499–503. [PubMed] [Google Scholar]
- Boissier MC, Assier E, Falgarone G, Bessis N. Shifting the imbalance from Th1/Th2 to Th17/treg: the changing rheumatoid arthritis paradigm. Joint Bone Spine. 2008;75:373–375. doi: 10.1016/j.jbspin.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Bonaccorso S, Marino V, Puzella A, Pasquini M, Biondi M, Artini M, Almerighi C, Verkerk R, Meltzer H, Maes M. Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J Clin Psychopharmacol. 2002;22:86–90. doi: 10.1097/00004714-200202000-00014. [DOI] [PubMed] [Google Scholar]
- Borland LM, Michael AC. Voltammetric study of the control of striatal dopamine release by glutamate. J Neurochem. 2004;91:220–229. doi: 10.1111/j.1471-4159.2004.02708.x. [DOI] [PubMed] [Google Scholar]
- Bower JE, Ganz PA, Tao ML, Hu W, Belin TR, Sepah S, Cole S, Aziz N. Inflammatory biomarkers and fatigue during radiation therapy for breast and prostate cancer. Clin Cancer Res. 2009;15:5534–5540. doi: 10.1158/1078-0432.CCR-08-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD. Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry. 2008;63:1022–1029. doi: 10.1016/j.biopsych.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull SJ, Huezo-Diaz P, Binder EB, Cubells JF, Ranjith G, Maddock C, Miyazaki C, Alexander N, Hotopf M, Cleare AJ, Norris S, Cassidy E, Aitchison KJ, Miller AH, Pariante CM. Functional polymorphisms in the interleukin-6 and serotonin transporter genes, and depression and fatigue induced by interferon-alpha and ribavirin treatment. Mol Psychiatry. 2008;14:1095–104. doi: 10.1038/mp.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buntinx M, Moreels M, Vandenabeele F, Lambrichts I, Raus J, Steels P, Stinissen P, Ameloot M. Cytokine-induced cell death in human oligodendroglial cell lines: I. Synergistic effects of IFN-gamma and TNF-alpha on apoptosis. J Neurosci Res. 2004;76:834–845. doi: 10.1002/jnr.20118. [DOI] [PubMed] [Google Scholar]
- Cai W, Khaoustov VI, Xie Q, Pan T, Le W, Yoffe B. Interferon-alpha-induced modulation of glucocorticoid and serotonin receptors as a mechanism of depression. J Hepatol. 2005;42:880–887. doi: 10.1016/j.jhep.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, Coupaye M, Pelloux V, Hugol D, Bouillot JL, Bouloumie A, Barbatelli G, Cinti S, Svensson PA, Barsh GS, Zucker JD, Basdevant A, Langin D, Clement K. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54:2277–2286. doi: 10.2337/diabetes.54.8.2277. [DOI] [PubMed] [Google Scholar]
- Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, Miller AH. Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology. 2002a;26:643–652. doi: 10.1016/S0893-133X(01)00407-9. [DOI] [PubMed] [Google Scholar]
- Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry. 2002b;7:468–473. doi: 10.1038/sj.mp.4000995. [DOI] [PubMed] [Google Scholar]
- Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D, Miller AH. Interferon-alpha-induced changes in tryptophan metabolism. relationship to depression and paroxetine treatment. Biol Psychiatry. 2003a;54:906–914. doi: 10.1016/s0006-3223(03)00173-2. [DOI] [PubMed] [Google Scholar]
- Capuron L, Raison CL, Musselman DL, Lawson DH, Nemeroff CB, Miller AH. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003b;160:1342–1345. doi: 10.1176/appi.ajp.160.7.1342. [DOI] [PubMed] [Google Scholar]
- Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry. 2004;56:819–824. doi: 10.1016/j.biopsych.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Capuron L, Ravaud A, Miller AH, Dantzer R. Baseline mood and psychosocial characteristics of patients developing depressive symptoms during interleukin-2 and/or interferon-alpha cancer therapy. Brain Behav Immun. 2004;18:205–213. doi: 10.1016/j.bbi.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Capuron L, Pagnoni G, Demetrashvili M, Woolwine BJ, Nemeroff CB, Berns GS, Miller AH. Anterior cingulate activation and error processing during interferon-alpha treatment. Biol Psychiatry. 2005;58:190–196. doi: 10.1016/j.biopsych.2005.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Miller AH, Irwin M. Psychoneuroimmunology of depressive disorder: mechanisms and clinical implications. In: Ader R, editor. Psychoneuroimmunology. 4. Elsevier Academic Press; New York: 2007a. pp. 509–530. [Google Scholar]
- Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, Berns GS, Nemeroff CB, Miller AH. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology. 2007b;32:2384–2392. doi: 10.1038/sj.npp.1301362. [DOI] [PubMed] [Google Scholar]
- Capuron L, Su S, Miller AH, Bremner JD, Goldberg J, Vogt GJ, Maisano C, Jones L, Murrah NV, Vaccarino V. Depressive symptoms and metabolic syndrome: is inflammation the underlying link? Biol Psychiatry. 2008;64:896–900. doi: 10.1016/j.biopsych.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Moranis A, Combe N, Cousson-Gelie F, Fuchs D, De Smedt-Peyrusse V, Barberger-Gateau P, Laye S. Vitamin E status and quality of life in the elderly: influence of inflammatory processes. Br J Nutr. 2009;102:1390–1394. doi: 10.1017/S0007114509990493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Poitou C, Machaux-Tholliez D, Frochot V, Bouillot JL, Basdevant A, Laye S, Clement K. Relationship between adiposity, emotional status and eating behaviour in obese women: role of inflammation. Psychol Med. 2010 Oct;20:1–12. doi: 10.1017/S0033291710001984. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Castanon N, Bluthe RM, Dantzer R. Chronic treatment with the atypical antidepressant tianeptine attenuates sickness behavior induced by peripheral but not central lipopolysaccharide and interleukin-1beta in the rat. Psychopharmacology (Berl) 2001;154:50–60. doi: 10.1007/s002130000595. [DOI] [PubMed] [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K, Viguerie N, Poitou C, Carette C, Pelloux V, Curat CA, Sicard A, Rome S, Benis A, Zucker JD, Vidal H, Laville M, Barsh GS, Basdevant A, Stich V, Cancello R, Langin D. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. Faseb J. 2004;18:1657–1669. doi: 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- Cope AP, Liblau RS, Yang XD, Congia M, Laudanna C, Schreiber RD, Probert L, Kollias G, McDevitt HO. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J Exp Med. 1997;185:1573–1584. doi: 10.1084/jem.185.9.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope AP, Londei M, Chu NR, Cohen SB, Elliott MJ, Brennan FM, Maini RN, Feldmann M. Chronic exposure to tumor necrosis factor (TNF) in vitro impairs the activation of T cells through the T cell receptor/CD3 complex; reversal in vivo by anti-TNF antibodies in patients with rheumatoid arthritis. J Clin Invest. 1994;94:749–760. doi: 10.1172/JCI117394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- D’Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211–221. doi: 10.1111/j.1749-6632.2009.05133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci U S A. 2007;104:1319–1324. doi: 10.1073/pnas.0610362104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry. 2008;65:409–415. doi: 10.1001/archpsyc.65.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R. Cytokine-induced sickness behavior: where do we stand? Brain Behav Immun. 2001;15:7–24. doi: 10.1006/brbi.2000.0613. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Bluthe RM, Laye S, Bret-Dibat JL, Parnet P, Kelley KW. Cytokines and sickness behavior. Ann N Y Acad Sci. 1998;840:586–590. doi: 10.1111/j.1749-6632.1998.tb09597.x. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Wollman EE, Vitkovic L, Yirmiya R. Cytokines, stress, and depression. Conclusions and perspectives. Adv Exp Med Biol. 1999;461:317–329. doi: 10.1007/978-0-585-37970-8_17. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- Delgado PL, Price LH, Miller HL, Salomon RM, Aghajanian GK, Heninger GR, Charney DS. Serotonin and the neurobiology of depression. Effects of tryptophan depletion in drug-free depressed patients. Arch Gen Psychiatry. 1994;51:865–874. doi: 10.1001/archpsyc.1994.03950110025005. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, Burke HM, Epel ES, Mellon SH, Rosser R, Reus VI, Wolkowitz OM. Low serum IL-10 concentrations and loss of regulatory association between IL-6 and IL-10 in adults with major depression. J Psychiatr Res. 2009;43:962–969. doi: 10.1016/j.jpsychires.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Dilger RN, Johnson RW. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol. 2008;84:932–939. doi: 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Wang J. Cytokine effects on CNS biogenic amines. Neuroimmunomodulation. 1995;2:319–328. doi: 10.1159/000097211. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Wang J, Ando T. Effects of cytokines on cerebral neurotransmission. Comparison with the effects of stress. Adv Exp Med Biol. 1999;461:117–127. doi: 10.1007/978-0-585-37970-8_8. [DOI] [PubMed] [Google Scholar]
- Eilat E, Mendlovic S, Doron A, Zakuth V, Spirer Z. Increased apoptosis in patients with major depression: A preliminary study. J Immunol. 1999;163:533–534. [PubMed] [Google Scholar]
- Eisenberger NI, Lieberman MD. Why rejection hurts: a common neural alarm system for physical and social pain. Trends Cogn Sci. 2004;8:294–300. doi: 10.1016/j.tics.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Emery CF, Fondow MD, Schneider CM, Christofi FL, Hunt C, Busby AK, Needleman BJ, Melvin WS, Elsayed-Awad HM. Gastric bypass surgery is associated with reduced inflammation and less depression: a preliminary investigation. Obes Surg. 2007;17:759–763. doi: 10.1007/s11695-007-9140-0. [DOI] [PubMed] [Google Scholar]
- Evans DL, Charney DS, Lewis L, Golden RN, Gorman JM, Krishnan KR, Nemeroff CB, Bremner JD, Carney RM, Coyne JC, Delong MR, Frasure-Smith N, Glassman AH, Gold PW, Grant I, Gwyther L, Ironson G, Johnson RL, Kanner AM, Katon WJ, Kaufmann PG, Keefe FJ, Ketter T, Laughren TP, Leserman J, Lyketsos CG, McDonald WM, McEwen BS, Miller AH, Musselman D, O’Connor C, Petitto JM, Pollock BG, Robinson RG, Roose SP, Rowland J, Sheline Y, Sheps DS, Simon G, Spiegel D, Stunkard A, Sunderland T, Tibbits P, Jr, Valvo WJ. Mood disorders in the medically ill: scientific review and recommendations. Biol Psychiatry. 2005;58:175–189. doi: 10.1016/j.biopsych.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Fagiolo U, Cossarizza A, Santacaterina S, Ortolani C, Monti D, Paganelli R, Franceschi C. Increased cytokine production by peripheral blood mononuclear cells from healthy elderly people. Ann N Y Acad Sci. 1992;663:490–493. doi: 10.1111/j.1749-6632.1992.tb38712.x. [DOI] [PubMed] [Google Scholar]
- Fagiolo U, Cossarizza A, Scala E, Fanales-Belasio E, Ortolani C, Cozzi E, Monti D, Franceschi C, Paganelli R. Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol. 1993;23:2375–2378. doi: 10.1002/eji.1830230950. [DOI] [PubMed] [Google Scholar]
- Floyd RA. Neuroinflammatory processes are important in neurodegenerative diseases: an hypothesis to explain the increased formation of reactive oxygen and nitrogen species as major factors involved in neurodegenerative disease development. Free Radic Biol Med. 1999;26:1346–1355. doi: 10.1016/s0891-5849(98)00293-7. [DOI] [PubMed] [Google Scholar]
- Forsythe LK, Wallace JM, Livingstone MB. Obesity and inflammation: the effects of weight loss. Nutr Res Rev. 2008;21:117–133. doi: 10.1017/S0954422408138732. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Frenois F, Moreau M, O’Connor J, Lawson M, Micon C, Lestage J, Kelley KW, Dantzer R, Castanon N. Lipopolysaccharide induces delayed FosB/DeltaFosB immunostaining within the mouse extended amygdala, hippocampus and hypothalamus, that parallel the expression of depressive-like behavior. Psychoneuroendocrinology. 2007;32:516–531. doi: 10.1016/j.psyneuen.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H, Seishima M. The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem. 2006;139:655–662. doi: 10.1093/jb/mvj072. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. Faseb J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol Allergy Clin North Am. 2009;29:321–337. doi: 10.1016/j.iac.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, Yirmiya R. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry. 2008;13:717–728. doi: 10.1038/sj.mp.4002055. [DOI] [PubMed] [Google Scholar]
- Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology. 2010;151:1622–1632. doi: 10.1210/en.2009-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–397. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- Haas HS, Schauenstein K. Neuroimmunomodulation via limbic structures--the neuroanatomy of psychoimmunology. Prog Neurobiol. 1997;51:195–222. doi: 10.1016/s0301-0082(96)00055-x. [DOI] [PubMed] [Google Scholar]
- Hamidi M, Drevets WC, Price JL. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol Psychiatry. 2004;55:563–569. doi: 10.1016/j.biopsych.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Harbuz MS, Chover-Gonzalez AJ, Jessop DS. Hypothalamo-pituitary-adrenal axis and chronic immune activation. Ann N Y Acad Sci. 2003;(992):99–106. doi: 10.1111/j.1749-6632.2003.tb03141.x. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry. 2009a;66:407–414. doi: 10.1016/j.biopsych.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Dolan RJ, Critchley HD. Neural origins of human sickness in interoceptive responses to inflammation. Biol Psychiatry. 2009b;66:415–422. doi: 10.1016/j.biopsych.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickie I, Hickie C, Lloyd A, Silove D, Wakefield D. Impaired in vivo immune responses in patients with melancholia. Br J Psychiatry. 1993;162:651–657. doi: 10.1192/bjp.162.5.651. [DOI] [PubMed] [Google Scholar]
- Himmerich H, Milenovic S, Fulda S, Plumakers B, Sheldrick AJ, Michel TM, Kircher T, Rink L. Regulatory T cells increased while IL-1beta decreased during antidepressant therapy. J Psychiatr Res. 2010;44:1052–7. doi: 10.1016/j.jpsychires.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71:171–186. doi: 10.1097/PSY.0b013e3181907c1b. [DOI] [PubMed] [Google Scholar]
- Ida T, Hara M, Nakamura Y, Kozaki S, Tsunoda S, Ihara H. Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci Lett. 2008;432:232–236. doi: 10.1016/j.neulet.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Irwin MR, Gillin JC. Impaired natural killer cell activity among depressed patients. Psychiatry Res. 1987;20:181–182. doi: 10.1016/0165-1781(87)90010-2. [DOI] [PubMed] [Google Scholar]
- Irwin M, Costlow C, Williams H, Artin KH, Chan CY, Stinson DL, Levin MJ, Hayward AR, Oxman MN. Cellular immunity to varicella-zoster virus in patients with major depression. J Infect Dis. 1998;178(Suppl 1):S104–108. doi: 10.1086/514272. [DOI] [PubMed] [Google Scholar]
- Irwin MR, Miller AH. Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun. 2007;21:374–383. doi: 10.1016/j.bbi.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Irwin MR. Human psychoneuroimmunology: 20 years of discovery. Brain Behav Immun. 2008;22:129–139. doi: 10.1016/j.bbi.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Ivanova SA, Semke VY, Vetlugina TP, Rakitina NM, Kudyakova TA, Simutkin GG. Signs of apoptosis of immunocompetent cells in patients with depression. Neurosci Behav Physiol. 2007;37:527–530. doi: 10.1007/s11055-007-0047-y. [DOI] [PubMed] [Google Scholar]
- Jehn CF, Kuehnhardt D, Bartholomae A, Pfeiffer S, Krebs M, Regierer AC, Schmid P, Possinger K, Flath BC. Biomarkers of depression in cancer patients. Cancer. 2006;107:2723–2729. doi: 10.1002/cncr.22294. [DOI] [PubMed] [Google Scholar]
- Johnson BA, 3rd, Baban B, Mellor AL. Targeting the immunoregulatory indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy. 2009;1:645–661. doi: 10.2217/IMT.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent S, Bluthe RM, Kelley KW, Dantzer R. Sickness behavior as a new target for drug development. Trends Pharmacol Sci. 1992;13:24–28. doi: 10.1016/0165-6147(92)90012-u. [DOI] [PubMed] [Google Scholar]
- Kim YK, Na KS, Shin KH, Jung HY, Choi SH, Kim JB. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1044–1053. doi: 10.1016/j.pnpbp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Kitagami T, Yamada K, Miura H, Hashimoto R, Nabeshima T, Ohta T. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. 2003;978:104–114. doi: 10.1016/s0006-8993(03)02776-8. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25:154–159. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. 2008;105:751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronfol Z, Silva J, Jr, Greden J, Dembinski S, Gardner R, Carroll B. Impaired lymphocyte function in depressive illness. Life Sci. 1983;33:241–247. doi: 10.1016/0024-3205(83)90382-x. [DOI] [PubMed] [Google Scholar]
- Kronfol Z, House JD, Silva J, Jr, Greden J, Carroll BJ. Depression, urinary free cortisol excretion and lymphocyte function. Br J Psychiatry. 1986;148:70–73. doi: 10.1192/bjp.148.1.70. [DOI] [PubMed] [Google Scholar]
- Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22:370–379. doi: 10.1016/S0893-133X(99)00134-7. [DOI] [PubMed] [Google Scholar]
- Laye S. Polyunsaturated fatty acids, neuroinflammation and well being. Prostaglandins Leukot Essent Fatty Acids. 2010;82:295–303. doi: 10.1016/j.plefa.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Lee LF, Lih CJ, Huang CJ, Cao T, Cohen SN, McDevitt HO. Genomic expression profiling of TNF-alpha-treated BDC2.5 diabetogenic CD4+ T cells. Proc Natl Acad Sci U S A. 2008;105:10107–10112. doi: 10.1073/pnas.0803336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ramenaden ER, Peng J, Koito H, Volpe JJ, Rosenberg PA. Tumor necrosis factor alpha mediates lipopolysaccharide-induced microglial toxicity to developing oligodendrocytes when astrocytes are present. J Neurosci. 2008;28:5321–5330. doi: 10.1523/JNEUROSCI.3995-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010a;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xiao B, Qiu W, Yang L, Hu B, Tian X, Yang H. Altered expression of CD4(+)CD25(+) regulatory T cells and its 5-HT(1a) receptor in patients with major depression disorder. J Affect Disord. 2010b;124:68–75. doi: 10.1016/j.jad.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Lotrich FE, Ferrell RE, Rabinovitz M, Pollock BG. Risk for depression during interferon-alpha treatment is affected by the serotonin transporter polymorphism. Biol Psychiatry. 2009;65:344–348. doi: 10.1016/j.biopsych.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano AM, Mayberg HS, Giacobbe P, Hamani C, Craddock RC, Kennedy SH. Subcallosal cingulate gyrus deep brain stimulation for treatment-resistant depression. Biol Psychiatry. 2008;64:461–467. doi: 10.1016/j.biopsych.2008.05.034. [DOI] [PubMed] [Google Scholar]
- Luheshi GN, Bluthe RM, Rushforth D, Mulcahy N, Konsman JP, Goldbach M, Dantzer R. Vagotomy attenuates the behavioural but not the pyrogenic effects of interleukin-1 in rats. Auton Neurosci. 2000;85:127–132. doi: 10.1016/S1566-0702(00)00231-9. [DOI] [PubMed] [Google Scholar]
- Lutgendorf SK, Lamkin DM, DeGeest K, Anderson B, Dao M, McGinn S, Zimmerman B, Maiseri H, Sood AK, Lubaroff DM. Depressed and anxious mood and T-cell cytokine expressing populations in ovarian cancer patients. Brain Behav Immun. 2008;22:890–900. doi: 10.1016/j.bbi.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M, Scharpe S, Meltzer HY, Bosmans E, Suy E, Calabrese J, Cosyns P. Relationships between interleukin-6 activity, acute phase proteins, and function of the hypothalamic-pituitary-adrenal axis in severe depression. Psychiatry Res. 1993;49:11–27. doi: 10.1016/0165-1781(93)90027-e. [DOI] [PubMed] [Google Scholar]
- Majer M, Welberg LA, Capuron L, Pagnoni G, Raison CL, Miller AH. IFN-alpha-induced motor slowing is associated with increased depression and fatigue in patients with chronic hepatitis C. Brain Behav Immun. 2008;22:870–880. doi: 10.1016/j.bbi.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews K, Schwartz J, Cohen S, Seeman T. Diurnal cortisol decline is related to coronary calcification: CARDIA study. Psychosom Med. 2006;68:657–661. doi: 10.1097/01.psy.0000244071.42939.0e. [DOI] [PubMed] [Google Scholar]
- Matute C, Domercq M, Sanchez-Gomez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia. 2006;53:212–224. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- McNally L, Bhagwagar Z, Hannestad J. Inflammation, glutamate, and glia in depression: a literature review. CNS Spectr. 2008;13:501–510. doi: 10.1017/s1092852900016734. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem. 2008;107:1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer. 2005;104:788–793. doi: 10.1002/cncr.21234. [DOI] [PubMed] [Google Scholar]
- Miller AH. Mechanisms of cytokine-induced behavioral changes: Psychoneuroimmunology at the translational interface. Brain Behav Immun. 2008;23:149–58. doi: 10.1016/j.bbi.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau M, Andre C, O’Connor JC, Dumich SA, Woods JA, Kelley KW, Dantzer R, Lestage J, Castanon N. Inoculation of Bacillus Calmette-Guerin to mice induces an acute episode of sickness behavior followed by chronic depressive-like behavior. Brain Behav Immun. 2008;22:1087–1095. doi: 10.1016/j.bbi.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moron JA, Zakharova I, Ferrer JV, Merrill GA, Hope B, Lafer EM, Lin ZC, Wang JB, Javitch JA, Galli A, Shippenberg TS. Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J Neurosci. 2003;23:8480–8488. doi: 10.1523/JNEUROSCI.23-24-08480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12:988–1000. doi: 10.1038/sj.mp.4002006. [DOI] [PubMed] [Google Scholar]
- Muller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B, Spellmann I, Hetzel G, Maino K, Kleindienst N, Moller HJ, Arolt V, Riedel M. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11:680–684. doi: 10.1038/sj.mp.4001805. [DOI] [PubMed] [Google Scholar]
- Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS, Greiner K, Nemeroff CB, Miller AH. Paroxetine for the prevention of depression induced by high-dose interferon alfa. N Engl J Med. 2001;344:961–966. doi: 10.1056/NEJM200103293441303. [DOI] [PubMed] [Google Scholar]
- Myint AM, Leonard BE, Steinbusch HW, Kim YK. Th1, Th2, and Th3 cytokine alterations in major depression. J Affect Disord. 2005;88:167–173. doi: 10.1016/j.jad.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Heim CM, Thase ME, Klein DN, Rush AJ, Schatzberg AF, Ninan PT, McCullough JP, Jr, Weiss PM, Dunner DL, Rothbaum BO, Kornstein S, Keitner G, Keller MB. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci U S A. 2003;100:14293–14296. doi: 10.1073/pnas.2336126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 2009a;14:511–22. doi: 10.1038/sj.mp.4002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JC, Lawson MA, Andre C, Briley EM, Szegedi SS, Lestage J, Castanon N, Herkenham M, Dantzer R, Kelley KW. Induction of IDO by bacille Calmette-Guerin is responsible for development of murine depressive-like behavior. J Immunol. 2009b;182:3202–3212. doi: 10.4049/jimmunol.0802722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TW, Miller AH. Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann N Y Acad Sci. 2009;1179:86–105. doi: 10.1111/j.1749-6632.2009.04984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Pavon L, Sandoval-Lopez G, Eugenia Hernandez M, Loria F, Estrada I, Perez M, Moreno J, Avila U, Leff P, Anton B, Heinze G. Th2 cytokine response in major depressive disorder patients before treatment. J Neuroimmunol. 2006;172:156–165. doi: 10.1016/j.jneuroim.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. 2010;219:25–32. doi: 10.1016/j.jneuroim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt D, Nagelmeier IE, Wilson HC, Raine CS. Glutamate uptake by oligodendrocytes: Implications for excitotoxicity in multiple sclerosis. Neurology. 2003;61:1113–1120. doi: 10.1212/01.wnl.0000090564.88719.37. [DOI] [PubMed] [Google Scholar]
- Plotkin SR, Banks WA, Kastin AJ. Comparison of saturable transport and extracellular pathways in the passage of interleukin-1 alpha across the blood-brain barrier. J Neuroimmunol. 1996;67:41–47. doi: 10.1016/0165-5728(96)00036-7. [DOI] [PubMed] [Google Scholar]
- Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun. 2007;21:727–735. doi: 10.1016/j.bbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160:1554–1565. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- Raison CL, Borisov AS, Broadwell SD, Capuron L, Woolwine BJ, Jacobson IM, Nemeroff CB, Miller AH. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66:41–48. doi: 10.4088/jcp.v66n0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Woolwine BJ, Demetrashvili MF, Borisov AS, Weinreib R, Staab JP, Zajecka JM, Bruno CJ, Henderson MA, Reinus JF, Evans DL, Asnis GM, Miller AH. Paroxetine for prevention of depressive symptoms induced by interferon-alpha and ribavirin for hepatitis C. Aliment Pharmacol Ther. 2007;25:1163–1174. doi: 10.1111/j.1365-2036.2007.03316.x. [DOI] [PubMed] [Google Scholar]
- Raison CL, Borisov AS, Majer M, Drake DF, Pagnoni G, Woolwine BJ, Vogt GJ, Massung B, Miller AH. Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry. 2009;65:296–303. doi: 10.1016/j.biopsych.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Borisov AS, Woolwine BJ, Massung B, Vogt G, Miller AH. Interferon-alpha effects on diurnal hypothalamic-pituitary-adrenal axis activity: relationship with proinflammatory cytokines and behavior. Mol Psychiatry. 2010a;15:535–547. doi: 10.1038/mp.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, Spivey JR, Saito K, Miller AH. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry. 2010b;15:393–403. doi: 10.1038/mp.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ. Gliogenesis and glial pathology in depression. CNS Neurol Disord Drug Targets. 2007;6:219–233. doi: 10.2174/187152707780619326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios C, Santamaria A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem Res. 1991;16:1139–1143. doi: 10.1007/BF00966592. [DOI] [PubMed] [Google Scholar]
- Rivest S, Lacroix S, Vallieres L, Nadeau S, Zhang J, Laflamme N. How the blood talks to the brain parenchyma and the paraventricular nucleus of the hypothalamus during systemic inflammatory and infectious stimuli. Proc Soc Exp Biol Med. 2000;223:22–38. doi: 10.1046/j.1525-1373.2000.22304.x. [DOI] [PubMed] [Google Scholar]
- Roitt I, Bostoff J, Male D. Immunology. 5. Mosby; New York: 1998. [Google Scholar]
- Rothwell NJ, Luheshi G, Toulmond S. Cytokines and their receptors in the central nervous system: physiology, pharmacology, and pathology. Pharmacol Ther. 1996;69:85–95. doi: 10.1016/0163-7258(95)02033-0. [DOI] [PubMed] [Google Scholar]
- Sanchez MM, Alagbe O, Felger JC, Zhang J, Graff AE, Grand AP, Maestripieri D, Miller AH. Activated p38 MAPK is associated with decreased CSF 5-HIAA and increased maternal rejection during infancy in rhesus monkeys. Mol Psychiatry. 2007;12:895–897. doi: 10.1038/sj.mp.4002025. [DOI] [PubMed] [Google Scholar]
- Schleifer SJ, Keller SE, Camerino M, Thornton JC, Stein M. Suppression of lymphocyte stimulation following bereavement. Jama. 1983;250:374–377. [PubMed] [Google Scholar]
- Schleifer SJ, Keller SE, Meyerson AT, Raskin MJ, Davis KL, Stein M. Lymphocyte function in major depressive disorder. Arch Gen Psychiatry. 1984;41:484–486. doi: 10.1001/archpsyc.1984.01790160070008. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303:1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- Schwarz MJ, Chiang S, Muller N, Ackenheil M. T-helper-1 and T-helper-2 responses in psychiatric disorders. Brain Behav Immun. 2001;15:340–370. doi: 10.1006/brbi.2001.0647. [DOI] [PubMed] [Google Scholar]
- Sephton SE, Sapolsky RM, Kraemer HC, Spiegel D. Diurnal cortisol rhythm as a predictor of breast cancer survival. J Natl Cancer Inst. 2000;92:994–1000. doi: 10.1093/jnci/92.12.994. [DOI] [PubMed] [Google Scholar]
- Sephton SE, Dhabhar FS, Keuroghlian AS, Giese-Davis J, McEwen BS, Ionan AC, Spiegel D. Depression, cortisol, and suppressed cell-mediated immunity in metastatic breast cancer. Brain Behav Immun. 2009;23:1148–55. doi: 10.1016/j.bbi.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Slavich GM, Way BM, Eisenberger NI, Taylor SE. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc Natl Acad Sci U S A. 2010;107:14817–14822. doi: 10.1073/pnas.1009164107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluzewska A, Sobieska M, Rybakowski JK. Changes in acute-phase proteins during lithium potentiation of antidepressants in refractory depression. Neuropsychobiology. 1997;35:123–127. doi: 10.1159/000119332. [DOI] [PubMed] [Google Scholar]
- Song C, Halbreich U, Han C, Leonard BE, Luo H. Imbalance between pro- and anti-inflammatory cytokines and between Th1 and Th2 cytokines in depressed patients: the effect of electroacupuncture or fluoxetine treatment. Pharmacopsychiatry. 2009;42:182–188. doi: 10.1055/s-0029-1202263. [DOI] [PubMed] [Google Scholar]
- Spiegel D, Giese-Davis J, Taylor CB, Kraemer H. Stress sensitivity in metastatic breast cancer: analysis of hypothalamic-pituitary-adrenal axis function. Psychoneuroendocrinology. 31:1231–1244. doi: 10.1016/j.psyneuen.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Miller AH, Trestman RL. Depression, the immune system, and health and illness. Findings in search of meaning. Arch Gen Psychiatry. 1991;48:171–177. doi: 10.1001/archpsyc.1991.01810260079012. [DOI] [PubMed] [Google Scholar]
- Su KP, Huang SY, Peng CY, Lai HC, Huang CL, Chen YC, Aitchison KJ, Pariante CM. Phospholipase A2 and cyclooxygenase 2 genes influence the risk of interferon-alpha-induced depression by regulating polyunsaturated fatty acids levels. Biol Psychiatry. 2010;67:550–557. doi: 10.1016/j.biopsych.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcigil L, Oktenli C, Musabak U, Bozkurt A, Cansever A, Uzun O, Sanisoglu SY, Yesilova Z, Ozmenler N, Ozsahin A, Sengul A. Pro- and anti-inflammatory cytokine balance in major depression: effect of sertraline therapy. Clin Dev Immunol. 2007;2007:76396. doi: 10.1155/2007/76396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweat V, Starr V, Bruehl H, Arentoft A, Tirsi A, Javier E, Convit A. C-reactive protein is linked to lower cognitive performance in overweight and obese women. Inflammation. 2008;31:198–207. doi: 10.1007/s10753-008-9065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuster-Ciesielska A, Slotwinska M, Stachura A, Marmurowska-Michalowska H, Dubas-Slemp H, Bojarska-Junak A, Kandefer-Szerszen M. Accelerated apoptosis of blood leukocytes and oxidative stress in blood of patients with major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:686–694. doi: 10.1016/j.pnpbp.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Taylor JL, Grossberg SE. The effects of interferon-alpha on the production and action of other cytokines. Semin Oncol. 1998;25:23–29. [PubMed] [Google Scholar]
- Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler JP, Schwartz MW. Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010;151:4109–4115. doi: 10.1210/en.2010-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton P, Pinteaux E, Gibson RM, Allan SM, Rothwell NJ. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J Neurochem. 2006;98:258–266. doi: 10.1111/j.1471-4159.2006.03872.x. [DOI] [PubMed] [Google Scholar]
- Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85:2059–2070. doi: 10.1002/jnr.21325. [DOI] [PubMed] [Google Scholar]
- Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, Lalla D, Woolley M, Jahreis A, Zitnik R, Cella D, Krishnan R. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29–35. doi: 10.1016/S0140-6736(05)67763-X. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, Martin D, Maier SF. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neurosci Lett. 1995;183:27–31. doi: 10.1016/0304-3940(94)11105-r. [DOI] [PubMed] [Google Scholar]
- Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Horl WH, Hengstschlager M, Muller M, Saemann MD. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112:1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes M. IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry. 2005;10:538–544. doi: 10.1038/sj.mp.4001600. [DOI] [PubMed] [Google Scholar]
- Wichers MC, Kenis G, Koek GH, Robaeys G, Nicolson NA, Maes M. Interferon-alpha-induced depressive symptoms are related to changes in the cytokine network but not to cortisol. J Psychosom Res. 2007;62:207–214. doi: 10.1016/j.jpsychores.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- Wong ML, Dong C, Maestre-Mesa J, Licinio J. Polymorphisms in inflammation-related genes are associated with susceptibility to major depression and antidepressant response. Mol Psychiatry. 2008;13:800–812. doi: 10.1038/mp.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HQ, Rassoulpour A, Schwarcz R. Kynurenic acid leads, dopamine follows: a new case of volume transmission in the brain? J Neural Transm. 2007;114:33–41. doi: 10.1007/s00702-006-0562-y. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, Tylavsky FA, Newman AB. The metabolic syndrome, inflammation, and risk of cognitive decline. Jama. 2004;292:2237–2242. doi: 10.1001/jama.292.18.2237. [DOI] [PubMed] [Google Scholar]
- Ye SM, Johnson RW. Increased interleukin-6 expression by microglia from brain of aged mice. J Neuroimmunol. 1999;93:139–148. doi: 10.1016/s0165-5728(98)00217-3. [DOI] [PubMed] [Google Scholar]
- Yirmiya R. Endotoxin produces a depressive-like episode in rats. Brain Res. 1996;711:163–174. doi: 10.1016/0006-8993(95)01415-2. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Weidenfeld J, Pollak Y, Morag M, Morag A, Avitsur R, Barak O, Reichenberg A, Cohen E, Shavit Y, Ovadia H. Cytokines, “depression due to a general medical condition,” and antidepressant drugs. Adv Exp Med Biol. 1999;461:283–316. doi: 10.1007/978-0-585-37970-8_16. [DOI] [PubMed] [Google Scholar]
- You T, Sonntag WE, Leng X, Carter CS. Lifelong caloric restriction and interleukin-6 secretion from adipose tissue: effects on physical performance decline in aged rats. J Gerontol A Biol Sci Med Sci. 2007;62:1082–1087. doi: 10.1093/gerona/62.10.1082. [DOI] [PubMed] [Google Scholar]
- Zhang X, Dong F, Ren J, Driscoll MJ, Culver B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol. 2005;191:318–325. doi: 10.1016/j.expneurol.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA, Blakely RD. p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J Biol Chem. 2005;280:15649–15658. doi: 10.1074/jbc.M410858200. [DOI] [PubMed] [Google Scholar]
- Zhu CB, Blakely RD, Hewlett WA. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology. 2006;31:2121–2131. doi: 10.1038/sj.npp.1301029. [DOI] [PubMed] [Google Scholar]
- Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA. Interleukin-1 Receptor Activation by Systemic Lipopolysaccharide Induces Behavioral Despair Linked to MAPK Regulation of CNS Serotonin Transporters. Neuropsychopharmacology. 2010 Sep 8; doi: 10.1038/npp.2010.116. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielasek J, Hartung HP. Molecular mechanisms of microglial activation. Adv Neuroimmunol. 1996;6:191–122. doi: 10.1016/0960-5428(96)00017-4. [DOI] [PubMed] [Google Scholar]
- Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A, McCorkle R, Seligman DA, Schmidt K. The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun. 2001;15:199–226. doi: 10.1006/brbi.2000.0597. [DOI] [PubMed] [Google Scholar]