

Abstract
Background
The TlyA protein has a controversial function as a virulence factor in Mycobacterium tuberculosis (M. tuberculosis). At present, its dual activity as hemolysin and RNA methyltransferase in M. tuberculosis has been indirectly proposed based on in vitro results. There is no evidence however for TlyA relevance in the survival of tubercle bacilli inside host cells or whether both activities are functionally linked. A thorough analysis of structure prediction for this mycobacterial protein in this study shows the need for reevaluating TlyA's function in virulence.
Results
Bioinformatics analysis of TlyA identified a ribosomal protein binding domain (S4 domain), located between residues 5 and 68 as well as an FtsJ-like methyltranferase domain encompassing residues 62 and 247, all of which have been previously described in translation machinery-associated proteins. Subcellular localization prediction showed that TlyA lacks a signal peptide and its hydrophobicity profile showed no evidence of transmembrane helices. These findings suggested that it may not be attached to the membrane, which is consistent with a cytoplasmic localization. Three-dimensional modeling of TlyA showed a consensus structure, having a common core formed by a six-stranded β-sheet between two α-helix layers, which is consistent with an RNA methyltransferase structure. Phylogenetic analyses showed high conservation of the tlyA gene among Mycobacterium species. Additionally, the nucleotide substitution rates suggested purifying selection during tlyA gene evolution and the absence of a common ancestor between TlyA proteins and bacterial pore-forming proteins.
Conclusion
Altogether, our manual in silico curation suggested that TlyA is involved in ribosomal biogenesis and that there is a functional annotation error regarding this protein family in several microbial and plant genomes, including the M. tuberculosis genome.
Background
Tuberculosis (TB) is an infectious disease that mainly afflicts populations in third-world countries. Although most infected people will never develop an active form of the disease, the global TB death rate is substantial, being around two million people per year. The situation has been further exacerbated by the emergence and spread of extremely resistant Mycobacterium tuberculosis strains (XDR) [1] which, together with the lack of effective antibiotics, urges the development of new alternatives for controlling this worldwide public health threat. Consequently, most research efforts have focused on understanding M. tuberculosis biology with the aim of identifying new therapeutic and vaccine targets.
Current management of TB cases consists of a two-month intensive treatment comprising first-line drugs such as Rifampicin (RIF), Isoniazid (INH), Pyrazinamide (PZA) and Ethambutol (EMB)/Streptomycin (SM) which seeks to ensure that mono-resistant strains do not proliferate [2]. When RIF and INH are ineffective, standard treatment guidelines recommend a combination of second-line drugs [3]. Among these therapeutic choices, aminoglycosides such as Capreomycin (CMN) and Viomycin (VMN) are given in combination with other antibiotics to treat multidrug-resistant strains (MDR); they are also effective against non-replicating M. tuberculosis strains, as well as being useful in the treatment of latent TB infections [4]. CMN- and VMN-resistant bacilli are classified as XDR-TB strains and are a cause of major concern since these drugs have more toxic side effects and might result in higher death rates, especially among HIV-infected persons [1].
Controversially, M. tuberculosis tlyA gene product has been annotated both as a virulence factor, due to its ability to lyse red blood cells, as well as a protein involved in ribosome biogenesis [5,6].
TlyA has been assumed to induce hemolytic activity due to its high amino acid similarity (37.3%) to the pore-forming hemolysin/cytotoxin virulence determinant from Serpulina hyodysenteriae. Notably such activity was only experimentally demonstrated with bacterial lysates [6] and another study has even suggested that TlyA could be associated with hemolysin expression in Escherichia coli [7,8]. Additionally, recent study showed that a concentration-dependent hemolysis of rabbit and human erythrocytes to be induced on incubation with the recombinant Rv1694 protein (the putative M. tuberculosis TlyA) when over-expressed in E. coli [8]. The article reported hemolysis inhibition using specific antiserum against the same protein as well as the presence of the recombinant protein on E. coli cell wall; these data were supportive of TlyA's hemolytic property, combined with a preliminary in silico analysis. Rv1694 oligomers were also observed on lysed erythrocyte membranes, as well as the susceptibility of Rv1694-expressing E. coli to CMN [8]. Despite such thorough in vitro analysis in E. coli, the functional activities suggested for Rv1694 have still not been clearly demonstrated when using M. tuberculosis mutant strains.
By contrast with the proposed hemolytic function, Johansen et al., [5] have reported that M. tuberculosis H37Rv and Beijing TlyA confer susceptibility to CMN and VMN. These antibiotics are structurally similar and share the same mechanism of action, both inhibiting bacterial proliferation by blocking the peptidyl transferase reaction. VMN hinders A-site and P-site tRNA positioning on the 50S ribosomal subunit, therefore hampering mRNA translation [9]. CMN resistance arises due to mutations in the tlyA gene which encodes a RNA 2'-O-methyltransferase (2'-O-MTase) that methylates ribose moieties from nucleotide C1409 in 16S rRNA and C1920 in 23S rRNA. The absence of these rRNA methylations confers resistance against CMN and VMN given that binding sites for these antibiotics are delineated by the localization of such methylated riboses [5]. Additional evidence has been provided by mutant complementation, showing that CMN susceptibility is restored by introducing an active copy of the tlyA gene [10]. TlyA methylation clearly enhances CMN and VMN susceptibility and is responsible, at least in part, for their efficacy against mycobacterial infections as drug binding is impaired when the enzyme is not produced. Resistance thus emerges when TlyA activity is lost. These studies show the need for a reevaluation of TlyA's function in virulence [5,11].
Results
TlyA related sequences
The BLAST search for TlyA-related sequences in the TB and Genbank databases showed that although TlyA-related sequences are found mainly in prokaryotic organisms, homologous protein sequences are also present in eukaryotic organisms such as plants and algae (Figure 1A).
Figure 1.

Hemolysin A family (InterPro: IPR004538) taxonomic coverage of 864 proteins. (A). Taxonomy-tree nodes are placed on the inner circles and radial lines lead to the description for each node. Proteins annotated by taxonomic division are numbered [http://www.ebi.ac.uk/interpro/IEntry?ac=IPR004538]. (B). Multiple sequence alignment of TlyA protein homologs in Mycobacterium. Sequences are colored according to amino acid physicochemical properties; the catalytic tetrad is marked by black asterisks (K69-D154-K182-E238). Similarity values for each amino acid are represented by bars (pink). Organism names correspond to nomenclature found in the tuberculosis database [http://www.tbdb.org/] (CLC sequence viewer alignment representation).
These M. tuberculosis-related TlyA protein sequences are mainly classified as hemolysin A inside the FtsJ-like class based on domain similarity and have been annotated as such in Genbank, EMBL, UniProt and SWISS-PROT databases according to sequence similarity with Serpulina hyodysenteriae TlyA annotated as being a hemolysin. Paradoxically, although hemolysin A contains similar domain architecture to TlyA, it is predicted to be a rRNA methylase instead of a contact-dependent hemolysin (Table S1, Additional file 1).
TlyA amino acid sequences from M. tuberculosis H37Rv, H37Ra, CDC1551, M. bovis and M. bovis-BCG reference strain alignment showed a 100% identity with the Harleem, F11 and C M. tuberculosis strains (Figure 1B). The same conservation pattern was obtained when the corresponding nucleotide sequences were aligned (> 99% identity).
Similarity percentages found in the multiple amino acid alignment of Mycobacterium species different to M. tuberculosis, such as M. smegmatis (76%), M. abscessus (72%), M. leprae (78%), M. avium (79-80%) and M. vanbaalenii PYR-1 (76%), showed high TlyA conservation at genus level. A glutamine residue insertion was found at position 269 in the protein's C-terminal region in all the aforementioned species, while a 12-residue insertion was found only in M. ulcerans (81%) and M. marinum (81%), which are pathogenic mycobacteria (Figure 1B).
TlyA transcriptional unit revealed some rearrangements in the Mycobacterium IprJ-recN operon
From the synteny of the IprJ-recN operon from M. tuberculosis, M. bovis BCG, M. bovis, M. leprae, M. ulcerans and M. marinum (Figure 2) it was evident that almost all gene products, except for tlyA, are involved in intermediary metabolism, cell wall biosynthesis and signal transduction rather than virulence. Moreover, no trafficking proteins were found within the operon or near tlyA that could be suggestive of TlyA transportation either to the cell membrane or the extracellular milieu.
Figure 2.
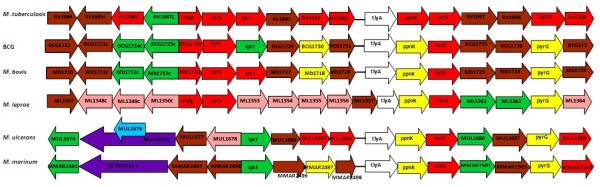
tlyA transcriptional unit gene syntheny. Genes are represented as arrows and are drawn according to transcriptional orientation and genome functional annotation. Arrow color representation for gene functional annotation is as follows: green for cell wall synthesis, red for signal transduction, brown for evolutionarily conserved, yellow for intermediary metabolism, white for tlyA, pink for pseudogenes, purple for PE/PPE and blue for IS/phages.
The IprJ-recN operon also had common gene organization in M. tuberculosis and M. bovis, while in M. leprae most genes have lost their function (i.e. have become pseudogenes). Such high rate of loss of function in genes involved in intermediary metabolism, cellular respiration and cell-wall biosynthesis may suggest that the loss of such genes confers an adaptive advantage on Mycobacterium during host cell infection or transmission [12]. No other genes reported to be involved in bacilli virulence were found within this operon.
TlyA three-dimensional structure
The three-dimensional structure of a putative hemolysin from Streptococcus thermophilus (PDB ID: 3HP7 chain A, at 1.53 Å resolution) was used as template for homology modeling. The obtained identity (37.2%, RMSD 0.44 Å, E-value 4.7e-26) suggested that the selected hemolysin structure was a suitable template for TlyA, and that the obtained modeled structure could reflect an experimentally-obtained M. tuberculosis TlyA structure.
Although this protein seemed to be a hemolysin, its domain architecture was also similar to RNA methyltransferase family, fibrillarin homologues and TlyA proteins. A consensus sequence was established which showed the presence of a common core, comprising six β-sheets, the first five of which were found parallel and the sixth anti-parallel between two layers of α-helices and random coiled regions. Such structural organization is commonly referred as to an AdoMet-dependent methyltransferase (MTase) fold [13,14].
TlyA has an overall globular fold and comprises two domains exhibiting different spatial arrangements. The smaller domain, named S4, is N-terminally located and is overlapped by the catalytic domain which exhibits structural similarity to various unrelated RNA-binding proteins, while the large catalytic domain, named FtsJ-like methyltransferase domain is common to 2'-O-MTases and exhibits a α/β fold with a deep pocket (Figure 3).
Figure 3.

Modeled spatial configuration built using UCSF Chimera. Ribbon (A) and space-filling (B) representations of TlyA catalytic tetrad (red), AdoMet binding motif GASTG90-94 (green) and S4 RNA binding domain (blue). Structural model was stored on Protein Model Database [PMDB: PM0076044, http://mi.caspur.it/PMDB/].
The S4 domain consists of 64 residues, starting at A-5 and ending at H-68. This domain is structurally formed by one β-sheet and three α-helices predominantly composed of positively charged residues (11 out of 47 residues) whose probable function is to mediate RNA binding and provide stability. In line with this, the RNABindR server identified three short sequences (1-MARRAR-6, 13-RRGLARSRQQ-22 and 31-KVR-33) inside the S4 domain that might be implicated in RNA binding (Figure 3). The S4 domain has been specifically identified in some bacterial and eukaryotic ribosomal proteins, pseudouridine synthases, RNA methylases, bacterial tyrosyl-tRNA synthetases and may also be involved in translation regulation [15].
The analysis of the protein's electrostatic surface showed that positively charged residues were localized towards the N-terminal extreme, forming bulges and long finger-like projections that probably extend into the rRNA core to stabilize its structure (Figure 4). In fact, charged residues like R and K exhibited the highest interface propensities, which is consistent with their ability to participate in interactions both with nucleotide bases and the negatively charged RNA phosphate backbone [16].
Figure 4.

Diagram of charge distribution on TlyA Connolly surface. Three-dimensional modeled structure according to positive (blue) and negative (red) charge distribution on the protein surface.
The FtsJ-like methyltransferase domain is located between residues 62 and 247 and includes the catalytic tetrad responsible for TlyA's enzymatic activity [14]. Moreover, the FtsJ-like methyltransferase domain exhibits a Rossmann fold composed of five parallel β-strand layers linked by two α-helices following an α/β secondary structure topological order [17].
Our comparative analysis of TlyA with the Fibrillarin family showed the presence of a GASTG AdoMet binding motif comprising residues 90-94 and a D located at position 112 juxtaposed to the above mentioned motif (Figure 3A), which might stabilize TlyA coenzyme binding [13]. AdoMet was located between β-strands 4 and 5 within the inner depression inside the catalytic pocket formed by GASTG residues' main-chain NH groups (Figure 3B). Moreover, substrate positioning might be driven by the Rossmann configuration fold and the pertinent AdoMet-binding/catalytic sites, suggesting substrate selectivity in the small ribosomal subunit [17]. Likewise, domain structure was in agreement with S-adenosyl-methionine (AdoMet)-dependent methyltransferases. AdoMet binding and affinity might indicate how an enzyme such as TlyA may methylate the two adjacent ribose rings on both ribosomal subunits. In this reaction S-adenosyl-methionine (SAM) acts as methyl donor, as it is converted into S-adenosyl-l-homocysteine (SAH); TlyA could thus be also classified as being an RNA-AdoMet-MTase class I [18].
This hypothesis is supported by the high structural similarity (98.1%) between TlyA and FtsJ RNA-MTase complexed with AdoMet (ligand-binding template PDB entry 1EJ0) (RMSD 2.98, Similarity score: 368.77 E-value: 1.30E-06) (Figure S1, Additional file 2). Two expected structural motifs were also identified in this structure which was formed by six buried residues between G-90 and F-96 which are pivotal for AdoMet interaction and therefore conserved in Mycobacterium TlyA proteins (Table 1).
Table 1.
Nest analysis
| Score | Residue range | Residue | Ramachandran region | Solvent accessibility | Cleft | Depth in cleft | Residue conservation |
|---|---|---|---|---|---|---|---|
| 5.50 | Thr93-Phe96 | Thr93 | RIGHT | 0.00% | 1 | 14.74 | 1.00 |
| Gly94 | LEFT | 0.22% | 1 | 15.55 | 1.00 | ||
| Gly95 | RIGHT | 2.72% | 1 | 16.88 | 1.00 | ||
| Phe96 | RIGHT | 1.03% | 1 | 19.99 | 1.00 | ||
| 4.67 | Gly90-Ser92 | Gly90 | LEFT | 4.04% | 1 | 14.84 | 1.00 |
| Ala91 | RIGHT | 0.00% | - | - | 1.00 | ||
| Ser92 | - | 0.00% | 1 | 18.32 | 1.00 |
Nest analysis of two structural motifs detected in TlyA functionally important regions for AdoMet binding region. Results were obtained on Profunc server [43].
The TlyA model was validated by using the RAMPAGE server which evaluates a protein's 3D-structure based on the stereochemistry quality of torsion angles and geometry [19]. The Ramachandran plot showed 95.1% feasibility (253 aa) in favorable positions and 3.8% (10 residues) in acceptable regions, with just 1.1% in an outlier region (3 residues), supporting the high quality of the 3D-model (Figure S2, Additional file 3).
This in silico structure analysis of M. tuberculosis proteins allowed a deeper understanding of TlyA's function and challenges the initially described function of TlyA-related proteins. Rather than its hypothetical function as a hemolysin, our data suggested functions as an MTase based on in silico prediction.
Predicting subcellular localization
Subcellular localization was predicted with TBpred [http://www.imtech.res.in/raghava/tbpred/] and the results suggested TlyA localization in the cytoplasmatic compartment (Table 2), also supporting the fact that TlyA acts as a methyltransferase [5]. A recent mycobacterial protein analysis led to ascertaining several predictors' reliability for subcellular localization, providing highly accurate results for Gpos-PLoc and PA-SUB v.2.5 and SignalP 2.0 as a confirmatory tool [20]. However, in this study, PA-SUB v.2.5 predicted TlyA as being an extracellular protein (72.84%), which could have been biased as the training strategy used involved results from the SWISS-PROT [21] database where TlyA is mis-annotated. Likewise, in agreement with BLAST scores for the query sequence (Rv1694), 26 out of 28 homologous proteins were designated as putative ribosomal RNA methyltransferases and RNA binding proteins. Only one submitted sequence seemed to be similar to hemolysin A (the S. hyodysenteriae sequence).
Table 2.
TlyA subcellular localization prediction through 9 available servers for prokaryotic proteins
| Tool | Subcellular localization | Value | Reliability Index | Method | Overall prediction accuracy | Ref. |
|---|---|---|---|---|---|---|
| TBpred | Cytoplasmic | 1.365 | Higher value above 0 or 1 | SVM/PSSM, MEME/MAST, HM. | 82.51, 80.39 and 86.62% | 24 |
| Gpos-PLoc | Cytoplasmic | ND | ND | By fusing PseAAC | > 80% | 25 |
| PSORTb | Cytoplasmic | 8.870 | ≥ 7.5 | SCL-BLAST, SVM, Motif and Profile Analysis | 96% | 26 |
| CELLO | Cytoplasmic | 3.809 | Higher value | SVM based on multiple n-peptide composition | 89% | 27 |
| LOCtree | Cytoplasmic | 8.000 | >3 | SVM | ND | 28 |
| SubLoc v1.0 | Cytoplasmic | 5.000 | 5 | SVM | 96% | 29 |
| PA-SUB | Extracellular | 72.84% | Higher percent | SVM | > 92% | 30 |
| MemType-2L | Non membrane protein | ND | ND | Pse-PSSM | 92.7 | 31 |
| TMBETA-SVM | Non membrane protein | 0.636 | ND | SVM | 92% | 32 |
ND: non defined value. Pse-PSSM: Pseudo Position-Specific Score Matrix. SVM: support vector machines. PSSM: Position-Specific Scoring Matrix. MEME/MAST: Multiple Em for Motif Elicitation/Motif Alignment and Search Tool. HM: Hybrid module. PseAAC: Pseudo Amino Acid Composition.
Evolutionary insights into tlyA genes
Evolutionary analysis of tlyA genes from 16 mycobacterial species, 8 species taxonomically related to Mycobacterium, 4 Adomet-RNA MTases and 7 PFP showed similar topology by both Neighbor-joining (NJ) and Maximum Parsimony (MP) methods, suggesting that the phylogenetic relationship observed was highly reliable (Figure 5).
Figure 5.

Evolutionary relationships of 34 taxon at amino acid level. Evolutionary relationships were inferred by the NJ method. The optimal tree had 5,628 branch length sum 5,000 replicates were carried out; bootstrap values are shown at each cluster.
The consensus tree showed the tlyA gene in M. tuberculosis H37Rv, CDC1551, C, Haarlem, Beijing, KZN and INH-RIF strains as well as in the M. bovis AF2122 and BCG strains to fit in a monophyletic cluster. The tlyA tree exhibited short genetic distances (less than 1%) for M. tuberculosis complex species, including those separated according to clinical considerations.
Phylogenetic analysis showed that Mycobacterium tlyA genes are closely related to those of Nocardia and Rhodococcus genus; such branching patterns were supported by high bootstrap values and agreed with the results obtained from 16S rDNA phylogeny. No differences were observed between antibiotic susceptible and resistant M. tuberculosis strains. Mycobacterium TlyA proteins fell into 4 groups; each group formed a well-delineated branch supported by high bootstrap values. M. abscessus was the most distant and TlyA-ancestral species from the Mycobacterium genus compared to the closest species to M. tuberculosis (M. ulcerans, M. marinum and M. leprae). Serpulina hyodysenteriae and Lawsonia intracellularis LsaA antigen sequences were included due to their sequence identity to M. tuberculosis TlyA (26.5%), sharing a common ancestor, although having experimentally-attributed different roles [6,22-24] (Figure 5).
Pattern branching of the phylogenetic tree showed that the TlyA protein formed a monophyletic cluster with AdoMet RNA MTases, suggesting a common evolutionary origin; these findings might also imply a similar functional role in translational machinery. PFP formed a distant cluster where diverse proteins were clustered on a branch with the most divergent proteins (Figure 5).
The synonymous and non-synonymous substitutions were calculated to identify the action of neutral, positive or purifying selection on tlyA genes; their synonymous, Pi(a), and non-synonymous diversity, Pi(s), was 0.672 and 0.132, respectively, corresponding to a 0.196 Pi(a)/Pi(s) rate which can be inferred as purifying selection acting on this gene among Mycobacterium species. This suggests that conservation at sequence level might show Mycobacterium tlyA gene homogeneity and show low inter-specific variation between the species compared in this study.
Discussion
The aim of the present study was to describe structural and phylogenetic features of M. tuberculosis TlyA, using bioinformatics tools. The structural and phylogenetic analysis shown here highlights the need for re-annotating this protein family and redefining it as an RNA-binding FtsJ-like methyltransferase in bioinformatics databases; this is based on the high degree of amino acid sequence similarity found in the Mycobacterium species analyzed here. It can be surmised that physical organization, domain spatial arrangement and protein folding on TlyA is structurally conserved.
Previous sequence analysis of 2'-O-MTase families has indicated that they use a similar catalytic mechanism and have inherited a common function from the same ancestral ribose 2'-O-MTase. Feder et al., suggested that TlyA is a 2'-O-MTase, identifying a catalytic tetrad consisting of lysine-69, aspartic acid-154, lysine-182 and glutamic acid in position 238 (K69-D154-K182-E238) [14]. Based on amino acid sequence alignment, our comparative analysis with the RrmJ family and other related RNA 2'-O-MTases [13,14,18] showed changes in D154-K182-E238 residue positions (Figure 1). However, despite being located in different sequence positions, the scaffold formed by these residues was maintained at the catalytic site in the modeled protein 3D structure without affecting protein function [25].
Despite the diversity of existing MTases, the majority maintain the same catalytic mechanism where K-182 binds to a hydrogen from the ribose 2-OH' group; this nucleophilic attack generates a transition state (SN2-like state). The lysine side chain shifts E-238 so that its negatively charged carboxyl group becomes available to promote the attack of the methyl donor [18]; furthermore, this conserved amino acid residue pattern was also observed in our evolutionary trace analysis.
TlyA three-dimensional structure resembled an RNA methyltransferase
The S. hyodysenteriae TlyA postulated mechanism for hemolysin action is said to be similar to that for pore-forming protein (PFP) mechanisms [6]; TlyA folding, however, is inconsistent with PFPs due its lack of membrane-binding domains, cholesterol-recognition and insertion motifs, and other characteristic features such as a conserved undecapeptide (ECTGLAWEWWR) near the C- or N- terminal ends. It also lacks repeat domains in the toxin (RTX) and calcium-binding glycine-rich motifs necessary for oligomerization [26]. This in silico structure analysis of M. tuberculosis proteins led to a deeper understanding of TlyA's role and challenged the initially described function for TlyA-related proteins.
A combination of bioinformatics analysis and experimental structure elucidation may suggest alternative functional roles for previously-annotated enzymes, as with (SAM)-dependent methyltransferase, phosphatase and N-acetyltransferase structures from M. tuberculosis [11,27,28]. Theoretical evidence has thus been provided for TlyA's role in RNA modification instead of it just being a hemolytic factor.
Subcellular localization suggested a cytoplasmatic localization consistent with ribosome-associated activity
Predicting subcellular localization is important since several characteristics can be deduced, such as protein function and genome annotation. It also aids experimental design for proteomics platforms, particularly for identifying new candidates for vaccine development and drug targets [29,30]. Protein sequence analysis did not reveal a signal peptide, suggesting that TlyA is not secreted; however, some authors have described TlyA as being a probable PFP [6], supported by the presence of over-expressed TlyA on the E. coli wall surface [8].
TlyA lacks transmembrane helices and β-barrels (2.877 value), suggesting that this protein might not be a membrane-embedded or contact-dependent hemolysin, as has previously been proposed [6,23]. TlyA also has a hydrophobic region in the C-terminal domain (residues 115-133) which may not form a membrane-spanning α-helix. Nonetheless, such a region might allow the adenine present in AdoMet to fit properly inside the catalytic pocket, thus suggesting a different role for these hydrophobic residues. Virulence-related functional annotation, based on all the above-mentioned data, remains controversial.
Conservation of tlyA genes and functions was expected according to our phylogenetic and evolutionary trace analyses, the initial hypothesis being that homologous proteins have related functions. However, the extent to which this is true has not been assessed in detail up to now; annotation based on protein homology might thus lead to finding unexpected variations in function.
Conclusions
The main concern of our work was to decipher structural information about the TlyA family by homology comparison. Our structural analysis and finding residue conservation within the active site supports recent experimental work [8] about TlyA's role as RNA 2`-O-MTase in M. tuberculosis. This is further supported by the domain composition also suggesting methyltransferase activity and RNA-binding. The distribution of charged residues also defines a likely RNA interaction instead of hemolytic activity, little similarity being exhibited with hemolysin proteins, perhaps representing inaccurate annotations in several databases, as observed with other protein families [31].
Since genome annotation is based on sequence comparison, it is the most commonly used approach for determining functional homology. Annotations are assigned if proteins surpass similarity cut-off and the propagation of original errors could be increased by each new entry. In such cases, protein function needs to be inferred from their common three-dimensional structures and manual curation.
Methods
Search for TlyA related sequences
Sequences homologous to M. tuberculosis TlyA (Rv1694; GenBank: CAA66941) in other Mycobacterium spp. were found by screening the TB-Database [http://www.tbdb.org/]. Protein and gene sequence similarity was analyzed by BLAST tools [32] for searching the NCBI non-redundant sequence database [http://www.ncbi.nlm.nih.gov/] and a homology comparison was made using BLASTp for the PDB database. The TlyA-like sequences so obtained were aligned using the CLUSTALW Multiple Sequence Alignment program [http://align.genome.jp/] [33] and manually edited in BIOEDIT [34]. The alignment was visualized using the CLC sequence alignment viewer v6.0 [http://www.clcbio.com] [32] to obtain similarity scores for each amino acid position. The M. tuberculosis IprJ-recN operon genes (IprJ, Rv1691, Rv1692, Rv1693, tlyA, ppnK and recN), where tlyA gene is located, were all searched within the M bovis, M bovis BCG, M. leprae, M. ulcerans and M. marinum genome databases for visual examination of the transcriptional unit.
Fold-recognition and domain analysis
This protein's domain composition was analyzed using the SMART-Simple Modular Architecture Research Tool [http://smart.embl-heidelberg.de] [35] in combination with the Pfam database [http://www.sanger.ac.uk/Software/Pfam/] [36].
An M. tuberculosis TlyA (Rv1694) secondary structure consensus was built based on the predictions obtained with SAM [37], PSIPRED [38] and JNet secondary structure prediction [39] servers. This consensus allowed poorly and highly structured regions to be compared for selecting the best tridimensional (3D) structure model according to structural homology and folding prediction.
Protein 3D-structure prediction
An M. tuberculosis TlyA structural model was obtained from its amino acid sequence by using SWISS MODEL [http://swissmodel.expasy.org/][40] and Protein Homology/analogY Recognition Engine (PHYRE) [http://www.sbg.bio.ic.ac.uk/phyre/] prediction servers [32,41]; the obtained models were classified according to identity percentages. Both, protein structure and function models (3HP7, 1QD7, 1Q8K, 3DOU, 2PLW, 1FJG, 1EJ0 PDB entries) were compared to the secondary consensus sequence obtained as described above. Structure refinement and minimization were carried out using UCSF CHIMERA program [42], and the resulting 3D-model was submitted to ProFunc server [http://www.ebi.ac.uk/thornton-srv/databases/profunc/][19,32,43] to predict biochemical functions and structural motifs. The obtained 3D-model was stereo-chemically evaluated on RAMPAGE server [19] which provides a score based on proline and glycine preferential positions according to a Ramachandran plot.
Subcellular localization prediction
The Phobius server [44] was used to predict the presence of the signal peptide and transmembrane α-helices within the M. tuberculosis TlyA amino acid sequence. TlyA's subcellular localization was predicted using the TBpred server [29], a specific tool for mycobacterial proteins. Results were compared to subcellular localization predictions obtained from Gpos-PLoc [30], PSORTb v2.0.4 [45], PA-SUB [46], CELLO v2.5 [47], LOCtree [48], SubLoc v1.0 [21], MemType-2L [49] and TMBETA-SVM [50]. The position of RNA binding residues was predicted using the RNAbind server [51].
β-barrel structures were predicted with PRED-TMBB [http://biophysics.biol.uoa.gr/PRED-TMBB/input.jsp] using a 2.965 threshold value [52].
Phylogenetic and evolutionary trace analysis
The molecular phylogenetic tree for TlyA was built with Neighbor Joining (NJ), using p-distance as substitution model, and Maximum Parsimony (MP) methods in MEGA Version 4.0, with 5,000 iterations for calculating bootstrap confidence levels [53]. Phylogenetic tree construction included the sequences for Mycobacterium TlyA proteins and/or putative cytotoxin/hemolysins reported in the TB-database for the following 11 mycobacterial species, including 14 different strains. Pertinent sequences were found in 21 M. tuberculosis-related species (Table S1, Additional file 1).
Evolutionary Trace Report Maker [54] and Evolutionary Trace Analysis [55] and DnaSP [56] were used for identifying residues under evolutionary pressure and substitution rates were determined using MEGA 4.0.
Authors' contributions
NEA, CV and LMS wrote the manuscript, validated the tools and carried out the data analysis and interpretation. CYS, MEP, MAP and AG contributed to the methodological design, supervised its development and critically revised the manuscript's content. All authors read and approved the final version of the manuscript.
Supplementary Material
Table S1. Descriptions of TlyA proteins used in the phylogenetic analysis. Protein sequences were gathered from the GenBank [http://www.ncbi.nlm.nih.gov/] by BLAST search [32].
Figure S1. Sequence alignment and secondary structure representation for TlyA and Ftsj-like RNA methyltransferase complexed with AdoMet, defined as the ligand-binding template [PDB:1EJ0] [31] showing the matched residues with the template. Alpha helices are represented by red spirals, beta strands by yellow arrows and random structures by straight lines. The red bar indicates the region with greater structural similarity (152 residues-long). Amino acid regions sharing the highest similarity are enclosed in boxes.
Figure S2. Ramachandran plot of TlyA three-dimensional modeled structure. Dispersion zones are shown in blue and orange representing proline and glycine favored and allowed regions (Figure obtained by RAMPAGE server).
{kind=link}
Contributor Information
Nelson E Arenas, Email: nearenass@unal.edu.co.
Luz M Salazar, Email: lmsalazarpu@unal.edu.co.
Carlos Y Soto, Email: cysotoo@unal.edu.co.
Carolina Vizcaíno, Email: cvizcaino@gmail.com.
Manuel E Patarroyo, Email: mepatarr@mail.com.
Manuel A Patarroyo, Email: mapatarr.fidic@gmail.com.
Arley Gómez, Email: arley.gomez@urosario.edu.co.
Acknowledgements
This article is dedicated to the memory of the Professor Leonardo Lareo, an example of work and dedication. Special thanks are due to Aylan Farid Arenas from Universidad del Quindío and Diego Garzón from FIDIC for their valuable ideas for molecular phylogenetic analysis and Jason Garry for reviewing the manuscript.
References
- Dorman SE, Chaisson RE. From magic bullets back to the magic mountain: the rise of extensively drug-resistant tuberculosis. Nat Med. 2007;13:295–298. doi: 10.1038/nm0307-295. [DOI] [PubMed] [Google Scholar]
- Tomioka H. Prospects for development of new antimycobacterial drugs. J Infect Chemother. 2000;6:8–20. doi: 10.1007/s101560050043. [DOI] [PubMed] [Google Scholar]
- Warner DF, Mizrahi V. Tuberculosis chemotherapy: the influence of bacillary stress and damage response pathways on drug efficacy. Clin Microbiol Rev. 2006;19:558–570. doi: 10.1128/CMR.00060-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felnagle EA, Rondon MR, Berti AD, Crosby HA, Thomas MG. Identification of the biosynthetic gene cluster and an additional gene for resistance to the antituberculosis drug capreomycin. Appl Environ Microbiol. 2007;73:4162–4170. doi: 10.1128/AEM.00485-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen SK, Maus CE, Plikaytis BB, Douthwaite S. Capreomycin binds across the ribosomal subunit interface using tlyA-encoded 2'-O-methylations in 16S and 23S rRNAs. Mol Cell. 2006;23:173–182. doi: 10.1016/j.molcel.2006.05.044. [DOI] [PubMed] [Google Scholar]
- Wren BW, Stabler RA, Das SS, Butcher PD, Mangan JA, Clarke JD, Casali N, Parish T, Stoker NG. Characterization of a haemolysin from Mycobacterium tuberculosis with homology to a virulence factor of Serpulina hyodysenteriae. Microbiology. 1998;144((Pt 5)):1205–1211. doi: 10.1099/00221287-144-5-1205. [DOI] [PubMed] [Google Scholar]
- Hsu T, Hutto DL, Minion FC, Zuerner RL, Wannemuehler MJ. Cloning of a beta-hemolysin gene of Brachyspira (Serpulina) hyodysenteriae and its expression in Escherichia coli. Infect Immun. 2001;69:706–711. doi: 10.1128/IAI.69.2.706-711.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Srivastava SS, Sneh A, Ahmed N, Krishnasastry MV. Molecular characterization of tlyA gene product, Rv1694 of Mycobacterium tuberculosis: a non-conventional hemolysin and a ribosomal RNA methyl transferase. BMC Biochem. 2010;11:35. doi: 10.1186/1471-2091-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolenko DN, Spiegel PC, Majumdar ZK, Hickerson RP, Clegg RM, Noller HF. The antibiotic viomycin traps the ribosome in an intermediate state of translocation. Nat Struct Mol Biol. 2007;14:493–497. doi: 10.1038/nsmb1243. [DOI] [PubMed] [Google Scholar]
- Maus CE, Plikaytis BB, Shinnick TM. Mutation of tlyA confers capreomycin resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2005;49:571–577. doi: 10.1128/AAC.49.2.571-577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JM, Arcus VL, Morton CJ, Parker MW, Baker EN. Crystal structure of a putative methyltransferase from Mycobacterium tuberculosis: misannotation of a genome clarified by protein structural analysis. J Bacteriol. 2003;185:4057–4065. doi: 10.1128/JB.185.14.4057-4065.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsolaki AG, Gagneux S, Pym AS, Goguet de la Salmoniere YO, Kreiswirth BN, Van Soolingen D, Small PM. Genomic deletions classify the Beijing/W strains as a distinct genetic lineage of Mycobacterium tuberculosis. J Clin Microbiol. 2005;43:3185–3191. doi: 10.1128/JCM.43.7.3185-3191.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujnicki JM, Droogmans L, Grosjean H, Purushothaman SK, Lapeyre B. In: Practical Bioinformatics. Bujnicki JM, editor. Vol. 15. Berlin: Springer-Verlag; 2004. Bioinformatics-Guided Identification and Experimental Characterization of Novel RNA Methyltransferases; pp. 139–168. full_text. [Google Scholar]
- Feder M, Pas J, Wyrwicz LS, Bujnicki JM. Molecular phylogenetics of the RrmJ/fibrillarin superfamily of ribose 2'-O-methyltransferases. Gene. 2003;302:129–138. doi: 10.1016/S0378-1119(02)01097-1. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. Novel predicted RNA-binding domains associated with the translation machinery. J Mol Evol. 1999;48:291–302. doi: 10.1007/PL00006472. [DOI] [PubMed] [Google Scholar]
- Lustig B, Arora S, Jernigan RL. RNA base-amino acid interaction strengths derived from structures and sequences. Nucleic Acids Res. 1997;25:2562–2565. doi: 10.1093/nar/25.13.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugl H, Fauman EB, Staker BL, Zheng F, Kushner SR, Saper MA, Bardwell JC, Jakob U. RNA methylation under heat shock control. Mol Cell. 2000;6:349–360. doi: 10.1016/S1097-2765(00)00035-6. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhang B, Shi PY. Flavivirus methyltransferase: a novel antiviral target. Antiviral Res. 2008;80:1–10. doi: 10.1016/j.antiviral.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Restrepo-Montoya D, Vizcaino C, Nino LF, Ocampo M, Patarroyo ME, Patarroyo MA. Validating subcellular localization prediction tools with mycobacterial proteins. BMC Bioinformatics. 2009;10:134. doi: 10.1186/1471-2105-10-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S, Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics. 2001;17:721–728. doi: 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003;12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffe M, Etienne G. The capsule of Mycobacterium tuberculosis and its implications for pathogenicity. Tuber Lung Dis. 1999;79:153–169. doi: 10.1054/tuld.1998.0200. [DOI] [PubMed] [Google Scholar]
- McCluskey J, Hannigan J, Harris JD, Wren B, Smith DG. LsaA, an antigen involved in cell attachment and invasion, is expressed by Lawsonia intracellularis during infection in vitro and in vivo. Infect Immun. 2002;70:2899–2907. doi: 10.1128/IAI.70.6.2899-2907.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AE, Orengo CA, Thornton JM. Evolution of protein function, from a structural perspective. Curr Opin Chem Biol. 1999;3:548–556. doi: 10.1016/S1367-5931(99)00007-1. [DOI] [PubMed] [Google Scholar]
- Tweten RK. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card GL, Peterson NA, Smith CA, Rupp B, Schick BM, Baker EN. The crystal structure of Rv1347c, a putative antibiotic resistance protein from Mycobacterium tuberculosis, reveals a GCN5-related fold and suggests an alternative function in siderophore biosynthesis. J Biol Chem. 2005;280:13978–13986. doi: 10.1074/jbc.M413904200. [DOI] [PubMed] [Google Scholar]
- Watkins HA, Baker EN. Structural and functional analysis of Rv3214 from Mycobacterium tuberculosis, a protein with conflicting functional annotations, leads to its characterization as a phosphatase. J Bacteriol. 2006;188:3589–3599. doi: 10.1128/JB.188.10.3589-3599.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid M, Saha S, Raghava GP. Support Vector Machine-based method for predicting subcellular localization of mycobacterial proteins using evolutionary information and motifs. BMC Bioinformatics. 2007;8:337. doi: 10.1186/1471-2105-8-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HB, Chou KC. Gpos-PLoc: an ensemble classifier for predicting subcellular localization of Gram-positive bacterial proteins. Protein Eng Des Sel. 2007;20:39–46. doi: 10.1093/protein/gzl053. [DOI] [PubMed] [Google Scholar]
- Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R. et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Cline M, Diekhans M, Grate L, Hughey R. Predicting protein structure using only sequence information. Proteins. 1999. pp. 121–125. [DOI] [PubMed]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Cuff JA, Barton GJ. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 2000;40:502–511. doi: 10.1002/1097-0134(20000815)40:3<502::AID-PROT170>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70:611–625. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Watson JD, Thornton JM. Protein function prediction using local 3D templates. J Mol Biol. 2005;351:614–626. doi: 10.1016/j.jmb.2005.05.067. [DOI] [PubMed] [Google Scholar]
- Kall L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FS. PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005;21:617–623. doi: 10.1093/bioinformatics/bti057. [DOI] [PubMed] [Google Scholar]
- Lu Z, Szafron D, Greiner R, Lu P, Wishart DS, Poulin B, Anvik J, Macdonell C, Eisner R. Predicting subcellular localization of proteins using machine-learned classifiers. Bioinformatics. 2004;20:547–556. doi: 10.1093/bioinformatics/btg447. [DOI] [PubMed] [Google Scholar]
- Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins. 2006;64:643–651. doi: 10.1002/prot.21018. [DOI] [PubMed] [Google Scholar]
- Nair R, Rost B. Mimicking cellular sorting improves prediction of subcellular localization. J Mol Biol. 2005;348:85–100. doi: 10.1016/j.jmb.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Chou KC, Shen HB. MemType-2L: a web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem Biophys Res Commun. 2007;360:339–345. doi: 10.1016/j.bbrc.2007.06.027. [DOI] [PubMed] [Google Scholar]
- Park KJ, Gromiha MM, Horton P, Suwa M. Discrimination of outer membrane proteins using support vector machines. Bioinformatics. 2005;21:4223–4229. doi: 10.1093/bioinformatics/bti697. [DOI] [PubMed] [Google Scholar]
- Terribilini M, Sander JD, Lee JH, Zaback P, Jernigan RL, Honavar V, Dobbs D. RNABindR: a server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res. 2007;35:W578–584. doi: 10.1093/nar/gkm294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagos PG, Liakopoulos TD, Spyropoulos IC, Hamodrakas SJ. A Hidden Markov Model method, capable of predicting and discriminating beta-barrel outer membrane proteins. BMC Bioinformatics. 2004;5:29. doi: 10.1186/1471-2105-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Mihalek I, Res I, Lichtarge O. Evolutionary trace report_maker: a new type of service for comparative analysis of proteins. Bioinformatics. 2006;22:1656–1657. doi: 10.1093/bioinformatics/btl157. [DOI] [PubMed] [Google Scholar]
- Innis CA, Shi J, Blundell TL. Evolutionary trace analysis of TGF-beta and related growth factors: implications for site-directed mutagenesis. Protein Eng. 2000;13:839–847. doi: 10.1093/protein/13.12.839. [DOI] [PubMed] [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Descriptions of TlyA proteins used in the phylogenetic analysis. Protein sequences were gathered from the GenBank [http://www.ncbi.nlm.nih.gov/] by BLAST search [32].
Figure S1. Sequence alignment and secondary structure representation for TlyA and Ftsj-like RNA methyltransferase complexed with AdoMet, defined as the ligand-binding template [PDB:1EJ0] [31] showing the matched residues with the template. Alpha helices are represented by red spirals, beta strands by yellow arrows and random structures by straight lines. The red bar indicates the region with greater structural similarity (152 residues-long). Amino acid regions sharing the highest similarity are enclosed in boxes.
Figure S2. Ramachandran plot of TlyA three-dimensional modeled structure. Dispersion zones are shown in blue and orange representing proline and glycine favored and allowed regions (Figure obtained by RAMPAGE server).