

Abstract
Molecular modeling of conformational changes occurring in the transmembrane (TM) region of the complement factor 5a receptor (C5aR) during receptor activation was performed by comparing two constitutively active mutants (CAMs) of C5aR, NQ (I124N/L127Q) and F251A, to those of the wild-type C5aR and NQ-N296A (I124N/L127Q/N296A), which have the wild-type phenotype. Modeling involved comprehensive sampling of various rotations of TM helices aligned to the crystal template of the dark-adapted rhodopsin along their long axes. By assuming that the relative energies of the spontaneously activated states of CAMs should be lower or at least comparable to energies characteristic for the ground states, we selected the plausible models for the conformational states associated with constitutive activation in C5aR. The modeling revealed that the hydrogen bonds between the side chains of D82–N119, S85–N119 and S131–C221 characteristic for the ground state were replaced by the hydrogen bonds D82–N296, N296–Y300 and S131–R134, respectively, in the activated states. Also, conformational transitions that occurred upon activation were hindered by contacts between the side chains of L127 and F251. The results rationalize the available data of mutagenesis in C5aR and offer the first specific molecular mechanism for the loss of constitutive activity in NQ-N296A. Our results contributed also to understanding the general structural mechanisms of activation in G-protein-coupled receptors lacking the “ionic lock”, R3.50 - E/D6.30. Importantly, these results were obtained by modeling approaches that deliberately simplify many elements in order to explore potential conformations of GPCRs involving large-scale molecular movements.
Keywords: GPCR, molecular modeling, constitutive activation, C5a receptor
INTRODUCTION
G-protein coupled receptors (GPCRs) comprise a vast protein family involved in variety of physiological functions. GPCRs are embedded in the cell membrane and include seven-helical transmembrane stretches (TM helices, TMs), the N- and C-terminal fragments and the extra- and intracellular loops connecting the TM helices. Agonist-binding activates GPCRs by triggering conformational movements that promote receptor interactions with corresponding G-proteins inside the cell. Some mutant GPCRs display constitutive activity, i.e., ligand-independent activity that produces a second messenger even in the absence of an agonist 1. Constitutively active mutants (CAMs) are known for many GPCRs, including rhodopsin, α1B adrenergic receptor, β2 adrenergic receptor, angiotensin receptor type 1, opioid and cholecystokinin receptors, and many others (see, e.g., a review 1). Since activation of GPCRs is believed to require conformational changes from their inactive ground states to their activated states, it is generally assumed that conformations of constitutively active mutants can mimic the activated conformations of GPCRs. Obviously, knowledge of molecular mechanisms of GPCR’s transition from the ground conformational state to the activated state would tremendously benefit the fields of molecular biophysics and pharmacology and especially drug design, since it has been estimated that almost 50% of the therapeutic compounds in use act on GPCRs 2.
Thus far seventeen crystal structures of six GPCRs are available in the PDB (see a minireview 3), and most capture the inactive ground conformational states of GPCRs. The possible exceptions are structures of photoactivated rhodopsin (the PDB entry 2I37 4), the β2 adrenergic receptor crystallized with the partial inverse agonist carazol (the PDB entry 2RH1 5), and of opsin (retinal-free rhodopsin) crystallized with the C-terminal fragment of α-transducin (the PDB entry 3DQB 6). Overall, the TM regions of all known crystal structures of GPCRs have been similar, with the rms values for the helical backbones not exceeding 2.0 Å – 2.5 Å (Cα atoms; see Ref. 7). This is also the case for the TM regions of the potentially active states of rhodopsin/opsin (2I37 and 3DQB) and β2 adrenergic receptor (2RH1), which differed from the ground state of rhodopsin (the PDB entry 1F88) by the rms values of 1.0 Å (for 2I37 4), 2.1 Å (3DQB 6) and 2.7 Å (2RH1 5). Likewise, the electron crystallography structure of the MI state of rhodopsin (the transition state preceding the activated MII state) showed only small scale molecular movements compared to the dark-adapted rhodopsin (not exceeding 2.0 Å for each TM helix) 8. However, the relatively sparse conformational changes observed in these structures contrasted earlier experimental data obtained in solution by site-directed spin labeling 9 and double electron-electron resonance technique 10. In these biophysical studies, the TM helical bundle of rhodopsin in the activated state significantly differed from that in the dark-adapted ground state. While the spin-labeling data is compatible with many aspects of the opsin-transducin peptide co-crystal structure (3DQB, the data is not shown), they were also consistent with a large-scale rotation of the TM6 along its long transmembrane axis by ca. 120 Å, as modeled in our previous study 11. Experimental estimations of transition times from the ground states to the activated structures (milliseconds for rhodopsin 12 or even seconds for β2 adrenoreceptor 13) also suggest, though indirectly, large scale molecular movements upon GPCR activation.
Future X-ray structures of GPCRs will undoubtedly provide higher definition snapshots of structures in the ground state and perhaps in the fully activated state of the GPCR. At the same time, these structures are not expected to provide the entire conformational trajectories associated with GPCR activation. As a complementary approach, computational modeling offers the opportunity to reveal movements in flexible regions of the extra- and intracellular loops or intermediate-by-intermediate conformational changes in the TM regions. However, current modeling techniques (such as straightforward MD simulations) for systems as large and complicated as GPCRs inserted in membrane (thousands interacting atomic centers, hundreds of water and lipid molecules) require huge state-of-the-art computer resources to cover trajectories only up to microseconds 14–16. Even this is not sufficient considering experimental estimations of the transition times from the ground to activated states (see above). Therefore, we developed a simplified modeling approach that deliberately sacrifices many details of the molecular GPCR system (rigid valence geometry of protein; no lipids; no water molecules, etc.) in order to sample comprehensively the conformational space of GPCRs. Previously, our approach established possible 3D structures of the resting state of the complement factor 5a receptor (C5aR) that included various structures of the extracellular and intracellular loops 17,18. Also, we have successfully developed the first detailed 3D models of the complex of C5aR with its natural ligand complement factor 5a (C5a) 19 and with the chemotaxis inhibitory protein of Staphylococcus aureus 20. The approach also successfully predicted novel constitutively active mutants of the angiotensin receptor type 1 21 and C5aR 22. Our approach also correctly restored the X-ray conformations of the extracellular loops in five GPCRs 23.
The present study applied the same simplified approach to modeling possible conformational changes occurring in the TM region of C5aR upon constitutive activation. C5aR is an important receptor in the physiology of immune defense, inflammation and human disease. It is a member of the rhodopsin-like family of GPCRs (family A, at least 700 members 24) with a sequence of 350 amino acid residues sharing 22% sequence identity with rhodopsin. C5aR has been thoroughly studied in our lab by random saturation mutagenesis (RSM) in yeast and mammalian systems yielding more than 150 mutants 17,18,25–28. We have found two mutants of C5aR, NQ (double mutant I124N/L127Q) and F251A, which were well-characterized as strong constitutively active mutants both in yeast and in mammalian systems 26,29; in addition, a weaker CAM, L127A, was predicted by molecular modeling and validated experimentally 22.
In this study, we also hypothesized that one aspect of receptor activation involves concerted rotations of various TM helices along their long axes. This hypothesis was based on experimental results of numerous site-directed mutagenesis studies in many GPCRs that were interpreted as indications of TM rotations relative to the ground state structures 45–57. For instance, a recent example of this type of study presented evidence for possible rotation of TM5 in the constitutively active mutant of the angiotensin receptor type 1 58. Besides, some TM rotations were characteristic also for the presumably activated state of opsin, 3DQB, the most pronounced being that of TM5 (see Table V below).
According to our general approach, we performed computational modeling of the possible conformational states of the TM regions for two CAMs, NQ and F251A, and for two C5aRs with wild-type phenotype, WT (i.e., C5aR itself) and the triple mutant NQ-N296A (I124N/L127Q/N296A) 29. The analyses involved comprehensive sampling of various rotations of TM helices aligned to the rhodopsin template (the PDB entry 1F88, the inactive dark-adapted rhodopsin). Regarding the appropriateness of rotational sampling for assembling possible TM configurations, numerous site-directed mutagenesis studies in many GPCRs support models for concerted rotations of various TM helices along their long axes upon activation 45–57. A recent example of this type of study presented evidence for possible rotation of TM5 in the constitutively active mutant of the angiotensin receptor type 1 58. By comparing the modeling results for CAMs and non-CAMs and by assuming that the relative energies of the spontaneously activated states of CAMs should be lower or at least comparable to energies characteristic for the inactive ground states, we determined the plausible conformational states associated with triggering constitutive activation in C5aR.
COMPUTATIONAL METHODS
Some modeling procedures employed in this study, as well as the force field used, are described in details in previous papers 11,19,30. Therefore, only significant differences in calculation protocols are outlined below more extensively; otherwise, only brief descriptions are given. Specific limitations due to simplifications in our approach are addressed also in the DISCUSSION section.
Force field
All energy calculations employed the ECEPP/2 force field with rigid valence geometry 31,32 and trans-conformations of Pro residues; residues Arg, Lys, Glu and Asp were regarded as charged species. Energy calculations were routinely performed with the value of the macroscopic dielectric constant ε of 2 (the standard value for ECEPP corresponding to a protein environment).
Building of TM region of C5aR and mutants
The procedure for building the TM bundle of the C5aR was the same as described earlier 28. The TM helical segments in C5aR were determined by homology to bovine rhodopsin and human β2 adrenergic receptor as follows: TM1, I38–A63 (the first, middle and last residue, respectively); TM2, N71–L84–Q98; TM3, A107–A122–V138; TM4, A150–W161–F172; TM5, E199–F211–F224; TM6, R236–F251–F267; and TM7, L281–Y290–Y300. (Note that the H8 helix of rhodopsin parallel to the intracellular membrane surface is absent in C5aR, see Ref. 28.) Each individual TM helix was first subjected to energy minimization starting from the all-helical backbone conformations (i.e., the values of all dihedral angles φ and ψ were initially of −60°). Some limitations on the φ and ψ values (−20° ≥ φ, ψ ≥ −100°) and the ω values (150° ≥ ω ≥ −150°) were placed during energy minimization to mimic, to some extent, limitations on intrahelical mobility of TM segments immobilized in the membrane. The resulting helical fragments were then aligned to the TM regions of the selected X-ray templates (1F88, 3DQB or 2RH1). Further modeling consisted of minimization of the sum of all intra- and interhelical interatomic energies in the multi-dimensional space of parameters assigned to each helix in the bundle. Those included the “global” parameters (related to movements of individual helices as rigid bodies, i.e., translations along the coordinate axes X, Y, Z and rotations around these axes Tx, Ty, and Tz) and the “local” parameters (the dihedral angles of the side chains for all helices; the dihedral angles of the backbone were frozen). The coordinate system for the global parameters was described earlier 28. For C5aR mutants, the corresponding side chains were mutated without changing the dihedral angles of the backbone for the mutated residue. The starting point of computational search for possible conformational states of the TM regions (the reference structure) was determined by global parameters corresponding to the X-ray structure of the template 1F88 (all Tx rotations were assumed to be equal to 0°).
Typically, two types of energy calculations were performed. The “preliminary” energy calculations comprised energy minimization in the space of the global parameters along with packing and repacking of spatial arrangements for each side chain at each convergence step of energy minimization by an algorithm developed previously 33. “Full” energy calculations involved energy minimization not only within the space of global parameters, but also including the dihedral angles of the side chains. New repacking of spatial arrangements for each side chain at each convergence step was also performed at full energy calculations.
Rotational sampling of potential configurations of helical packing
Our search for possible conformational transitions in the TM regions of C5aR included sampling of possible rotations of TM helices along their long axes (“rotational sampling”). For this, we have developed a procedure to probe possible rotations of the seven TM helices with a grid step of 30° without actually sampling all 127 = 35,831,808 configurations. The procedure was based on three main considerations. First, in our particular case of CAMs, we are interested only in configurations of helices with relative energy values lower or at least comparable with those of the inactive ground states of the receptors. Second, all available X-ray structures of the TM regions of GPCRs showed that the direct interactions between TM helices are within the five “triplets” of helices: TM1-TM2-TM7 (TM127), TM2-TM3-TM7 (TM237), TM3-TM4-TM5 (TM345), TM3-TM5-TM6 (TM356) and TM3-TM6-TM7 (TM367). Therefore, if relative energy of any particular configuration of a triplet is noticeably higher than the configuration corresponding to the reference structure in the same triplet, this configuration may be removed from further consideration and be not included in the next level of calculations, i.e., calculations performed for packages of the four contacting helices, TM1237, TM2367, TM3567 and TM3456. After a new round of energy calculations and selection, the newly selected configurations are combined into packages of five contacting helices and so on until the entire TM bundle is built (see also Ref. 34). Third, it is not likely that relatively small molecular replacements leading to constitutive activity (as F251 → A251 in F251A or I124 → N124 and L127 → Q127 in NQ) would result in too large scale reorientations in the TM helical bundle. It could be safely assumed that rotations of the TM helices corresponding to the constitutively activated states would not differ from the reference structures (i.e., configurations with all TM rotations of 0°) by more than ±90°.
Sampling of the TM regions can be also limited by selecting only rotations precluding direct exposure of the charged residues in the middle sections of each TM helix to the lipid core of the membrane. This structural feature was observed in all crystal structures of GPCRs present in the PDB. For C5aR, the residues in question were D82, D133, R134 and R206, but not E199, R200, R236, K239, K242 and D282, since the latter ones are located close to the heterogeneous membrane boundaries and may interact with the EC loops, as well as with the charged heads of phospholipids. Likewise, it is reasonable to consider only TM rotations that orient the highly hydrophobic tryptophan residues in the middle sections of the TM helices towards the exterior of the TM bundles. Again, this was observed for all tryptophans in the crystal structures of GPCRs with a notable exception of the highly conserved Trp in TM6 (W255 in C5aR, W6.48 according to the universal nomenclature 35) that is oriented towards the interior of the bundle. The tryptophan residues assumed to be oriented towards the lipid membrane core in the case of C5aR were W60, W74, W154, W161 and W213.
Applying these limitations, rotational sampling in the TM regions of the C5a receptors was performed with the initial TM orientations of 0°, 30°, 60°, 90°, 270°, 300° and 330° for TM1; 0°, 270°, 300° and 330° for TM2; 0°, 30°, 60°, 270°, 300° and 330° for TM3; 0°, 30°, 60°, 90°, 300° and 330° for TM4; 0°, 30°, 300° and 330° for TM5; 0°, 30°, 60°, 90°, 270°, 300° and 330° for TM6; and 0°, 30°, 60°, 90°, 270°, 300° and 330° for TM7.
RESULTS
Computational modeling was performed for the TM regions of four receptors, namely C5aR (WT), NQ, NQ-N296A and F251A. For each receptor, systematic sampling of possible rotations of TM helices with the grid step of 30° was performed starting from the reference configuration corresponding to the crystal template 1F88.
Selection of TM configurations
The main steps of our modeling procedure are outlined in Table I, which contains the numbers of configurations of TM helical bundles selected for further consideration at each step of calculations. First, preliminary energy calculations were performed for all possible configurations of the five TM triplets for each receptor as described in COMPUTATIONAL METHODS. Obviously, some triplets were the same for different receptors (e.g., TM127 was the same for WT, NQ and F251A, but not for NQ-N296A). Two criteria determined the selection of triplet configurations for further consideration: 1) energies of the selected configurations should not exceed the energies of the reference structures (all TM rotations of 0°) by more than the arbitrary cut-off of 5 kcal/mol, and, 2) low-energy configurations must be connected to the reference structures by series of minimal rotations of 30°. For instance, in the TM127 bundle for WT, there were no low-energy configurations with orientations of TM7 = 330°; therefore, configurations with orientations of TM7 = 300° and 270° cannot be reached from the reference structure without crossing the high-energy barriers at TM7 = 330°. This limitation on TM7 orientation was extended also to TM127 for NQ and F251A; to TM237 for WT, NQ and F251A; and to TM367 for WT and NQ. Also, since orientation of TM3 = 330° was absent in low-energy configurations of TM367 for NQ-N296A, configurations with orientations of TM3 = 300° and 270° were removed from further consideration.
Table I.
Outline of step-by-step calculation procedure
| Method | TM bundle | WT | NQ | NQ-N296A | F251A |
|---|---|---|---|---|---|
| Preliminary energy calculations | TM127 | 32 | 32 | 68 | 32 |
| TM237 | 84 | 85 | 161 | 84 | |
| TM345 | 56 | 103 | 103 | 56 | |
| TM356 | 67 | 46 | 46 | 41 | |
| TM367 | 63 | 39 | 67 | 109 | |
| TM3567 | 89 | 98 | 169 | 195 | |
| Full energy calculations | TM3567 | 21 | 20 | 39 | 37 |
| TM237 | 58 | 51 | 113 | 58 | |
| TM23567 | 16 | 6 | 17 | 13 | |
| TM1234567 | 5 | 4 | 7 | 9 | |
At the next step, the configurations of the four-helical bundles, TM3567’s, were obtained by combining the selected configurations of TM356 and TM367. Preliminary energy calculations of the TM3567 bundles yielded different numbers of low-energy configurations for different receptors (see Table I). For each receptor, the same energy cut-off of not exceeding energy of the reference structure by more than 5 kcal/mol was applied. The selected configurations of TM3567’s were subjected to further full energy calculations, first for WT, and then to the other receptors. The results of full energy calculations for WT 3567 revealed that configurations with energies less than 25 kcal/mol relative to the configuration with the minimal energy (the arbitrary cut-off) were divided into two separated groups, one with TM6 rotations of 0°, 30°, 60° and 90°, and the other with TM6 rotations of 300° and 270°. These groups could not be connected through sequential series of 30° rotations. Therefore, TM6 rotations of 330°, 300° and 270° were excluded from consideration in the other receptors, which was reflected by the numbers of selected configurations listed in Table I.
Full energy calculations of TM3567’s revealed also that energies of the reference configurations of TM3567’s (the initial orientations of all TM helices of 0°) in all cases were significantly higher than those for the configurations with minimal energies. Specifically, relative energies of the reference configurations were ca. 12 kcal/mol for WT, 23 kcal/mol for F251A, 52 kcal/mol for NQ, and 49 kcal/mol for NQ-N296A. At the same time, some TM3567 configurations, which differed from the reference configuration only by minimal TM rotations of ±30°, possessed lower energies. This finding indicated that the models of the ground states for the TM bundles of the C5a receptors likely differ from the reference configuration adopted for the rotational sampling of the TM configurations (the X-ray structure of the ground state of bovine rhodopsin, 1F88). Instead, configurations with deviations from the reference structure by minimal TM rotations of ±30° can be regarded as more likely models for the ground states of the corresponding C5a receptors (this assumption is discussed in more details below). To retain such configurations for further steps of the modeling procedure, we relaxed the energy cut-offs to 35 kcal/mol for NQ and NQ-N296A and to 25 kcal/mol for WT and F251A relative to the minimal energies of all calculated configurations of the corresponding TM3567’s.
Full energy calculations were then performed for configurations of the TM237 triplets selected earlier by preliminary energy calculations. Table I lists the numbers of low-energy configurations (those with energies lower than those of the reference structure) selected for further combination with the selected configurations of TM3567’s. The five-membered TM23567 bundle represents the core in the entire TM packages of GPCRs, since each TM in this bundle (TM2, TM3, TM5, TM6 and TM7) directly interacts with at least three other TM helices, while the two TM helices not included in TM23567, TM1 and TM4, interact with only two other TMs. Full energy calculations of the TM23567 bundles for each receptor produced sets of low-energy conformations (see Table I) selected by the energy cut-off of 15 kcal/mol; this cut-off ensured that the presumed ground state configurations remained among the selected ones. Then, the lowest-energy configurations of TM1 and TM4 adjustable for each of the selected TM23567 configurations for every receptor were selected by the results of full energy calculations of corresponding configurations of TM127’s and TM345’s, respectively. These sets of orientations of TM1 and TM4 were combined with all selected TM23567 configurations to form possible configurations of the entire seven-helical TM bundles. Full energy calculations revealed the final sets of configurations of the TM bundles.
Probable models of the ground states for the C5a receptors
As was mentioned above, configurations that differ from the reference configuration adopted for rotational sampling (the crystal template 1F88) by minimal TM rotations of ±30° may be of lower energy than the reference configuration with all TM rotations of 0o. Additional full energy calculations performed for various configurations of the entire seven-helical TM bundles with combinations of rotations TM2, TM3, TM4, TM5 and TM6 = 0°±30° confirmed this assumption. For all four receptors, configurations with all TM rotations of 0° (configurations Rh) possess energies significantly higher than those of some configurations differing only by small rotations of ±30° (see Table II).
Table II.
Configurations corresponding to the ground states of the C5a receptors
| Receptor, TM configuration | TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | Energy, kcal/mol | |
|---|---|---|---|---|---|---|---|---|---|
| WT | Rh | 0 | 0 | 0 | 0 | 0 | 0 | 0 | −540 |
| A1 | 0 | 330 | 30 | 30 | 330 | 30 | 30 | −557 | |
| A2 | 0 | 330 | 30 | 330 | 330 | 30 | 30 | −551 | |
| A3 | 0 | 0 | 0 | 330 | 0 | 0 | 0 | −564 | |
| A4 | 0 | 330 | 30 | 330 | 330 | 30 | 0 | −545 | |
| NQ | Rh | 0 | 0 | 0 | 0 | 0 | 0 | 0 | −513 |
| A1 | 0 | 330 | 30 | 30 | 330 | 30 | 30 | −546 | |
| A2 | 0 | 330 | 30 | 330 | 330 | 30 | 0 | −552 | |
| A3 | 0 | 330 | 30 | 330 | 330 | 30 | 30 | −553 | |
| A4 | 0 | 0 | 0 | 330 | 0 | 0 | 0 | −540 | |
| A5 | 0 | 330 | 0 | 330 | 330 | 30 | 30 | −544 | |
| A6 | 0 | 330 | 30 | 30 | 330 | 30 | 0 | −538 | |
| A7 | 0 | 330 | 0 | 330 | 330 | 30 | 0 | −536 | |
| NQ-N296A | Rh | 0 | 0 | 0 | 0 | 0 | 0 | 0 | −508 |
| A1 | 0 | 330 | 30 | 330 | 330 | 30 | 0 | −556 | |
| A2 | 330 | 330 | 30 | 30 | 330 | 30 | 30 | −539 | |
| F251A | Rh | 0 | 0 | 0 | 0 | 0 | 0 | 0 | −506 |
| A1 | 0 | 330 | 30 | 30 | 330 | 30 | 0 | −539 | |
| A2 | 0 | 330 | 0 | 330 | 330 | 30 | 0 | −531 | |
| A3 | 0 | 330 | 30 | 330 | 330 | 30 | 0 | −540 | |
| A4 | 0 | 330 | 30 | 330 | 330 | 30 | 30 | −542 | |
Table II lists all configurations with rotations TM2, TM3, TM4, TM5 and TM6 = 0°±30° possessing relative energies not exceeding the minimal energy found for this set of configurations in each receptor by more than the arbitrary cut-off of 20 kcal/mol (configurations Ai). (Obviously, the actual values of rotational angles around the long axes of TM helices obtained by energy calculations differed slightly from the round values corresponding to the starting TM configurations, shown in Table II and, further, in Tables III and V.) Taking into account the deliberate limitations of our modeling procedure (considering only TM bundles without connecting loops; simple force field; absence of explicit membrane environment, water, ions, etc.) it would be unreasonable to select only one single configuration as the most probable model of the ground state for each receptor. Rather, the sets of all low-energy configurations Ai collectively may be regarded as plausible possibilities for the ground state models for the given receptor. Geometrically, the Ai configurations are close to each other (the inter-structure rms values varying from 0.54 Å to 1.29 Å for WT), but show greater differences relative to the template 1F88 structure (the rms values of 2.9 – 3.0 Å).
Table III.
Final set of low-energy configurations
| Receptor, TM configuration | TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | Energy, kcal/mol | Connections | |
|---|---|---|---|---|---|---|---|---|---|---|
| WT | B | 30 | 300 | 0 | 0 | 0 | 0 | 30 | −571 | A1/A2/A4/C/E |
| C | 30 | 270 | 0 | 0 | 0 | 0 | 0 | −571 | B/E | |
| D | 30 | 0 | 60 | 0 | 0 | 60 | 0 | −569 | A1/A2/A4 | |
| E | 30 | 300 | 0 | 330 | 330 | 30 | 0 | −565 | A2/A4/B/C | |
| NQ | B | 30 | 300 | 60 | 0 | 0 | 30 | 30 | −561 | A1/A2/A3/A6/C/D |
| C | 0 | 330 | 60 | 30 | 30 | 30 | 30 | −557 | B/D/3DQB | |
| D | 30 | 300 | 60 | 30 | 30 | 30 | 30 | −548 | B/C | |
| NQ-N296A | B | 300 | 300 | 60 | 0 | 0 | 30 | 30 | −555 | A2/C |
| C | 300 | 300 | 60 | 30 | 30 | 30 | 30 | −556 | B | |
| D | 300 | 300 | 60 | 30 | 300 | 30 | 30 | −567 | A2 | |
| F251A | B | 0 | 330 | 30 | 30 | 300 | 30 | 30 | −556 | A1/E |
| C | 30 | 300 | 60 | 0 | 0 | 60 | 0 | −536 | A1/A3/A4/D | |
| D | 0 | 330 | 60 | 30 | 30 | 30 | 30 | −551 | C/3DQB | |
| E | 30 | 330 | 30 | 30 | 300 | 30 | 60 | −530 | B | |
| F | 30 | 300 | 0 | 330 | 330 | 30 | 0 | −562 | A2/A3/A4/G | |
| G | 30 | 270 | 0 | 330 | 330 | 30 | 0 | −556 | F | |
Configurations Ai, therefore, may collectively serve as more adequate models for the ground states of the TM bundles of the C5a receptors than configurations Rh, which more closely corresponds to the ground state of rhodopsin (the rms value of 2.4 Å for WT). This observation is not an artifact of the modeling procedure, since application of the same procedure to the dark-adapted rhodopsin, on the contrary, showed that deviations of each of individual TM helices from the Rh configuration by ±30° resulted in configurations with higher energies. It was observed for cis-retinal-containing rhodopsin by our earlier modeling, where only preliminary energy calculations were performed and electrostatic interactions between atoms were not taken into account 11, and also by additionally performed full energy calculations in this study (the data not shown).
Also, we have performed calculations for the TM region of opsin according to the same protocol as described above for C5aR (the reference structure was that of 3DQB). The reference configuration was present among the low energy structures of TM3567 (according to full energy calculations), but it cannot be connected to any other low energy structure through minimal TM rotations of ±30° without crossing high energy barriers defined as the trajectory points above the energy cut-off of 35 kcal/mol. In other words, contrary to the C5aR case, the 3DQB configuration becomes the only possibility for the TM region of opsin at the level of TM3567 already. (However, other TM configurations may be allowed for rhodopsin, which features cis/trans retinal attached to K296.) This result provides additional validation of our procedure for identifying the native packing pattern of a GPCR.
Structural features associated with constitutive activity
Table III lists all configurations of TM bundles found by rotational sampling and possessing energies not exceeding the minimal energies found for the probable ground states of the given receptor (i.e., for configuration A3 for WT, A3 for NQ, A1 for NQ-N296A and A4 for F251A, see Table II) by more than 20 kcal/mol. All listed configurations of every receptor can be connected to each other and to the ground state configurations Ai (see Table II) by series of minimal TM rotations of ±30°; direct connections between each pair of configurations are shown in the last column of Table III.
According to our main assumption, TM configurations associated with constitutive activity would be those with energies lower or at least comparable to those of the ground state structures (configurations Ai in Table II) and characteristic only to constitutively active mutants NQ and F251A, but not for WT or NQ-N296A. We assumed also that these configurations should be connected to the ground states through series of minimal TM rotations (of ±30°) without crossing high-energy barriers, i.e., avoiding configurations with energies higher than that of the ground state.
Keeping in mind that we have thoroughly sampled only the TM23567 bundles (the suitable rotations of TM1 and TM4 were adjusted to selected configurations of TM23567’s), Table III was examined as to combinations of rotations of TM2, TM3, TM5, TM6 and TM7 that would be present in the set of configurations found for NQ and F251A, and not for WT and NQ-N296A. There was only one combination found by rotational sampling satisfying this requirement, namely that corresponding to configuration C for NQ and D for F251A (shown in bold in Table III). Therefore, these configurations may be regarded as the structures associated with constitutive activity (the CAM configurations).
Major structural differences between the ground states (Ai) and CAM configurations can be summarized by patterns of contacts between the side chains that may form hydrogen bonds, either directly, or through mediation of the water molecules (which were not involved directly in our modeling). The contacts with the O…H distances less than 3.5 Å between the side chains in the ground state configurations Ai for all four receptors and in the CAM configurations of NQ and F251A are listed in Table IV. (Table IV, however, does not contain the contact between E199 and R200 in TM5 or information on absence of the contact between D133 and R134 in TM3, both found for all configurations listed in Tables II and III.)
Table IV.
Possible hydrogen bonds between the side chains in TM regions
| Hydrogen bonding | Ground states | CAMs | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | NQ | NQ-N296A | F251A | ||||||||||||||||
| A1 | A2 | A3 | A4 | A1 | A2 | A3 | A4 | A5 | A6 | A7 | A1 | A2 | A1 | A2 | A3 | A4 | NQ | F251A | |
| N71 – D133 | |||||||||||||||||||
| D82 – S85 | * | * | * | * | |||||||||||||||
| D82 – C86 | * | * | * | * | * | * | * | ||||||||||||
| D82 – N119 | * | * | * | * | * | * | * | * | * | * | * | * | * | ||||||
| D82 – C293 | * | * | * | * | * | * | * | ||||||||||||
| D82 – N296 | * | * | * | * | * | * | * | * | * | ||||||||||
| S85 – C86 | * | ||||||||||||||||||
| S85 – N119 | * | * | * | * | * | * | * | * | * | * | * | * | |||||||
| N119 – S123 | * | * | |||||||||||||||||
| Y121 – R206 | * | * | |||||||||||||||||
| N124 – Q127 | * | * | * | * | |||||||||||||||
| N124 – W255 | * | ||||||||||||||||||
| S131 – R134 | * | * | * | * | * | ||||||||||||||
| S131 – C221 | * | * | * | * | * | * | * | * | * | * | * | ||||||||
| S171 – R206 | * | ||||||||||||||||||
| R200 – S266 | * | ||||||||||||||||||
| R206 –Y258 | |||||||||||||||||||
| R206 – Q259 | * | ||||||||||||||||||
| W255 – Q259 | * | * | * | * | * | * | * | * | * | * | * | * | * | ||||||
| N296 – Y300 | * | * | * | * | * | * | * | * | |||||||||||
The contacts found by modeling in the various ground state configurations vary from receptor to receptor; however, there are some contacts that were observed most often for all four receptors (see Table IV). These contacts may be regarded as the most characteristic for the ground state configurations of the receptors. These interactions include hydrogen bonding between the β-carboxyl of D82 in TM2 and the β-amido group of N119 in TM3; between the hydroxyl of S85 in TM2 and the β-amido group of N119 in TM3; between the hydroxyl group of S131 in TM3 and the sulfhydryl group of C221 in TM5; and between the γ-carbonyl of Q259 and the Nε hydrogen in W255, both in TM6. Figure 1 presents a sketch of the representative model of the ground state of WT (configuration A4) showing the discussed contacts between the side chains.
Figure 1.

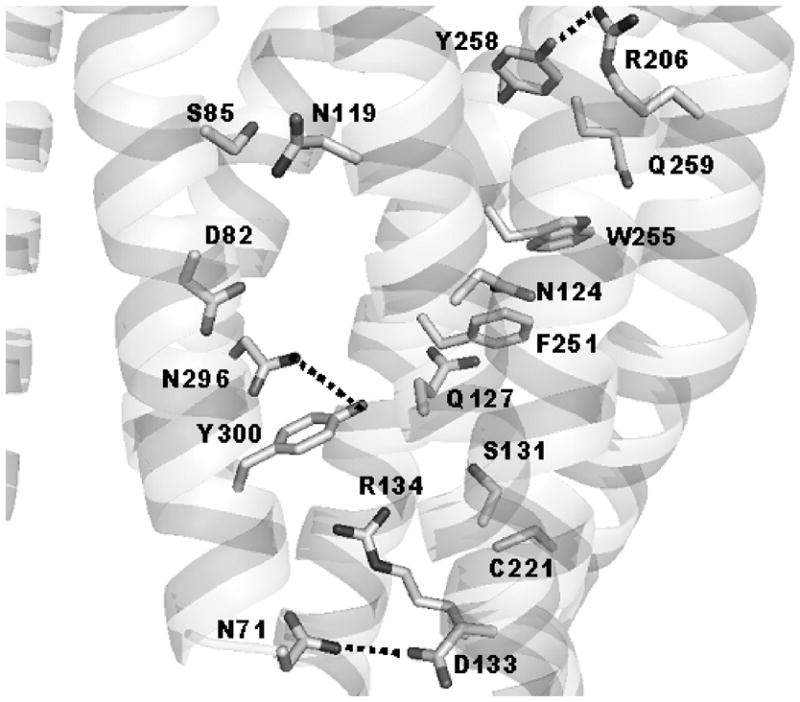
Three persistent potential contacts through hydrogen bonding between side chains were observed for the CAM configurations found by rotational sampling for both NQ and F251A (see Table IV). There were contacts between the β-carboxyl of D82 in TM2 and the β-amido group of N296 in TM7; between the hydroxyl of S131 and the guanidine group of R134, both in TM3; and between the β-amido group of N296 in TM7 and the hydroxyl of Y300, both in TM7. These contacts were sometimes observed also in the ground states of the receptors, but rather sparsely compared to the contacts D82 – N119, S85 – N119, S131 – C221 and W255 – Q259 that were characteristic for the ground states (Table IV). Figure 2 illustrates the system of hydrogen bonding in the CAM configuration of the NQ receptor.
Figure 2.

While configurations Ai represent the ground states of the TM regions of the C5a receptors, the CAM configurations found by rotational sampling may be regarded as the models for the activated state. Comparison of Figs 1 and 2 suggests that main conformational changes occurring in the TM regions of C5aR upon activation would break the contacts between N119 and S85 (and D82) due to rotation of TM3 and, at the same time, enhance the contact between D82 and N296. Accordingly, interactions involving N119 could stabilize the ground state of C5aR, and interactions involving N296 could stabilize the possible activated state. Additional stabilization of the CAM configurations may be also provided by interaction N296 – Y300. These findings provide a molecular mechanism for the fact that the N296A mutation suppresses the constitutive activity when substituted into the constitutively active mutant NQ 29. Another notable conformational re-ordering upon activation would be breaking the hydrogen bonding between S131 and C221 and movement of the side chain of R134 (R3.50 in the universal classification 35) towards S131 resulting in the hydrogen bond between the two side chains. Interestingly, conformational movement of R3.50 towards the protein core upon activation of the β2 adrenergic receptor by formoterol was also suggested by a very recent study 36.
A hydrogen bond between W255 and Q259 represents another potential interaction broken in the activated state. For NQ, one reason for this is orientation of the side chain of F251, which corresponds to the trans conformation (χ1 angle ca. 180o) in the CAM configuration and is involved in stacking interactions with the W255 side chain (see Figure 2). In fact, all configurations listed in Table III for NQ and NQ-N296 featured the trans conformations of the F251 side chain. However, for some configurations of WT, namely B, C and E, the gauche(−) conformation (χ1 angle ca. −60°) of the F251 side chain was more characteristic (this specific conformation is depicted also in the sketch in see Figure 1). As shown by our earlier modeling, steric hindrance of the voluminous side chains of I124 and, especially, L127 in TM3 prevents the side chain of F251 from rotation towards the trans conformation in the Rh configuration of WT 22. At the same time, the side chains of N124 and Q127 in NQ and NQ-N296A are more flexible and accommodate the trans conformation of the side chain of F251 in the ground state.
Our results suggest plausible conformational trajectories from the ground states to the CAM states in NQ (configurations A1–3,6 ↔ configuration B ↔ configuration C) and in F251A (configurations A1,3,4 ↔ configuration C ↔ configuration D). The resulting models also follow plausible specific details associated with molecular mechanisms of activation (see Figure 3). For instance, in the process of activation in NQ (e.g., along the possible trajectory A3 ↔ B ↔ C), the important hydrogen bond between the β-amido group of N296 and the β-carboxyl of D82 existing in A3 was broken in the intermediate state B and reappeared in C. In contrast, the hydrogen bond between the β-amido group of N199 and the β-carboxyl of D82 was broken in B and did not restore in C. The hydroxyl of S131 formed three different hydrogen bonds along the trajectory, namely, with the sulfhydryl of C221 in A3, with the γ-amido group of Q127 in B, and with the guanidino group of R134 in C, respectively. The hydrogen bond between the γ-carbonyl of Q259 and the Nε hydrogen in W255 was preserved only in A3 ↔ B transition, while the hydrogen bond between the β-amido group of N296 and the hydroxyl of Y300 was conserved during the entire trajectory.
Figure 3.

Validation: mutants N119S, R10 and R306
We have deduced the possible CAM configuration of the TM region of C5aR by comparing computational results for WT, NQ, NQ-N296A and F251A. The parameters of the modeling procedure were calibrated for distinguishing NQ and F251A (CAMs) from WT and NQ-N296A (non-CAMs). The ability of this procedure to distinguish CAMs from non-CAMs in other C5aR mutants would serve as an important validation for the results of our study.
Our results suggested the importance of the hydrogen bond between the β-amido group of N119 and the hydroxyl of S85 and the β-carboxyl of D82 in stabilizing the ground state of the receptor. The very recent studies on site-directed mutagenesis of C5aR performed in our lab independently 60 showed significant constitutive activity of the N119S mutant, where these interactions may be weakened. Also, as was noted above, the RSM technique found that the mutant R306 (F48Y/L49V/V50A/L55I/L57V) is a CAM, whereas the mutant with similar replacements in TM1, R10 (F48Y/L49V/V52L/G54A), is a non-CAM.
For the three mutants, N119S, R306 and R10, full energy calculations were performed for collection of all configurations found for WT, NQ, NQ-N296A and F251A in Table II (the ground states) and Table III (other possible states). Configurations satisfying the same criteria as applied above (those with energies not exceeding the minimal energies found for the ground states by the specified energy cut-off and connected to each other and to the ground state configurations by series of minimal TM rotations) are listed in Table VI, which allows one to follow possible trajectories from the ground states to the CAM state. For N119S, the minimal energy trajectory would be A2 (0) ↔ A3 (14) ↔ B (8) ↔ CAM (13); here numbers in brackets are energy values (kcal/mol) relative to the minimal energy found for the ground states (labeled by the asterisks in Table VI). Clearly, these results determine N119S as a CAM with the low-energy path from the ground state to the CAM state possessing the relative energy of 13 kcal/mol (within the energy cut-off of 20 kcal/mol assumed above for WT, NQ, NQ-N296A and F251A). For R306, the corresponding trajectory would be A2 (0) ↔ A1 (14) ↔ B (−4) ↔ CAM (21). In this case, the relative energy of the CAM state was on the border of the cut-off of 20 kcal/mol, but for the similar mutant R10 it was significantly higher (36 kcal/mol, see Table VI). Thus, our procedure correctly differentiated R306 (CAM) from R10 (non-CAM). The difference in relative energies of the CAM states is due to the strong hydrogen bonding of the hydroxyl of Y290 and the backbone carbonyl of F44 available in R306, but not in R10, since the lesser voluminous V52 side chain in R306 (L52 in R10) allows small movement of the Y52 side chain, which, in turn, facilitates rearrangement of the Y290 side chain leading to this hydrogen bonding (the data not shown).
Table VI.
Configurations of TM regions of N119S, R10 and R306
| Receptor, TM configuration | TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | Energy, kcal/mol | Connections | |
|---|---|---|---|---|---|---|---|---|---|---|
| N119S | A1 | 0 | 330 | 30 | 330 | 330 | 30 | 30 | −546 | A2/A3/B |
| A2 | 0 | 0 | 0 | 330 | 0 | 0 | 0 | −566* | A1/A3 | |
| A3 | 0 | 330 | 30 | 330 | 330 | 30 | 0 | −552 | A1/A2/B | |
| B | 30 | 0 | 60 | 0 | 0 | 60 | 0 | −558 | A1/A3/CAM | |
| CAM | 0 | 330 | 60 | 30 | 30 | 30 | 30 | −553 | B | |
| R306 | A1 | 0 | 330 | 30 | 330 | 330 | 30 | 30 | −531 | A2/A3/B/C/D |
| A2 | 0 | 0 | 0 | 330 | 0 | 0 | 0 | −545* | A1/A3 | |
| A3 | 0 | 330 | 30 | 330 | 330 | 30 | 0 | −529 | A1/A2/B/C/D | |
| B | 30 | 0 | 60 | 0 | 0 | 60 | 0 | −549 | A1/A3/CAM | |
| C | 30 | 300 | 60 | 0 | 0 | 30 | 30 | −541 | A1/A3/CAM | |
| D | 30 | 300 | 60 | 0 | 0 | 60 | 0 | −524 | A1/A3/CAM | |
| CAM | 0 | 330 | 60 | 30 | 30 | 30 | 30 | −524 | B/C/D | |
| R10 | A1 | 0 | 330 | 30 | 30 | 330 | 30 | 30 | −557* | A2/A3/B/C |
| A2 | 0 | 330 | 30 | 30 | 330 | 30 | 0 | −537 | A1/A3/B/C | |
| A3 | 330 | 330 | 30 | 30 | 330 | 30 | 30 | −548 | A1/A2 | |
| B | 30 | 0 | 60 | 0 | 0 | 60 | 0 | −556 | A1/A3/CAM | |
| C | 30 | 300 | 60 | 0 | 0 | 60 | 0 | −543 | A1/A3/CAM | |
| CAM | 0 | 330 | 60 | 30 | 30 | 30 | 30 | −521 | B/C | |
The same procedure was used for predictions of the novel possible CAMs of C5aR. For instance, in the series N119X (X = D, H, W, L and A), the relative energies of the CAM states were found of 5 kcal/mol for N119W, 32 for N119D, 39 for N119H, 26 for N119L and 27 for N119A. Accordingly, our modeling procedure predicts N119W as a possible CAM, N119D and N119H as non-CAMs, and is less specific in regard of N119L and N119A, thus presenting a testable hypothesis for further verifying/disproving by further site-directed mutagenesis.
DISCUSSION
Approach: simplifications
This study employed an approach that was deliberately simplified in many elements in order to model possible large-scale movements in TM regions of GPCRs. Such simplifications imply certain limitations on precision of the modeling results.
The ECEPP force field used in our study has been developed specifically for modeling peptides and proteins more than twenty-five years ago. Nevertheless, this simple force field reproduced the distribution of experimental φ, ψ points on a Ramachandran map of the Ac-Ala-OMe (the typical benchmark for validation of various force fields) with accuracy comparable to that of the very recent QM/MM and polarizable force field approaches surpassing more sophisticated GROMOS and OPLS force field (see the recent review 37). The ECEPP force field was also implemented in the ICM program package successfully used for predictions of protein structures in the CASP and CAPRI competitions 38 as well as for recent prediction of ligand docking mode to the human adenosine A2A receptor 39. Other simplifications include calculating electrostatic interactions with the macroscopic dielectric constant of 2; rigid valence geometry and dihedral angles of TM backbones (during helix packing); absence of the loops connecting TM helices and lipids interacting with the protein core. It is reasonable to assume that these simplifications in modeling approach will affect the accuracy of reproducing the experimental structures of the TM regions of GPCRs. However, despite these limitations, our approach reproduced the X-ray structure of the TM region of the dark-adapted rhodopsin with an accuracy of the rms value of 2.5 Å, which is quite comparable to other approaches that used experimentally derived constraints on GPCR structures (2.9 Å 40), statistical inter-TM residue potentials (3.2 Å 41), de novo predictions by PREDICT (2.9 Å, 42) and MembStuck (3.1 Å 43), or refined threading approach (2.1 Å 44).
Nevertheless, energies corresponding to the TM configurations obtained in this study should be regarded as estimates rather than precise values. The accuracy of these estimates may be evaluated by comparing energies calculated for the geometrically close groups of configurations Ai corresponding to the possible ground states of the C5a receptors (see Table I). The rounded average energies calculated for Ai’s and their standard deviations were −553 ± 7 kcal/mol for WT, −544 ± 7 kcal/mol for NQ, −547 ± 12 kcal/mol for NQ-N296A, and −538 ± 5 kcal/mol for F251A. Accordingly, TM configurations differing in energies less than these standard deviations cannot be reliably ranked in the order of their energies; but the larger energy cut-off of 20 kcal/mol employed for selection of low-energy configurations in Tables II, III and V seems quite acceptable.
Also, it should be noted that due to limitations of our approach, geometrical and energetic differences between TM configurations differing by rotations of less than ca. 30° could be difficult to distinguish (see the Ai structures discussed in the above paragraph), which makes the choice of rotation grid less than 30° not reasonable.
Modeling results rationalize the data of mutagenesis for C5aR
Molecular modeling based on rotational TM sampling performed in the present study delineated several important elements in the structural mechanism of C5aR constitutive activation. First, it demonstrated that the activated state is stabilized by interactions involving the side chain of N296, specifically, by hydrogen bonding with D82. Second, it was shown that interactions involving the side chain of N119 stabilize the ground state of the receptor by hydrogen bonding with S85 and D82. Third, the modeling demonstrated that interactions between S131 and either C221 or R134 stabilize, respectively, either the ground state or the activated state of the receptors. And, fourth, the activated state accommodates the trans conformation of the side chain of F251, while in the ground state of WT this conformation may be hindered by unfavorable interaction with the side chains of I124 and L127.
These results are consistent with the fact that all constitutively active mutants of C5aR found by random saturation mutagenesis (RSM) in the TM helices included mutations of either L127, or F251 (the only exception was R306 mutant, where all mutations occurred in TM1 25). Though NQ (I124N/L127Q) was the most pronounced CAM both in the yeast and mammalian systems, some strong CAMs obtained in the RSM scans of TM3, such as R35 and R37 26, did not feature the additional mutations of I124. On the other hand, single mutations of L127 (as in L127Q 26 or in L127A 22) led to only weak CAMs. Abolishing the voluminous side chain of F251 in the F251A mutant yielded the strong CAM 26, whereas combining the mutations of L127 and F251 in NQ-F251N resulted in weaker basal activity than that of NQ 22.
While the importance of possible contact between L127 and F251 for receptor activation could be suggested on the basis of the initial rhodopsin-like 3D model of C5aR in the resting state (configuration Rh) 22, restoring of the wild-type phenotype in the NQ-N296A mutant did not have an adequate explanation other than general observation that N296 locates in the TM region of C5aR close to F251, which, in turn, is close to L127 29,59. Our current results in this study provide a plausible molecular mechanism for the role of N296A mutation in the NQ-N296A and highlight a role for N296 in stabilization of the activated state of wild-type C5aR by the hydrogen bond between the side chains of D82 and N296. According to our results, both the ground and activated states of the receptor are stabilized also by hydrogen bonding between N296 and Y300. Obviously, these hydrogen bonds cannot be maintained in NQ-N296A, or in the N296A mutant that was nonfunctional 29.
Possible alternative models of the activated state: 2RH1 and 3DQB
The X-ray structures of opsin co-crystallized with transducin peptide (3DQB) and β2 adrenergic receptor (2RH1) were earlier suggested as prototypes for the activated states of GPCRs 5,6. We have separately modeled two specific configurations of the TM region of the C5a receptors corresponding to these templates (configurations 3DQB and 2RH1) to evaluate whether they may serve as the alternative models of the CAM states.
The results of the full energy calculations for the two structures are listed in Table V. Only one structure, namely 3DQB, satisfied our general requirement that the activated states should be configurations with energies lower or comparable to those of the ground states for mutants NQ and F251A, but not for WT or NQ-N296A. It makes configuration 3DQB (but not 2RH1) an alternative possibility for the configuration of the activated state of C5aR. The corresponding trajectories of activation would be then A1–3,6 ↔ B ↔ C ↔ 3DQB for NQ and A1,3,4 ↔ C ↔ D ↔ 3QDB for F251A. Note that in both cases configuration 3QDB is connected to the ground states only via the CAM configurations suggested by rotational sampling (C for NQ and D for F251A). In fact, the TM rotations characteristic for configuration 3DQB were not that different from the CAM configurations, the maximal difference being in the rotation of TM3 by ca. 25° (compare Tables III and V).
Table V.
Configurations 3DQB and 2RH1
| Receptor, TM configuration | TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | Energy, kcal/mol | Connections | |
|---|---|---|---|---|---|---|---|---|---|---|
| WT | 3DQB | 0 | 0 | 35 | 0 | 45 | 20 | 0 | −548 | - |
| 2RH1 | 0 | 15 | 0 | 310 | 0 | 0 | 0 | −521 | A3 | |
| NQ | 3DQB | 0 | 0 | 35 | 0 | 45 | 20 | 0 | −562 | C |
| 2RH1 | 0 | 15 | 0 | 310 | 0 | 0 | 0 | −489 | A3 | |
| NQ-N296A | 3DQB | 0 | 0 | 35 | 0 | 45 | 20 | 0 | −521 | - |
| 2RH1 | 0 | 15 | 0 | 310 | 0 | 0 | 0 | −524 | - | |
| F251A | 3DQB | 0 | 0 | 35 | 0 | 45 | 20 | 0 | −541 | D |
| 2RH1 | 0 | 15 | 0 | 310 | 0 | 0 | 0 | −502 | - | |
The alternative models for the activated states of NQ and F251A represented by configurations 3DQB displayed different systems of hydrogen bonding between the side chains. Though the hydrogen bonding between the side chains of N296 and Y300 was present both in the CAM and 3DQB configurations, the 3DQB configurations featured also the potential hydrogen bonds between the β-amido group of N71 in TM2 and the β-carboxyl of D133 in TM3, as well as between the guanidino group of R206 in TM5 and the hydroxyl of Y258 in TM6, all of them were not present in the CAM configurations (see also Figure 4). Most importantly, the crucial hydrogen bonding between the side chains of D82 and N296 present in the CAM configurations was broken in configurations 3DQB, which means that the 3DQB model of the activated state of C5aR would not explain the loss of constitutive activity in NQ-N296A. Nevertheless, in our view, additional experimental data are still needed to further evaluate the 3DQB configurations as possible alternative models for the activated states of the constitutively active mutants of C5aR.
Figure 4.
Possible generalization of the modeling results over GPCR family
Our results singled out residues involved in residue-residue interactions that are especially important in conformational transition from the resting to the activated state of C5aR (see Table IV and Figures 1 and 2). Those were D822.50, S852.53, N1193.35, L1273.43, S1313.47, R1343.50, C2215.57, F2516.44, W2556.48, Q2596.49, N2967.49 and Y3007.50 (superscripts denote positions of the residues in TM helices according to the commonly used nomenclature 35). The modeling results were obtained without any a priori assumptions on functional importance of various residues in C5aR. Nevertheless, seven out of the twelve residues (shown in the bold font) are highly evolutionary conserved throughout the rhodopsin-like family of GPCRs 61. The presence of the conserved residues supports the generalization of the modeling results for the rhodopsin-like family of GPCRs.
For instance, the functional importance of interaction between D822.50 and N2967.49 in C5aR suggested by our modeling is also well-documented for many other GPCRs. 94% of GPCRs contain D in position 2.50 and 96% contain N or D in position 7.49 61. Several studies investigated mutations of these residues in various receptors introducing single D2.50 ↔N and N7.49 ↔D mutations as well as the reciprocal D2.50 ↔N/N7.49 ↔D mutation, which, in many cases, was capable of restoring functional activity impaired by the D2.50 ↔N or N7.49 ↔D mutations (see e.g., Ref 62 and references therein).
An additional structural feature revealed by our modeling was that interaction of the side chains of L1273.43 and F2516.44 influenced the transition from the ground to the activated states in WT C5aR. It is reasonable to expect that such transitions in other GPCRs could similarly be limited by unfavorable interactions of the side chains in positions 3.43 (90% of L, I or V 61) and 6.44 (88% F or Y 61). In C5aR, mutations in position 3.43 (such as L127Q) or in position 6.44 (such as F251A) led to constitutive active mutants; to our knowledge, residues in these cognate positions have not been mutated in other GPCRs with the exception of the muscarinic receptor, where mutations of F451 yielded constitutively active mutants 63. Also, though residue N1193.35 is not conserved, mutations of the cognate residue N1113.35 in the angiotensin receptor type 1 and the CXCR4 chemokine receptor led to the most potent constitutive active mutants for these receptors (see e.g., Ref. 21 and references therein).
Our results complement the concept of the “ionic lock” in GPCRs between highly conserved residues R3.50 and E/D6.30. The concept was originated by the data on mutations of highly conserved residue E6.30 in β2 adrenergic receptor that yielded several strong constitutively active mutants 64. In conjunction with the observation that the side chain of R3.50 was involved in the salt bridges with D3.49 and E6.30 in the X-ray structure of dark-adapted rhodopsin, it strongly suggested that breaking the ionic lock between R3.50 and E/D6.30 would lead to GPCR activation by allowing movement of the cytoplasmic part of TM6 away from TM3. Indeed, constitutively active mutants were obtained by mutations of E/D6.30 in the α1B adrenergic receptor 65, the 5-HT2A receptor 49, the TSH receptor 66, the LH receptor 67 and the FSH receptor 68. On the other hand, mutations of D3.49, which likely strengthen the R3.50 – E/D6.30 salt bridge by eliminating a negatively charged side chain competing with E/D6.30 for interaction with R3.50, also confer constitutive activity in α1B adrenergic receptor 69, the H2 receptor 70, β2 adrenergic receptor 71, the μ-opioid receptor 72 and the angiotensin receptor type 1 73. The very recent study on constitutive activation on histamine receptors also disputes the functional significance of the ionic lock of the R3.50 - E/D6.30 type 74.
For C5aR, the ionic lock of the R3.50 - E/D6.30 type does not exist since there is no negatively charged residue in the cytoplasmic part of TM6. This potential element of structural mechanism for activation would not exist also in many other GPCRs. Generally, at position 6.30 only 32% of GPCRs feature residues E or D, whereas 34% contain the positively charged residues K or R 61. Our modeling of the C5aR predicts that the side chain of R1343.50 rearranges in the activated state and forms a hydrogen bond with the side chain of S1313.47. Since 63% of GPCRs contain either S or T in position 3.47 and 96% contain R in position 3.50 61, this interaction between R3.50 – S/T3.47 may be a more general alternative to the ionic lock of the R3.50 - E/D6.30 type in GPCRs.
CONCLUDING REMARKS
Our simplified modeling approach applied to CAM and non-CAM C5a receptors revealed the likely conformational transitions in the TM regions of these receptors upon constitutive activation. Based on rotational TM sampling, structural mechanisms of C5aR activation can be inferred to involve a concerted rotation of several TM helices around their long axes. Our results showed also that, first, this rotation in the wild type receptor is hindered by interaction between the side chains of L127 in TM3 and F251 in TM6; second, that the ground state of C5aR is stabilized by hydrogen bonding between the side chains of N119 in TM3 and S85 (and D82) in TM2 as well as by hydrogen bonding between the side chains of S131 in TM3 and C221 in TM5; and, third, that the activated states of the constitutively active mutants, NQ and F251A, are stabilized by the hydrogen bond between the side chains of D82 in TM2 and N296 in TM7. We have shown also that the ground states of C5a receptors may be slightly different from that of the dark-adapted rhodopsin. These results were obtained by the de novo rotational TM sampling, where the only information employed a priori was that NQ and F251A were constitutively active mutants, and WT and NQ-N296A were not. Additionally, we have considered also the alternative models of the activated state of C5aR based on the X-ray structures of opsin.
The results of molecular modeling provided rationalization of the available data of mutagenesis in C5aR and offered the first specific molecular mechanism for the loss of the constitutively active phenotype in NQ upon addition of the N296A mutation. The procedure was also validated by correctly determining the N119S mutant as CAM and by distinguishing mutant R306 as CAM and very similar mutant R10 as non-CAM. Since the most important interactions revealed by molecular modeling in the C5a receptors occurred between residues that are evolutionary conserved in the rhodopsin-like family of GPCRs, the results provided also insights into the general structural mechanisms of GPCR activation. Our modeling emphasized the significance of the interaction between residues D2.50 and N7.49, which has been validated as important in many GPCRs. The modeling also provided a molecular rationale for the known constitutive activity in GPCRs that are mutated in positions 3.43 and 6.44, as well as in 3.35 (L127A, F251A, and N119S in the C5aR, respectively). Our modeling results contributed also to understanding the possible interactions of R3.50 upon activation in GPCRs that lack the “ionic lock”, R3.50 - E/D6.30. Finally, our calculations showed that while the ground states of the TM region of C5aR were connected to some other possible low-energy TM configurations through minimal TM rotations, the X-ray configuration of opsin remained isolated from other low-energy structures. In our view, it could indicate that different GPCRs possess different structural mechanisms of activation.
Acknowledgments
Grant sponsor: NIH; Grant numbers GM 71634 and GM63720.
References
- 1.Parnot C, Miserey-Lenkei S, Bardin S, Corvol P, Clauser E. Lessons from constutitively active mutants of G protein-coupled receptors. Trends in Endocrin Metabol. 2002;13:336–343. doi: 10.1016/s1043-2760(02)00628-8. [DOI] [PubMed] [Google Scholar]
- 2.Drews J. Drug discovery: A historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 3.Mustafi D, Palczewski K. Topology of class A G protein-coupled receptors: insights gained from crystal structures of rhodopsins, adrenergic and adenosine receptors. Mol Pharmacol. 2009;75(1):1–12. doi: 10.1124/mol.108.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci U S A. 2006;103(44):16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455(7212):497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 7.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322(5905):1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. Electron crystallography reveals the structure of metarhodopsin I. Embo J. 2004;23(18):3609–3620. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hubbell WL, Altenbach C, Khorana HG. Rhodopsin Structure, Dynamics and Activation. Adv Protein Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- 10.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci U S A. 2008;105(21):7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nikiforovich GV, Marshall GR. 3D Model for Meta-II Rhodopsin, An Activated G-Protein Coupled Receptor. Biochemistry. 2003;42:9110–9120. doi: 10.1021/bi034586o. [DOI] [PubMed] [Google Scholar]
- 12.Borhan B, Souto ML, Imai H, Shichida Y, Nakanishi K. Movement of retinal along the visual transduction path. Science. 2000;288(5474):2209–2212. doi: 10.1126/science.288.5474.2209. [DOI] [PubMed] [Google Scholar]
- 13.Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK. Agonist-induced conformational changes in the G-protein-coupling domain of the β2 adrenergic receptor. Proc Natl Acad Sci U S A. 2001;98:5997–6002. doi: 10.1073/pnas.101126198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grossfield A, Feller SE, Pitman MC. Convergence of molecular dynamics simulations of membrane proteins. Proteins. 2007;67(1):31–40. doi: 10.1002/prot.21308. [DOI] [PubMed] [Google Scholar]
- 15.Dror RO, Arlow DH, Borhani DW, Jensen MO, Piana S, Shaw DE. Identification of two distinct inactive conformations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc Natl Acad Sci U S A. 2009;106(12):4689–4694. doi: 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K, Pitman MC, Reggio PH. A lipid pathway for ligand binding is necessary for a cannabiniod G protein-coupled receptor. J Biol Chem. 2010 doi: 10.1074/jbc. M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto ML, Narzinski K, Nikiforovich GV, Baranski TJ. A comprehensive structure-function map of the intracellular surface of the human C5a receptor. II. Elucidation of G protein specificity determinants. J Biol Chem. 2007;282(5):3122–3133. doi: 10.1074/jbc.M607683200. [DOI] [PubMed] [Google Scholar]
- 18.Klco JM, Nikiforovich GV, Baranski TJ. Genetic Analysis of the First and Third Extracellular Loops of the C5a Receptor Reveals an Essential WXFG Motif in the First Loop. J Biol Chem. 2006;281:12010–12019. doi: 10.1074/jbc.M600548200. [DOI] [PubMed] [Google Scholar]
- 19.Nikiforovich GV, Marshall GR, Baranski TJ. Modeling Molecular Mechanisms of Binding of the Anaphylotoxin C5a to the C5a Receptor. Biochemistry. 2008;47:3117–3130. doi: 10.1021/bi702321a. [DOI] [PubMed] [Google Scholar]
- 20.Nikiforovich GV, Baranski TJ. Structural models for the complex of chemotaxis inhibitory protein of Staphylococcus aureus with the C5a receptor. Biochem Biophys Res Commun. 2009;390:481–484. doi: 10.1016/j.bbrc.2009.09.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikiforovich GV, Mihalik B, Catt KJ, Marshall GR. Molecular mechanisms of constitutive activity: mutations at position 111 of the angiotensin AT1 receptor. J Pept Res. 2005;66:236–248. doi: 10.1111/j.1399-3011.2005.00293.x. [DOI] [PubMed] [Google Scholar]
- 22.Sen S, Baranski TJ, Nikiforovich GV. Conformational Movement of F251A Contributes to the Molecular Mechanism of Constitutive Activation in the C5a receptor. Chemical Biology & Drug Design. 2008;71:197–204. doi: 10.1111/j.1747-0285.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- 23.Nikiforovich GV, Taylor CM, Marshall GR, Baranski TJ. Modeling the possible conformations of the extracellular loops in G-protein-coupled receptors. Proteins: Structure, Function, and Genetics. 2010;78:271–285. doi: 10.1002/prot.22537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gether U. Uncovering Molecular Mechanisms Involved in Activation of G Protein-Coupled Receptors. Endocrin Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 25.Geva A, Lassere TB, Lichtarge O, Pollitt SK, Baranski TJ. Genetic mapping of the human C5a receptor: Identification of transmembrane amino acids critical for receptor function. J Biol Chem. 2000;275:35393–35401. doi: 10.1074/jbc.M005602200. [DOI] [PubMed] [Google Scholar]
- 26.Baranski TJ, Herzmark P, Lichtarge O, Gerber BO, Trueheart J, Meng EC, Iiri T, Sheikh SP, Bourne HR. C5a receptor activation. Genetic identification of critical residues in four transmembrane helices. J Biol Chem. 1999;274(22):15757–15765. doi: 10.1074/jbc.274.22.15757. [DOI] [PubMed] [Google Scholar]
- 27.Klco JM, Wiegand CB, Narzinski K, Baranski TJ. Essential role for the second extracellular loop in C5a receptor activation. Nature Struct Mol Biol. 2005;12:320–326. doi: 10.1038/nsmb913. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto ML, Narzinski K, Kiser PD, Nikiforovich GV, Baranski TJ. A comprehensive structure-function map of the intracellular surface of the human C5a receptor. I. Identification of critical residues. J Biol Chem. 2007;282(5):3105–3121. doi: 10.1074/jbc.M607679200. [DOI] [PubMed] [Google Scholar]
- 29.Whistler JL, Gerber BO, Meng E, Baranski TJ, von Zastrow M, Bourne HR. Constitutive Activation and Endocytosis of the Complement Factor 5a Receptor: Evidence for Multiple Activated Conformations of a G Protein-Coupled Receptor. Traffic. 2002;3:866–877. doi: 10.1034/j.1600-0854.2002.31203.x. [DOI] [PubMed] [Google Scholar]
- 30.Nikiforovich GV, Marshall GR. Modeling Flexible Loops in the Dark-Adapted and Activated States of Rhodopsin, a Prototypical G-Protein-Coupled Receptor. Biophys J. 2005;89:3780–3789. doi: 10.1529/biophysj.105.070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunfield LG, Burgess AW, Scheraga HA. Energy Parameters in Polypeptides. 8. Empirical Potential Energy Algorithm for the Conformational Analysis of Large Molecules. J Phys Chem. 1978;82:2609–2616. [Google Scholar]
- 32.Nemethy G, Pottle MS, Scheraga HA. Energy Parameters in Polypeptides. 9. Updating of Geometrical Parameters, Nonbonded Interactions, and Hydrogen Bond Interactions for the Naturally Occurring Amino Acids. J Phys Chem. 1983;87:1883–1887. [Google Scholar]
- 33.Nikiforovich GV, Hruby VJ, Prakash O, Gehrig CA. Topographical Requirements for Delta-Selective Opioid Peptides. Biopolymers. 1991;31(8):941–955. doi: 10.1002/bip.360310804. [DOI] [PubMed] [Google Scholar]
- 34.Nikiforovich GV, Marshall GR. 3D modeling of the activated states of constitutively active mutants of rhodopsin. Biochem Biophys Res Comm. 2006;345:430–437. doi: 10.1016/j.bbrc.2006.04.058. [DOI] [PubMed] [Google Scholar]
- 35.Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60(1):1–19. [PubMed] [Google Scholar]
- 36.Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi H-J, Thian FS, Kobilka TS, Puglisi JD, Weis WI, Pardo L, Prosser RS, Mueller L, Kobilka BK. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463 doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nikiforovich GV, Marshall GR. Computational Approaches in Peptide and Protein Design: An Overview. In: Jensen K, editor. Peptide and Protein Design for Biopharmaceutical Applications. NY: John Wiley & Sons; 2009. pp. 5–48. [Google Scholar]
- 38.Fernandez-Recio J, Abagyan R, Totrov M. Improving CAPRI predictions: optimized desolvation for rigid-body docking. Proteins. 2005;60(2):308–313. doi: 10.1002/prot.20575. [DOI] [PubMed] [Google Scholar]
- 39.Katritch V, Rueda M, Lam PC, Yeager M, Abagyan R. GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins. 2010;78(1):197–211. doi: 10.1002/prot.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pogozheva ID, Przydzial MJ, Mosberg HI. Homology modeling of opioid receptor-ligand complexes using experimental constraints. AAPS Journal. 2005;7(2):E434–448. doi: 10.1208/aapsj070243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sale K, Faulon JL, Gray GA, Schoeniger JS, Young MM. Optimal bundling of transmembrane helices using sparse distance constraints. Protein Sci. 2004;13(10):2613–2627. doi: 10.1110/ps.04781504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shacham S, Marantz Y, Bar-Haim S, Kalid O, Warshaviak D, Avisar N, Inbal B, Heifetz A, Fichman M, Topf M, Naor Z, Noiman S, Becker OM. PREDICT modeling and in-silico screening for G-protein coupled receptors. Proteins. 2004;57(1):51–86. doi: 10.1002/prot.20195. [DOI] [PubMed] [Google Scholar]
- 43.Vaidehi N, Floriano WB, Trabanino R, Hall SE, Freddolino P, Choi EJ, Zamanakos G, Goddard WA., 3rd Prediction of structure and function of G protein-coupled receptors. Proc Natl Acad Sci U S A. 2002;99(20):12622–12627. doi: 10.1073/pnas.122357199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, DeVries ME, Skolnick J. Structure Modeling of All Identified G Protein-Coupled Receptors in the Human Genome. PLoS Computational Biology. 2006;2(2):e13. doi: 10.1371/journal.pcbi.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gether U, Lin S, Ghanouni P, Ballesteros JA, Weinstein H, Kobilka BK. Agonists induce conformational changes in transmembrane domains III and VI of the beta2 adrenoceptor. Embo J. 1997;16(22):6737–6747. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jensen AD, Guarnieri F, Rasmussen SG, Asmar F, Ballesteros JA, Gether U. Agonist-induced conformational changes at the cytoplasmic side of transmembrane segment 6 in the beta 2 adrenergic receptor mapped by site-selective fluorescent labeling. J Biol Chem. 2001;276(12):9279–9290. doi: 10.1074/jbc.M004871200. [DOI] [PubMed] [Google Scholar]
- 47.Sheikh SP, Vilardarga JP, Baranski TJ, Lichtarge O, Iiri T, Meng EC, Nissenson RA, Bourne HR. Similar structures and shared switch mechanisms of the beta2-adrenoceptor and the parathyroid hormone receptor. Zn(II) bridges between helices III and VI block activation. J Biol Chem. 1999;274(24):17033–17041. doi: 10.1074/jbc.274.24.17033. [DOI] [PubMed] [Google Scholar]
- 48.Sylte I, Bronowska A, Dahl SG. Ligand induced conformational states of the 5-HT(1A) receptor. European Journal of Pharmacology. 2001;416(1–2):33–41. doi: 10.1016/s0014-2999(01)00860-3. [DOI] [PubMed] [Google Scholar]
- 49.Shapiro DA, Kristiansen K, Weiner DM, Kroeze WK, Roth BL. Evidence for a model of agonist-induced activation of 5-hydroxytryptamine 2A serotonin receptors that involves the disruption of a strong ionic interaction between helices 3 and 6. J Biol Chem. 2002;277(13):11441–11449. doi: 10.1074/jbc.M111675200. [erratum appears in J Biol Chem 2002 May 17;277(20):18244] [DOI] [PubMed] [Google Scholar]
- 50.Khorana HG. Molecular Biology of Light Transduction by the Mammalian Photoreceptor, Rhodopsin. J Biomol Struct Dynamics 2000;Conversation. 11:1–16. doi: 10.1080/07391102.2000.10506598. [DOI] [PubMed] [Google Scholar]
- 51.Elling CE, Thirstrup K, Holst B, Schwartz TW. Conversion of agonist site to metal-ion chelator site in the beta(2)-adrenergic receptor. Proc Natl Acad Sci U S A. 1999;96(22):12322–12327. doi: 10.1073/pnas.96.22.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holst B, Elling CE, Schwartz TW. Partial agonism through a zinc-Ion switch constructed between transmembrane domains III and VII in the tachykinin NK(1) receptor. Mol Pharmacol. 2000;58(2):263–270. doi: 10.1124/mol.58.2.263. [DOI] [PubMed] [Google Scholar]
- 53.Porter JE, Perez DM. Characteristics for a salt-bridge switch mutation of the alpha(1b) adrenergic receptor. Altered pharmacology and rescue of constitutive activity. J Biol Chem. 1999;274(49):34535–34538. doi: 10.1074/jbc.274.49.34535. [DOI] [PubMed] [Google Scholar]
- 54.Gerber BO, Meng EC, Dotsch V, Baranski TJ, Bourne HR. An activation switch in the ligand binding pocket of the C5a receptor. J Biol Chem. 2001;276(5):3394–3400. doi: 10.1074/jbc.M007748200. [DOI] [PubMed] [Google Scholar]
- 55.Donnelly D, Maudsley S, Gent JP, Moser RN, Hurrell CR, Findlay JB. Conserved polar residues in the transmembrane domain of the human tachykinin NK2 receptor: functional roles and structural implications. Biochemical Journal. 1999;339(Pt 1):55–61. [PMC free article] [PubMed] [Google Scholar]
- 56.Govaerts C, Lefort A, Costagliola S, Wodak SJ, Ballesteros JA, Van Sande J, Pardo L, Vassart G. A conserved Asn in transmembrane helix 7 is an on/off switch in the activation of the thyrotropin receptor. J Biol Chem. 2001;276(25):22991–22999. doi: 10.1074/jbc.M102244200. [DOI] [PubMed] [Google Scholar]
- 57.Miura S, Karnik SS. Constitutive Activation of Angiotensin II Type 1 Receptor Alters the Orientation of Transmembrane Helix-2. J Biol Chem. 2002;277:24299–24305. doi: 10.1074/jbc.M202743200. [DOI] [PubMed] [Google Scholar]
- 58.Domazet I, Martin SS, Holleran BJ, Morin M-E, Lacasse P, Lavigne P, Escher E, Leduc R, Guillemette G. The fifth transmembrane domain of angiotensin II type I receptor participates in the formation of the ligand binding pocket and undergoes a counterclockwise rotation upon receptor activation. J Biol Chem. 2009;284:31953–31961. doi: 10.1074/jbc.M109.051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hagemann IS, Nikiforovich GV, Baranski TJ. Comparison of the retinitis pigmentosa mutations in rhodopsin with a functional map of the C5a receptor. Vision Res. 2006;46(27):4519–4531. doi: 10.1016/j.visres.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 60.Rana S, Baranski TJ. The Third Extracellular Loop (EC3)-N terminus Interaction is Important for 7TM Receptor Function: Implications for an Activation Microswitch Region. J Biol Chem. 2010 doi: 10.1074/jbc. M1110.129213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mirzadegan T, Benko G, Filipek S, Palczewski K. Sequence Analyses of G-Protein Coupled Receptors: Similarities to Rhodopsin. Biochemistry. 2003;42:2769–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nikiforovich GV, Zhang M, Yang Q, Jagadeesh G, Chen HC, Hunyady L, Marshall GR, Catt KJ. Interactions between conserved residues in transmembrane helices 2 and 7 during angiotensin AT1 receptor activation. Chemical Biology & Drug Design. 2006;68(5):239–249. doi: 10.1111/j.1747-0285.2006.00444.x. [DOI] [PubMed] [Google Scholar]
- 63.Spalding TA, Burstein ES, Henderson SC, Ducote KR, Brann MR. Identification of a ligand-dependent switch within a muscarinic receptor. J Biol Chem. 1998;273(34):21563–21568. doi: 10.1074/jbc.273.34.21563. [DOI] [PubMed] [Google Scholar]
- 64.Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276(31):29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 65.Greasley PJ, Fanelli F, Rossier O, Abuin L, Cotecchia S. Mutagenesis and modelling of the alpha(1b)-adrenergic receptor highlight the role of the helix 3/helix 6 interface in receptor activation. Mol Pharmacol. 2002;61(5):1025–1032. doi: 10.1124/mol.61.5.1025. [DOI] [PubMed] [Google Scholar]
- 66.Parma J, Duprez L, Van Sande J, Cochaux P, Gervy C, Mockel J, Dumont J, Vassart G. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;365(6447):649–651. doi: 10.1038/365649a0. [DOI] [PubMed] [Google Scholar]
- 67.Kosugi S, Mori T, Shenker A. The role of Asp578 in maintaining the inactive conformation of the human lutropin/choriogonadotropin receptor. J Biol Chem. 1996;271(50):31813–31817. doi: 10.1074/jbc.271.50.31813. [DOI] [PubMed] [Google Scholar]
- 68.Montanelli L, Van Durme JJ, Smits G, Bonomi M, Rodien P, Devor EJ, Moffat-Wilson K, Pardo L, Vassart G, Costagliola S. Modulation of ligand selectivity associated with activation of the transmembrane region of the human follitropin receptor. Molecular Endocrinology. 2004;18(8):2061–2073. doi: 10.1210/me.2004-0036. [DOI] [PubMed] [Google Scholar]
- 69.Scheer A, Fanelli F, Costa T, De Benedetti PG, Cotecchia S. The activation process of the alpha1 β-adrenergic receptor: potential role of protonation and hydrophobicity of a highly conserved aspartate. Proc Natl Acad Sci U S A. 1997;94(3):808–813. doi: 10.1073/pnas.94.3.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alewijnse AE, Timmerman H, Jacobs EH, Smit MJ, Roovers E, Cotecchia S, Leurs R. The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol Pharmacol. 2000;57(5):890–898. [PubMed] [Google Scholar]
- 71.Rasmussen SG, Jensen AD, Liapakis G, Ghanouni P, Javitch JA, Gether U. Mutation of a highly conserved aspartic acid in the beta2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol Pharmacol. 1999;56(1):175–184. doi: 10.1124/mol.56.1.175. [DOI] [PubMed] [Google Scholar]
- 72.Li J, Huang P, Chen C, de Riel JK, Weinstein H, Liu-Chen LY. Constitutive activation of the mu opioid receptor by mutation of D3.49(164), but not D3.32(147): D3.49(164) is critical for stabilization of the inactive form of the receptor and for its expression. Biochemistry. 2001;40(40):12039–12050. doi: 10.1021/bi0100945. [DOI] [PubMed] [Google Scholar]
- 73.Gaborik Z, Jagadeesh G, Zhang M, Spat A, Catt KJ, Hunyady L. The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. Endocrinology. 2003;144(6):2220–2228. doi: 10.1210/en.2002-0135. [DOI] [PubMed] [Google Scholar]
- 74.Schneider EH, Schnell D, Strasser A, Dove S, Seifert R. Impact of the DRY Motif and the Missing “Ionic Lock” on Constitutive Activity and G-Protein Coupling of the Human Histamine H4 Receptor. Journal of Pharmacology and Experimental Therapeutics. 2010 doi: 10.1124/jpet.1109.163220. [DOI] [PubMed] [Google Scholar]