

Abstract
Integrase (IN) represents a clinically validated target for the development of antivirals against human immunodeficiency virus (HIV). Inhibitors with a novel structure core are essential for combating resistance associated with known IN inhibitors (INIs). We have previously disclosed a novel dual inhibitor scaffold of HIV IN and reverse transcriptase (RT). Here we report the complete structure-activity relationship (SAR), molecular modeling and resistance profile of this inhibitor type on IN inhibition. These studies support an antiviral mechanism of dual inhibition against both IN and RT and validate 3-hydroxypyrimidine-2,4-diones as an IN inhibitor scaffold.
Introduction
HIV infection and the establishment of proviral latency depend critically on the insertion of viral DNA into host genome.1–3 This integration process comprises two distinct steps: 3′ processing (3′P) in which a sequence-specific endonucleolytic cleavage of the viral DNA generates a CA-OH at the 3′ end; and strand transfer (ST) where the processed viral DNA is inserted into the host genome through a transesterification reaction using the 3′ CA-OH as the nucleophile.4 The virally encoded enzyme IN catalyzes both reactions; thus represents an important target for HIV chemotherapeutic intervention. IN as an antiviral target is rendered particular attractive by the lack of host cellular counterpart. On the other hand, IN uses the same active site (DD35E motif) for both steps where either a viral DNA (in 3′P) or a cellular DNA (in ST) serves as the endogenous substrate. Therefore, inhibitors of IN could also benefit from a potentially high genetic barrier to resistance development. In the past two decades, IN-targeted antiviral research has led to the identification of various inhibitor types, of which the most prominent scaffolds all feature a diketoacid (DKA) functionality or its heterocyclic bioisosteres.5–9 These chemotypes can accommodate highly selective ST inhibition and have played an essential role in unraveling the pharmacophore requirements for IN binding.10–15 Extensive research on these chemotypes culminated with a number of investigational drugs (Figure 1), including raltegravir (1)16, 17, the first and only INI presently approved for clinical use; elvitegravir (2)18, a promising candidate in phase III development; and S/GSK-1349572 (3)19, 20, a second-generation IN inhibitor in phase IIb trials.21 In the meantime, HIV integration also involves a cellular co-factor LEDGF/p75.22–24 Targeting the protein-protein interaction between IN and LEDGF/p75 represents an emerging approach to HIV chemotherapeutic intervention.25 Collectively inhibitors targeting IN can substantially enhance the current highly active anti-retroviral therapy (HAART), the standard chemotherapy for HIV/AIDS.26 However, mutations conferring resistance to these inhibitors have been generated in cell culture and emerged in clinical studies27–29, suggesting that INIs will be confronted with the same resistance issue that has plagued HIV/AIDS chemotherapy. Continued search for structurally novel INIs to combat resistance is therefore urgently needed. Previously we have reported that N-3 hydroxylation of pyrimidine-2,4-diones yielded inhibitors dually active against HIV reverse transcriptase (RT) and IN (4, figure 1).30 Since this scaffold represents a novel chemotype for INIs, further SAR efforts on IN inhibition are warranted.
Figure 1.

Structures of best known INIs (1–3) and a novel INI (4).
HIV IN is a 32-kDa protein encoded by viral pol gene consisting of three functional domains31: the N-terminal domain (NTD) with a conserved “HH-CC” zinc-binding motif; the catalytic core domain (CCD) containing the key D64-D116-E152 catalytic triad; and the C-terminal domain (CTD) important for DNA binding. Crystal structures of each single domain as well as double domains have been resolved32–34, albeit without the binding of viral DNA substrate. Detailed understanding on HIV IN catalysis requires structural information on a DNA-bound full-length IN complex, or an intasome, which currently remains elusive due to major technical barriers35 in obtaining stable HIV intasome. Recently disclosed crystal structures of a homologous prototype foamy virus (PFV) intasome36, 37 have revealed the requisite catalytic molecular network comprising IN, viral and/or host DNA substrates and the inhibitor, allowing a largely improved understanding on the mechanism of catalysis and inhibition of retroviral IN. Homologous models38, 39 constructed based on these PFV crystal structures have provided valuable details into HIV IN mechanism of action and formed the basis for structure-based INI design. Significantly these models corroborate the minimal pharmacophore embedded in major INIs: a chelating triad capable of binding two Mg2+ ions, and a hydrophobic benzyl moiety. Our novel IN inhibitors are constructed around a pyrimidine-2,4-dione ring, a pharmacophore core that has found wide applications in medicinal chemistry.40–43 Central to our design is to introduce an OH group on the N-3 position which, along with the two flanking carbonyl groups, would yield a chelating triad of C(O)N(OH)C(O) that mimics a DKA functionality (Figure 2, chelating domain). On the other hand, the hydrophobic benzyl moiety can be easily installed via an N-1 alkylation, and the mono- or di-halogenation of this terminal benzyl group will allow us to explore the halogen effect IN inhibition (Figure 2, aromatic domain). Furthermore, to ensure that this benzyl group can fit in the hydrophobic cavity, its spatial placement over the chelating triad has to be optimized. This can be achieved through a linker of different length (Figure 2, linker domain). Finally, the overall hydrophobicity of the inhibitor could be modified by altering the C5 and C6 substituents (Figure 2, hydrophobic domain).
Figure 2.
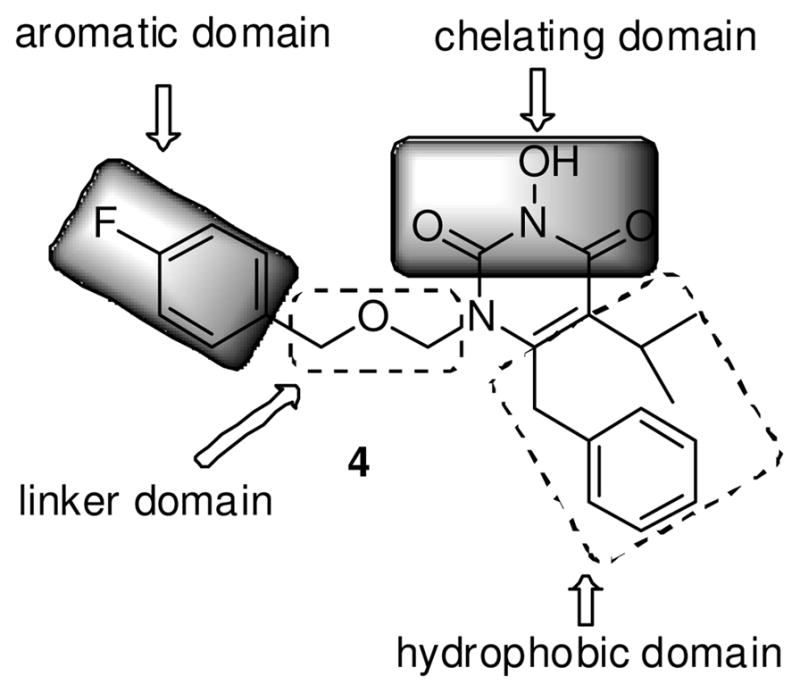
SAR design of lead compound 4. Major structural domains are highlighted.
Results and Discussion
Chemistry
The chemical synthesis of aromatic and chelating domains starts with a known 5,6-disubstituted pyrimidine-2,4-dione 544, 45 (Scheme 1). Subsequent selective N-1 alkylation of 5 via a bis-silylated pyrimidine intermediate with an in situ generated chloromethyl ether allows the incorporation of the aromatic domain with an ether linkage (6–10, Scheme 1). As previously reported 30, the key N-3 hydroxylation was achieved via a base-mediated meta-chloroperbenzoic acid (mCPBA) oxidation. To assess the significance of this hydroxyl group on IN inhibition, an N-3 amino analogue (15) as well as analogues with an N-3 methyl or methoxy group were also synthesized (10 and 14, Scheme 1).
Scheme 1.

a Synthesis of aromatic and chelating domains
aReagents and conditions: a) CH3C(OTMS)=NTMS (BSA), substituted benzyl alcohol, (HCHO)n, TMSCl, TBAI ( cat.), CH2Cl2, rt, 70–90%; b) Cs2CO3, THF, MeI, 40 °C, 53–63%; c) NaH, m-CPBA, THF, rt, 53–85%; d) NaH, MSH, THF, rt, 54%.
The synthesis of the linker domain is described in Scheme 2. Analogues with an ether linkage were easily prepared through a selective N-1 alkylation with a chloromethyl ether (Scheme 2, B). The requisite alcohols (19–21) were synthesized from commercial carboxylic acids via a direct reduction with LAH. The construction of all-carbon tethers in the linker domain, however, proved much more challenging. The BSA-assisted selective N-1 alkylation of pyrimidine-2,4-diones generally involves a chloromethyl ether as the alkylating agent. Same reaction with benzyl bromide was effected only under forcing conditions (microwave, high temperature and long reaction time; see Scheme 2, C). Generic alkyl halides fail to undergo alkylation with the bis-silylated pyrimidine intermediate, and base-mediated direct alkylation normally leads to a mixture of region-isomers. As it turned out, the introduction of all-carbon chain into N-1 requires multiple protection and deprotection steps (Scheme 2, D). In this event, the N-1 position was first protected with an easily introduced ethoxymethyl group, which allowed the protection of N-3 position with an acetophenone group. The N-1 ether linkage was then cleaved under acidic condition to yield the N-3 protected intermediate 35. Base-mediated direct alkylation led to the incorporation of the carbon tethers followed by a Mg mediated N-3 deprotection to yield key N-1 alkylated intermediates 39–41. All N-1 alkylated pyrimidine-2,4-diones were then oxidized with mCPBA to produce desired N-3 hydroxylated compounds (Scheme 2).
Scheme 2.

a Synthesis of linker domain
aReagents and conditions: a) LiAlH4, THF, rt, 67–72%; b) CBr4, PPh3, THF, rt, 73–80%; c) CH3C(OTMS)=NTMS (BSA), CH2Cl2; 19–21, (HCHO)n, TMSCl, TBAI ( cat.), rt, 83–89%; d) NaH, m-CPBA, THF, rt, 62–72%; e) BSA, benzyl bromide, chlorobenzene, microwave, 160 °C, 50%; f) BSA, chloromethyl ethyl ether, TBAI ( cat.), CH2Cl2, rt, 89%; g) K2CO3, PhCOCH2Br, MeCN, reflux, 84%; h) TFA:H2O (9:1), reflux, 76%; i) Cs2CO3, 4-F-Ph(CH2)nBr, DMF, 80 °C, 31–63%; j) Mg, AcOH, MeOH, rt, 48–57%.
The synthesis of hydrophobic domain with variable C-5 and C-6 substituents (Scheme 3, A) was achieved through a similar N-3 hydroxylation of N-1 alkylated intermediates (45–50 and 57–63). Compounds with a slightly different scaffold, the 5,6-benzopyrimidine-2,4-diones (75–76), were also prepared to probe the effect of a fused benzene ring on IN inhibition. These analogues were synthesized via a route depicted in Scheme 3, C.
Scheme 3.

a Synthesis of hydrophobic domain
aReagents and conditions: a) NaH, m-CPBA, THF, rt 32–72%; b) (i) CDI/toluene, reflux; then NH2OBn, reflux; (ii) NaOH/EtOH, reflux, 53%; c) benzyl bromide or 4-Flurobenzyl bromide, Cs2CO3, DMF, 80 °C, 58–67%; d) HBr (48% in H2O), reflux, 77–78%.
SAR on IN Inhibition
All synthetic analogues were first tested against recombinant HIV-1 IN using a gel assay. Both 3′P and ST functions were evaluated. As clearly shown in Tables 1–3, our scaffold tends to selectively inhibit ST as none of the analogues demonstrated inhibitory activity against 3′P.
Table 1.
Effects of aromatic and chelating domains on IN inhibition
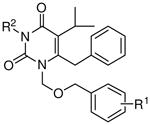 | ||||
|---|---|---|---|---|
| Compd | R1 | R2 | IN IC50 (μM)a |
|
| 3′P | STb | |||
| 11c | H | OH | >111 | 21 ± 2 |
| 12 | 2,4-F | OH | >111 | 4.8 ± 0.7 |
| 13 | 3-Cl-2-F | OH | >111 | 2.3 ± 0.6 |
| 4c | 4-F | OH | >111 | 3.5 ± 0.6 |
| 9 | 4-F | H | >111 | >111 |
| 10 | 4-F | Me | >111 | >111 |
| 14 | 4-F | OMe | >111 | >111 |
| 15c | H | NH2 | >111 | >111 |
Concentration inhibiting enzymatic function by 50%.
Mean value ± standard deviation from triplicate experiments.
Ref 30.
Table 3.
Effects of hydrophobic domain on IN inhibition
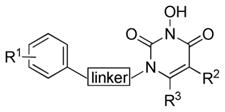 | ||||||
|---|---|---|---|---|---|---|
| Compd | R1 | linker | R2 | R3 | IN IC50 (μM)a |
|
| 3′P | STb | |||||
| 11c | H | CH2OCH2 | iPr | benzyl | >111 | 21 ± 2 |
| 51 | H | CH2OCH2 | iPr | 4-Fbenzyl | >111 | 7.3 ± 0.8 |
| 52 | F | CH2OCH2 | iPr | 4-Fbenzyl | >111 | 5.5 ± 0.7 |
| 53 | H | CH2OCH2 | Et | benzyl | >111 | 8.2 ± 1.0 |
| 54 | H | CH2OCH2 | Me | H | >111 | 75 ± 7 |
| 55 | H | CH2OCH2 | benzyl | Et | >111 | 36 ± 6 |
| 56 | F | CH2OCH2 | benzyl | Et | >111 | 11 ± 0.8 |
| 64 | H | CH2 | iPr | 4-Fbenzyl | >111 | 32 ± 6 |
| 65 | F | CH2 | iPr | 4-Fbenzyl | >111 | 73 ± 12 |
| 66 | H | CH2 | iPr | benzyl | >111 | 28 ± 2 |
| 67 | H | CH2 | Et | benzyl | >111 | >111 |
| 68 | F | CH2 | Et | benzyl | >111 | 24 ± 3 |
| 69 | H | CH2 | Me | H | >111 | >111 |
| 70 | F | CH2 | Me | H | >111 | >111 |
| 75 | H | CH2 | benzo (fused) | >111 | >111 | |
| 76 | F | CH2 | benzo (fused) | >111 | >111 | |
Concentration inhibiting enzymatic function by 50%.
Mean value ± standard deviation from triplicate experiments.
Ref 30.
Aromatic and Chelating Domains
We have shown previously30 that removing the phenyl ring at the end of the N-1 pendant resulted in a significant loss of inhibition against IN, and that without the OH group the N(3)H analogue is completely inactive against IN. These findings corroborate the pharmacophore hypothesis that HIV-1 IN inhibition requires minimally a chelating domain and an aromatic domain. In this study, additional analogues were synthesized to further study these two domains. As shown in Table 1 and Figure 3, halogen substitution of the N-1 benzyl seems to significantly benefit IN binding. A fluorination at the para position (inhibitor 4) leads to a 6-fold improvement in IN inhibition over the unsubstituted compound 11. Further improvement in activity was only marginal with a 3-chloro-2-fluoro substituted analogue 13, whereas a 2,4-difluoro compound 12 showed slightly reduced inhibition when compared to 4 (Table 1). On the other hand, replacing the N-3 OH group with H, Me, OMe or even NH2 yielded compounds completely devoid of inhibitory activity against IN (9–10 and 14–15, Table 1), confirming the critical requirement of the N-3 OH for chelating Mg2+. The lack of potency with the amino analogue 15 also reflects an inferior chelating ability of NH2 for Mg2+ when compared to OH.30
Figure 3.
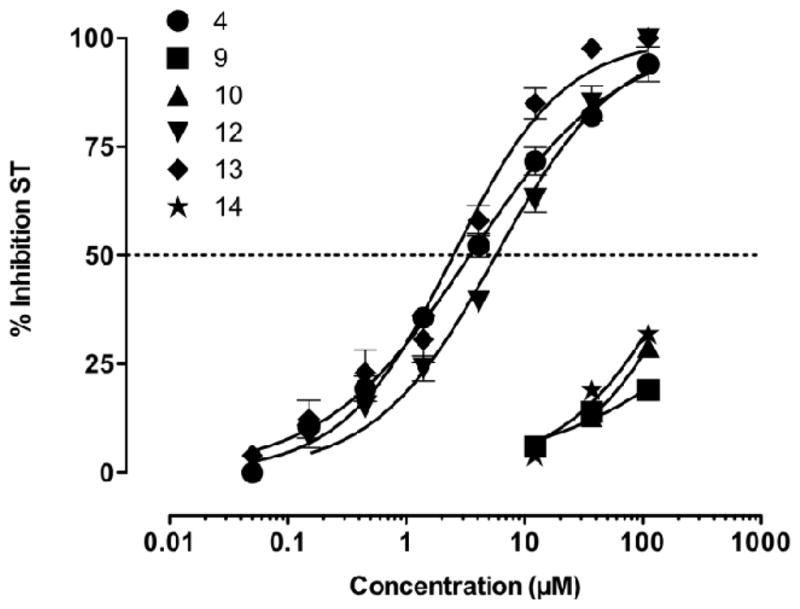
Dose response curves of compounds 4, 9–10 and 12–14.
The linker domain
Having established the chelating triad and the N-1 benzyl group as the two major structural determinants of our novel INI scaffold, our SAR efforts were then centered around the linker domain as it dictates the angle and distance between the chelator and benzyl group. Toward this end, we have designed linkers of different nature and length. Compounds 4 and 28–30 all have an ether linkage with incremental methylene groups (Table 2). Interestingly, these analogues all exhibited inhibitory activity in ST assay at low micromolar range, and demonstrated a discernible binding dependence on the length of the linker. As shown in Table 2, the 4-atom linker (CH2)2OCH2 (inhibitor 28) conferred the highest inhibition within this series, and a reduced inhibition was achieved with compounds having a shorter (4) or longer linker (29–30). A similar albeit more sensitive trend was observed with compounds having an all carbon N-1 linker. In this case, short linkages, such as CH2 (32) and (CH2)2 (42), appeared to pose severe binding difficulties while the 3- and 4-atom linkers (CH2)3 (43) and (CH2)4 (44) conferred a drastically improved inhibition against ST (Table 2). These findings strongly imply that the optimal IN inhibition entails a linker of 3–4 atoms. The nature of the linker (ether linkage vs all-carbon linkage) was also found to substantially impact IN inhibition as inhibitors 43 and 44 demonstrated significantly better inhibition than compound 28. Detailed docking studies on the inhibitor binding may provide a molecular basis for this observation.
Table 2.
Effects of linker domain on IN inhibition
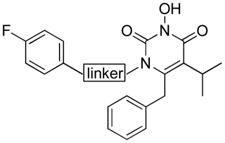 | |||
|---|---|---|---|
| Compd | Linker | IN IC50 (μM)a |
|
| 3′P | STb | ||
| 4c | CH2OCH2 | >111 | 3.5 ± 0.6 |
| 28 | (CH2)2OCH2 | >111 | 1.6 ± 0.2 |
| 29 | (CH2)3OCH2 | >111 | 3.2 ± 0.5 |
| 30 | (CH2)4OCH2 | >111 | 4.5 ± 0.4 |
| 32 | CH2 | >111 | 22 ± 0.5 |
| 42 | (CH2)2 | >111 | 46 ± 6 |
| 43 | (CH2)3 | >111 | 0.44 ± 0.1 |
| 44 | (CH2)4 | >111 | 0.56 ± 0.04 |
Concentration inhibiting enzymatic function by 50%.
Mean value ± standard deviation from triplicate experiments.
Ref 30.
The hydrophobic domain
Synthetic analogues in these domains vary mainly in the size of the C-5 alkyl group (iPr, Et and Me) and fluorine substitution on the C-6 benzyl. These changes impact most likely the steric hindrance and the overall hydrophobicity of these inhibitors. Interestingly, a C-5 ethyl group was found to confer significantly better inhibition than isopropyl group (53 vs 11, Table 3), indicating that less steric hindrance at C-5 could benefit IN binding. However, a reversed effect was observed in compounds with an N-1 linker of CH2, wherein a C-5 ethyl substitution (67) caused a complete loss of inhibition when compared to a C-5 isopropyl substitution (66). On the other hand, swapping C5 and C6 substituents of inhibitor 53 seems to harm IN binding (55 vs 53) as the IC50 value increased by 4-fold. Notably, a para-fluoro substitution at the C-6 benzyl clearly favors IN inhibition (53 vs 11, 56 vs 55). With a more dramatic change, a C-5 and C-6 fused benzene ring was found to render compounds 75–76 completely inactive against IN inhibition (Table 3). These findings indicate that the hydrophobic domain affects IN binding in a less comprehensible way and changes in this domain impact the activity most likely in concert with the linker domain.
Modeling
The common mode of inhibitory binding was explored through molecular docking using a homologous HIV IN model38 constructed based on the crystal structure of prototype foamy virus (PFV) IN.36 Significantly, the new inhibitor 43 fits perfectly into the IN binding site through two major binding domains also featured in raltegravir (1): the chelation of two Mg2+ ions, and the placement of the 4-F-benzyl group into the protein-DNA interfacial hydrophobic pocket (Figure 4). The high degree of similarity shown between the binding of 1 and 43 confirms that our new molecular scaffold can effectively engage with HIV IN. Interestingly, while residue Y143 contributes siginificantly to binding of 1 through a π-stacking interaction (Figure 4, A), the same interaction was not observed with our inhibitor 43 (Figure 4, B), suggesting that our inhibitor could be less susceptible to resistance associated with Y143 mutation. It is also noteworthy that the linker domain is situated right in the hydrophobic cleft between residue P145 and viral DNA nucleobases. Therefore, an ether linkage may provide a less favorable hydrophobic interaction than an all-carbon linker, thus the observed reduced inhibition with the ether linkage (Table 2, 43 Vs 28).
Figure 4.

Docking of raltegravir (1, A) and inhibitor 43 (B) in HIV-1 IN CCD in complex with Mg2+ and DNA. The 3-N hydroxyl group simultaneously chelates to both Mg2+ ions while allowing the placement of the benzyl group into the protein-DNA interfacial hydrophobic pocket.
Further validation was achieved by correlating the SAR of docked inhibitors 4, 11, 28, 29, 30, 32, 42, 43, 44, 66 as shown in Figure 5. The outstanding correlation observed between these two sets of IC50 data (exp. vs calc., correlation coefficiency = 0.82) supports our docking model with a common mode of binding as a valid platform for HIV IN inhibitor design.
Figure 5.

Strong correlation of docked 3-hydroxypyrimidine-2,4,-diones analogues supports a common mode of binding for HIV IN STI design
Antiviral Assay
To evaluate the antiviral activity of newly synthesized INIs, all compounds with an IC50 of 10 μM or lower from IN ST assay were tested against HIV-1IIIB in CEM-SS cells using an assay based on viral cytopathic effect (CPE). In this assay, the reduction of CPE was used to indicate the antiviral activity of a compound. All selected compounds were first screened under a single concentration (10 μM). Inhibitors with excellent CPE reduction and cell viability from this screening assay were further tested in a dose response fashion, from which the EC50 and CC50 were determined. Table 4 summarizes the antiviral testing results.
Table 4.
Anti-HIV-1 activity of IN inhibitors
| Compd | Single Concentration (10 μM) | Dose Response | |||
|---|---|---|---|---|---|
| CPE Reduction (%)a | Cell Viability (%) | EC50 (μM)b | CC50 (μM)c | TId | |
| 4e | 100 | 100 | 0.024 | >20 | >833 |
| 11e | 100 | 100 | 0.0080 | >20 | >2,500 |
| 12 | 100 | 100 | 2.1 | >100 | >47 |
| 13 | 100 | 100 | 0.95 | >100 | >105 |
| 28 | 27 | 83 | NDf | ND | -- |
| 29 | 30 | 92 | ND | ND | -- |
| 30 | 12 | 88 | ND | ND | -- |
| 43 | 12 | 97 | ND | ND | -- |
| 44 | 6.0 | 94 | ND | ND | -- |
| 49 | 48 | 100 | ND | ND | -- |
| 50 | 10 | 92 | ND | ND | -- |
| 51 | 100 | 100 | 1.1 | >20 | >18 |
Reduction of viral cytopathic effect, indicating inhibitory activity of a compound.
Concentration inhibiting virus replication by 50%.
Concentration resulting in 50% cell death.
Therapeutic index, defined by CC50/EC50.
Ref 30.
Not determined.
Remarkably, compounds 43 and 44, the two most potent INIs from IN ST assay with submicromolar IC50s, have shown only marginal activity against HIV-1 at 10 μM (Table 4). This finding strongly suggests that despite their ability to confer exceptional IN inhibition in biochemical assays, all-carbon linkers lack the proper physicochemical properties for the compounds to reach their target in cell culture. By contrast, with the exception of compounds having a para-fluorine at the C-6 benzyl group (49–50, Table 4), INIs with an ether linkage of CH2OCH2 (4, 11–13 and 51) showed antiviral activity at the low micromolar to low nanomolar range without appreciable cytotoxicity (Table 4). Longer ether linkages that produced excellent anti-IN activity for compounds 28–30 did not generate similar activity in the antiviral assay, further demonstrating that additional CH2s in the linker domain hinder antiviral activity.
Resistance
To establish the resistance profile of our inhibitors, we first tested two representative inhibitors 4 and 13 against HIV IN mutants. In this study, major mutants associated with raltegravir resistance23, 24, namely a G140S/Q148H double mutant and two single mutants Y143R and N155H were employed. The results are summarized in Table 5. Notably the G140S/Q148H double mutant which causes high resistance to raltegravir (1) yielded a much lower fold resistance to 4 and 13, whereas the two single mutants confer comparable resistance to 4, 13 and 1, suggesting a potentially common binding mode for these inhibitors.
Table 5.
Fold-resistance of inhibitors 4 and 13 against IN mutants
| IN Mutants | Fold-resistancea, b |
||
|---|---|---|---|
| 4c | 13 | 1 | |
| G140S/Q148H | 10/13 | 14/17 | 148/139 |
| Y143R | 7/4 | 7/5 | 24/28 |
| N155H | 5/6 | 5/7 | 10/8 |
Fold-resistance is defined by IC50(mutant)/IC50 (WT).
Values from two independent experiments.
Ref 30.
Based on these biochemical data, inhibitors 4 and 13 were further evaluated in cell culture against raltegravir-resistant HIV strains. Unlike the initial antiviral assay wherein HIV-1 IIIB was used in CEM-SS cells, the resistance studies were carried out with NL4-3 based viral strains in MT-4 cells. Selected strains contain major mutations corresponding to the IN mutants (Table 6 Vs Table 5). As shown in Table 6, all three viral strains show a high level resistance to raltegravir (1). Interestingly, although likely within experimental variations, the small fold-resistances observed with inhibitor 4 (Table 6) correlate closely with resistance observed with IN mutants in biochemical assays (Table 5). The testing of inhibitor 13 was complicated by its toxicity observed in MT-4 cells. Notably the same compound did not show toxicity in CEM-SS cells (TC50 > 100 μM, Table 4). On the other hand, antiviral assay with two HIV-1NL4-3 strains containing major mutations associated with NNRTI resistance, K103N or Y181C, revealed a fold-resistance of 16 and 48, respectively, to inhibitor 4.46 These results suggest that the overall antiviral activity of 4 could reflect a concerted yet not necessarily balanced inhibition to both RT and IN.
Table 6.
Fold-resistance of inhibitors 4 and 13 against raltegravir-resistant HIV-1 strains
In summary, through complete SAR and docking studies we have demonstrated that the N-3 OH group and the 4-F-benzyl moiety on the N-1 side chain are the key structural determinants for IN inhibition, and that the linker domain sits in a hydrophobic cleft between DNA nucleobases and P145. Therefore, both the nature and the length of the linker greatly impact IN binding, with the optimal IN inhibition entailing a 3–4 atom all-carbon linkage. However, studies in cell culture revealed that inhibitors with an all-carbon linker generally lack antiviral activity, presumably due to unfavored physicochemical properties. Resistance studies with raltegravir-resistant IN mutants as well as HIV-1NL4-3 viral strains demonstrated low level resistances toward inhibitors 13 and 4, likely due to their anti-RT activity. In the end, compounds (4, 11 and 13) featuring a CH2OCH2 linker demonstrate low micromolar IN inhibition and nanomolar anti-HIV activity, and are suitable for further development.
Experimental
Chemistry
General Procedures
All commercial chemicals were used as supplied unless otherwise indicated. Dry solvents (THF, Et2O, CH2Cl2 and DMF) were dispensed under argon from an anhydrous solvent system with two packed columns of neutral alumina or molecular sieves. Flash chromatography was performed on a Teledyne Combiflash RF-200 with RediSep columns (silica) and indicated mobile phase. All reactions were performed under inert atmosphere of ultra-pure argon with oven-dried glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Mass data were acquired on an Agilent TOF II TOS/MS spectrometer capable of ESI and APCI ion sources. Analysis of sample purity was performed on a Varian Prepstar SD-1 HPLC system with a Varian Microsorb-MW 100-5 C18 column (250mm × 4.6 mm): solvent A = H2O, solvent B = MeCN; flow rate = 1.0 mL/min; Method: linear 30–95% (B) over 25 min. All tested compounds have a purity ≥ 96%.
6-Benzyl-1-(benzyloxymethyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (6).44
To a suspension of paraformaldehyde (144 mg, 4.80 mmol) in TMSCl (2.0 mL), was added benzyl alcohol (518 mg, 4.80 mmol) at room temperature. The reaction mixture was stirred until a clear solution was formed. The solution was concentrated under reduced pressure to give benzyl chloromethyl ether as oil, which was taken to the next step without further purification. To a suspension of pyrimidine 5 (976 mg 4.00 mmol) in 16 mL of anhydrous DCM was added BSA (2.16 mL, 8.80 mmol) at room temperature. The resulting mixture was stirred until a clear solution was achieved. The freshly prepared benzyl chloromethy ether was added to this solution followed by the addition of a catalytic amount of TBAI. The reaction mixture was kept overnight and then quenched by adding a saturated aqueous solution of NaHCO3. The aqueous phase was extracted with CH2Cl2 (20 mL × 3). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The resultant residue was subjected to combiflash (Hex/EtOAc, 2:1) to afford compound 6 (1.25 g, 86 %) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.28 (m, 6H), 7.20 (m, 2H), 7.00 (d, J = 7.8 Hz, 2H), 5.14 (s, 2H), 4.59 (s, 2H), 4.10 (s, 2H), 2.79 (septet, J = 6.6 Hz, 1H), 1.21 (d, J = 6.6 Hz, 6H); MS (ESI+) m/z: 365.17 (M+1).
6-Benzyl-1-((2,4-difluorobenzyloxy)methyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (7)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 88 %; 1HNMR (600 MHz, CDCl3) δ 9.32 (s, 1H), 7.34 (m, 3H), 7.26 (t, J = 7.8 Hz, 1H), 7.07 (d, J = 7.8 Hz, 2H), 6.86 (t, J = 9.0 Hz, 1H), 6.81 (d, J = 9.0 Hz, 1H), 5.20 (s, 2H), 4.64 (s, 2H), 4.16 (s, 2H), 2.87 (m, 1H), 1.28 (d, J = 6.6 Hz, 6H); MS (ESI−) m/z: 399.42 (M-1).
6-Benzyl-1-((3-chloro-2-fluorobenzyloxy)methyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (8)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 90 %; 1 HNMR (600 MHz, CDCl3) δ 8.96 (b, 1H), 7.36 (m, 3H), 7.33 (t, J = 7.8 Hz, 2H), 7.10 (m, 3H), 5.21 (s, 2H), 4.69 (s, 2H), 4.16 (s, 2H), 2.87 (m, 1H), 1.28 (d, J = 6.6 Hz, 6H); MS (ESI−) m/z: 415.87 (M-1).
6-Benzyl-1-((4-fluorobenzyloxy)methyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (9)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 89 %; 1HNMR (600 MHz, CDCl3) δ 8.97 (b, 1H), 7.34 (t, J = 7.2 Hz, 2H), 7.30 (m, 3H), 7.07 (d, J = 7.8 Hz, 2H), 7.03 (m, 2H), 5.20 (s, 2H), 4.61 (s, 2H), 4.16 (s, 2H), 2.88 (septet, J = 7.2 Hz, 1H), 1.28 (d, J = 7.2 Hz, 6H); MS (ESI+) m/z: 383.18 (M+1).
6-Benzyl-1-((4-fluorophenethoxy)methyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (25)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 89 %; 1HNMR (600 MHz, CDCl3) δ 8.89 (b, 1H), 7.27 (t, J = 7.2 Hz, 2H), 7.20 (m, 1H), 7.08(m, 2H), 6.99 (d, J = 7.8 Hz, 2H), 6.91 (t, J = 8.4 Hz, 2H), 5.04 (s, 2H), 3.94 (s, 2H), 3.71 (t, J = 6.6 Hz, 2H), 2.74 (m, 3H), 1.20 (d, J = 6.6 Hz, 6H); MS (ESI−) m/z: 395.45 (M-1).
6-Benzyl-1-((3-(4-fluorophenyl)propoxy)methyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (26)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 83 %; 1HNMR (600 MHz, CDCl3) δ 8.97 (b, 1H), 7.29 (t, J = 7.8 Hz, 2H), 7.21 (t, J = 7.8 Hz, 1H), 7.05 (m, 4H), 6.89 (t, J = 9.0 Hz, 2H), 5.05 (s, 2H), 4.11 (s, 2H), 3.50 (t, J = 6.0 Hz, 2H), 2.87 (septet, J = 7.2 Hz, 1H), 2.56 (t, J = 7.8 Hz, 2H), 1.78 (m, 2H), 1.23 (d, J = 6.6 Hz, 6H); MS (ESI−) m/z: 409 (M-1).
6-Benzyl-1-((4-(4-fluorophenyl)butoxy)methyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (27)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 84 %; 1HNMR (600 MHz, CDCl3) δ 10.01 (b, 1H), 7.34 (t, J = 7.8 Hz, 2H), 7.26 (t, J = 7.2 Hz, 1H), 7.10 (m, 4H), 6.94 (d, J = 9.0 Hz, 2H), 5.23 (s, 2H), 4.16 (s, 2H), 3.58 (t, J = 6.6 Hz, 2H), 2.86 (septet, J = 7.2 Hz, 1H), 2.58 (t, J = 7.2 Hz, 2H), 1.63 (m, 2H), 1.56 (m, 2H), 1.27 (d, J = 7.2 Hz, 6H); MS (ESI−) m/z: 423.51 (M-1).
6-Benzyl-1-(ethoxymethyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (33)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 89 %; 1HNMR (600 MHz, CDCl3) δ 8.68 (b, 1H), 7.29 (t, J = 7.8 Hz, 2 H), 7.22 (m, 2H), 7.06 (d, J = 7.8 Hz, 1H), 5.05 (s, 2H), 4.11 (s, 2H), 3.56 (q, J = 7.2 Hz, 2H), 2.81 (septet, J = 7.2 Hz, 1H), 1.22 (d, J = 6.6 Hz, 6H), 1.11 (t, J = 7.2 Hz, 3H); MS (ESI−) m/z: 301.37 (M-1).
1-(Benzyloxymethyl)-5-methylpyrimidine-2,4(1H,3H)-dione (48)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 82 %; 1HNMR (600 MHz, CDCl3) δ 9.21 (b, 1H), 7.34 (m, 5H), 7.10 (s, 1H), 5.21 (s, 2H), 4.66 (s, 2H), 4.62 (s, 2H), 1.91 (s, 3H); MS (ESI−) m/z: 245.10 (M-1).
5-Benzyl-1-(benzyloxymethyl)-6-ethylpyrimidine-2,4(1H,3H)-dione (49)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 78 %; 1HNMR (600 MHz, CDCl3) δ 9.64 (s, 1H), 7.31 (m, 4H), 7.24 (m, 3H), 6.99 (m, 3H), 5.43 (s, 2H), 4.67 (s, 2H), 4.08 (s, 2H), 2.73 (q, J = 6.6 Hz, 2H), 1.06 (t, J = 6.6 Hz, 3H); MS (ESI−) m/z: 349.62 (M-1).
5-Benzyl-6-ethyl-1-((4-fluorobenzyloxy)methyl)pyrimidine-2,4(1H,3H)-dione (50)
This compound was prepared as a white solid following the procedure described for the preparation of 6. Yield: 80 %; 1 HNMR (600 MHz, CDCl3) δ 9.51 (s, 1H), 7.29 (t, J = 7.2 Hz, 4H), 7.20 (d, J = 6.6 Hz, 2H), 7.00 (t, J = 6.6 Hz, 2H), 5.41 (s, 2H), 4.62 (s, 2H), 4.08 (s, 2H), 2.72 (q, J = 6.6 Hz, 2H), 1.07 (t, J = 6.6 Hz, 3H); MS (ESI−) m/z: 367.15 (M-1).
6-Benzyl-1-((4-fluorobenzyloxy)methyl)-5-isopropyl-3-methylpyrimidine-2,4(1H,3H)-dione (10)
A mixture of compound 9 (60 mg, 0.16 mmol), methyl iodide (27 mg, 0.19 mmol) and Cs2CO3 (61 mg, 0.19 mmol) in 2 mL of THF was stirred under 40 °C for 3 h, and then allowed to cool to room temperature. The reaction mixture was quenched by adding 5 mL of H2O and extracted with EtOAc (10 mL × 3). The combined organic phases were dried over Na2SO4, and then concentrated under reduced pressure. The resultant residue was purified with combiflash to give compound 10 (40 mg, 63%) as clean oil: 1HNMR (600 MHz, CDCl3) δ 7.25 (t, J = 7.8 Hz, 2H), 7.21 (m, 3H), 6.99 (d, J = 7.2 Hz, 2H), 6.95 (t, J = 8.4 Hz, 2H), 5.16 (s, 2H), 4.53 (s, 2H), 4.09 (s, 2H), 3.28 (s, 3H), 2.87 (septet, J = 7.2 Hz, 1H), 1.22 (d, J = 7.2 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 163.2, 161.8, 161.5, 152.6, 146.0, 135.4, 133.2 (d, J = 3.3 Hz), 129.6 (d, J = 7.8 Hz), 129.2, 127.3 (d, J = 7.4 Hz), 118.9, 115.4 (d, J = 21.2 Hz), 73.7, 71.0, 33.4, 28.2, 27.9, 20.4; MS (ESI−) m/z: 395.45 (M-1).
6-Benzyl-1-((4-fluorobenzyloxy)methyl)-5-isopropyl-3-methoxypyrimidine-2,4(1H,3H)-dione (14)
This compound was prepared following the procedure described for the preparation of 10. Yield: 53 %; 1HNMR (600 MHz, CDCl3) δ 7.27 (t, J = 8.4 Hz, 2H), 7.24 (m, 3H), 6.99 (d, J = 7.2 Hz, 2H), 6.96 (t, J = 8.4 Hz, 2H), 5.14 (s, 2H), 4.55 (s, 2H), 4.08 (s, 2H), 3.94 (s, 3H), 2.85 (septet, J = 7.2 Hz, 1H), 1.22 (d, J = 7.2 Hz, 6H); MS (ESI−) m/z: 411.45 (M-1).
3-Amino-6-benzyl-1-(benzyloxymethyl)-5-isopropylpyrimidine-2,4(1H,3H)-dione (15).30
To a solution of 6 (60 mg, 0.16 mmol) in l.0 mL of THF was added NaH (31 mg, 0.82 mmol) at 0 °C. This reaction mixture was allowed to warm to room temperature over 1h., and then cooled to 0 °C followed by the addition of O-(Mesitylsulfonyl)hydroxylamine47 (MSH, 66 mg, 0.32 mmol). After stirring for 0.5 h, this reaction mixture was stirred at rt overnight. The reaction was quenched by adding 10 mL of H2O; the aqueous phase was then extracted with ethyl acetate (10 mL × 3). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The resultant residue was subjected to combiflash (Hex/EtOAc, 1:1) to give compound 15 (22 mg, 54 % based on consumed start material) as a white solid: 1HNMR (600 MHz, CDCl3) δ 7.33-7.26 (m, 8H), 7.04-7.03 (d, J = 7.2 Hz, 2H), 5.24 (s, 2H), 4.66 (s, 2H), 4.19 (s, 2H), 2.90 (septet, J = 7.2 Hz, 1H), 1.29 (d, J = 6.6 Hz, 6H); HRMS (ESI−) calcd. for C22H25N3O3 [M−H]− 380.1969, found 380.1976 (E = −1.9 ppm).
6-Benzyl-1-((4-fluorobenzyloxy)methyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (4).47
To a solution of 9 (100 mg, 0.26 mmol) in 6 mL of THF was added NaH (31 mg, 1.30 mmol) at 0 °C. This reaction mixture was allowed to warm to room temperature over 1 h., and then cooled to 0 °C followed by the addition of m-CPBA (135 mg, 0.79 mmol). This reaction mixture was allowed to warm to room temperature and stirred overnight. After the reaction was quenched by adding 10 mL of H2O, the aqueous phase was acidified with 1N HCl to pH = 7, and then extracted with ethyl acetate (10 mL × 3). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The resultant residue was subjected to combiflash (Hex/EtOAc, 1:1) to give compound 4 (38 mg, 72 % based on consumed start material) as a white solid. 1HNMR (600 MHz, CDCl3) δ 7.33-7.26 (m, 5H), 7.04-6.99 (m, 4H), 5.03 (s, 2H), 4.64 (s, 2H), 4.19 (s, 2H), 2.90 (septet, J = 7.2 Hz, 1H), 1.29 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 163.3, 162.4, 161.6, 152.1, 148.3, 135.2, 133.1(d, J = 3.3 Hz), 129.7 (d, J = 7.8 Hz), 129.2, 127.2(d, J = 6.8 Hz), 119.9, 115.4 (dd, J = 21.2 Hz), 72.8, 70.9, 33.5, 28.2, 20.4; HRMS (ESI−) calcd. for C22H23FN2O4 [M−H]− 399.1715, found 399.1705 (E = 2.4 ppm).
6-Benzyl-1-(benzyloxymethyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (11)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 53 %; 1HNMR (600 MHz, CDCl3) δ 7.32-7.30 (m, 7H), 7.02 (J = 7.2 Hz, 2H), 5.30 (s, 2H), 4.67 (s, 2H), 4.19 (s, 2H), 2.90 (septet, J = 7.2 Hz, 1H), 1.28 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 162.2, 151.9, 148.4, 137.3, 135.3, 129.1, 128.5, 128.0, 127.8, 127.3, 127.2, 119.8, 73.0, 71.8, 33.5, 28.2, 20.4; HRMS (ESI−) calcd. for C22H24N2O4 [M−H]− 381.1809, found 381.1803 (E = 1.5 ppm).
6-Benzyl-1-((2,4-difluorobenzyloxy)methyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (12)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 73 %; 1HNMR (600 MHz, CDCl3) δ 7.18 (m, 1H). 7.20 (m, 2H), 7.14 (t, J = 7.2 Hz, 1H), 6.97 (d, J = 7.8 Hz, 2H), 6.73 (td, J = 1.8, 9.6 Hz, 1H), 6.67 (td, J = 1.8, 9.6 Hz, 1H), 5.13 (s, 2H), 4.55 (s, 2H), 4.04 (s, 2H), 2.75 (m, 1H), 1.13 (d, J = 6.6 Hz, 6H); 13C NMR (150, CDCl3) δ 163.8 (d, J = 11.7 Hz), 162.1(dd, J = 7.8, 19.5 Hz), 160.4 (d, J = 11.7 Hz), 148.3, 135.2, 131.5 (d, J = 7.8 Hz), 129.2, 127.2, 120.4 (d, J = 15.6 Hz), 120.0, 111.2 (dd, J = 3.9, 21.2 Hz), 104.1 (t, J = 25.2 Hz), 72.7, 64.9, 33.4, 28.3, 20.3; HRMS (ESI−) calcd. for C22H22F2N2O4[M−H]− 415.1475, found 415.1473 (E = 0.45 ppm).
6-Benzyl-1-((3-chloro-2-fluorobenzyloxy)methyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (13)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 85 %; 1HNMR (600 MHz, CD3OD) δ 7.32 (t, J = 6.6 Hz, 1H), 7.23 (m, 3H), 7.18 (t, J = 7.2 Hz, 1H), 7.05 (t, J = 7.8 Hz, 1H), 7.00 (d, J = 7.8 Hz, 2H), 5.29 (s, 2H), 4.67 (s, 2H), 4.11 (s, 2H), 2.87 (m, 1H), 1.22 (d, J = 7.2 Hz, 6H); 13C NMR (150, CDCl3) 163.7 (d, J = 11.7 Hz), 162.1 (t, J = 11.7 Hz), 160.4 (t, J = 11.7 Hz), 159.7, 144.2, 135.5, 131.8 (d, J = 11.7 Hz), 129.1, 127.3(t, J = 16.2 Hz), 120.5 (d, J = 11.1 Hz), 118.2, 116.6, 111.1 (dd, J = 3.5, 21.3 Hz), 103.9 (t, J = 25.1 Hz), 72.9, 65.5, 33.4, 28.3, 20.4; HRMS (ESI+) calcd. for C22H22ClFN2O4 [M−H]− 431.1179, found 431.1181 (E = −0.83 ppm).
6-Benzyl-1-((4-fluorophenethoxy)methyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (28)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 66 %; 1HNMR (600 MHz, CDCl3) δ 7.34 (t, J = 7.2 Hz, 2H), 7.29 (d, J = 7.2 Hz, 1H), 7.14 (m, 2H), 7.03 (d, J = 7.8 Hz, 2H), 6.97 (t, J = 8.4 Hz, 2H), 5.19 (s, 2H), 4.04 (s, 2H), 3.82 (t, J = 6.6 Hz, 2H), 2.88 (septet, J = 7.2 Hz, 1H), 2.81 (t, J = 6.6 Hz, 2H), 1.28 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 162.5 (d, J = 16.2 Hz), 160.7, 155.5, 152.1, 148.4, 135.4, 134.2 (d, J = 2.9 Hz), 130.3 (d, J = 7.8 Hz), 129.2, 127.2, 119.8, 115.2 (d, J = 21.2 Hz), 72.9, 70.0, 35.1, 33.3, 28.2, 20.4; HRMS (ESI+) calcd. for C23H25FN2O4 [M+H]+ 413.1871, found 413.1868 (E = 0.76 ppm).
6-Benzyl-1-((3-(4-fluorophenyl)propoxy)methyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (29)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 71 %; 1 HNMR (600 MHz, CDCl3) δ 7.33 (m, 2H), 7.26 (m, 1H), 7.07 (m, 2H), 7.01 (m, 2H), 6.95 (m, 2H), 5.19 (s, 2H), 4.18 (s, 2H), 3.58 (m, 2H), 2.89 (septet, J = 7.2 Hz, 1H), 2.60 (m, 2H), 1.82 (m, 2H), 1.29 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 162.9, 162.1, 160.5, 152.4, 148.5, 137.2 (d, J = 3.3 Hz), 135.4, 129.7 (d, J = 7.8 Hz), 129.2, 127.3 (d, J = 7.8 Hz), 119.9, 115.2 (d, J = 20.6 Hz), 73.0, 68.4, 33.5, 31.3, 31.1, 28.2, 20.4; HRMS (ESI+) calcd. for C24H27FN2O4 [M+H]+ 427.2028, found 427.2026 (E = 0.38 ppm).
6-Benzyl-1-((4-(4-fluorophenyl)butoxy)methyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (30)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 64 %; 1HNMR (600 MHz, CDCl3) δ 7.34 (t, J = 7.2 Hz, 2H), 7.28 (d, J = 7.2 Hz, 1H), 7.11 (dd, J = 1.8, 7.8 Hz, 2H), 7.07 (d, J = 7.8 Hz, 2H), 6.96 (t, J = 8.4 Hz, 2H), 5.21 (s, 2H), 4.19 (s, 2H), 3.60 (t, J = 6.0 Hz, 2H), 2.91 (septet, J = 7.2 Hz, 1H), 2.58 (t, J = 7.2 Hz, 2H), 1.63 (m, 2H), 1.58 (m, 2H), 1.29 (d, J = 6.6 Hz, 6H); 13C NMR (150, CDCl3) δ 162.7, 162.0, 160.4, 152.2, 148.5, 137.8 (d, J = 3.3 Hz), 135.4, 129.7 (d, J = 7.8 Hz), 129.2, 127.3 (d, J = 7.8 Hz), 119.8, 115.0 (d, J = 20.7 Hz), 73.1, 69.3, 34.7, 33.4, 29.2, 28.9, 28.1, 20.3; HRMS (ESI+) calcd. for C25H29FN2O4 [M+H]+ 441.2184, found 441.2164 (E = 4.6 ppm).
6-Benzyl-1-(4-fluorobenzyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (32)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 72 %; 1HNMR (600 MHz, CDCl3) δ 7.30 (t, J = 7.2 Hz, 2H), 7.05-7.03 (m, 3H), 6.98 (d, J = 7.2 Hz, 2H), 6.94 (m, 2H), 4.88 (s, 2H), 3.83 (s, 2H), 2.81 (septet, J = 7.2 Hz, 1H), 1.22 (d, J = 6.6 Hz, 6H,); HRMS (ESI+) calcd. for C21H21FN2O3 [M+H]+ 369.1609, found 369.1608 (E = 0.26 ppm).
6-Benzyl-1-(4-fluorophenethyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (42)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 62 %; 1HNMR (600 MHz, CDCl3) δ 7.26–7.24 (m, 2H), 7.21 (m, 1H), 6.97(m, 2H), 6.89-6.88 (m, 2H), 6.85-6.84 (m, 2H), 3.79 (s, 2H), 3.73 (t, J = 7.8 Hz, 2H), 2.82 (septet, J = 7.2 Hz, 1H), 2.68 (t, J = 7.8 Hz, 2H), 1.29 (d, J = 6.6 Hz, 6H); HRMS (ESI+) calcd. for C23H23FN2O3 [M+H]+ 383.1765, found 383.1768 (E = 0.38 ppm).
6-Benzyl-1-(3-(4-fluorophenyl)propyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (43)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 72 %; 1HNMR (600 MHz, CDCl3) δ 7.21 (m, 3H), 6.94 (m, 2H), 6.87 (t, J = 8.4 Hz, 2H), 6.75 (d, J = 7.8 Hz, 2H), 3.71 (s, 2H), 3.58 (t, J = 7.2 Hz, 2H), 2.78 (septet, J = 7.2 Hz, 1H), 2.46 (t, J = 7.2 Hz, 2H), 1.79 (m, 2H), 1.29 (d, J = 6.6 Hz, 6H); HRMS (ESI+) calcd. for C23H25FN2O3[M+H]+ 397.1922, found 397.1924 (E = −0.51 ppm).
6-Benzyl-1-(4-(4-fluorophenyl)butyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (44)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 65 %; 1HNMR (600 MHz, CDCl3) δ 7.34 (t, J = 7.2 Hz, 2H), 7.28 (d, J = 7.8 Hz, 1H), 7.05 (m, 4H), 6.94 (t, J = 8.4 Hz, 2H), 3.92 (s, 2H), 3.73 (m, 2H), 2.87 (septet, J = 7.2 Hz, 1H), 2.51 (t, J = 7.2 Hz, 2H), 1.56 (m, 2H), 1.51 (m, 2H), 1.27 (d, J = 7.2 Hz, 6H); HRMS (ESI+) calcd. for C24H27FN2O3[M+H]+ 411.2078, found 411.2081 (E = −0.62 ppm).
1-(Benzyloxymethyl)-6-(4-fluorobenzyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (51)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 74%; 1HNMR (600 MHz, CDCl3) δ 7.32-7.27 (m, 5H), 7.00-6.98 (m, 4H), 5.28 (s, 2H), 4.67 (s, 2H), 4.14 (s, 2H), 2.81 (septet, J = 7.2 Hz, 1H), 1.27 (d, J = 6.6 Hz, 6H); HRMS (ESI−) calcd. for C22H23FN2O4[M−H]− 399.1715, found 399.1706 (E = 2.1 ppm).
6-(4-Fluorobenzyl)-1-((4-fluorobenzyloxy)methyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (52)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 72%; 1HNMR (600 MHz, CDCl3) δ 7.28-7.27 (m, 6H), 7.00 (d, J = 7.2 Hz, 2H), 5.24 (s, 2H), 4.61 (s, 2H), 4.12 (s, 2H), 2.83 (septet, J = 7.2 Hz, 1H), 1.25 (d, J = 6.6 Hz, 6H); HRMS (ESI−) calcd. for C22H22F2N2O4[M−H]− 417.1620, found 417.1620 (E = 0.10 ppm).
6-Benzyl-1-(benzyloxymethyl)-5-ethyl-3-hydroxypyrimidine-2,4(1H,3H)-dione (53)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 70 %; 1HNMR (600 MHz, CDCl3) δ 7.33-7.27 (m, 8H), 7.03 (d, J = 7.8 Hz, 2H), 5.26 (s, 2H), 4.65 (s, 2H), 4.15 (s, 2H), 2.51 (q, J = 7.8 Hz, 2H), 1.06 (t, J = 7.2 Hz, 3H); HRMS (ESI−) calcd. for C21H22N2O4[M−H]− 367.1652, found 367.1641 (E = 3.1 ppm).
1-(Benzyloxymethyl)-3-hydroxy-5-methylpyrimidine-2,4(1H,3H)-dione (54)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 37 %; 1 HNMR (600 MHz, CDCl3) δ 7.32-7.28 (m, 5H), 7.06 (s, 1H), 5.25 (s, 2H), 4.60 (s, 2H), 1.93 (s, 3H); HRMS (ESI−) calcd. for C13H14N2O4 [M−H]− 263.1026, found 263.1023 (E = 1.3 ppm).
5-Benzyl-1-(benzyloxymethyl)-6-ethyl-3-hydroxypyrimidine-2,4(1H,3H)-dione (55)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 53%; 1HNMR (600 MHz, CDCl3) δ 7.22 (m, 4H), 7.19 (m, 3H), 7.12 (m, 3H), 5.42 (s, 2H), 4.61 (s, 2H), 3.76 (s, 2H), 2.68 (q, J = 7.2 Hz, 2H), 0.99 (t, J = 7.2 Hz, 3H); HRMS (ESI−) calcd. for C21H22N2O4 [M−H]− 365.1507, found 365.1484 (E = 6.2 ppm).
5-Benzyl-6-ethyl-1-((4-fluorobenzyloxy)methyl)-3-hydroxypyrimidine-2,4(1H,3H)-dione (56)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 59%; 1HNMR (600 MHz, CDCl3) δ 7.13 (m, 3H), 7.07 (m, 4H), 6.83 (t, J = 6.6 Hz, 2H), 5.28 (s, 2H), 4.48 (s, 2H), 3.67 (s, 2H), 2.72 (q, J = 6.6 Hz, 2H), 1.07 (t, J = 6.6 Hz, 3H); HRMS (ESI−) calcd. for C21H21FN2O4[MH]− 383.1485, found 383.1394 (E = 4.8 ppm).
1-Benzyl-6-(4-fluorobenzyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (64)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 70 %; 1HNMR (600 MHz, CDCl3) δ 7.33 (t, J = 7.2 Hz, 2H), 7.30 (m, 1H), 7.11 (d, J = 7.2 Hz, 2H), 7.06-7.04 (m, 2H), 7.02-7.00 ( m, 2H), 4.98 (s, 2H), 3.86 (s, 2H), 2.84 (septet, J = 7.2 Hz, 1H), 1.29 (d, J = 6.6 Hz, 6H); HRMS (ESI+) calcd. for C21H21FN2O3 [M+H]+ 369.1609, found 369.1606 (E = −1.9 ppm).
1,6-Bis(4-fluorobenzyl)-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (65)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 70 %; 1HNMR (600 MHz, CDCl3) δ 7.10-7.08 (m, 2H), 7.06-7.04 (m, 2H), 7.02-6.99 (m, 4H), 4.94 (s, 2H), 3.89 (s, 2H), 2.84 (septet, J = 7.2 Hz, 1H), 1.28 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 163.1 (d, J = 31.2 Hz), 162.2, 161.4 (d, J = 31.2 Hz), 151.8, 148.3, 132.2 (d, J = 3.3 Hz), 130.4 (d, J = 2.9 Hz), 128.6 (d, J = 7.8 Hz), 127.6 (d, J = 8.4 Hz), 119.7, 116.4 (d, J = 21.8 Hz), 116.1 (d, J = 21.8 Hz), 46.6, 33.6, 28.2, 20.4; HRMS (ESI+) calcd. for C21H20F2N2O3 [M+H]+ 387.1515, found 387.1502 (E = 3.3 ppm).
1,6-Bibenzyl-3-hydroxy-5-isopropylpyrimidine-2,4(1H,3H)-dione (66)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 53 %; 1HNMR (600 MHz, CDCl3) δ 7.39-7.34 (m, 4H), 7.32-7.30 (m, 2H), 7.12 (d, J = 7.2 Hz, 2H), 7.06 ( d, J = 7.8 Hz, 2H), 5.00 (s, 2H), 3.91 (s, 2H), 2.90 (septet, J = 7.2 Hz, 1H), 1.29 (d, J = 7.2 Hz, 6H); HRMS (ESI+) calcd. for C21H22N2O3 [M+H]+ 351.1703, found 351.1689 (E = 4.1 ppm).
1,6-Bibenzyl-5-ethyl-3-hydroxypyrimidine-2,4(1H,3H)-dione (67)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 43 %; 1HNMR (600 MHz, CDCl3) δ 7.31-7.27 (m, 3H), 7.26-7.23 (m, 3H), 7.04 (d, J = 6.6 Hz, 2H), 7.00 (d, J = 6.6 Hz, 2H), 4.91 (s, 2H), 3.80 (s, 2H), 2.47 (q, J = 7.2 Hz, 2H), 1.00 (t, J = 7.2 Hz, 3H); HRMS (ESI+) calcd. for C20H20N2O3 [M+H]+ 337.1547, found 337.1534 (E = 3.8 ppm).
6-Benzyl-5-ethyl-1-(4-fluorobenzyl)-3-hydroxypyrimidine-2,4(1H,3H)-dione (68)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 48 %; 1HNMR (600 MHz, CDCl3) δ 7.38-7.35 (m, 2H), 7.32 (m, 1H), 7.15-7.13 (m, 2H), 7.07 (d, J = 7.8 Hz, 2H), 7.04 (m, 2H), 4.94 (s, 2H), 3.88 (s, 2H), 2.52 (q, J = 7.2 Hz, 2H), 1.07 (t, J = 7.2 Hz, 3H); HRMS (ESI+) calcd. for C20H19FN2O3 [M+H]+ 355.1452, found 355.1442 (E = 3.0 ppm).
1-Benzyl-3-hydroxy-5-methylpyrimidine-2,4(1H,3H)-dione (69)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 32 %; 1HNMR (600 MHz, CDCl3) δ 7.35-7.31 (m, 3H), 7.28 (d, J = 7.2 Hz, 2H), 6.94 (s, 1H), 4.94 (s, 2H), 1.90 (s, 3H); HRMS (ESI+) calcd. for C12H12N2O3 [M+H]+ 233.0921, found 233.0926 (E = −2.3 ppm).
1-(4-Fluorobenzyl)-3-hydroxy-5-methylpyrimidine-2,4(1H,3H)-dione (70)
This compound was prepared as a white solid following the procedure described for the preparation of 4. Yield: 41 %; 1 HNMR (600 MHz, CDCl3) δ 7.30-7.27 (m, 2H), 7.08-7.05 (m, 2H), 6.96 (s, 1H), 4.85 (s, 2H), 1.89 (s, 3H); HRMS (ESI−) calcd. for C12H11FN2O3[M−H]− 251.0826, found 251.0822 (E = 1.8 ppm).
Biology
In Vitro Integrase Catalytic Assays
Expression and purification of the recombinant IN in Escherichia coli were performed as previously reported28, 48 with addition of 10% glycerol to all buffers. Preparation of oligonucleotide substrates has been described.49 Integrase reactions were performed in 10 μL with 400 nM of recombinant IN, 20 nM of 5′-end [32P]-labeled oligonucleotide substrate and inhibitors at various concentrations. Solutions of 10% DMSO without inhibitors were used as controls. Reactions were incubated at 37 °C (60 minutes) in buffer containing 50 mM MOPS, pH 7.2, 7.5 mM MgCl2, and 14.3 mM 2-mercaptoethanol. Reactions were stopped by addition of 10 μL of loading dye (10 mM EDTA, 98% deionized formamide, 0.025% xylene cyanol and 0.025% bromophenol blue). Reactions were then subjected to electrophoresis in 20% polyacrylamide–7 M urea gels. Gels were dried and reaction products were visualized and quantitated with a Typhoon 8600 (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Densitometric analyses were performed using ImageQuant from Molecular Dynamics Inc. The concentrations at which enzyme activity was reduced by 50% (IC50) were determined using “Prism” software (GraphPad Software, San Diego, CA) for nonlinear regression to fit dose-response data to logistic curve models.
HIV-1 Assay
The HIV Cytoprotection assay used CEM-SS cells and the IIIB strain of HIV-1. Briefly virus and cells were mixed in the presence of test compound and incubated for 6 days. The virus was pre-titered such that control wells exhibit 70 to 95% loss of cell viability due to virus replication. Therefore, antiviral effect or cytoprotection was observed when compounds prevent virus replication. Each assay plate contained cell control wells (cells only), virus control wells (cells plus virus), compound toxicity control wells (cells plus compound only), compound colorimetric control wells (compound only) as well as experimental wells (compound plus cells plus virus). Cytoprotection and compound cytotoxicity were assessed by MTS (CellTiter® 96 Reagent, Promega, Madison WI) and the EC50 (concentration inhibiting virus replication by 50%), CC50 (concentration resulting in 50% cell death) and a calculated TI (therapeutic index CC50/EC50) were provided. Each assay included the HIV RT inhibitor AZT as a positive control.
Modeling and docking
All modeling study was carried out using schrodinger modeling suite50 based on our previous homology model38 of HIV-1 IN CCD-DNA complex using Glide with standard precision protocol.51 The Mg2+ ions and the interfacial hydrophobic pocket between the HIV-1 IN and DNA were defined as required constraints. The van der Waals radii of non-polar atoms for each of the ligands were scaled by a factor of 0.8 to account for structure variability to specific ligand binding. Validation of the docking protocol was carried out by re-docking both raltegravir and elvitegravir into the ligand binding site with structure comparison to the earlier reported mode of binding in PFV IN complex.36 To further validate our structural model for HIV IN inhibitor design, correlation of the experimental and predicted IC50s for a common mode of binding was performed for the most active compounds (4, 11, 28, 29, 30, 32, 42) based on linear reposnse method52 implemented within Liason.50
Supplementary Material
Acknowledgments
This research was supported by the Center for Drug Design at the University of Minnesota, the Center for Cancer Research, Intramural Program of the National Cancer Institute, and an NIH grant from IATAP (Intramural AIDS Targeted Antiviral Program). We thank Roger Ptak at Southern Research Institute for the antiviral assay and the Minnesota Supercomputing Institute for computing resources.
Abbreviations List
- IN
integrase
- HIV
human immunodeficiency virus
- INI
integrase inhibitor
- SAR
structure-activity-relationship
- 3′P
3′ processing
- ST
strand transfer
- DKA
diketoacid
- HAART
highly active antiretroviral therapy
- AIDS
acquired immunodeficiency syndrome
- RT
reverse transcriptase
- NTD
N-terminal domain
- CCD
catalytic core domain
- CTD
C-terminal domain
- PFV
prototype foamy virus
- mCPBA
meta-chloroperbenzoic acid
- CPE
cytopathic effect
Footnotes
Supporting Information Available: Additional experimental details and NMR, MS, and HPLC data for 31, 34–41, 57–63 and 72–76. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Bagasra O. A unified concept of HIV latency. Expert Opinion on Biological Therapy. 2006;6:1135–1149. doi: 10.1517/14712598.6.11.1135. [DOI] [PubMed] [Google Scholar]
- 2.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JAM, Baseler M, Lloyd AL, Nowak MA, Fauci AS. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 4.Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA integration: Mechanism of viral DNA cleavage and DNA strand transfer. Cell. 1991;67:1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- 5.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discovery. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 6.Cotelle P. Patented HIV-1 integrase inhibitors (1998–2005) Recent Pat Anti-Infect Drug Discovery. 2006;1:1–15. doi: 10.2174/157489106775244082. [DOI] [PubMed] [Google Scholar]
- 7.Henao-Mejia J, Goez Y, Patino P, Rugeles MT. Diketo acids derivatives as integrase inhibitors: the war against the acquired immunodeficiency syndrome. Recent Pat Anti-Infect Drug Discovery. 2006;1:255–265. doi: 10.2174/157489106777452700. [DOI] [PubMed] [Google Scholar]
- 8.Dubey S, Satyanarayana YD, Lavania H. Development of integrase inhibitors for treatment of AIDS: An overview. Eur J Med Chem. 2007;42:1159–1168. doi: 10.1016/j.ejmech.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 9.Marchand C, Maddali K, Metifiot M, Pommier Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr Top Med Chem. 2009;9:1016–1037. doi: 10.2174/156802609789630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dayam R, Sanchez T, Neamati N. Diketo Acid Pharmacophore. 2. Discovery of Structurally Diverse Inhibitors of HIV-1 Integrase. J Med Chem. 2005;48:8009–8015. doi: 10.1021/jm050837a. [DOI] [PubMed] [Google Scholar]
- 11.Dayam R, Sanchez T, Clement O, Shoemaker R, Sei S, Neamati N. β-Diketo acid pharmacophore hypothesis. 1. Discovery of a novel class of HIV-1 integrase inhibitors. J Med Chem. 2005;48:111–120. doi: 10.1021/jm0496077. [DOI] [PubMed] [Google Scholar]
- 12.Deng J, Sanchez T, Neamati N, Briggs JM. Dynamic Pharmacophore Model Optimization: Identification of Novel HIV-1 Integrase Inhibitors. J Med Chem. 2006;49:1684–1692. doi: 10.1021/jm0510629. [DOI] [PubMed] [Google Scholar]
- 13.Barreca ML, Ferro S, Rao A, De Luca L, Zappala M, Monforte AM, Debyser Z, Witvrouw M, Chimirri A. Pharmacophore-Based Design of HIV-1 Integrase Strand-Transfer Inhibitors. J Med Chem. 2005;48:7084–7088. doi: 10.1021/jm050549e. [DOI] [PubMed] [Google Scholar]
- 14.Mustata GI, Brigo A, Briggs JM. HIV-1 integrase pharmacophore model derived from diverse classes of inhibitors. Bioorg Med Chem Lett. 2004;14:1447–1454. doi: 10.1016/j.bmcl.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 15.Carlson HA, Masukawa KM, Rubins K, Bushman FD, Jorgensen WL, Lins RD, Briggs JM, McCammon JA. Developing a Dynamic Pharmacophore Model for HIV-1 Integrase. J Med Chem. 2000;43:2100–2114. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 16.Cocohoba J, Dong BJ. Raltegravir: the first HIV integrase inhibitor. Clin Ther. 2008;30:1747–1765. doi: 10.1016/j.clinthera.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J Med Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 18.Shimura K, Kodama EN. Elvitegravir: a new HIV integrase inhibitor. Antiviral Chem Chemother. 2009;20:79–85. doi: 10.3851/IMP1397. [DOI] [PubMed] [Google Scholar]
- 19.Vandekerckhove L. GSK-1349572, a novel integrase inhibitor for the treatment of HIV infection. Curr Opin Invest Drugs. 2010;11:203–212. [PubMed] [Google Scholar]
- 20.Min S, Song I, Borland J, Chen S, Lou Y, Fujiwara T, Piscitelli SC. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob Agents Chemother. 2009;54:254–258. doi: 10.1128/AAC.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prada N, Markowitz M. Novel integrase inhibitors for HIV. Expert Opin Investig Drugs. 2010;19:1087–1098. doi: 10.1517/13543784.2010.501078. [DOI] [PubMed] [Google Scholar]
- 22.Cherepanov P, Ambrosio ALB, Rahman S, Ellenberger T, Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci U S A. 2005;102:17308–17313. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hombrouck A, De Rijck J, Hendrix J, Vandekerckhove L, Voet A, De Maeyer M, Witvrouw M, Engelborghs Y, Christ F, Gijsbers R, Debyser Z. Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal tethering of HIV. PLoS Pathog. 2007;3:418–430. doi: 10.1371/journal.ppat.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelman A. Host cell factors and HIV-1 integration. Future HIV Ther. 2007;1:415–426. [Google Scholar]
- 25.Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol. 2010;6:442–448. doi: 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- 26.Yeni P. Update on HAART in HIV. J Hepatol. 2006;44:S100–S103. doi: 10.1016/j.jhep.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 27.Metifiot M, Marchand C, Maddali K, Pommier Y. Resistance to integrase inhibitors. Viruses. 2010;2:1347–1366. doi: 10.3390/v2071347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metifiot M, Maddali K, Naumova A, Zhang X, Marchand C, Pommier Y. Biochemical and Pharmacological Analyses of HIV-1 Integrase Flexible Loop Mutants Resistant to Raltegravir. Biochemistry. 2010;49:3715–3722. doi: 10.1021/bi100130f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.da Silva D, Van Wesenbeeck L, Breilh D, Reigadas S, Anies G, Van Baelen K, Morlat P, Neau D, Dupon M, Wittkop L, Fleury H, Masquelier B. HIV-1 resistance patterns to integrase inhibitors in antiretroviral-experienced patients with virological failure on raltegravir-containing regimens. J Antimicrob Chemother. 2010;65:1262–1269. doi: 10.1093/jac/dkq099. [DOI] [PubMed] [Google Scholar]
- 30.Tang J, Maddali K, Dreis CD, Sham YY, Vince R, Pommier Y, Wang Z. N-3 Hydroxylation of Pyrimidine-2,4-diones Yields Dual Inhibitors of HIV Reverse Transcriptase and Integrase. ACS Med Chem Lett. 2011;2:63–67. doi: 10.1021/ml1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esposito D, Craigie R. HIV integrase structure and function. Advances in Virus Research. 1999;52:319–333. doi: 10.1016/s0065-3527(08)60304-8. [DOI] [PubMed] [Google Scholar]
- 32.Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, Davies DR. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc Natl Acad Sci USA. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 34.O’Brien C. HIV Integrase Structure Catalyzes Drug Search. Science. 1994;266:1946. doi: 10.1126/science.7801119. [DOI] [PubMed] [Google Scholar]
- 35.Alian A, Griner SL, Chiang V, Tsiang M, Jones G, Birkus G, Geleziunas R, Leavitt AD, Stroud RM. Catalytically-active complex of HIV-1 integrase with a viral DNA substrate binds anti-integrase drugs. Proc Natl Acad Sci U S A. 2009;106:8192–8197. doi: 10.1073/pnas.0811919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hare S, Vosb AM, Claytonb RF, Thuringb JW, Cummingsb MD, Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci USA. 2010;107:20057–20062. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang J, Maddali K, Pommier Y, Sham YY, Wang Z. Scaffold rearrangement of dihydroxypyrimidine inhibitors of HIV integrase: Docking model revisited. Bioorg Med Chem Lett. 2010;20:3275–3279. doi: 10.1016/j.bmcl.2010.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc Natl Acad Sci U S A. 2010;107:15910–15915. doi: 10.1073/pnas.1002346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka H, Hayakawa H, Haraguchi K, Miyasaka T, Walker RT, De Clercq E, Baba M, Stammers DK, Stuart DI. HEPT: from an investigation of lithiation of nucleosides towards a rational design of non-nucleoside reverse transcriptase inhibitors of HIV-1. Adv Antiviral Drug Design. 1999;3:93–144. [Google Scholar]
- 41.Pontikis R, Benhida R, Aubertin AM, Grierson DS, Monneret C. Synthesis and Anti-HIV Activity of Novel N-1 Side Chain-Modified Analogs of 1-[(2-Hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) J Med Chem. 1997;40:1845–1854. doi: 10.1021/jm960765a. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka H, Takashima H, Ubasawa M, Sekiya K, Inouye N, Baba M, Shigeta S, Walker RT, Clercq ED, Miyasaka T. Synthesis and Antiviral Activity of 6-Benzyl Analogs of 1-[(2-Hydroxyethoxy)methyl]-5-(phenylthio)thymine (HEPT) as Potent and Selective Anti-HIV-1 Agents. J Med Chem. 1995;38:2860–2865. doi: 10.1021/jm00015a008. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka H, Baba M, Hayakawa H, Sakamaki T, Miyasaka T, Ubasawa M, Takashima H, Sekiya K, Nitta I, Shigeta S, Walker RT, Balzarini J, Clercq ED. A new class of HIV-1 specific 6-substituted acyclouridine derivatives: synthesis and anti-HIV-1 activity of 5- or 6-substituted analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) J Med Chem. 1991;34:349–357. doi: 10.1021/jm00105a055. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Bennett EM, Wilson DJ, Salomon C, Vince R. Rationally Designed Dual Inhibitors of HIV Reverse Transcriptase and Integrase. J Med Chem. 2007;50:3416–3419. doi: 10.1021/jm070512p. [DOI] [PubMed] [Google Scholar]
- 45.Danel K, Larsen E, Pedersen EB, Vestergaard BF, Nielsen C. Synthesis and Potent Anti-HIV-1 Activity of Novel 6-Benzyluracil Analogs of 1-[(2-Hydroxyethoxy)methyl]-6-(phenylthio)thymine. J Med Chem. 1996;39:2427–2431. doi: 10.1021/jm9600499. [DOI] [PubMed] [Google Scholar]
- 46.These assays were done in MAGI R5 cells with NNRTI TNK-651 as a control. The observed fold-resistances for the control were 22 and 72 for K103N and Y181C mutants.
- 47.Bernardes GJL, Chalker JM, Errey JC, Davis BG. Facile Conversion of Cysteine and Alkyl Cysteines to Dehydroalanine on Protein Surfaces: Versatile and Switchable Access to Functionalized Proteins. J Am Chem Soc. 2008;130:5052–5053. doi: 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- 48.Leh H, Brodin P, Bischerour J, Deprez E, Tauc P, Brochon JC, LeCam E, Coulaud D, Auclair C, Mouscadet JF. Determinants of Mg2+-dependent activities of recombinant human immunodeficiency virus type 1 integrase. Biochemistry. 2000;39:9285–9294. doi: 10.1021/bi000398b. [DOI] [PubMed] [Google Scholar]
- 49.Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. Preferential inhibition of the magnesium-dependent strand transfer reaction of HIV-1 integrase by α-hydroxytropolones. Mol Pharmacol. 2006;69:1454–1460. doi: 10.1124/mol.105.020321. [DOI] [PubMed] [Google Scholar]
- 50.Maestro v9.1, G v, Macromodel v9.8, Liason v56 version 9.1. Schrodinger, LLC; New York: [Google Scholar]
- 51.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1139–1149. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 52.Hansson T, Aqvist J. Estimation of binding free energies for HIV proteinase inhibitors by molecular dynamics simulations. Protein Eng. 1995;8:1137–1144. doi: 10.1093/protein/8.11.1137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.