

Abstract
A broad range of applications requires access to water-soluble, bioconjugatable porphyrins. Branched alkyl groups attached at the branching site to the porphyrin meso position are known to impart high organic solubility. Such “swallowtail” motifs bearing a polar group (hydroxy, dihydroxyphosphoryl, dihydroxyphosphoryloxy) at the terminus of each branch have now been incorporated at a meso site in trans-AB-porphyrins. The incorporation of the swallowtail motif relies on rational synthetic methods whereby a 1,9-bis(N-propylimino)dipyrromethane (bearing a bioconjugatable tether at the 5-position) is condensed with a dipyrromethane (bearing a protected 1,5-dihydroxypent-3-yl unit at the 5-position). The two hydroxy groups in the swallowtail motif of each of the resulting zinc porphyrins can be transformed to the corresponding diphosphate or diphosphonate product. A 4-(carboxymethyloxy)phenyl group provides the bioconjugatable tether. The six such porphyrins reported here are highly water-soluble (≥20 mM at room temperature in water at pH 7) as determined by visual inspection, UV–vis absorption spectroscopy, or 1H NMR spectroscopy. Covalent attachment was carried out in aqueous solution with the unprotected porphyrin diphosphonate and a monoclonal antibody against the T-cell receptor CD3ε. The resulting conjugate performed comparably to a commercially available fluorescein isothiocyanate-labeled antibody with Jurkat cells in flow cytometry and fluorescence microscopy assays. Taken together, this work enables preparation of useful quantities of water-soluble, bioconjugatable porphyrins in a compact architecture for applications in the life sciences.
INTRODUCTION
A large and growing number of applications in the life sciences require porphyrinic macrocycles that are water-soluble and are suited for conjugation in a variety of formats. The applications encompass flow cytometry, cellular and whole-organism imaging, sensing, photodynamic therapy, biomimetic catalysis, and radical scavenging. The success of these applications relies on a host of factors, including (1) significant solubility in aqueous saline solutions, thereby avoiding intermolecular aggregation (and excited-state quenching), (2) minimal nonspecific binding to cellular components, (3) incorporation of a single reactive group for conjugation, thereby avoiding cross-linking and the formation of product mixtures, and (4) robust synthesis affording ample quantities for experimentation.
The large hydrophobic face of a porphyrinic macrocycle presents a challenge to water solubilization. Traditional approaches to achieve water solubility of porphyrins have largely relied on uroporphyrin (1–5), meso-tetrakis[4-(N-methyl)-pyridinium]porphyrin (6), and meso-tetrakis(4-sulfophenyl)-porphyrin (6) (Chart 1). More recent approaches have entailed attachment of the following motifs to the porphyrin via a meso-aryl group: oligo(ethylene glycol) (OEG) chains (7, 8), OEG dendrimers (9), glycosyl units (10), alkyl polyamine chains (11), branched polycarboxy units (12), or malonate groups (13). Solubilizing groups that do not incorporate an aryl group include 1,2-dihydroxyethyl at the porphyrin meso position (14) and a variety of substituents attached to the β-position of a sapphyrin (15). A lengthy review of water-soluble porphyrins has been prepared by Hambright (6). Each solubilizing group has merit; however, many are not compatible with bioconjugation reactions, and others are so large (mass >10-times that of the porphyrin) as to limit the scope of desired experiments.
Chart 1.

The presence of a bioconjugatable group is essential for attachment of water-soluble porphyrins to substances ranging from nanoparticles to biological targeting agents. Indeed, porphyrinic molecules have been attached to diverse molecular entities such as peptides (16–19), estrogen (20), acridine (21), bovine serum albumin (22), antibodies (23–27), transferrin (28), epidermal growth factor (29, 30), low-density lipoprotein (31), nucleic acids (32), and polymers such as polylysine (33) or poly(vinyl alcohol) (29, 30). In some cases, the bioconjugation is carried out in organic solution, whereas in others aqueous media are employed. Regardless of medium, the choice of bioconjugatable group must enable conjugation to be carried out without interference with the functional groups that afford water solubility.
To meet the objectives stated above requires a water solubilization unit that is (1) compact, (2) synthetically accessible, preferably amenable for use in the building-block synthesis of meso-substituted porphyrins, and (3) compatible with bio-conjugation procedures. Anionic substituents are particularly attractive in the water solubilization motif to minimize non-specific binding to cellular components. A chief challenge is to solubilize the rather hydrophobic porphyrinic macrocycle so as to suppress cofacial interaction, a likely mechanism for aggregation and precipitation. We recently found that branched-alkyl groups (e.g., tridec-7-yl) known as “swallowtail” substituents are very effective at imparting a high level of organic solubility to porphyrins (Figure 1) (34, 35). The hydrocarbon chains of the swallowtail motif project over both faces of the macrocycle, suppressing π–π interactions between porphyrin rings (34). Such hydrocarbon swallowtails were shown earlier to impart excellent solubility in organic media to perylene dyes, which otherwise are quite insoluble (36). We expected that swallowtail moieties modified with polar end groups could render porphyrins water-soluble. The swallowtail groups do not include a meso-aryl substituent and hence are compact, helping limit the overall molecular weight of the porphyrinic compound.
Figure 1.
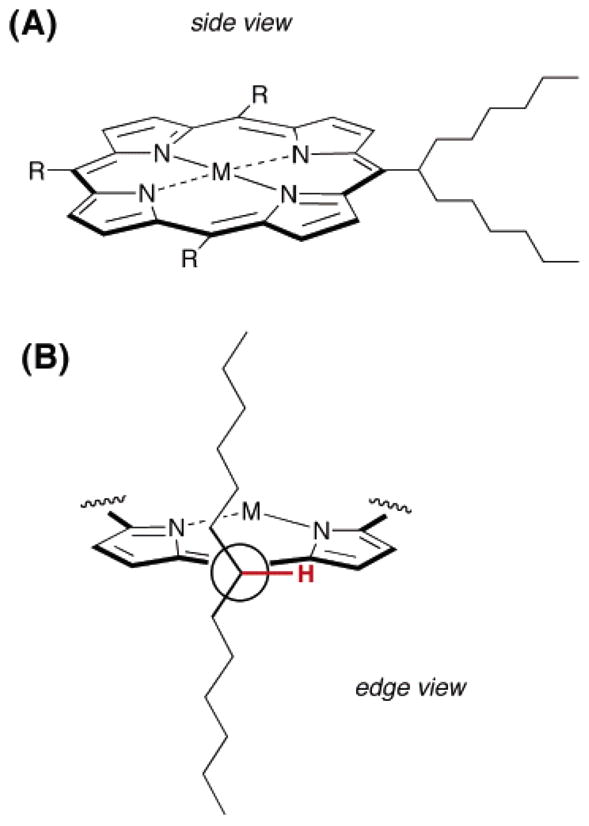
(A) Porphyrin bearing one swallowtail substituent and (B) Newman projection showing the alkyl chains of the swallowtail substituent above and below the plane of the porphyrin.
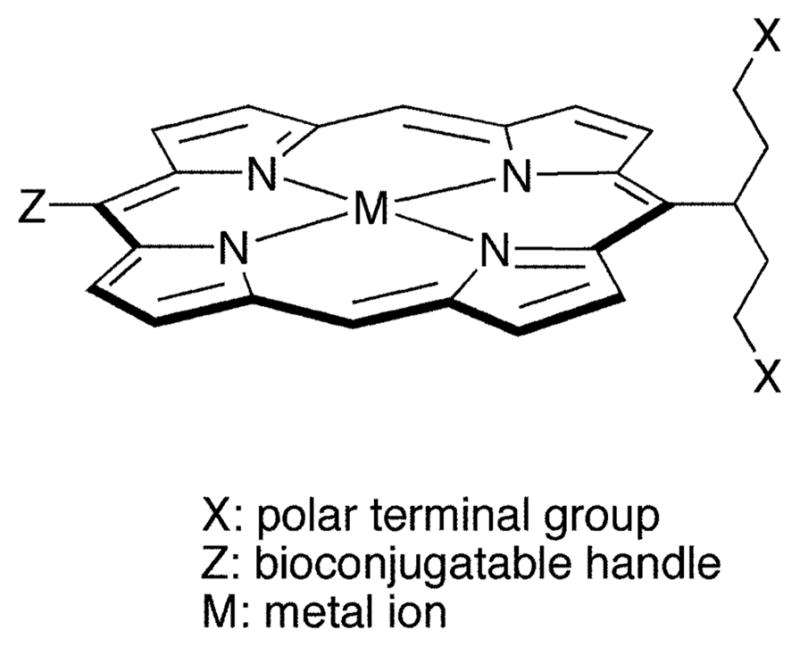
In this paper, we report the synthesis of trans-AB-porphyrins bearing one conjugatable group and one swallowtail motif. The presence of two small meso substituents affords a minimalist design suited for diverse applications. The swallowtail motif is a pent-3-yl group bearing a polar group (dihydroxyphosphoryl, dihydroxyphosphoryloxy) at each terminus. The conjugatable group is either 4-bromophenyl or 4-(carboxymethyloxy)phenyl. The solubility of the porphyrins has been examined in aqueous media. The bromo substituent is not directly useful for bioconjugation but can be substituted in Pd-mediated reactions. In addition, the porphyrin bearing the bromo substituent can serve as a benchmark for assessing the extent to which the swallowtail motifs are responsible for water solubilization without the polar, ionizable carboxylic acid. The favorable properties of the porphyrin allowed the facile preparation of soluble nonaggre-gated protein conjugates. The biological activity of the conjugated porphyrin was established by the stringent method of intracellular fluorescent staining using a monoclonal antibody against the epsilon invariant chain of the human T-cell receptor CD3, and assessing the performance by flow cytometry and fluorescence microscopy. This work establishes the foundation for the rational synthesis of porphyrins for applications where water solubility and bioconjugation are required.
EXPERIMENTAL PROCEDURES
General Methods
1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded in CDCl3 unless noted otherwise. Bulb-to-bulb distillation was performed using a standard-size Kugelrohr apparatus. Absorption spectra and fluorescence spectra were collected at room temperature in CH2Cl2 unless noted otherwise. Infrared absorption spectra were recorded as thin films. Hydrophobic porphyrins were analyzed in neat form by laser desorption mass spectrometry (LD-MS) (37). The water-soluble porphyrins were analyzed by direct infusion of water/methanol (40:60) solutions by atmospheric pressure electrospray ionization mass spectrometry (ESI-MS). Both in LD-MS and ESI-MS analyses, positive ions were detected unless noted otherwise. Melting points are uncorrected. Solvents were dried according to standard procedures. LDA was generated in situ (38). The mouse IgG1 anti-human CD3 intracellular ε-chain (clone UCHT) (39) and the same antibody derivatized with a fluorescein-isothiocyanate (FITC) label, were obtained from BD Biosciences (San Jose, CA). All other materials were used as received from commercial sources. Molecular mechanics calculations were carried out using PCMODEL for Windows version 7.50.00 (Serena Software).
Chromatography
Preparative chromatography was performed using silica (230–400 mesh) or alumina (80–200 mesh). Thin-layer chromatography was performed on silica or alumina. Samples were visualized by UV light (254 and 365 nm), Br2 vapor, or KMnO4/K2CO3. Dipyrromethanes and compounds 2–4 and 6a were analyzed by GC as described previously (40). Reversed-phase preparative column chromatography was carried out using C-18-coated silica and eluants based on water admixed with methanol. Analytical RP-HPLC was carried out using an ODS C-18 column (5 μm, 125 mm × 4 mm), flow rate = 1 mL/min, detection at 254, 410, and 417 nm, and the following elution program with solvents A (water containing 0.1% TFA) and B (acetonitrile containing 0.1% TFA): 0–2 min, 0% B; 2–20 min, 0→90% B; 20–23 min, 90% B.
2-(tert-Butyldimethylsilyloxy)ethyl Bromide (2)
Following a published procedure (41), 2-bromoethanol (10.0 mL, 141 mmol) was added to a mixture of imidazole (12.5 g, 184 mmol) and tert-butyldimethylsilyl chloride (21.1 g, 140 mmol) in anhydrous DMF (25 mL). The reaction mixture was stirred at room temperature for 12 h. Water and diethyl ether were added. The phases were separated. The aqueous phase was extracted with diethyl ether. The organic phase was washed with water and brine. The solution was dried (Na2SO4). Evaporation of the solvent followed by bulb-to-bulb distillation (40–45 °C, 0.05 mmHg) yielded a colorless liquid (32.5 g, 97%): IR (film, νmax cm−1) 2951, 2859, 1471; 1H NMR δ 0.07 (s, 6H), 0.89 (s, 9H), 3.36–3.41 (m, 2H), 3.85–3.90 (m, 2H); 13C NMR δ −5.06, 18.49, 26.04, 33.45, 63.74; EI-MS 137/139, 181/183; Anal. (C8H19BrOSi) C, H.
3-Cyano-1,5-bis(tert-butyldimethylsilyloxy)pentane (3)
A solution of HMPA (12.0 mL) and LDA (33.5 mmol) in dry THF (39 mL) at −78 °C under argon was treated with acetonitrile (1.75 mL, 33.5 mmol). The solution was stirred for 30 min. Then, 2 (6.82 g, 29.0 mmol) in THF (30 mL) was added dropwise. Stirring was continued for 2 h, after which a second portion of LDA (33.5 mmol in 39 mL THF) was added. The solution was stirred for 30 min, after which 2 (6.82 g, 29.0 mmol) in THF (30 mL) was added dropwise. The reaction was allowed to proceed for 2 h. Saturated aqueous NH4Cl was added, and the mixture was allowed to reach room temperature. Diethyl ether was added, the phases were separated, and the aqueous layer was extracted with diethyl ether. The organic phase was washed with water and brine, dried over Na2SO4, and concentrated. Chromatography [silica, petroleum ether/diethyl ether (40: 1)] afforded a colorless liquid (7.79 g, 77%): IR (film, νmax cm−1) 2953, 2859, 1738, 1472; 1H NMR δ 0.07 (s, 12H), 0.89 (s, 18H), 1.76–1.83 (m, 4H), 3.03–3.13 (m, 1H), 3.73–3.83 (m, 4H); 13C NMR δ −5.24, 18.45, 24.82, 26.09, 35.32, 60.08, 122.15; EI-MS 115, 142/144, 156/157, 182/184, 198; Anal. (C18H39NO2Si2) C, H, N.
3-Formyl-1,5-bis(tert-butyldimethylsilyloxy)pentane (4)
Following a published procedure (41), a solution of 3 (3.65 g, 10.2 mmol) in dry toluene (52 mL) at −78 °C under argon was treated with DIBALH (12 mmol, 8.3 mL, 1.5 M solution in toluene). The reaction was allowed to proceed for 1 h. Water (2.6 mL) was added, and the mixture was allowed to reach room temperature. Aqueous NaOH (2.6 mL, 4.0 M solution) was added, and stirring was continued for 15 min. Water (7.8 mL) was added, and the suspension was stirred for a further 15 min. The reaction mixture was dried with a large quantity of Na2SO4. The dried solution was concentrated at reduced pressure and chromatographed [silica, petroleum ether/diethyl ether (20:1)], affording a colorless liquid (2.70 g, 73%): IR (film, νmax cm−1) 2955, 2859, 1729, 1708, 1472; 1H NMR δ 0.01 (s, 12H), 0.87 (s, 18H), 1.64–1.74 (m, 2H), 1.85–1.96 (m, 2H), 2.50 (m, 1H), 3.57–3.68 (m, 4H), 9.65 (d, J = 2.4 Hz, 1H); 13C NMR δ −5.27, 18.45, 26.09, 32.15, 46.40, 60.80, 204.74; EI-MS 171/172, 141, 97, 75; Anal. (C18H40O3Si2) C, H; C calcd, 59.94; found, 58.15.
5-[1,5-Bis(tert-butyldimethylsilyloxy)pent-3-yl]dipyrromethane (5)
Following a standard procedure (40), aldehyde 4 (4.61 g, 12.8 mmol) was dissolved in dry pyrrole (89.1 mL, 1.28 mmol), and the solution was flushed with argon for 10 min. InCl3 (285 mg, 1.28 mmol) was added, and the reaction was allowed to proceed for 3 h. The reaction was quenched by addition of powdered NaOH (1.54 g, 38.5 mmol). The mixture was stirred for 45 min. The mixture was filtered. The filtrate was concentrated at reduced pressure. The residue was chromatographed [silica, ethyl acetate/CH2Cl2/hexanes (1:2:7)] affording a viscous, pale yellow liquid (5.87 g, 96%): IR (film, νmax cm−1) 3427, 1639; 1H NMR δ 0.09 (s, 12H), 0.94 (s, 18H), 1.35–1.44 (m, 2H), 1.71–1.78 (m, 2H), 2.41–2.43 (m, 1H), 3.64–3.77 (m, 4H), 4.38 (d, J = 4.2 Hz, 1H), 6.03 (app s, 2H), 6.13–6.16 (m, 2H), 6.65–6.66 (m, 2H), 8.45–8.55 (br, 2H); 13C NMR δ −5.08, 18.65, 26.29, 35.13, 35.37, 40.41, 62.18, 107.03, 108.27, 116.45, 132.25; LD-MS obsd 475.6; FAB-MS obsd 476.3255, calcd 476.3254 (C26H48N2O2Si2); Anal. C, H, N.
5-(1,5-Dihydroxypent-3-yl)dipyrromethane (5-(OH)2)
A solution of 5 (3.81 g, 8.01 mmol) in THF (30 mL) was treated with TBAF (4.61 g, 17.6 mmol). The reaction was allowed to proceed until the starting material could not be detected by TLC [alumina, CH2Cl2/MeOH (97:3)]. The solvent was evaporated. The residue was dissolved in a mixture of ethyl acetate and water. The aqueous layer was extracted with ethyl acetate. The organic phase was washed with water and dried (Na2SO4). The solvent was removed, and the residue was chromatographed [neutral alumina, CH2Cl2/MeOH (2→10%)] to afford an off-white viscous oil (1.60 g, 89%): 1H NMR δ 1.43–1.50 (m, 2H), 1.68–1.79 (m, 2H), 2.39–2.43 (m, 3H), 3.59–3.75 (m, 4H), 4.21 (d, J = 5.4 Hz, 1H), 6.04 (d, J = 0.9 Hz, 2H), 6.13–6.16 (m, 2H), 6.65–6.66 (m, 2H), 8.40–8.55 (br, 2H); 13C NMR δ 25.88, 34.86, 35.60, 41.79, 61.25, 106.77, 108.38, 117.03, 131.84; FAB-MS obsd 248.1528, calcd 248.1525 (C14H20N2O2); Anal. C calcd, 67.71; found, 66.59; H calcd, 8.12; found, 8.62; N calcd, 11.28; found, 10.34.
5-[1,5-Bis(dimethoxyphosphoryloxy)pent-3-yl]dipyrromethane (5-(P)2)
A solution of 5-(OH)2 (950 mg, 4.24 mmol) in CH2Cl2 (16 mL) was treated with DMAP (1.18 g, 9.67 mmol) followed by slow addition of a solution of dimethyl chlorophosphate (1.06 mL, 9.83 mmol) in CH2Cl2 (11 mL). The reaction mixture was stirred at room temperature for 8 h. The crude mixture was diluted with CH2Cl2 and water. The aqueous layer was extracted with CH2Cl2. The organic layer was washed with water. The organic layer was dried (Na2SO4), concentrated, and chromatographed [silica, CH2Cl2/MeOH (2→5%)], affording a pale yellow oil (1.19 g, 61%): IR (film, νmax cm−1) 3320, 1567; 1H NMR δ 1.57–1.64 (m, 2H), 1.84–1.90 (m, 2H), 2.19 (m, 1H), 3.72 (s, 6H), 3.76 (s, 6H), 4.02 (q, J = 8.4 Hz, 4H), 4.28 (d, J = 5.1 Hz, 1H), 6.00–6.01 (m, 2H), 6.10–6.13 (m, 2H), 6.67–6.69 (m, 2H), 8.79 (br, 2H); 13C NMR δ 31.16, 32.54, 32.62, 35.21, 40.76, 54.55, 54.65, 66.29, 106.85, 108.32, 117.22, 130.88; FAB-MS obsd 464.1480, calcd 464.1477 (C18H30N2O8P2); Anal. C, H, N. Data for 5-(P/OH). TLC analysis of the crude mixture revealed the presence of a more polar component, which was isolated as an off-white oil: IR (film, νmax cm−1) 3320, 1566, 1450; 1H NMR δ 1.45–1.87 (m, 4H), 2.44–2.46 (m, 1H), 3.66 (t, J = 5.1 Hz, 2H), 3.75 (s, 3H), 3.78 (s, 3H), 3.95–4.19 (m, 2H), 4.26 (d, J = 4.8 Hz, 1H), 6.03 (app s, 2H), 6.14 (app s, 2H), 6.68 (app s, 2H), 8.64 (br, 2H); 13C NMR δ 33.07, 34.84, 35.23, 41.33, 54.67, 54.73, 61.02, 66.65, 66.71, 106.65, 106.96, 108.26, 108.34, 111.76, 117.08, 117.15, 131.27, 131.69; EI-MS 157/158, 230/231; FAB-MSobsd356.2, calcd356.2(C16H25N2O5P); Anal.(C16H25N2O5P•0.5CH3-OH) C, H, N.
4-(tert-Butoxycarbonylmethoxy)benzaldehyde (6a)
A solution of 4-hydroxybenzaldehyde (2.44 g, 20.0 mmol) in dry acetonitrile (8.0 mL) was treated with powdered, dried K2CO3 (3.04 g, 22.0 mmol) and NaI (304 mg, 2.00 mmol). The mixture was refluxed under argon for 30 min. tert-Butyl bromoacetate (1.48 mL, 1.95 g, 10.0 mmol) was added dropwise, and the reflux was continued for 12 h. Water and CH2Cl2 were added, and the phases were separated. The aqueous layer was extracted with CH2Cl2. The organic phase was washed with water. The organic layer was dried (Na2SO4). Evaporation of the solvent and chromatography of the oily residue [silica, ethyl acetate/hexanes (3:7)] afforded a white, crystalline solid (2.18 g, 92%): mp 56–57 °C; IR (film, νmax cm−1) 1762, 1752, 1685, 1600; 1H NMR δ 1.48 (s, 9H), 4.60 (s, 2H), 6.99 (d, J = 8.7 Hz, 2H), 7.84 (d, J = 8.7 Hz, 2H), 9.89 (s, 1H); 13C NMR δ 28.25, 65.76, 83.17, 115.08, 130.82, 132.18, 163.00, 167.37, 190.98; EI-MS 105/107, 135, 193/194, 236/237; FAB-MS obsd 237.1120, calcd 237.1127 [(M + H)+, M = C13H16O4]; Anal. C, H.
5-[4-(tert-Butoxycarbonylmethoxy)phenyl]dipyrromethane (7a)
Following a standard procedure (40), aldehyde 6a (2.07 g, 8.78 mmol) was dissolved in dry pyrrole (56.0 mL, 880 mmol), and the solution was flushed with argon for 10 min. InCl3 (666 mg, 0.800 mmol) was added, and the reaction was allowed to proceed for 3 h. The reaction was quenched by addition of powdered NaOH (977 mg, 24.4 mmol). The mixture was stirred for 45 min. The mixture was filtered. The filtrate was concentrated at reduced pressure. Chromatography [silica, ethyl acetate/CH2Cl2/hexanes (1:2:7)] afforded a pale yellow oil (2.73 g, 88%): IR (film, νmax cm−1) 1744, 1608; 1H NMR δ 1.49 (s, 9H), 4.49 (s, 2H), 5.42 (s, 1H), 5.90 (app s, 2H), 6.15 (d, J = 2.7 Hz, 2H), 6.69 (app s, 2H), 6.83 (d, J = 9.0 Hz, 2H) 7.12 (d, J = 9.0 Hz, 2H), 7.93 (br, 2H); 13C NMR δ 28.30, 43.35, 65.96, 82.66, 107.32, 108.61, 114.91, 117.40, 129.70, 132.96, 135.33, 157.13, 168.32; EI-MS 145, 229/230, 295/296, 352; FAB-MS obsd 352.1800, calcd 352.1787 (C21H24N2O3); Anal. H, N; C calcd, 71.57; found, 71.00.
5-[4-(tert-Butoxycarbonylmethoxy)phenyl]-1,9-diformyldipyrromethane (8a)
Following a standard procedure (42), a solution of 7a (2.66 g, 7.55 mmol) in DMF (7.53 mL) was cooled to 0 °C under argon, and phosphorus oxychloride (1.48 mL, 16.2 mmol) was added. The mixture was allowed to reach room temperature. Stirring was continued for 1 h. The solution was poured into aqueous sodium hydroxide (80 mL of 10 wt % solution) and was extracted into ethyl acetate (5 × 80 mL). The organic phases were combined, washed with water and brine, dried (Na2SO4), and concentrated. Chromatography [neutral alumina, CH2Cl2/MeOH (0.5→1%)] gave a pale brown solid (1.36 g, 45%): mp 84–85 °C (dec); IR (film, νmax cm−1) 1754, 1599; 1H NMR δ 1.47 (s, 9H), 4.48 (s, 2H), 5.52 (s, 1H), 6.03 (d, J = 3.9 Hz, 2H), 6.81–6.85 (m, 4H), 7.15 (d, J = 8.4 Hz, 2H), 9.21 (s, 2H), 10.20–10.80 (br, 2H); 13C NMR δ 28.48, 43.79, 65.94, 82.90, 111.64, 115.23, 12.36, 129.79, 132.90, 141.88, 162.82, 168.13, 179.17; FAB-MS obsd 408.1668, calcd 408.1685 (C23H24N2O5); Anal. H, N; C calcd, 67.63; found, 66.43.
Dibutyl[5-[4-(tert-butoxycarbonylmethoxy)phenyl]-1,9-diformyl-5,10-dihydrodipyrrinato]tin(IV) (8aSnBu2)
Following a standard method (43), a solution of 8a (1.79 g, 4.39 mmol) in CH2Cl2 (3.6 mL) was treated with TEA (1.83 mL) and dibutyltin dichloride (1.33 g, 4.38 mmol). The solution was stirred at room temperature for 1 h. The reaction mixture was concentrated at reduced pressure. Chromatography [silica, CH2Cl2/TEA (99:1)] gave a dark orange oil (0.79 g, 28%): IR (film, νmax cm−1) 1753, 1599; 1H NMR δ 0.70–0.79 (m, 6H), 1.10–1.57 (m, 23H), 4.46 (s, 2H), 5.46 (s, 1H), 6.11 (d, J = 3.3 Hz, 2H), 6.79 (d, J = 3.3 Hz, 2H), 7.03–7.06 (m, 2H), 9.15 (s, 2H); 13C NMR δ 13.71, 13.78, 24.14, 24.56, 26.26, 26.50, 27.29, 27.33, 28.27, 66.01, 22.64, 82.60, 115.04, 124.11, 129.42, 136.85, 138.11, 152.52, 157.11, 168.21, 178.79; FAB-MS obsd 641.2004, calcd 641.2037 [(M + H)+, M = C31H40N2O5Sn]; Anal. C, H, N.
5-[4-(tert-Butoxycarbonylmethoxy)phenyl]-1,9-bis(N-propylimino)dipyrromethane (9a)
A solution of 8a (1.49 g, 3.64 mmol) in THF (12 mL) was treated with propylamine (6.0 mL). The solution was stirred at room temperature for 1 h. The volatile components were evaporated. The crude product was dried at reduced pressure, affording a pale brown solid (quantitative): mp 119–121 °C; IR (film, νmax cm−1) 1755, 1639; 1H NMR δ 0.88 (t, J = 7.5 Hz, 6H), 1.48 (s, 9H), 1.55–1.62 (m, 4H), 3.37 (t, J = 6.6 Hz, 4H), 4.47 (s, 2H), 5.36 (s, 1H), 5.88 (d, J = 3.6 Hz, 2H), 6.32 (d, J = 3.6 Hz, 2H), 6.81 (d, J = 8.7 Hz, 2H), 7.09 (d, J = 8.7 Hz, 2H), 7.88 (s, 2H); 13C NMR δ 12.04, 24.51, 28.29, 43.76, 62.90, 66.01, 82.58, 109.31, 114.55, 114.96, 129.68, 130.39, 134.24, 136.73, 151.68, 157.21, 168.26, 20.98; FAB-MS obsd 491.3026, calcd. 491.3022 [(M + H)+, M = C29H38N4O3]; Anal. C, H, N.
Zn(II) 5-[1,5-Bis(tert-butyldimethylsilyloxy)pent-3-yl]-15-[4-(tert-butoxycarbonylmethoxy)phenyl]porphyrin (Zn10a)
A solution of 9a (132 mg, 0.269 mmol) and 5 (142 mg, 0.298 mmol) in toluene (30.0 mL) was treated with Zn(OAc)2 (550 mg, 3.00 mmol). The mixture was refluxed for 18 h open to the air. The toluene was evaporated. The residue was chromatographed (silica, CH2Cl2) to give a purple solid (62.0 mg, 24%): 1H NMR δ −0.19 (s, 12H), 0.75 (s, 18H), 1.62 (s, 9H), 3.03–3.12 (m, 2H), 3.25–3.37 (m, 2H), 3.56–3.71 (m, 4H), 4.80 (s, 2H), 5.93 (m, 1H), 7.29–7.33 (m, 2H), 8.12–8.15 (m, 2H), 9.10–9.12 (m, 2H), 9.38–9.40 (m, 2H), 9.46–9.50 (m, 2H), 9.91 (d, J = 4.8 Hz, 1H), 10.04 (d, J = 5.1 Hz, 1H), 10.25 (s, 2H); LD-MS obsd 908.4; FAB-MS obsd 908.3694, calcd 908.3707 (C49H64N4O5Si2Zn); λabs (log ε) 408 (4.64), 538 (3.68) nm; λem (λexc 408 nm) 580, 634 nm.
Zn(II) 5-(4-tert-Butoxycarbonylmethoxyphenyl)-15-(1,5-dihydroxypent-3-yl)porphyrin (Zn11a)
A solution of Zn10a (48.7 mg, 0.0536 mmol) was dissolved in THF containing TBAF (3.0 mL of 1.0 M solution, water content ~5%). The reaction mixture was cooled in an ice–water bath, and the reaction was allowed to proceed for 1 h. Then, the mixture was poured into ethyl acetate, and the organic phase was washed with water. The aqueous layer was extracted with ethyl acetate. The organic phase was washed with water and dried (Na2SO4). The sample was concentrated. Anhydrous zinc acetate (200 mg, 1.09 mmol) was added, and the mixture was stirred at room temperature for 15 min. Chromatography [neutral alumina, CH2Cl2/MeOH (2→10%)] afforded a bright purple solid (31.8 mg, 78%): 1H NMR (THF-d8) δ 1.53 (s, 9H), 2.95–2.99 (m, 2H), 3.19–3.24 (m, 2H), 3.48–3.52 (m, 4H), 4.74 (s, 2H), 5.71 (m, 1H), 7.21 (d, J = 7.8 Hz, 2H), 8.05 (d, J = 7.8 Hz, 2H), 8.95 (s, 2H), 9.24 (s, 2H), 9.32–9.33 (m, 2H), 9.69–9.70 (m, 1H), 9.80–9.81 (m, 1H), 10.06 (s, 2H); LD-MS obsd (−) 678.7; FAB-MS obsd 680.1963, calcd 680.1977 (C37H36N4O5Zn); λabs (log ε) 411 (5.22), 540 (4.34) nm; λem (λexc 411 nm) 583, 635 nm.
Zn(II) 5-(4-tert-Butoxycarbonylmethoxyphenyl)-15-[1,5-bis(dimethoxyphosphoryloxy)pent-3-yl]porphyrin (Zn12a)
A solution of 9a (211 mg, 0.431 mmol) and 5c (200 mg, 0.431 mmol) in toluene (46 mL) was treated with anhydrous zinc acetate (0.810 g, 4.43 mmol). The mixture was refluxed open to the air for 18 h. The mixture was concentrated to dryness. The crude product was dried and chromatographed [silica, CH2Cl2/EtOAc (0→20%) then CH2Cl2/methanol (0→2%)], affording a deep red solid (87.0 mg, 22%): 1H NMR δ 1.63 (s, 9H), 2.81–3.00 (m, 14H), 3.19–2.28 (m, 2H), 3.33–3.37 (m, 4H), 4.80 (s, 2H), 5.48–5.55 (m, 1H), 7.28–7.31 (m, 2H), 8.11–8.16 (m, 2H), 9.05–9.06 (m, 2H), 9.27–9.38 (m, 3H), 9.41–9.42 (m, 1H), 9.55–9.61 (m, 2H), 10.08 (s, 1H), 10.16 (s, 1H); LD-MS obsd 896.8; FAB-MS obsd 896.1970, calcd 896.1930 (C41H46N4O11P2Zn); λabs (log ε) 412 (4.96), 541 (4.12) nm; λem (λexc 412 nm) 591, 638 nm.
5-[4-(Carboxymethoxy)phenyl]-15-[1,5-bis(dimethoxyphosphoryloxy)pent-3-yl]porphyrin (12a)
A solution of Zn12a (41.9 mg, 0.0468 mmol) in CH2Cl2 (1.5 mL) was treated with TFA (1.5 mL). The solution was stirred at room temperature for 3 h. The volatile components were evaporated. The dark green residue was dissolved in CH2Cl2. The solution was washed with water. The aqueous phase was extracted with CH2Cl2. The organic phase was washed with water, dried (Na2SO4), and concentrated, affording a dark green solid (29.6 mg, 76%): 1H NMR δ 3.05–3.33 (m, 16H), 3.81–4.03 (m, 4H), 4.71 (s, 2H), 5.76 (m, 1H), 7.38 (d, J = 8.7 Hz, 2H), 8.02 (d, J = 8.7 Hz, 2H), 8.98–8.99 (m, 2H), 9.31–9.35 (m, 2H), 9.44–9.51 (m, 2H), 9.63–9.65 (m, 2H), 9.81–9.83 (m, 2H), 10.27 (s, 2H); LD-MS 778.6 (M + H)+, 801.7 (M + Na)+, 815.7 (M + K)+; FAB-MS obsd 779.2247, calcd 779.2247 [(M + H)+, M = C37H40N4O11P2]; λabs 407, 505 nm; λem (λexc 407 nm) 635, 699 nm.
Cu(II) 5-(4-(Carboxymethoxy)phenyl)-15-[1,5-bis(dimethoxyphosphoryloxy)pent-3-yl]porphyrin (Cu12a)
A solution of 12a (16 mg, 0.021 mmol) in CHCl3/CH3OH (5.0 mL, 9:1 v/v) was treated with Cu(OAc)2•H2O (42 mg, 0.21 mmol). The solution was stirred at room temperature for 12 h. Water was added to the reaction mixture. The phases were separated. The aqueous layer was extracted with CH2Cl2. The organic phase was washed with water, dried (Na2SO4), and concentrated, affording a dark orange solid (17 mg, 98%): LD-MS obsd 862.7, calcd 862.1 [(M + Na)+, M = C37H38CuN4O11P2]; λabs (log ε) 405 (4.47), 530 (2.50) nm.
5-(4-(Carboxymethoxy)phenyl)-15-[1,5-bis(dihydroxyphosphoryloxy)pent-3-yl]porphyrin (13a)
A thoroughly dried sample of 12a (11.8 mg, 0.0151 mmol) was dissolved in dry CH2Cl2 (2.0 mL) and then treated with TMS–Br (100 μL, 0.760 mmol). The reaction mixture was stirred at room temperature for 3 h under argon. The volatile components were evaporated at reduced pressure. The residue was dissolved in MeOH (3.0 mL). The solution was stirred for 1 h at room temperature. The volatile components were evaporated. The residue was dissolved in MeOH (3 mL). The solution was stirred for 1 h. The sample was concentrated. The solid residue was dissolved in ~0.5 mL of dilute aqueous NaOH (0.1 M) and was diluted to ~3 mL with water. The resulting solution was chromatographed [silica C-18, water/MeOH (0→50%)] to afford a dark red solid (8.20 mg, 76%): 1H NMR (D2O) δ 3.61 (m, 4H), 3.95 (m, 2H), 4.33 (m, 2H), 5.04 (s, 2H), 5.89 (m, 1H), 7.57 (d, J = 7.8 Hz, 2H), 7.94–7.97 (m, 2H), 8.91 (m, 2H), 9.28–9.34 (m, 2H), 9.78–9.82 (m, 2H), 10.24–10.33 (m, 4H); ESI-MS obsd (+) 362.1 (M + 2H)2+, 723.1 (M + H)+, 745.1 (M + Na)+, (−) 721.1 (M − H)−, calcd 722.15 (M = C33H32N4O11P2); λabs 402, 506 nm; λem (λexc 402 nm) 628, 688 nm; HPLC tR = 9.83 min.
Cu(II) 5-(4-(Carboxymethoxy)phenyl)-15-[1,5-bis(dihydroxyphosphoryloxy)pent-3-yl]porphyrin (Cu13a)
A thoroughly dried sample of Cu12a (28.7 mg, 0.034 mmol) was dissolved in dry CH2Cl2 (1.5 mL) and then treated with TMS–Br (111 μL, 0.84 mmol). The reaction mixture was stirred at room temperature for 3 h under argon. The volatile components were evaporated at reduced pressure. The residue was dissolved in MeOH (3.0 mL). The solution was stirred for 1 h at room temperature. The mixture was concentrated. The solid residue was dissolved in ~0.5 mL of dilute aqueous NaOH (0.1 M) and was diluted to ~3 mL with water. Chromatography [silica C-18, water/MeOH (0→50%)] afforded an orange solid (12.2 mg, 12.2%, 46%): ESI-MS obsd (−) 782.4, 391.0, calcd 784.0, 391.5 [(M − H)−, (M − 2H)2−, M = C33H30CuN4O11P2], also obsd 796.2 (M + methylene)+; λabs 402, 530 nm.
5-(4-(tert-Butoxycarbonylmethoxy)phenyl)-15-(1,5-dibromopent-3-yl)porphyrin (14a)
A sample of Zn11a was thoroughly dried by treatment overnight under vacuum (0.05 mmHg) at room temperature. Following a standard method (44), a solution of the dried sample of Zn11a (35.6 mg, 57 μmol) in dry CH2Cl2 (12 mL) was treated with CBr4 (53.5 mg, 0.16 mmol). The solution was cooled in an ice–water bath for 10 min. Tri-phenylphosphine (84 mg, 0.32 mmol) was added. The solution was allowed to warm to room temperature. The reaction was allowed to proceed at room temperature for 12 h. Water was added, and the phases were separated. The aqueous phase was extracted with CH2Cl2. The organic phase was washed with water. The organic layer was dried (Na2SO4). Chromatography (silica, CH2Cl2) afforded a dark green solid (38.7 mg, 91%): 1H NMR δ −2.86 (s, 1H), −2.82 (s, 1H), 1.64 (s, 9H), 3.24–3.39 (m, 6H), 3.65–3.75 (m, 2H), 4.85 (s, 2H), 5.83 (m, 1H), 7.35 (d, J = 8.1 Hz, 2H), 8.18 (d, J = 8.1 Hz, 2H), 9.08–9.11 (m, 2H), 9.37–9.60 (m, 5H), 9.98 (d, J = 4.2 Hz, 1H), 10.30 (s, 2H); LD-MS obsd 745.1; FAB-MS obsd 742.1166, calcd 742.1154 (C37H36Br2N4O3); λabs (log ε) 407 (5.00), 503 (4.15) nm; λem (λexc 407 nm) 635, 701 nm.
5-(4-(tert-Butoxycarbonylmethoxy)phenyl)-15-[1,5-bis-(dimethoxyphosphoryl)pent-3-yl]porphyrin (15a)
A sample of 14a (38.7 mg, 0.0521 mmol) was dissolved in P(OCH3)3 (10.0 mL). The solution was refluxed under argon for 1.5 days. The P(OCH3)3 was evaporated at reduced pressure. The residue was chromatographed [silica, CH2Cl2/MeOH (0→5%)] to yield a dark purple solid (24.6 mg, 65%): 1H NMR δ 1.64 (s, 9H), 3.15 (m, 2H), 3.32–3.36 (m, 2H), 3.43 (s, 3H), 3.47 (s, 6H), 3.50 (s, 3H), 3.81–3.87 (m, 4H), 4.84 (s, 2H), 5.44 (m, 1H), 7.35 (d, J = 8.1 Hz, 2H), 8.17 (d, J = 8.1 Hz, 2H), 9.10 (s, 2H), 9.40 (s, 2H), 9.50–9.51 (m, 2H), 9.73–9.74 (m, 2H), 10.30–10.31 (m, 2H); LD-MS obsd 803.7; FAB-MS obsd 803.2983, calcd 803.2975 (C41H48N4O9P2); λabs (log ε) 407 (4.90), 504 nm; λem (λexc 407 nm) 636 nm, 702 nm. Varying amounts of a less polar purple solid (15a-Br) were isolated along with small amounts of the starting material 14a (limited characterization). Data for 15a-Br: 1H NMR δ 0.86–0.95 (m, 1H), 1.65 (s, 9H), 1.91–2.00 (m, 1H), 3.13–3.40 (m, 6H), 3.44 (s, 3H), 3.48 (s, 3H), 3.55–3.85 (m, 2H), 4.83 (s, 2H), 5.51–5.61 (m, 1H), 7.34 (d, J = 8.1 Hz, 2H), 8.17 (d, J = 8.1 Hz, 2H), 9.08–9.10 (m, 2H), 9.37–9.41 (m, 2H), 9.49–9.51 (m, 2H), 9.67–9.68 (m, 1H), 9.84–9.85 (m, 1H), 10.30 (s, 2H); LD-MS obsd 773.6, calcd 773.2 [(M + H)+, M = C39H42-BrN4O6P2]; λabs 407, 502 nm; λem (λexc 407 nm) 634, 702 nm.
5-(4-(Carboxymethoxy)phenyl)-15-[1,5-bis(dihydroxyphosphoryl)pent-3-yl]porphyrin (16a)
A solution of 15a (19 mg, 0.024 mmol) in anhydrous CHCl3 (2 mL) was flushed with argon for 10 min. Bromotrimethylsilane (400 μL, 3.03 mmol) was added, and the solution was refluxed for 4 h. The reaction mixture was allowed to cool to room temperature. The volatile components were evaporated. The residue was dissolved in MeOH (3 mL). The solution was stirred at room temperature for 1 h. The volatile components were evaporated. Aqueous NaOH (0.3 mL, 0.1 M) was added. The mixture was diluted to ~3 mL with distilled water. The resulting solution was chromatographed (C-18 silica, H2O/MeOH gradient) to yield a red solid (9.8 mg, 58%): 1H NMR (D2O) δ 1.31–1.36 (m, 2H), 2.10–2.15 (m, 2H), 3.16–3.62 (m, 4H), 5.03 (s, 2H), 5.60–5.72 (m, 1H), 7.55–7.60 (m, 2H), 7.82–7.96 (m, 2H), 8.82–8.94 (m, 2H), 9.32–9.40 (m, 2H), 9.84–9.93 (m, 2H), 10.35–10.48 (m, 4H); ESI-MS obsd 691.1 (M + H)+, 346.1 (M + 2H)2+, 713.2 (M + Na)+, calcd 690.2 (M = C33H32N4O9P2); λabs (H2O) 402, 506 nm; λem (λexc 402 nm) 627, 688 nm; HPLC tR = 9.37 min.
Zn(II) 5-(4-(Carboxymethoxy)phenyl)-15-[1,5-bis(dihydroxyphosphoryl)pent-3-yl]porphyrin (Zn16a) (by in Situ Metalation of 16a)
A solution of 15a (10.4 mg, 0.013 mmol) in anhydrous CHCl3 (2 mL) was flushed with argon for 10 min. Bromotrimethylsilane (300 μL, 2.27 mmol) was added, and the solution was refluxed for 4 h. The reaction mixture was allowed to cool to room temperature. The volatile components were evaporated. The residue was dissolved in MeOH (2 mL). The solution was stirred at room temperature for 1 h. The solvent was evaporated. Aqueous NaOH (0.3 mL, 15 wt %) was added. The sample was diluted with distilled water (3 mL). The mixture was treated with Zn(OAc)2 (55 mg, 0.301 mmol) for 1 h. A second portion of aqueous NaOH (0.3 mL, 15 wt %) was added. The resulting solution was chromatographed [C-18 silica, water/MeOH (0→50%)] to yield a bright purple solid (9.1 mg, 93%): 1H NMR (D2O) δ 1.17–1.30 (m, 2H), 2.01–2.15 (m, 2H), 3.38–3.49 (m, 4H), 5.05 (s, 2H), 5.72 (s, 1H), 7.74 (d, J = 6.9 Hz, 2H), 8.37 (d, J = 6.9 Hz, 2H), 9.33 (app s, 2H), 9.71 (app s, 2H), 9.85–9.95 (m, 2H), 10.36–10.38 (m, 1H), 10.44–10.46 (m, 1H), 10.57 (s, 1H), 10.60 (s, 1H); ESI-MS obsd 376.9 (M + 2H)2+, 753.0 (M + H)+, 774.9 (M + Na)+, calcd 752.0779 (M = C33H30N4O9P2Zn); λabs (H2O) 409, 544 nm; λem (λexc 409 nm) 590, 641 nm; HPLC tR = 9.83 (27%), 12.10 (73%) min.
5-(4-Bromophenyl)dipyrromethane (7b)
This compound was previously prepared by a less efficient method (45). Following a standard procedure (40), 4-bromobenzaldehyde (5.55 g, 30.0 mmol) was dissolved in dry pyrrole (208 mL, 3.00 mol), and the solution was flushed with argon for 10 min. InCl3 (666 mg, 3.00 mmol) was added, and the reaction was allowed to proceed for 90 min. The reaction was quenched by addition of powdered NaOH (3.60 g, 90.0 mmol). The mixture was stirred for 45 min. The mixture was filtered. The filtrate was concentrated at reduced pressure. Chromatography [silica, ethyl acetate/CH2Cl2/hexanes (1:2:7)] afforded a pale yellow solid (8.29 g, 92%): mp 120–122 °C; IR (film, νmax cm−1) 1487; 1H NMR δ 5.43 (s, 1H), 5.88–5.89 (m, 2H), 6.15–6.16 (m, 2H), 6.70–6.71 (m, 2H), 7.06–7.08 (m, 2H), 7.42–7.44 (m, 2H), 7.88–7.93 (br, 2H); 13C NMR δ 43.66, 107.65, 108.80, 112.61, 117.73, 121.08, 130.36, 131.92, 141.40; EI-MS 145, 234/236, 300/302; FAB-MS obsd 300.0257, calcd 300.0262 (C15H13BrN2); Anal. C, H, N.
5-(4-Bromophenyl)-1,9-diformyldipyrromethane (8b)
Following a standard procedure (42), a solution of 7b (1.51 g, 5.00 mmol) in DMF (5.00 mL) was cooled to 0 °C under argon, and phosphorus oxychloride (980 μL) was added. The mixture was allowed to reach room temperature. Stirring was continued for 1 h. The solution was poured into aqueous sodium hydroxide (50 mL of 10 wt % solution) and was extracted into ethyl acetate (5 × 50 mL). The organic phases were combined, washed with water and brine, dried (Na2SO4), and chromatographed [neutral alumina, CH2Cl2/MeOH (0.5→1%)] to afford a pale brown solid (1.43 g, 80%): mp 79–80 °C (dec); IR (film, νmax cm−1) 1645, 1485; 1H NMR δ 5.54 (s, 1H), 6.03–6.05 (m, 2H), 6.85–6.87 (m, 2H), 7.17 (d, J = 7.1 Hz, 2H), 7.46 (d, J = 7.1 Hz, 2H), 9.18 (s, 2H), 10.60–10.70 (br, 2H); 13C NMR δ 44.14, 112.07, 121.89, 122.68, 130.48, 132.27, 132.93, 138.63, 141.61, 179.37; FAB-MS obsd 356.0173, calcd 356.0160 (C17H13BrN2O2); Anal. C, H, N.
Dibutyl[5-(4-bromophenyl)-1,9-diformyl-5,10-dihydrodipyrrinato]tin(IV) (8bSnBu2)
Following a standard procedure (43), a solution of 8b (1.55 g, 4.35 mmol) in CH2Cl2 (3.5 mL) was treated with TEA (1.82 mL) and dibutyltin dichloride (1.32 g, 4.34 mmol). The solution was stirred at room temperature for 1 h. The reaction mixture was concentrated at reduced pressure. Chromatography [silica, CH2Cl2/TEA (99:1)] gave a crystalline pink solid (0.410 g, 16%): mp 115–117 °C; IR (film, νmax cm−1) 1601; 1H NMR δ 0.70–0.80 (m, 6H), 1.12–1.59 (m, 12H), 5.49 (s, 1H), 6.13 (d, J = 3.9 Hz, 2H), 7.00 (d, J = 8.1 Hz, 2H), 7.06 (d, J = 3.9 Hz, 2H), 7.39 (d, J = 8.1 Hz, 2H), 9.17 (s, 2H); 13C NMR δ 13.72, 13.79, 24.16, 24.70, 26.25, 27.27, 27.37, 44.83, 112.62, 115.63, 121.09, 124.12, 130.01, 132.05, 138.24, 142.88, 151.45, 179.09; FAB-MS obsd 589.0500, calcd. 589.0513 [(M + H)+, M = C25H29BrN2O2Sn]; Anal. C, H, N.
5-(4-Bromophenyl)-1,9-bis(N-propylimino)dipyrromethane (9b)
Following a standard procedure (42), a solution of 8b (488 mg, 1.37 mmol) in THF (4.6 mL) was treated with propylamine (2.25 mL, 27.4 mmol). The solution was stirred at room temperature for 1 h. The volatile components were evaporated, and the crude product was dried at reduced pressure, affording a pale brown solid: mp 136–138 °C; IR (film, νmax cm−1) 1634, 1485; 1H NMR δ 0.85–0.89 (m, 6H), 1.52–1.55 (m, 4H), 3.31–3.39 (m, 4H), 5.36 (s, 1H), 5.88 (app s, 2H), 6.34 (app s, 2H), 7.05 (d, J = 7.1 Hz, 2H), 7.40 (d, J = 7.1 Hz, 2H), 7.90 (s, 2H), 8.80–9.05 (br, 2H); 13C NMR δ 12.04, 24.53, 44.00, 62.77, 109.44, 114.62, 121.10, 130.35, 130.69, 131.74, 136.07, 140.59, 151.84; FAB-MS obsd 439.1503, calcd 439.1497 [(M + H)+, M = C23H27BrN4]; Anal. C, H, N.
Zn(II) 5-(4-Bromophenyl)-15-[1,5-bis(tert-butyldimethylsilyloxy)pent-3-yl] porphyrin (Zn10b)
A solution of 9b (132 mg, 0.269 mmol) and 5a (142 mg, 0.298 mmol) in toluene (30.0 mL) was treated with Zn(OAc)2 (550 mg, 3.00 mmol). The mixture was refluxed for 18 h open to the air. The mixture was concentrated, and the resulting residue was chromatographed (silica, CH2Cl2) to give a purple solid (62.0 mg, 24%): 1H NMR δ −0.07 (s, 12H), 0.96 (s, 18H), 3.13–3.15 (m, 2H), 3.37–3.44 (m, 2H), 3.67–3.81 (m, 4H), 6.03 (m, 1H), 7.82–7.90 (m, 4H), 8.78–8.80 (m, 2H), 9.04–9.10 (m, 2H), 9.39–9.42 (m, 1H), 9.48–9.50 (m, 2H), 9.93–9.97 (m, 2H), 10.05–10.14 (m, 2H); 13C NMR δ −0.11, 23.45, 23.80, 31.50, 43.96, 50.83, 67.50, 111.18, 111.46, 116.12, 123.12, 127.61, 129.08, 135.31, 136.46, 137.19, 137.34, 141.40, 147.07, 152.87, 154.27, 154.68, 155.94, 155.12, 155.52, 157.57, 174.92; LD-MS obsd 853.7; FAB-MS obsd 856.2232, calcd 856.2182 (C43H53BrN4O2Si2Zn); λabs 409, 539 nm; λem (λexc 409 nm) 578, 633 nm.
Zn(II) 5-(4-Bromophenyl)-15-(1,5-dihydroxypent-3-yl)porphyrin (Zn11b)
A sample of Zn10b (54 mg, 0.063 mmol) was dissolved in dry THF (1.26 mL) containing 1.0 M TBAF (1.3 mmol, 10.0 equiv), and the reaction mixture was stirred overnight at room temperature. The THF was evaporated. The residue was dissolved in ethyl acetate. The solution was washed with water. The aqueous layer was extracted with ethyl acetate. The organic phase was washed with water and brine, dried over Na2SO4, and concentrated. LD-MS indicated demetalation (obsd m/z 563), whereupon the solid was dissolved in CHCl3/MeOH (10 mL, 9:1) and Zn(OAc)2 (195 mg, mmol) was added. The mixture was stirred at room temperature for 1 h. The mixture was concentrated and chromatographed [neutral alumina, CH2Cl2/methanol (2→10%)], affording a bright red solid (34.5 mg, 87%): 1H NMR (THF-d8) δ 3.04–3.10 (m, 2H), 3.24–3.28 (m, 2H), 3.28–3.62 (m, 4H), 5.96 (m, 1H), 7.95 (d, J = 8.1, 2H), 8.14 (d, J = 8.1 Hz, 2H), 8.98–8.99 (m, 2H), 9.37–9.38 (m, 2H), 9.42–9.46 (m, 2H), 9.93 (d, J = 4.5 Hz, 1H), 10.05 (d, J = 4.5 Hz, 1H), 10.19–10.20 (m, 2H); LD-MS obsd 627.2 (M − H)−; FAB-MS obsd 628.0424, calcd 628.0452 (C31H25-BrN4O2Zn); λabs (CH2Cl2/MeOH, 97.5:2.5) (log ε) 408 (4.89), 538 nm; λem (λexc 408 nm) 589, 643 nm.
Zn(II) 5-(4-Bromophenyl)-15-[1,5-bis(dimethoxyphosphoryloxy)pent-3-yl]porphyrin (Zn12b)
Method A
A solution of Zn11b (30.0 mg, 0.048 mmol) in dry pyridine (1.0 mL) was cooled in an ice–water bath. Dimethyl chlorophosphate (168 μL, 225 mg, 1.56 mmol) was added, and stirring was continued with cooling for 2 h. The reaction mixture was allowed to reach room temperature. The reaction was allowed to proceed for a further 12 h. The sample was poured into CH2Cl2 and was washed with brine. The aqueous layer was extracted with CH2Cl2. The organic phase was washed with brine and dried (Na2SO4). LD-MS analysis of the crude mixture indicated partial demetalation (obsd m/z 782), whereupon the residue was dissolved in CHCl3/MeOH (15 mL, 9:1) and Zn(OAc)2 (230 mg, 1.26 mmol) was added. The mixture was stirred at room temperature for 2 h. Chromatography [silica, CH2Cl2/methanol (1→2%)] afforded a deep red solid (12.5 mg, 30%): 1H NMR δ 2.75 (s, 3H), 2.78 (s, 3H), 2.84 (s, 3H), 2.88 (s, 3H), 3.18 (m, 6H), 2.75–2.88 (m, 2H), 5.42 (m, 1H), 7.90 (d, J = 8.1 Hz, 2H), 8.07 (d, J = 8.1 Hz, 2H), 9.00–9.02 (m, 2H), 9.26–9.62 (m, 6H), 10.09 (s, 1H), 10.21 (s, 1H); LD-MS obsd 842.5; FAB-MS obsd 844.0444, calcd 844.0405 (C35H35-BrN4O8P2Zn); λabs (log ε) 411 (4.92), 541 (4.01) nm; λem (λexc 411 nm) 587, 636 nm.
Method B
A solution of 9b (247 mg, 0.50 mmol) and 5c (247 mg, 0.53 mmol) in toluene (58 mL) was treated with anhydrous zinc acetate (1.03 g, 5.65 mmol). The mixture was refluxed open to the air for 18 h. The mixture was concentrated, dried, and chromatographed [silica, CH2Cl2/methanol (0→2%)], affording a deep red solid (95.5 mg, 21%) with the same physical properties (1H NMR, LD-MS, λabs) as the sample obtained by method A.
5-(4-Bromophenyl)-15-[1,5-bis(dimethoxyphosphoryloxy)-pent-3-yl]porphyrin (12b)
A solution of Zn12b (50.6 mg, 0.0600 mmol) in CH2Cl2 (2 mL) was treated with TFA (2 mL). The solution was stirred at room temperature for 30 min. The volatile components were evaporated. The dark green residue was dissolved in CH2Cl2. The solution was washed with water. The aqueous phase was extracted with CH2Cl2. The organic phases were combined and washed with water. The organic layer was dried (Na2SO4). Chromatography [silica, CH2Cl2/MeOH (97:3)] afforded a deep purple solid (46.0 mg, 98%): 1H NMR δ 3.22–3.24 (m, 2H), 3.42–3.51 (m, 2H), 3.90–3.99 (m, 14H), 4.01–4.11 (m, 2H), 5.75–5.92 (m, 1H), 7.94–7.96 (m, 2H), 8.11–8.13 (m, 2H), 9.00–9.05 (m, 2H), 9.38–9.43 (m, 2H), 9.45–9.52 (m, 2H), 9.67–9.68 (m, 1H), 9.84–9.85 (m, 1H), 10.30 (s, 1H), 10.32 (s, 1H); LD-MS obsd (−) 782.8; FAB-MS obsd 782.1257, calcd 782.1270 (C35H37BrN4O8P2); λabs 405, 504 nm; λem (λexc 405 nm) 635, 700 nm.
Cu(II) 5-(4-Bromophenyl)-15-[1,5-bis(dimethoxyphosphoryloxy)pent-3-yl]porphyrin (Cu12b)
A solution of 12b (16.5 mg, 0.0211 mmol) in CHCl3/CH3OH (5 mL, 9:1 v/v) was treated with Cu(OAc)2•H2O (42.0 mg, 0.231 mmol). The solution was stirred at room temperature for 12 h. The volatile components were evaporated, and the residue was suspended in a small volume of CH2Cl2. Chromatography [silica, CH2-Cl2/MeOH (95:5)] afforded an orange solid (17.6 mg, 99%): LD-MS obsd 846.7 (M + H)+; FAB-MS obsd 843.0443, calcd 843.0410 (C35H35BrCuN4O8P2); λabs 405, 504 nm.
5-(4-Bromophenyl)-15-(1,5-bis[dihydroxyphosphoryloxy)-pent-3-yl]porphyrin (13b)
A thoroughly dried sample of 12b (23 mg, 0.031 mmol) was dissolved in dry CH2Cl2 (1.0 mL) and then treated with TMS–Br (25 μL, 0.19 mmol). The reaction mixture was stirred at room temperature for 3 h under argon. The volatile components were evaporated at reduced pressure. The residue was dissolved in MeOH (3.0 mL). The solution was stirred for 1 h at room temperature. The methanol was evaporated. The resulting solid was dissolved in aqueous NaOH (0.3 mL, 0.1 M) and diluted to ~3 mL with water. The solution was chromatographed [silica C-18, water/MeOH (0→50%)] to afford a red solid (9.6 g, 43%): 1H NMR (D2O) δ 3.63–3.68 (m, 4H), 3.67–3.99 (m, 2H), 4.30–4.35 (m, 2H), 5.91–5.96 (m, 1H), 7.40 (d, J = 6.3 Hz, 2H), 7.75 (d, J = 6.3 Hz, 2H), 8.44–8.51 (m, 2H), 8.99–9.02 (br, 1H), 9.13–9.16 (br, 1H), 9.79–9.87 (m, 2H), 10.15–10.18 (m, 1H), 10.33–10.40 (m, 3H); ESI-MS obsd (+) 726.3 (M + H)+, 748.8 (M + Na)+, (−) 724.9 (M − H)−, calcd 726.1 (M = C31H29-BrN4O8P2); λabs (H2O) 400, 504 nm; λem (λexc 400 nm) 624, 686 nm; HPLC tR = 13.80 min.
Cu(II) 5-(4-Bromophenyl)-15-[1,5-bis(dihydroxyphosphoryloxy)pent-3-yl]porphyrin (Cu13b)
A thoroughly dried sample of Cu12b (16.5 mg, 0.019 mmol) was dissolved in dry CH2Cl2 (800 μL) and then treated with TMS–Br (18 μL, 0.136 mmol). The reaction mixture was stirred at room temperature for 3 h under argon. The solvents were evaporated at reduced pressure. The residue was dissolved in MeOH (3.0 mL). The solution was stirred for 1 h at room temperature. The methanol was evaporated. The resulting solid was dissolved in aqueous NaOH (0.3 mL, 0.1 M) and diluted to ~3 mL with water. The solution was concentrated and chromatographed [silica C-18, water/MeOH (0→50%)] to afford a red solid (10.1 mg, 68%): ESI-MS (−) obsd 786.1 (M − H)−, calcd 787.0 (M = C31H27-BrCuN4O8P2), also obsd 800.1 (M + methylene)+, 814.2 (M + 2methylene)+; λabs (H2O) 400, 528 nm.
5-(4-Bromophenyl)-15-(1,5-dibromopent-3-yl)porphyrin (14b)
A sample of Zn11b was thoroughly dried by treatment overnight under vacuum (0.05 mmHg) at room temperature. Following a standard method (44), a solution of the dried sample of Zn11b (52 mg, 83 μmol) in dry CH2Cl2 (20 mL) was treated with CBr4 (84 mg, 0.25 mmol). The solution was cooled in an ice–water bath for 10 min. Triphenylphosphine (132 mg, 0.504 mmol) was added. The solution was allowed to warm to room temperature. Water was added, and the phases were separated. The aqueous phase was extracted with CH2Cl2. The organic phase was washed with water. The organic layer was dried (Na2SO4). Chromatography (silica, CH2Cl2) afforded a dark green solid (44.5 mg, 78%): 1H NMR δ −2.90 (s, 1H), −2.85 (s, 1H), 0.86–0.95 (m, 2H), 3.17–3.39 (m, 6H), 3.65–3.74 (m, 2H), 5.81–5.94 (m, 1H), 7.94 (d, J = 8.1 Hz, 2H), 8.11 (d, J = 8.1 Hz, 2H), 9.02–9.06 (m, 2H), 9.36–9.42 (m, 2H), 9.45–9.47 (m, 1H), 9.51–9.52 (m, 1H), 9.58–9.60 (m, 1H), 9.99 (d, J = 4.8 Hz, 1H), 10.30 (s, 2H); LD-MS 688.1; FAB-MS obsd 690.9700, calcd 690.9708 (C31H25N4Br3); λabs (log ε) 406 (4.96), 503 (4.01) nm; λem (λexc 407 nm) 633, 700 nm.
5-(4-Bromophenyl)-15-[1,5-bis(dimethoxyphosphoryl)pent-3-yl]porphyrin (15b)
A solution of 14b (33 mg, 0.047 mmol) was dissolved in P(OCH3)3 (8.0 mL). The solution was refluxed under argon for 2.5 days. The P(OCH3)3 was evaporated. The residue was chromatographed [silica, CH2Cl2/MeOH (0→5%)] to yield a dark purple solid (26 mg, 72%): 1H NMR δ −2.93 (s, 1H), −2.85 (s, 1H), 1.35–1.42 (m, 2H), 1.86–2.03 (m, 2H), 3.12–3.50 (m, 16H), 5.30–5.46 (m, 1H), 7.95 (d, J = 8.1 Hz, 2H), 8.12 (d, J = 8.1 Hz, 2H), 9.04–9.05 (m, 2H), 9.41–9.51 (m, 4H), 9.74–9.75 (m, 2H), 10.31–10.33 (m, 2H); LD-MS obsd 749.1 (M − H)−; FAB-MS obsd 751.1454, calcd 751.1450 [(M + H)+, M = C35H37BrN4O6P2); λabs 406, 503 nm; λem (λexc 406 nm) 633, 699 nm. Varying amounts of a less polar purple solid (15b-Br) were isolated along with small amounts of the starting material 14b (limited characterization). Data for 15b-Br: 1H NMR δ 0.74–0.99 (m, 2H), 3.14–3.41 (m, 6H), 3.45 (s, 3H), 3.49 (s, 3H), 3.69–3.80 (m, 2H), 5.63–5.63 (m, 1H), 7.95 (d, J = 7.5 Hz, 2H), 8.12 (d, J = 7.5 Hz, 2H), 9.04–9.05 (m, 2H), 9.39–9.42 (m, 2H), 9.49–9.52 (m, 2H), 9.69 (d, J = 4.2 Hz, 1H), 9.86 (d, J = 4.2 Hz, 1H), 10.31–10.32 (m, 2H); LD-MS obsd 718.3, calcd 720.1 (C33H31Br2N4O3P); λabs 406, 502 nm; λem (λexc 406 nm) 633, 699 nm.
5-(4-Bromophenyl)-15-[1,5-bis(dihydroxyphosphoryl)pent-3-yl]porphyrin (16b)
A solution of 15b (11 mg, 0.015 mmol) in anhydrous CHCl3 (2 mL) under argon was treated with TMS–Br (300 μL, 2.27 mmol). The sample was refluxed for 4 h. The mixture was concentrated, and the residue was dissolved in MeOH (3 mL). The mixture was stirred for 1 h. Evaporation of the solvent yielded a dark green solid, which was dissolved in dilute aqueous NaOH. Chromatography (C-18 silica, water/MeOH gradient) yielded a dark red solid (6.1 mg, 60%): 1H NMR (D2O) δ 0.88–0.97 (m, 2H), 1.70–1.84 (m, 2H), 2.96–3.08 (m, 2H), 3.09–3.24 (m, 2H), 5.21–5.33 (m, 1H), 6.80–6.87 (br, 2H), 7.26–7.38 (br, 2H), 7.91–7.95 (m, 2H), 8.36–8.40 (br, 1H), 8.52–8.61 (br, 1H), 9.35 (br, 1H), 9.48 (br, 1H), 9.62 (br, 1H), 9.82 (br, 1H), 9.95 (s, 1H), 10.03 (s, 1H); LD-MS obsd 693.8, ESI-MS obsd 695.0 (M + H)+, 717.0 (M + Na)+, 348.0 (M + 2H)2+, calcd 694.1 (M = C31H29BrN4O6P2); λabs (H2O) 400, 504 nm; λem (λexc 400 nm) 625, 687 nm; HPLC tR = 13.48 min.
Zn(II) 5-(4-Bromophenyl)-15-[1,5-bis(dihydroxyphosphoryl)pent-3-yl]porphyrin (Zn16b)
A solution of 15b (18.9 mg, 0.025 mmol) in anhydrous CHCl3 (2 mL) under argon was treated with TMS–Br (200 μL, 1.51 mmol). The reaction mixture was refluxed for 4 h. The volatile components were evaporated, and the residue was dissolved in MeOH (3 mL). The mixture was stirred for 1 h. The mixture was treated with aqueous NaOH (0.2 mL of 15 wt % solution). Zn(OAc)2•2H2O (125 mg, 0.57 mmol) was added, and stirring was continued for 1 h. The mixture was concentrated, treated with 0.1 mL of aqueous NaOH followed by 3 mL of water, and chromatographed [C-18 silica, water/MeOH (0→50%)], affording a deep purple solid (7.7 mg, 41%): 1H NMR (D2O) δ 0.72 (dd, J1 = 13.2 Hz, J2 = 15.6 Hz, 2H), 1.64 (dd, J1 = 14.4 Hz, J2 = 14.1 Hz, 2H), 2.96–3.22 (br, 4H), 5.36 (br, 1H), 7.65–7.74 (m, 4H), 8.76 (m, 2H), 9.17 (m, 1H), 9.49 (m, 1H), 9.53 (m, 1H), 10.01 (m, 1H), 10.09 (m, 1H), 10.14 (m, 1H), 10.20 (m, 1H); ESI-MS obsd 755.7 (M + H)+, 380.2 (M + 2H)2+, calcd 756.0 (M = C31H27BrN4O6P2Zn); λabs 409, 542 nm; λem (λexc 409 nm) 589, 640 nm; HPLC tR = 9.86 (17%), 15.51 (83%) min.
meso-Tetrakis[1,5-bis(tert-butyldimethylsilyloxy)pent-3-yl]-porphyrin (17)
Following a standard procedure (34), a solution of 4 (360 mg, 1.00 mmol) and pyrrole (70.0 μL, 1.00 mmol) in CHCl3 (100 mL) was treated with NaCl (1.46 g, 25.0 mmol). The mixture was flushed with argon for 10 min. The reaction was initiated with BF3•OEt2 (41 μL, 0.30 mmol). Stirring was continued overnight. DDQ (172 mg, 0.758 mmol) was added, and the reaction mixture was stirred for 1 h. The solvent was removed at reduced pressure. Chromatography [silica, hexanes/CH2Cl2 (1:1)] yielded a dark purple solid (41 mg, 10%): 1H NMR δ −2.46 (br, 2H), −0.15 (s, 48H), 0.86 (s, 72H), 2.88–2.92 (m, 8H), 3.11–3.16 (m, 8H), 3.61–3.69 (m, 16H), 5.57 (m, 4H), 9.49 (m, 4H), 9.66 (m, 4H); 13C NMR δ −0.00, 23.72, 31.45, 35.18, 45.22, 50.33 (br), 67.33, 116.15, 174.86; LD-MS 1631.9; FAB-MS obsd 1631.0, calcd 1631.1 (C88H166N4O8Si8); λabs 421, 522 nm; λem (λexc 421 nm) 665, 732 nm.
Zn(II) meso-Tetrakis[1,5-bis(tert-butyldimethylsilyloxy)-pent-3-yl]porphyrin (Zn17)
A solution of 17 (20 mg, 0.012 mmol) in CHCl3/MeOH (5 mL, 9:1) was treated with Zn(OAc)2• 2H2O (100 mg, 0.5 mmol). The reaction mixture was stirred at room temperature for 2 h. The volatile components were evaporated. Chromatography (silica, CH2Cl2) afforded a dark red solid (15.6 mg, 78%): 1H NMR δ −0.19 (s, 48H), 0.84 (s, 72H), 2.92–2.98 (m, 8H), 3.17–3.21 (m, 8H), 3.61–3.68 (m, 16H), 5.65 (m, 4H), 9.49 (m, 4H), 9.66–9.68 (m, 4H); LD-MS 1686.9; FAB-MS obsd 1695.0, calcd 1692 (C88H164N4O8Si8-Zn); λabs 422, 556 nm; λem (λexc 422 nm) 606, 657 nm.
16a–anti-CD3ε
A mixture of 16a (1.50 mg, 2.17 μmol) and EDCI (4.13 mg, 21.6 μmol) in PBS (50 μL) was added to a solution of IgG1 anti-human CD3 intracellular ε-chain (clone UCHT) (39) (125 μg in 50 μL PBS). The reaction was allowed to proceed at room temperature for 2 h. PBS (2.4 mL) was added to the reaction mixture, and the conjugate was isolated by chromatography (PD-10 size exclusion column, 1 mL fractions, PBS). The antibody conjugate was characterized by sodium dodecyl sulfate–polyacrylamide gel electrophoresis in a 5% nonreducing gel giving a single red fluorescent band migrating at 150 kDa. Absorption spectroscopy comparing predominant absorption by the antibody (λ280 nm) and porphyrin absorption (λ410 nm) indicated an average labeling efficiency of approximately two porphyrins per antibody.
Biological Experiments
Jurkat cells (human T-cell lymphoma, ATCC no. TIB-152) and murine Lewis lung carcinoma (LLC) cells (ATCC no. CRL-1642) were cultured in RPMI with L-glutamine and NaHCO3 supplemented with 10% heat inactivated fetal bovine serum and penicillin (100 U/mL) and streptomycin (100 μg/mL) (all from Sigma, St Louis, MO) at 37°C in a 5% CO2 humidified atmosphere in 75 cm3 tissue culture flasks (BD Falcon, Franklin Lakes, NJ). On the day of the experiment, when cells reached 80% confluence, the cells were washed with phosphate-buffered saline (PBS). The LLC cells were harvested with 2 mL of 0.25% trypsin–EDTA solution (Sigma), centrifuged, and counted with a hemocytometer, then resuspended in Cytofix/Cytoperm solution (BD Biosciences) and incubated for 30–40 min at 4 °C to perme-abilize the membranes. The cells were washed twice in perm/wash buffer (BD Biosciences) and incubated at room temperature for 20–30 min with porphyrin conjugate 16a–anti-CD3ε or a control antibody, FITC-conjugated anti-CD3ε IgG1 or FITC-conjugated IgG1 isotype (Sigma). Control cells were incubated with nothing. After incubation, cells were washed twice with perm/wash buffer. Flow cytometry analysis was performed using a FACSCalibur flow cytometry instrument (BD Biosciences) with laser excitation at 488 nm. Fluorescence emission was counted in several channels including FL1 (530 ± 30 nm) and FL3 (670-nm long pass) channels. For fluorescence microscopy, cells were seeded on glass slides by centrifugation and then analyzed with an Axiophot microscope (Carl Zeiss Inc, Thornwood, NY) fitted with a Spot camera and RT software, v 3.5 (Diagnostic Instruments, Stirling Heights, MI). Images were captured with a 40× objective and filter sets: FITC excitation 480–500 nm, emission 520–580 nm; rhodamine excitation 480–500 nm, emission 635-nm long pass.
RESULTS AND DISCUSSION
1. Synthesis
The target porphyrins are of the trans-AB-type, bearing one swallowtail substituent, one bioconjugatable group, and no substituents at the flanking meso positions (Chart 2). We recently developed two rational routes to trans-AB-porphyrins: (1) reaction of a 1,9-bis(N,N-dimethylaminomethyl)-dipyrromethane and a dipyrromethane in the presence of zinc acetate in ethanol followed by oxidation with DDQ (46) and (2) reaction of a 1,9-bis(imino)dipyrromethane and a dipyrromethane in the presence of zinc acetate in ethanol (42). Both routes directly afford zinc porphyrins; the latter method was employed herein. The dipyrromethanes bearing appropriate substituents for preparing the target porphyrins are available via a one-flask reaction of an aldehyde and excess pyrrole (39).
Chart 2.
A. Dipyrromethanes
The synthesis of a dipyrromethane bearing a swallowtail diol is shown in Scheme 1. The synthesis begins with 2-bromoethanol. Some early intermediates in the synthesis have been described in the literature but without complete characterization data (41). The complete synthesis is described as follows.
Scheme 1.
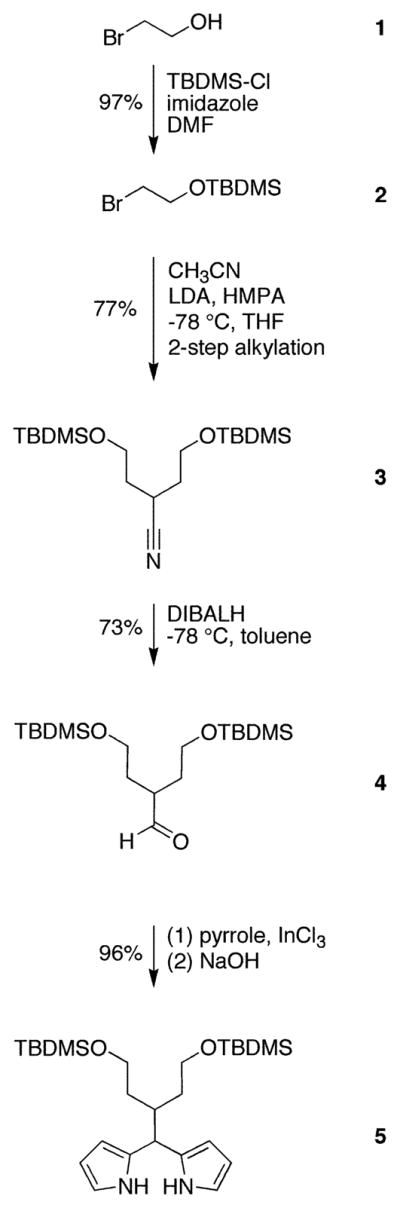
2-Bromoethanol (1) was protected by reaction with tert-butyldimethylsilyl chloride (TBDMS–Cl) in a mixture of imidazole and DMF. Aqueous workup followed by bulb-to-bulb distillation afforded the TBDMS-protected species 2 in 97% yield. A swallowtail nitrile was described by de Groot as a side-product in the alkylation of acetonitrile (41). We modified the de Groot method by changing the ratios of reactants and by performing sequential alkylation in a two-step one-flask process. Thus, treatment of acetonitrile with LDA in the presence of HMPA followed by alkylating agent 2 and repetition of this deprotonation/alkylation sequence afforded the dialkylated nitrile as the major product. Silica column chromatography gave 3 in 77% yield, along with mono- and trialkylated nitriles and a small amount of recovered 2. Reduction of 3 with DIBALH at −78 °C in toluene gave 4 in 73% yield. Aldehyde 4 reacted with excess pyrrole in the presence of InCl3 to give the 5-substituted dipyrromethane 5 in 96% yield as a pale yellow, viscous oil.
The bioconjugatable site was constructed from 4-hydroxy-benzaldehyde by O-alkylation with tert-butyl bromoacetate. The alkylation proceeded smoothly in anhydrous acetonitrile in the presence of potassium carbonate and a catalytic amount of sodium iodide, affording 6a as a white, crystalline solid in excellent yield. The aldehyde 6a was reacted with excess pyrrole in the presence of InCl3 followed by silica column chromatography to give dipyrromethane 7a as a pale yellow, viscous liquid in 88% yield (Scheme 2). Vilsmeier formylation of 7a followed by basic workup and chromatography on neutral alumina gave the 1,9-diformyldipyrromethane 8a. Yields were generally low (25–45%), presumably because of the sensitivity of the tert-butyl ester to the formylation conditions. For characterization purposes, the dibutyltin complex, 8aSnBu2, was prepared by derivatization of 8a with dibutyltin dichloride in a standard reaction (43). A similar sequence of reactions led from 4-bromo-benzaldehyde (6b) via the dipyrromethane 7b (45) to 1,9-diformyldipyrromethane 8b. Yields were comparable to those obtained for the tert-butyl ester derivatives, with the exception of the Vilsmeier formylation, which proceeded in excellent yield. Treatment of 8a or 8b with excess propylamine at room temperature quantitatively afforded the bis-imine 9a or 9b, respectively.
Scheme 2.

B. Porphyrin Diphosphates
The condensation of the swallowtail dipyrromethane 5 and the bis(imino)dipyrromethane 9a or 9b was carried out in the presence of a 10-fold excess of anhydrous zinc acetate in refluxing dry toluene open to the air (Scheme 3). In the original publication (42), ethanol was the solvent of choice for porphyrin formation. Ethanol proved to be an excellent solvent for the synthesis of the 4-bromophenyl-substituted zinc porphyrin (Zn10b), affording the desired product in 24–27% yield. However, under the same conditions only very small amounts of Zn10a were isolated. The main side reaction was hydrolysis of the tert-butyl ester. Also, some transesterification by the solvent occurred (either directly or via the free acid), as shown by LD-MS analysis of the crude reaction mixture and by 1H NMR analysis of the pure porphyrin. Therefore, small-scale trial reactions were carried out in a number of solvents to find a substitute for ethanol. The use of chloroform resulted both in very low yields and in extensive scrambling (i.e., formation of other porphyrin products owing to fragmentation of dipyrromethane units and subsequent undesired recombination processes). Upon reaction in dry toluene, scrambling was not observed, and both Zn10a and Zn10b could be isolated after straightforward silica column chromatography. Treatment of Zn10a or Zn10b with TBAF in THF cleaved the TBDMS protecting groups and also caused some demetalation of the zinc porphyrin, in each case affording a mixture of the zinc chelate and the free base porphyrin as shown by LD-MS. Remetalation with zinc acetate followed by chromatography on neutral alumina gave the zinc porphyrin diol Zn11a or Zn11b in >80% yield. It was important to limit the reaction time for conversion of Zn10a to Zn11a, because prolonged exposure under these conditions resulted in hydrolysis of the tert-butyl ester group.
Scheme 3.

Porphyrin diols Zn11a and Zn11b are highly polar and are only sparingly soluble in most organic solvents. Reaction of Zn11a with dimethyl chlorophosphate in anhydrous pyridine gave no Zn12a, whereas similar reaction with Zn11b gave a very low yield of Zn12b. The poor yields are presumably due to the combination of low solubility and low reactivity on the part of the porphyrin diol starting materials. We assumed that incorporation of the dimethoxyphosphoryl groups at an earlier stage could provide a more efficient process.
Our attempts to introduce the phosphate groups to aldehyde 4 were unsuccessful. Phosphorylation of the hydroxy groups of aldehyde 4 appeared attractive, but deprotection of 4 afforded the monocyclic hemiacetal and the bicyclic acetal of the corresponding 3-formylpentane-1,5-diol as an approximately 1:2 equilibrium mixture. None of the free diol was observed by 1H NMR spectroscopy. Therefore, the phosphate groups were introduced at the dipyrromethane stage (Scheme 4). Treatment of the TBDMS-protected swallowtail dipyrromethane 5 with TBAF in THF at room temperature resulted in cleavage of the protecting groups. Aqueous–organic workup followed by chromatography on neutral alumina gave the dipyrromethane diol 5-(OH)2 as a pale yellow oil in 89% yield. The dipyrromethane diol 5-(OH)2 was reacted with dimethyl chlorophosphate in anhydrous CH2Cl2 to give the bis(dimethoxyphosphoryloxy)dipyrromethane 5-(P)2 as a yellow oil in 61% yield. The monophosphate derivative 5-(P/OH) was also isolated (20–30% yield) and could be transformed to 5-(P)2. Alternatively, 5-(P/OH) could prove useful if a second functional group is to be introduced into the swallowtail moiety.
Scheme 4.

Reaction of 5-(P)2 with bis-imine 9a or 9b in anhydrous toluene in the presence of zinc acetate furnished the corresponding zinc porphyrin Zn12a or Zn12b in ~15% yield (Scheme 5). The spectroscopic yields of Zn12a and Zn12b were higher (30–45%) than the isolated yields. Two problems were encountered in handling the porphyrin diphosphate compounds Zn12a and Zn12b: (1) hydrolysis of the phosphates during purification, and (2) hydrolysis of the phosphates during “standard” conditions for methyl ester hydrolysis (47, 48). A lengthy series of experiments indicated that the loss of phosphate was facilitated by the presence of the centrally coordinated zinc atom (see Supporting Information for experiments and results). In summary, zinc porphyrins bearing phosphate-terminated swallowtail groups were not obtained for the pent-3-yl groups examined. Accordingly, we turned our attention to free base and copper analogues of the porphyrin diphosphates. Copper is strictly four-coordinate and was not expected to facilitate phosphate hydrolysis.
Scheme 5.

The free base porphyrins 12a and 12b were prepared by demetalation of Zn12a and Zn12b, respectively, by treatment with a 1:1 mixture of TFA and CH2Cl2. The demetalation was essentially complete within 15 min (as determined by absorption spectroscopy), but Zn12a was allowed to react further to allow for the quantitative cleavage of the tert-butyl ester. The resulting free base porphyrins 12a and 12b maintained the phosphate protecting groups although 12a contained a free carboxylic acid. Treatment of 12a,b with copper acetate afforded the corresponding copper porphyrins (Cu12a,b). Compounds Cu12a and Cu12b were characterized by LD-MS, FAB-MS, and absorption spectroscopy (but not 1H NMR spectroscopy because of the broadened spectra of copper porphyrins).
Cleavage of the phosphate protecting groups in the free base porphyrins (12a,b) and copper porphyrins (Cu12a,b) was carried out using TMS–Br. Porphyrin 12b reacted smoothly with a slight excess of TMS–Br in dry CH2Cl2 at room temperature. Subsequent treatment with aqueous NaOH and reversed-phase column chromatography yielded the desired porphyrin diphosphate 13b in excellent yield. The carboxylate-functionalized 12a required a larger excess of TMS–Br, whereupon the desired fully deprotected carboxy-porphyrin diphosphate 13a was obtained in 76% yield, along with a second fraction containing fully deprotected 13a and monomethyl-13a. Increasing the amount of TMS–Br, the reaction time, or the temperature resulted in conversion of 12a to the dibromo-substituted 14a.
It is noteworthy that the copper complexes were only partially deprotected by the same amount of TMS–Br as used for the corresponding free base porphyrins. ESI-MS showed the di-methyl and monomethyl Cu13a,b as the major products. Increasing the amount of TMS–Br resulted in almost complete demetalation and a reduction in the deprotection yield. Small quantities of Cu13a and Cu13b could be isolated, but the samples were only partially characterized. Moreover, neither diol 11a nor 11b (nor copper chelates thereof) was observed by LD-MS or ESI-MS during the deprotection reaction, by contrast with the case of the zinc chelates (Zn12a,b and Zn13a,b) where central metal atom coordination promoted hydrolysis. In summary, the free base and copper porphyrin diphosphates were prepared without the complications of loss of phosphate encountered with the corresponding zinc chelates.
C. Porphyrin Diphosphonates
For in vivo studies, the phosphate groups may be susceptible to cleavage by phosphatases. Therefore, porphyrin diphosphonates, which are stable to phosphatases, were prepared from the porphyrin diols Zn11a and Zn11b (Scheme 6). Montforts has reported the conversion of a porphyrin alcohol to the corresponding bromide upon reaction with CBr4 and triphenylphosphine (44). Similar treatment of a thoroughly dried sample of Zn11a or Zn11b in anhydrous CH2Cl2 with CBr4 at 0 °C followed by excess triphenylphosphine afforded the corresponding free base di-bromo-porphyrin 14a or 14b in excellent yield. During the chromatographic purification of the products, a very nonpolar bluish-green compound was isolated, in addition to the desired 14a or 14b. This compound was not characterized due to lack of material (<1 mg) and its instability, but absorption spectroscopy suggested that it was a chlorin species (λabs 414, 641 nm). It is possible that the triphenylphosphine could reduce one of the porphyrin pyrrole rings, giving rise to the chlorin. Refluxing 14a or 14b in neat P(OCH3)3 under an inert atmosphere for 36 h afforded 15a or 15b in moderate to good yield as a dark red solid. The reaction also produced the intermediate monobromo, monophosphonate porphyrins (15a-Br, 15b-Br). Although conditions have been optimized for the production of 15a and 15b, it is possible that the intermediates could be obtained in good yields and serve as starting points for the introduction of two different polar groups into one molecule.
Scheme 6.

The cleavage of the phosphonate protecting groups (and the tert-butyl ester for 15a) was investigated by treatment of 15a or 15b with TMS–Br beginning with the conditions we had employed earlier for cleaving porphyrin phosphonates (49, 50). The phosphonate groups are more robust than phosphate groups (vide supra) and will tolerate use of excess reagent at higher temperature for prolonged reaction times. The use of excess TMS–Br in refluxing chloroform under argon gave good results, affording the free acids after methanolysis of the silyl intermediates. The resulting green solids were insoluble in a number of organic solvents and water. Addition of small quantities of dilute aqueous base (NaOH or NaHCO3) rendered the porphyrins water-soluble. Treatment of the basic porphyrin solutions (prior to purification) with zinc acetate resulted in formation of the corresponding zinc chelates. Purification of each free base or zinc porphyrin was achieved by reversed-phase column chromatography upon elution with water/methanol. Removal of the solvent followed by freeze-drying of the aqueous samples yielded the desired porphyrin (16a, 16b, Zn16a, and Zn16b) in good to excellent yield.
2. Characterization and Water Solubility
All of the hydrophobic porphyrins were analyzed by LD-MS, FAB-MS, 1H NMR spectroscopy (except copper chelates), and absorption and emission spectroscopy. In certain cases OH, NH, and COOH signals were not observed owing to broadening, exchange with the solvent, or coincidence with the solvent peak. All of the deprotected porphyrin diphosphates and porphyrin diphosphonates were characterized by 1H NMR spectroscopy, ESI-MS, and absorption and emission spectroscopy. Each ESI-MS spectrum exhibited a strong peak due to the parent molecule (protonated and sodium adduct in the positive-ion mode; deprotonated in the negative-ion mode), with no peaks owing to partially deprotected porphyrins. Each deprotected free base porphyrin sample was found to be homogeneous upon examination by reversed-phase HPLC, while the zinc chelates eluted as two separate peaks. The front peak (minor) had the same tR as the corresponding free base porphyrin. Because the elution mixture contains TFA, partial demetalation during chromatography is likely. The free base porphyrins were not observed by ESI-MS, 1H NMR spectroscopy, or absorption or emission spectroscopies (see Experimental Procedures section).
All of the porphyrin diphosphates and porphyrin diphosphonates examined herein were readily soluble in water across a wide pH range and exhibited sufficient solubility to enable 1H NMR measurements in D2O. Although good-quality 1H NMR spectra were obtained at room temperature, the resolution increased substantially upon increasing the temperature to 60 °C. The higher temperature also enabled observation of the OCH2 signal in the carboxylic acid, which at room temperature was coincident with the water signal. gCOSY experiments carried out on the porphyrin diphosphonates enabled the partial assignment of the signals.
Determination of the upper limit of water solubility was not possible given the amount of material in hand and the relatively high solubility encountered. Indeed, in each case examined, 8–10 mg of pure porphyrin (13a, 13b, 16a, 16b, Zn16a, Zn16b) completely dissolved in 500 μL of distilled water, consistent with a concentration of ≥20 mM. The aqueous solutions are stable for days at room temperature when shielded from the light. Precipitation was not observed either with the naked eye or by absorption spectroscopy.
A standard method for the detection of aggregation of porphyrin solutions is based on the comparison of absorption spectra at different concentrations. In the absence of aggregation, the absorption spectrum recorded at concentration c1 and path length l1 should be unaltered upon 10-fold dilution and simultaneous increase of l1 to 10l1. For this purpose, a stock solution of each of the least polar water-soluble porphyrin (16b) and the most polar water-soluble porphyrin (13a) was prepared. The concentration ([16b] ≈ 10 mM, [13a] = 13.4 mM) was such that A ≈ 0.6 for the visible absorption band near 505 nm. Then aliquots of both samples were diluted by 10-fold and 100-fold, and the absorption spectrum was recorded for each sample. For the most concentrated sample of 16b, peak broadening in the 450–700 nm region indicated some aggregation at a concentration of ~10 mM. The absorption spectra of the 1 mM and the 0.1 mM samples were not broadened, consistent with a nonaggregated sample at these concentrations (Figure 2A). By contrast, no band broadening was observed for 13a over a concentration of 0.13–13.4 mM (Figure 2B).
Figure 2.

(A) Absorption spectra of porphyrin 16b: (blue) c ≈ 10 mM, l = 0.1 mm; (black) c ≈ 1 mM, l = 1 mm; (red) c ≈ 0.1 mM, l = 10 mm. (B) Absorption spectra of porphyrin 13a: (blue) c = 13 mM, l = 0.1 mm; (black) c = 1.3 mM, l = 1 mm; (red) c = 0.13 mM, l = 10 mm.
3. Structural Studies
The success of the hydrocarbon swallowtails in the lipophilization of extended aromatic systems has been explained by projection of the alkyl groups above and below the plane of the macrocycle, which prevents π–π stacking and aggregation (Figure 1). Molecular mechanics calculations were carried out on a series of swallowtail porphyrins wherein the swallowtail substituents were hydrophobic or terminated with polar substituents, including neutral or ionic groups. In all cases, the calculated geometries showed the projection of the substituents above and below the plane of the macrocycle. The geometry calculated for an ionic-functionalized swallowtail porphyrin is shown in Figure 3.
Figure 3.
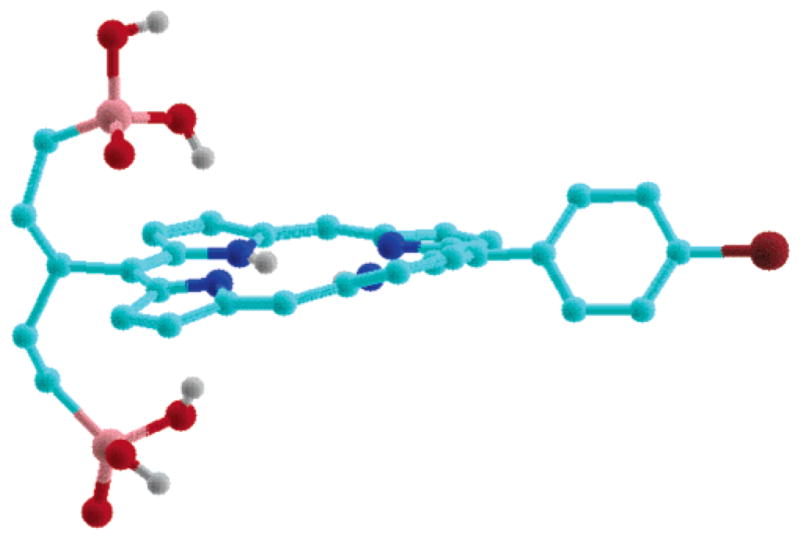
MMX-calculated gas-phase geometry of porphyrin 16b bearing dimethoxyphosphoryl end groups. Hydrogens on carbons are omitted for clarity.
We carried out 1H NMR studies (1 and 2D NMR, VT NMR) of a series of swallowtail porphyrins that were synthesized in this work as well as a porphyrin bearing a single hydrocarbon swallowtail group (34) to gain further insight concerning conformation of the swallowtail groups. An A4-swallowtail porphyrin (17) was synthesized from 4 upon condensation with pyrrole via a standard procedure (34) followed by oxidation with DDQ. The corresponding chelate, Zn17, was prepared by metalation with zinc acetate (Scheme 7). The results are as follows.
Scheme 7.
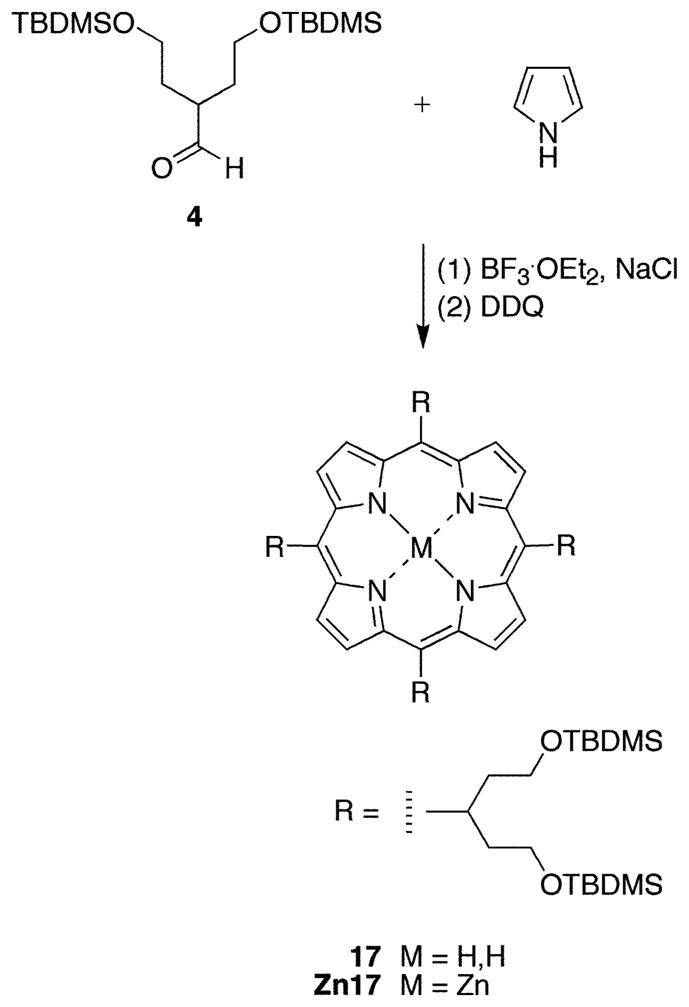
The 1H NMR spectrum of each A4-swallowtail porphyrin in CDCl3 (17 and Zn17) contains two broad apparent singlets (~9.67 and ~9.49 ppm) in the aromatic region each with integration of 4. For an A4-porphyrin with four magnetically equivalent substituents, the eight β-protons would resonate as one singlet in the aromatic region with an integration of eight. The signal corresponding to the CH protons of the swallowtail motifs (the branch site C1′H) appears as a broad, structureless multiplet (~5.56 ppm). NOESY experiments revealed that the two types of aromatic protons giving rise to the broad apparent singlets were closely juxtaposed and also that the one giving rise to the 9.67 ppm signal is close to C1′H (see Supporting Information). The spectra are consistent with restricted motion of the swallowtail substituent at the site of attachment to the porphyrin.
At room temperature in toluene-d8, the spectrum of 17 was similar to that recorded in CDCl3, although the signal at higher field consisted of two overlapping broad singlets. Increasing the temperature resulted in the broadening of the two aromatic signals (9.60 and 9.93 ppm). At ~70 °C, the two signals collapsed into one broad singlet (9.77 ppm), which eventually became a sharp singlet (9.72 ppm, 90 °C) with an integration of 8 (Figure 4). A similar but less pronounced sharpening was observed for the broad singlet at −1.72 ppm (NH) and the multiplet of C1′H (5.88 ppm) (see Supporting Information). The spectra at elevated temperature are consistent with a dynamic averaging on the NMR time scale of the conformation at the swallowtail branching site. On the basis of a coalescence temperature of 60 or 70 °C, an energy of activation of 68 or 70 kJ/mol is calculated for rotation of the swallowtail motif about the porphyrin meso carbon and the branch carbon C1′.
Figure 4.

Changes in the β-proton signals in porphyrin 17 with increasing temperature.
The trans-AB-porphyrins examined (Zn10b and Zn12b) have nominally lower symmetries than the A4-porphyrins 17 and Zn17. A trans-AB-porphyrin with simple A and B substituents is expected to exhibit signals in the aromatic region stemming from the four distinct porphyrin β-positions and the unsubstituted meso position. However, the 1H NMR spectra of all swallowtail trans-AB-porphyrins (Zn10a,b–Zn12a,b, 12a,b–16a,b, and Zn16a,b) studied in this work were more complex. Typically, two separate meso-proton signals were observed (at ~10.0 and 10.1 ppm). The four β-protons on the pyrrole units flanking the swallowtail substituents gave four doublets: a pair around 9.7 and 9.5 ppm with different coupling constants, and a pair at ~9.30 ppm (overlapping). In one case examined (Zn12b), increasing the temperature again simplified the spectrum, whereupon (1) the two meso-proton signals collapsed into one singlet (~100 °C, δ 10.03 ppm) and (2) the doublets corresponding to the two protons flanking the swallowtail group broadened out and moved closer to each other, although in the available temperature range (up to 100 °C) the doublets remained separate signals. Similarly to the A4-porphyrin, NOESY experiments on Zn12b showed the proximity between C1′H and one of either swallowtail-flanking β-pyrrolic protons. Two of the four C2′H2 protons are pointing toward the other swallowtail-flanking β-proton (see Supporting Information). NOESY experiments carried out on a porphyrin bearing a single all-hydrocarbon swallowtail group (34) indicated a similar configuration.
These results suggest that the swallowtail alkyl groups not only preferentially occupy positions out-of-plane with the porphyrin ring but at room temperature are hindered in their rotation around the carbon–carbon single bond between the porphyrin meso position and the swallowtail branching site (i.e., C5–C1′H). The C1′H is more or less in the plane of the porphyrin macrocycle. This conformation results in the two sets of β-protons and the two different meso-proton signals. At increased temperature, the rotational barrier is more easily surmounted and the time-averaged C2 symmetry of the porphyrin is restored. This geometry undoubtedly helps suppress cofacial interaction between porphyrin molecules at room temperature, thereby increasing solubility. The results of the NMR experiments are consistent with previous observations obtained via EPR spectroscopy (34) and provide additional evidence concerning the conformation of the swallowtail porphyrins in solution.
4. Bioconjugation Studies
The availability of water-soluble porphyrins each bearing a single bioconjugatable tether opens a number of applications in the life sciences. The porphyrin carboxylate 16a was used to label a monoclonal antibody (IgG). The labeling was carried out in aqueous solution using the water-soluble carbodiimide reagent EDCI. Purification was achieved in aqueous solution via size exclusion chromatography. Absorption spectroscopy of the purified conjugate (16a-anti–CD3ε) indicated an average loading of two porphyrins per antibody. It is noteworthy that no precipitation was observed during conjugation or purification or on standing during the three-week period over which biological experiments were carried out.
The performance of the conjugate was examined for staining an intracellular epitope using both flow cytometry and fluorescence microscopy. The use of dye-tagged monoclonal antibodies for staining both extracellular and intracellular epitopes is well established. In the present case, we employed the human T-cell lymphoma cell line, Jurkat cells, for which monoclonal antibodies (and FITC-labeled analogues) against the CD3ε chain are available. The flow cytometry results upon intracellular staining are shown in Figure 5. Panel A demonstrates that Jurkat cells bind anti-CD3ε but do not bind IgG1 isotype control, thus confirming the expression of intracellular CD3ε on Jurkat cells and the lack of Fc receptors that can nonspecifically bind IgG molecules via Fc regions. Panel B demonstrates that the epithelial cancer cells LLC do not bind FITC–anti-CD3ε, consistent with their known lack of T-cell receptors. Panel C shows that Jurkat cells bind porphyrin conjugate 16a–anti-CD3ε in an analogous fashion to the commercially available FITC–anti-CD3ε, thereby showing that porphyrin conjugation did not adversely affect the antigen recognition domain of the antibody. The TCR-negative LLC cells are also negative to 16a–anti-CD3ε, thereby demonstrating the lack of any non-bound porphyrin present in the conjugate preparation that could have also bound nonspecifically to receptor-negative cells.
Figure 5.
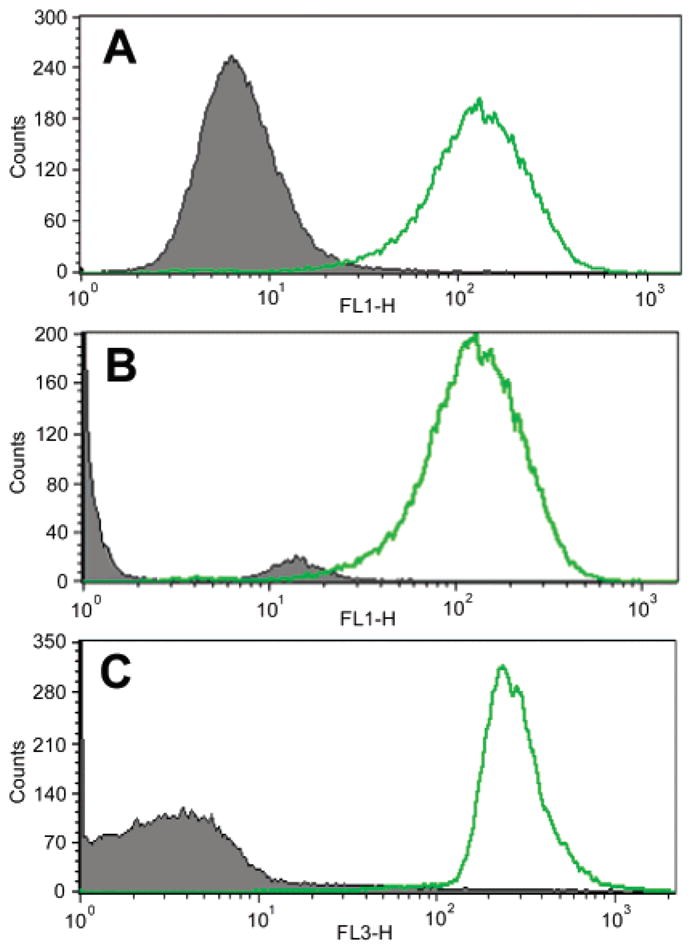
FACS analysis histograms: (A) green fluorescence (FL1) obtained with Jurkat cells stained with FITC–anti-CD3ε (open curve) or FITC isotype IgG1 (shaded curve); (B) green fluorescence obtained with Jurkat cells (open curve) or LLC cells (shaded curve) stained with FITC–anti-CD3ε; (C) red fluorescence (FL3) obtained with Jurkat cells (open curve) or LLC cells (shaded curve) stained with 16a–anti-CD3ε.
The specificity and functionality of the porphyrin conjugate 16a–anti-CD3ε was assessed by fluorescence microscopy as shown in Figure 6. Panels A and B show the green fluorescence staining of Jurkat cells with FITC–anti-CD3ε. Panels C and D show the red fluorescence staining of Jurkat cells with the porphyrin conjugate 16a–anti-CD3ε. Note that while the pattern of porphyrin staining is equally specific for target cells as the FITC counterpart, the subcellular pattern is somewhat different and appears to be more concentrated in the middle of the cell for unknown reasons. Panels E and F show that the receptor-negative LLC cells give no red staining with 16a–anti-CD3ε, a result that is consistent with the FACS data reported above.
Figure 6.
Fluorescence microscopy. The top panel shows Jurkat cells stained with FITC–anti-CD3ε under (A) bright field illumination or (B) green fluorescence detection. The middle panel shows Jurkat cells stained with 16a–anti-CD3ε under (C) bright field illumination or (D) red fluorescence detection. The bottom panel shows LLC cells stained with 16a–anti-CD3ε under (E) bright field illumination or (F) red fluorescence detection. The scale bar is 50 μm.
OUTLOOK
A compact water-solubilizing motif has been developed for use with porphyrinic macrocycles. The water-solubilizing motif consists of a short, branched alkyl chain (“swallowtail”) with polar end groups (phosphate, phosphonate) and does not include any aryl moieties. Incorporation of only one such swallowtail motif (based on a 1,5-disubstituted pent-3-yl group) at the meso position suffices to impart high aqueous solubility (≥20 mM) to a trans-AB-porphyrin, where the other meso substituent consists of a bioconjugatable group. The alkyl groups project above and below the hydrophobic porphyrinic macrocycle, thereby suppressing cofacial aggregation. The versatile synthetic chemistry for manipulating swallowtail substituents should facilitate their use as compact water-solubilizing motifs with a variety of porphyrinic macrocycles. The ability to carry out bioconjugations in aqueous solution is exceptionally valuable for use with diverse biomolecules. The requirement for only one swallowtail motif to achieve water solubility opens up a great deal of latitude in the design of porphyrinic-based molecular architectures for diverse applications in the life sciences.
Supplementary Material
Acknowledgments
This work was supported by the NIH (Grant GM36238 to J.S.L. and Grant CA/AI838801 to M.R.H.). Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the NSF.
Footnotes
Supporting Information Available: Description of experiments concerning the hydrolytic lability of zinc porphyrin diphosphates, VT-NMR data for Zn12b and 17, NOESY data for Zn12b and 17, and evidence concerning water solubility of Zn16b. This material is available free of charge via the Internet at http://pubs.acs.org.
LITERATURE CITED
- 1.Mauzerall D. The photoreduction of porphyrins: Structure of the products. J Am Chem Soc. 1962;84:2437–2445. [Google Scholar]
- 2.Mauzerall D. The photoreduction of uroporphyrin: Effect of pH on the reaction with EDTA. J Phys Chem. 1962;66:2531–2533. [Google Scholar]
- 3.Mauzerall D. Spectra of molecular complexes of porphyrins in aqueous solution. Biochemistry. 1965;4:1801–1810. [Google Scholar]
- 4.Carapellucci PA, Mauzerall D. Photosynthesis and porphyrin excited-state redox reactions. Ann New York Acad Sci. 1975;244:214–238. doi: 10.1111/j.1749-6632.1975.tb41533.x. [DOI] [PubMed] [Google Scholar]
- 5.Feitelson J, Mauzerall D. Enthalpy and electrostriction in the electron-transfer reaction between triplet zinc uroporphyrin and ferricyanide. J Phys Chem B. 2002;106:9674–9678. [Google Scholar]
- 6.Hambright P. Chemistry of water soluble porphyrins. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Volume 3: Inorganic, Organometallic and Coordination Chemistry. Chapter 18. Academic Press; San Diego, CA: 2000. pp. 129–210. [Google Scholar]
- 7.Grahn MF, Giger A, McGuinness A, de Jode ML, Stewart JCM, Ris H-B, Altermatt HJ, Williams NS. mTHPC polymer conjugates: The in vivo photodynamic activity of four candidate compounds. Lasers Med Sci. 1999;14:40–46. doi: 10.1007/s101030050019. [DOI] [PubMed] [Google Scholar]
- 8.Hornung R, Fehr MK, Walt H, Wyss P, Berns MW, Tadir Y. PEG-m-THPC-mediated photodynamic effects on normal rat tissues. Photochem Photobiol. 2000;72:696–700. doi: 10.1562/0031-8655(2000)072<0696:pmtmpe>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 9.Zingg A, Felber B, Gramlich V, Fu L, Collman JP, Diederich F. Dendritic iron(II) porphyrins as models for hemoglobin and myoglobin: Specific stabilization of O2 complexes in dendrimers with H-bond-donor centers. Helv Chim Acta. 2002;85:333–351. [Google Scholar]
- 10.Schell C, Hombrecher HK. Synthesis and investigation of glycosylated mono- and diarylporphyrins for photodynamic therapy. Bioorg Med Chem. 1999;7:1857–1865. doi: 10.1016/s0968-0896(99)00133-9. [DOI] [PubMed] [Google Scholar]
- 11.Lamarche F, Sol V, Huang YM, Granet R, Guilloton M, Krausz P. Synthesis and biological evaluation of polyamine-porphyrin conjugates as potential agents in photodynamic therapy (PDT) J Porphyrins Phthalocyanines. 2002;6:130–134. [Google Scholar]
- 12.Subbarayan M, Shetty SJ, Srivastava TS, Noronha OPD, Samuel AM, Mukhtar H. Water-soluble 99mTc-labeled dendritic novel porphyrins tumor imaging and diagnosis. Biochem Biophys Res Commun. 2001;281:32–36. doi: 10.1006/bbrc.2001.4289. [DOI] [PubMed] [Google Scholar]
- 13.Jee J-E, Eigler S, Hampel F, Jux N, Wolak M, Zahl A, Stochel G, van Eldik R. Kinetic and mechanistic studies on the reaction of nitric oxide with a water-soluble octa-anionic iron(III) porphyrin complex. Inorg Chem. 2005;44:7717–7731. doi: 10.1021/ic050924t. [DOI] [PubMed] [Google Scholar]
- 14.Cornia M, Valenti C, Capacchi S, Cozzini P. Synthesis, characterisation and conformational studies of lipophilic, amphiphilic and water-soluble C-glyco-conjugated porphyrins. Tetrahedron. 1998;54:8091–8106. [Google Scholar]
- 15.Král V, Davis J, Andrievsky A, Kralová J, Synytsya A, Poucková P, Sessler JL. Synthesis and biolocalization of water-soluble sapphyrins. J Med Chem. 2002;45:1073–1078. doi: 10.1021/jm0104320. [DOI] [PubMed] [Google Scholar]
- 16.Bisland SK, Singh D, Gariépy J. Potentiation of chlorin e6 photodynamic activity in vitro with peptide-based intracellular vehicles. Bioconjugate Chem. 1999;10:982–992. doi: 10.1021/bc990020u. [DOI] [PubMed] [Google Scholar]
- 17.de Luca S, Tesauro D, di Lello P, Fattorusso R, Saviano M, Pedone C, Morelli G. Synthesis and solution characterization of a porphyrin-CCK8 conjugate. J Pept Sci. 2001;7:386–394. doi: 10.1002/psc.337. [DOI] [PubMed] [Google Scholar]
- 18.Chaloin L, Bigey P, Loup C, Marin M, Galeotti N, Piechaczyk M, Heitz F, Meunier B. Improvement of porphyrin cellular delivery and activity by conjugation to a carrier peptide. Bioconjugate Chem. 2001;12:691–700. doi: 10.1021/bc000125t. [DOI] [PubMed] [Google Scholar]
- 19.Sibrian-Vazquez M, Jensen TJ, Fronczek FR, Hammer RP, Vicente MGH. Synthesis and characterization of positively charged porphyrin–peptide conjugates. Bioconjugate Chem. 2005;16:852–863. doi: 10.1021/bc050057g. [DOI] [PubMed] [Google Scholar]
- 20.James DA, Swamy N, Paz N, Hanson RN, Ray R. Synthesis and estrogen receptor binding affinity of a porphyrin-estradiol conjugate for targeted photodynamic therapy of cancer. Bioorg Med Chem Lett. 1999;9:2379–2384. doi: 10.1016/s0960-894x(99)00390-x. [DOI] [PubMed] [Google Scholar]
- 21.Karagianis G, Reiss JA. Preparation and characterization of porphyrin-acridine conjugates as bifunctional antitumor agents. Aust J Chem. 1995;48:1693–1705. [Google Scholar]
- 22.Sutton JM, Clarke OJ, Fernandez N, Boyle RW. Porphyrin, chlorin, and bacteriochlorin isothiocyanates: Useful reagents for the synthesis of photoactive bioconjugates. Bioconjugate Chem. 2002;13:249–263. doi: 10.1021/bc015547x. [DOI] [PubMed] [Google Scholar]
- 23.Bedel-Cloutour CH, Maneta-Peyret L, Pereyre M, Bezian JH. Synthesis of a monoclonal antibody-indium-111-porphyrin conjugate. J Immunol Methods. 1991;144:35–41. doi: 10.1016/0022-1759(91)90227-7. [DOI] [PubMed] [Google Scholar]
- 24.Bhalgat MK, Roberts JC, Mercer-Smith JA, Knotts BD, Vessella RL, Lavallee DK. Preparation and biodistribution of copper-67-labeled porphyrins and porphyrin-A6H immunoconjugates. Nucl Med Biol. 1997;24:179–185. doi: 10.1016/s0969-8051(96)00215-6. [DOI] [PubMed] [Google Scholar]
- 25.Birchler M, Viti F, Zardi L, Spiess B, Neri D. Selective targeting and photocoagulation of ocular angiogenesis mediated by a phage-derived human antibody fragment. Nat Biotechnol. 1999;17:984–988. doi: 10.1038/13679. [DOI] [PubMed] [Google Scholar]
- 26.Vrouenraets MB, Visser GWM, Loup C, Meunier B, Stigter M, Oppelaar H, Stewart FA, Snow GB, van Dongen GAMS. Targeting of a hydrophilic photosensitizer by use of internalizing monoclonal antibodies: A new possibility for use in photodynamic therapy. Int J Cancer. 2000;88:108–114. doi: 10.1002/1097-0215(20001001)88:1<108::aid-ijc17>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 27.Oseroff AR, Ara G, Ohuoha D, Aprille J, Bommer JC, Yarmush ML, Foley J, Cincotta L. Strategies for selective cancer photochemotherapy: Antibody-targeted and selective carcinoma cell photolysis. Photochem Photobiol. 1987;46:83–96. doi: 10.1111/j.1751-1097.1987.tb04740.x. [DOI] [PubMed] [Google Scholar]
- 28.Cavanaugh PG. Synthesis of chlorin e6-transferrin and demonstration of its light-dependent in vitro breast cancer cell killing ability. Breast Cancer Res Treat. 2002;72:117–130. doi: 10.1023/a:1014811915564. [DOI] [PubMed] [Google Scholar]
- 29.Gijsens A, De Witte P. Photocytotoxic action of EGF-PVA-Sn(IV)chlorin e6 and EGF-dextran-Sn(IV)chlorin e6 in-ternalizable conjugates on A431 cells. Int J Oncol. 1998;13:1171–1177. doi: 10.3892/ijo.13.6.1171. [DOI] [PubMed] [Google Scholar]
- 30.Gijsens A, Missiaen L, Merlevede W, de Witte P. Epidermal growth factor-mediated targeting of chlorin e6 selectively potentiates its photodynamic activity. Cancer Res. 2000;60:2197–2202. [PubMed] [Google Scholar]
- 31.Schmidt-Erfurth U, Diddens H, Birngruber R, Hasan T. Photodynamic targeting of human retinoblastoma cells using covalent low-density lipoprotein conjugates. Br J Cancer. 1997;75:54–61. doi: 10.1038/bjc.1997.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frau S, Bernadou J, Meunier B. Nuclease activity and binding characteristics of a cationic “manganese porphyrin–bis(benzimidazole) dye (Hoechst 33258)” conjugate. Bioconjugate Chem. 1997;8:222–231. doi: 10.1021/bc970007e. [DOI] [PubMed] [Google Scholar]
- 33.Soukos NS, Hamblin MR, Hasan T. The effect of charge on cellular uptake and phototoxicity of polylysine chlorin e6 conjugates. Photochem Photobiol. 1997;65:723–729. doi: 10.1111/j.1751-1097.1997.tb01916.x. [DOI] [PubMed] [Google Scholar]
- 34.Thamyongkit P, Speckbacher M, Diers JR, Kee HL, Kirmaier C, Holten D, Bocian DF, Lindsey JS. Swallowtail porphyrins: Synthesis, characterization and incorporation into porphyrin dyads. J Org Chem. 2004;69:3700–3710. doi: 10.1021/jo049860e. [DOI] [PubMed] [Google Scholar]
- 35.Thamyongkit P, Lindsey JS. Synthesis of swallowtail-substituted multiporphyrin rods. J Org Chem. 2004;69:5796–5799. doi: 10.1021/jo049348t. [DOI] [PubMed] [Google Scholar]
- 36.Langhals H, Ismael R, Yürük O. Persistent fluorescence of perylene dyes by steric inhibition of aggregation. Tetrahedron. 2000;56:5435–5441. [Google Scholar]
- 37.Srinivasan N, Haney CA, Lindsey JS, Zhang W, Chait BT. Investigation of MALDI-TOF mass spectrometry of diverse synthetic metalloporphyrins, phthalocyanines, and multi-porphyrin arrays. J Porphyrins Phthalocyanines. 1999;3:283–291. [Google Scholar]
- 38.Marshall JA, Andrews RC, Lebioda L. Synthetic studies on cembranolides. Stereoselective total synthesis of isolobo-phytolide. J Org Chem. 1987;52:2378–2388. [Google Scholar]
- 39.Callard RE, Smith CM, Beverley PC. Phenotype of human T helper and suppressor cells in an in vitro specific antibody response. Eur J Immunol. 1982;12:232–236. doi: 10.1002/eji.1830120312. [DOI] [PubMed] [Google Scholar]
- 40.Laha JK, Dhanalekshmi S, Taniguchi M, Ambroise A, Lindsey JS. A scalable synthesis of meso-substituted dipyrromethanes. Org Process Res Dev. 2003;7:799–812. [Google Scholar]
- 41.Vader J, Sengers H, de Groot A. The stereoselective syntheses of substituted furo[2,3b]furans (part II) Tetrahedron. 1989;45:2131–2142. [Google Scholar]
- 42.Taniguchi M, Balakumar A, Fan D, McDowell BE, Lindsey JS. Imine-substituted dipyrromethanes in the synthesis of porphyrins bearing one or two meso substituents. J Porphyrins Phthalocyanines. 2005;9:554–574. [Google Scholar]
- 43.Tamaru S-I, Yu L, Youngblood WJ, Muthukumaran K, Taniguchi M, Lindsey JS. A tin-complexation strategy for use with diverse acylation methods in the preparation of 1,9-diacyldipyrromethanes. J Org Chem. 2004;69:765–777. doi: 10.1021/jo035622s. [DOI] [PubMed] [Google Scholar]
- 44.Wedel M, Walter A, Montforts F-P. Synthesis of metalloporphyrins and metallochlorins for immobilization on electrode surfaces. Eur J Org Chem. 2001:1681–1687. [Google Scholar]
- 45.Lee CH, Lindsey JS. One-flask synthesis of meso-substituted dipyrromethanes and their application in the synthesis of trans-substituted porphyrin building blocks. Tetrahedron. 1994;50:11427–11440. [Google Scholar]
- 46.Fan D, Taniguchi M, Yao Z, Dhanalekshmi S, Lindsey JS. 1,9-Bis(N,N-dimethylaminomethyl)dipyrromethanes in the synthesis of porphyrins bearing one or two meso substituents. Tetrahedron. 2005;61:10291–10302. [Google Scholar]
- 47.Zhang D, Poulter CD. Biosynthesis of archaebacterial ether lipids. Formation of ether linkages by prenyltransferases. J Am Chem Soc. 1993;115:1270–1277. [Google Scholar]
- 48.Xu Y, Prestwich GD. Concise synthesis of acyl migration-blocked 1,1-difluorinated analogues of lysophosphatidic acid. J Org Chem. 2002;67:7158–7161. doi: 10.1021/jo0203037. [DOI] [PubMed] [Google Scholar]
- 49.Muthukumaran K, Loewe RS, Ambroise A, Tamaru S-I, Li Q, Mathur G, Bocian DF, Misra V, Lindsey JS. Porphyrins bearing arylphosphonic acid tethers for attachment to oxide surfaces. J Org Chem. 2004;69:1444–1452. doi: 10.1021/jo034945l. [DOI] [PubMed] [Google Scholar]
- 50.Loewe RS, Ambroise A, Muthukumaran K, Padmaja K, Lysenko AB, Mathur G, Li Q, Bocian DF, Misra V, Lindsey JS. Porphyrins bearing mono or tripodal benzyl-phosphonic acid tethers for attachment to oxide surfaces. J Org Chem. 2004;69:1453–1460. doi: 10.1021/jo034946d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.