

Abstract
Protegrin-1 (PG-1) is an 18 residues long, cysteine-rich β-sheet antimicrobial peptide (AMP). PG-1 induces strong cytotoxic activities on cell membrane and acts as a potent antibiotic agent. Earlier we reported that its cytotoxicity is mediated by its channel-forming ability. In this study, we have examined the amyloidogenic fibril formation properties of PG-1 in comparison with a well-defined amyloid, the amyloid-β (Aβ1–42) peptide. We have used atomic force microscopy (AFM) and thioflavin-T staining to investigate the kinetics of PG-1 fibrils growth and molecular dynamics simulations to elucidate the underlying mechanism. AFM images of PG-1 on a highly hydrophilic surface (mica) show fibrils with morphological similarities to Aβ1–42 fibrils. Real-time AFM imaging of fibril growth suggests that PG-1 fibril growth follows a relatively fast kinetics compared to the Aβ1–42 fibrils. The AFM results are in close agreement with results from thioflavin-T staining data. Furthermore, the results indicate that PG-1 forms fibrils in solution. Significantly, in contrast, we do not detect fibrillar structures of PG-1 on an anionic lipid bilayer 2-dioleoyl-sn-glycero-3-phospho-L-serine/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; only small PG-1 oligomers can be observed. Molecular dynamics simulations are able to identify the presence of these small oligomers on the membrane bilayer. Thus, our current results show that cytotoxic AMP PG-1 is amyloidogenic and capable of forming fibrils. Overall, comparing β-rich AMPs and amyloids such as Aβ, in addition to cytotoxicity and amyloidogenicity, they share a common structural motif, and are channel forming. These combined properties support a functional relationship between amyloidogenic peptides and β-sheet-rich cytolytic AMPs, suggesting that amyloids channels may have an antimicrobial function.
Introduction
Antimicrobial peptides (AMPs) are small peptides with a strong antibiotic activity against a wide range of microorganisms, including enveloped viruses, fungi, and bacteria. They usually exert their effect by killing the invading bacteria directly (bactericidal), rather than by inhibiting their proliferation as bacteriostatic antibiotics (1–3). Their ability to selectively target bacterial cells stems from the difference in the membrane composition of bacterial and mammalian cells, such as lipopolysaccharides (LPS, also known as endotoxin) and peptidoglycans, and differing transmembrane potentials (4,5). AMPs form five different subgroups based on their amino acid composition and structure (1,2,6–9). In the cationic subgroup of disulfide-bonded, β-sheet forming peptides, which includes protegrin-1 (PG-1), the presence of positively charged amino acid residues allows their strong interaction with the negatively charged lipid headgroups present in the outer leaflets of bacterial cell membranes. Upon this initial contact with a bacterial cell, cytotoxicity primarily takes place by disrupting the bacterial membrane via pore formation (1,8,10–16). Other cysteine-rich cationic AMP known as defensin also forms pores, similar to those formed by PG-1 (17). The AMPs positive charge disfavors their interaction with the electrically neutral outer leaflet of mammalian cells, because negatively charged headgroups are primarily located in the inner leaflet of the mammalian cell membrane (4,5,8). Significantly, a number of AMPs of the cationic α-helix forming family have been reported to form fibrillar aggregates with amyloid-like properties in the presence of lipid bilayers (18–22).
PG-1 is an 18 residues (RGGRLCYCRRRFCVCVGR) cysteine-rich β-hairpin AMP with ∼2 kDa molecular mass (6,23). Four cysteine residues, producing disulfide S–S bonds at the 6Cys-15Cys and 8Cys-13Cys locations, provide PG-1 the ability to form β-sheets (1,13,23,24). The peptide has six arginine residues that allow PG-1 to interact via strong electrostatic interactions with surfaces of lipid bilayers composed of negatively charged headgroups. The hydropathy index of PG-1 is −0.25, which is relatively close to the value of 0.2 for the Alzheimer's β-amyloids (Aβ), Aβ1–42 peptide (25,26).
PG-1's structural analogy with Aβ, in particular with respect to β-sheet formation, makes PG-1 a good candidate to possess some of the properties observed for amyloids (13,24). Aβ fibrils have been the subject of intense studies (27–30). The Aβ morphology is highly polymorphic with a rugged oligomeric and fibril energy landscape (31), and they form heterogeneous ion channels with a central pore (32–43). Recently, it has been found that the Aβ1–42 and Aβ1–40 peptides exert antimicrobial activity comparable to that of the α-helical LL-37 AMP (44). We and others have shown that the β-forming PG-1 peptide exhibits amyloid-like ion channel behavior in model lipid bilayers (10,13,24). These channels are likely to precede the final disruption of lipid bilayers via massive pore formations (11,15,16). The structural similarities between AMPs and Aβ peptides provide a starting point to understand common patterns in the behavior of these systems.
In this study, we present evidence for the amyloidogenic property of the PG-1 peptide. We employed atomic force microscopy (AFM) to investigate the conformational adaptability of PG-1 peptides in different environments, e.g., on the surfaces of the highly hydrophilic mica and an anionic lipid bilayer composed of 2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE). Molecular dynamics (MD) simulations provided the morphology of PG-1 aggregates on the anionic bilayer. We further used thioflavin-T (ThT) staining to investigate whether the PG-1 peptides form a fibril in solution, and performed MD simulations to confirm the stability of preassembled PG-1 fibrils in water. To understand the morphological basis of the β-sheet fibrils of PG-1, we compared its structures with the structures of Aβ1–42 fibrils on the same mica surface and lipid bilayers. Our observations illustrate that antimicrobial peptides share a common structural motif with amyloids when they are exposed to similar environments. The similarity between β-rich AMPs and amyloids leads us to propose a functional relatedness between them.
Materials and Methods
Materials
PG-1 peptides were synthesized in Dr. Carl Saxinger's laboratory at the National Cancer Institute (National Cancer Institute-Frederick, MD) (24). For storage, the peptides were dissolved in 0.01% acetic acid to 1 mg/mL concentration and aliquoted. Aliquots were stored at −20°C and thawed only once before use. Aβ1–42 peptides were purchased from AnaSpec (Fremont, CA). These were dissolved in 1% NH4OH (Sigma-Aldrich, St. Louis, MO) solutions, aliquoted to concentrations of 1 mg/mL and stored at −20°C for ∼30 days before use. Molecular Biology Grade water used for solution preparation and Dulbecco's phosphate buffered saline without Ca2+ and Mg2+ used for AFM imaging were from Fisher Scientific (Pittsburgh, PA). Phospholipids, DOPS and POPE were purchased from Avanti polar lipids (Alabaster, AL).
AFM imaging
AFM images were acquired using a 5.30 Nanoscope controller (Veeco, Santa Barbara, CA) with an Extender electronics module. Imaging in liquid was performed in phosphate-buffered saline (PBS) at room temperature using a fluid cell (Veeco). Before use, the fluid cell was cleaned in detergent for ∼20 min and rinsed vigorously in a continuous flow of ultrapure water (Millipore, Billerica, MA). Oxide-sharpened Si3N4 cantilevers (Veeco) with nominal spring constants (kn) of 0.12 N/m were used. Images in liquid were acquired in tapping mode at scan frequencies of 0.5–1.0 Hz and drive amplitudes below 100 mV. The cantilever oscillation frequency was 5–10 kHz. In air, images were acquired in contact mode at forces of 1–5 nN. Image analysis was performed using the Veeco software. Some of the AFM images were low-pass filtered to remove noise.
Sample preparation
Freshly dissolved PG-1 peptides, with concentrations ranging from 200 to 300 μg/mL in PBS, were allowed to absorb on freshly cleaved mica for ∼20–30 min before imaging. For imaging Aβ1–42 fibril formation in fluid, the mica surface was first imaged in PBS and subsequently Aβ1–42 peptides were added to the fluid cell to a final concentration of 300 μg/mL. Both measurements were carried out at room temperature. For imaging Aβ1–42 fibrils in air, a 1 mg/mL aliquot of Aβ1–42 peptides was incubated for 72 h at 37°C. A 10 μL droplet with fibrils was deposited on mica for ∼2 min, after which it was rinsed with ultrapure water to remove salt residues and other weakly adsorbed materials.
ThT staining
Fibrillogenesis was measured using ThT fluorescence measurements. Briefly, ThT was added to 1 mg/mL of PG-1 at a final concentration of 20 μM. 100 μL of this mixture was deposited in 6 wells (replicates) in an opaque black walled 96-well plate (Nunc) and incubated at room temperature for 6 h. Fluorescence was recorded (Ex/Em: 450/482 nm) on a spectrofluorometer (Spectra Max XS, Molecular Devices, Sunnyvale, CA). Aβ1–42 at 100 μM concentrations was used as a positive control; ThT mixed with PBS was used as a blank. Blank fluorescence was subtracted from all treatments and plotted against time.
MD simulations
The PG-1 fibrils were made with 10 identical PG-1 β-hairpins, initially arranged to form a linear β-sheet. Depending on the β-hairpin arrangement, the PG-1 fibril was assembled into two distinct β-sheet motifs; antiparallel (turn-next-to-tail) and parallel (turn-next-to-turn) β-sheets in a multimeric NCCN packing mode (see Fig. S1 in the Supporting Material) (45). We used the prerelaxed β-hairpin structure obtained from preliminary simulations of the solution NMR structure of a PG-1 monomer (46) in the lipid environment, as described in previous simulation protocols (13,24). The 10-mer PG-1 fibril was minimized with a rigid body motion for the peptides to enhance the formation of intermolecular backbone hydrogen bonds (H-bonds) within a β-sheet, and then deposited on the surface of DOPS/POPE bilayer (mole ratio 1:2). Because the simulation method closely follows the protocol previously described for PG-1 monomer (47), dimer (48), octamer channel (13), and decamer channel (24), in this study we only describe briefly key parameters used for the decameric PG-1 fibril simulations.
We constructed a unit cell containing a 10-mer PG-1 fibril, lipids, salts, and waters; overall almost 160,000 atoms. For the lipid bilayer, 480 lipids (160 DOPS and 320 POPE lipids) constitute the lateral cell. The lateral dimension of the mixed lipid bilayer was chosen to be 119.1 × 119.1 Å2. We used the values of 65.3 Å2 per DOPS (49) and 56.0 Å2 per POPE (50) at 303 K. TIP3P waters were added and relaxed through a series of minimization and dynamics. The system contains MgCl2 and NaCl at the same concentration of 50 mM to satisfy a total cation concentration of ∼100 mM. In addition to the decameric PG-1 fibril simulations on the lipid bilayer, we also performed the simulations for the decameric PG-1 fibrils in the water box. A series of minimization and dynamics for the PG-1 fibril/water system were performed with the same simulations parameters used for the PG-1 fibril simulations.
The CHARMM program (51) was used to construct the set of starting points and to relax the systems to a production-ready stage. In the preequilibrium stages, the initial configurations were gradually relaxed, with the peptides held rigid. A series of dynamic cycles were performed with the harmonically restrained peptides in the fibrils, and then the harmonic restraints were gradually diminished with the full Ewald electrostatics calculation and constant temperature (Nosé-Hoover) thermostat/barostat at 303 K. For production runs to 50 ns, the NAMD code (52) on a Biowulf cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov) was used for the starting point. Averages were taken after 10 ns discarding the initial equilibrarion period.
Results
PG-1 fibril morphologies on mica
AFM images of PG-1 peptides adsorbed on mica surfaces reveal several fibrillar structures (Fig. 1 A). The typical height (h) of these structures ranges between 3 and 4 nm and their typical width (w) is ∼5–10 nm after tip-induced broadening is corrected using the relationship ; wm is the measured width in AFM images with an AFM tip of radius (R) (27,53,54). A value of R = 30 nm was assumed. On the basis of the lowest heights measured in these experiments, the fibril appears to consist of multilayered β-sheets. The planar geometry of these fibrils correlates well with the geometry of fibrils reported for amyloids and proteins with amyloid-like behavior (27–29,53). Typical fibril lengths are in the range of 50–250 nm, but these values increased as fibrils grew during imaging (see below). Most of the fibrillar structures are linear without turns, although some of them have mild curvatures. Their orientations follow different directions on the surface and appear not to be directly correlated to the hexagonal symmetry of mica. Several times new smaller fibrils branched out in different directions from a single point of a preexisting fibril (see white arrow in Fig. 2 A).
Figure 1.
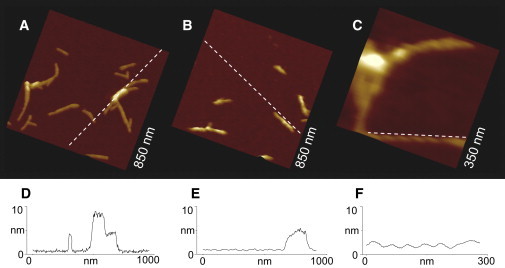
AFM height images of (A) PG-1 and (B) Aβ1–42 fibrils on mica. (C) Higher resolution image of a PG-1 fibril displaying a helical intrafibrillar structure. Corresponding cross sections for the (D) PG-1 and (E) Aβ1–42 fibrils. (F) Cross section for the helical intrafibrillar structure of PG-1 fibril.
Figure 2.
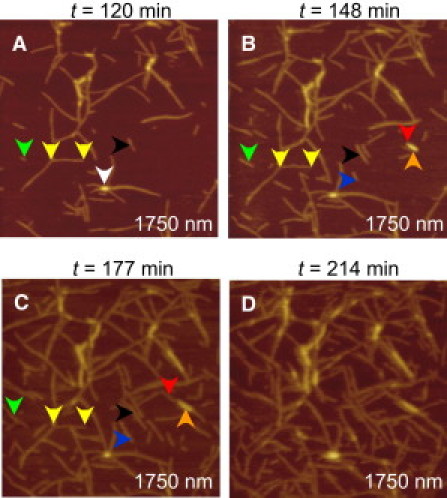
Dynamical changes in length and surface concentration of the PG-1 fibrils on mica. (A–D) Time sequential images were taken at the times indicated in the figure in a 1.75 μm × 1.75 μm area. The different colored arrows denote different types of fibril growth as explained in the text. Time zero corresponds to the time when the 200 μg/mL solution of PG-1 peptides came in contact with the mica surface.
AFM images of amyloid Aβ1–42 fibrils were acquired for comparison. Fig. 1 B shows the fibrilar structure of Aβ1–42 peptides incubated at 37°C for 72 h. The heights measured for these fibrils were in the same range as the height of the fibrillar structures observed for PG-1. The widths, w, of these fibrils were ∼10 nm, in agreement with previous reports. Typical lengths were 100–200 nm. Numerous fibrillar structures have been reported for amyloid peptides and have been broadly classified as filaments, protofibrils, type I fibrils, and type II fibrils. Although filaments and protofibrils have typical lengths <100 nm, lengths of fibrils are >100 nm. Based on these lengths, fibrillar structures in this article will be referred to as either filaments or fibrils and no strict distinction between filaments and protofibrils or type I and type 2 fibrils will be made.
In addition to the amyloid-like fibrillar structures observed for PG-1, a helical intrafibrillar structure was observed in some of these fibrils (Fig. 1 C), similar to those reported for the fibrillar structures for Aβ1–42 and Aβ1–40, as well as for other amyloids (27–29). The helical structure observed for the PG-1 fibrils in this work is right-handed, has a periodicity of ∼41 nm, and has the corrugation amplitude of ∼0.75 nm.
Kinetics of PG-1 fibrils growth
In situ AFM images of PG-1 fibrils on mica show a dynamic behavior with fibril length and surface concentration increasing significantly over a few hours (Fig. 2). Although the fibril density is ∼20 fibrils/μm2 ∼120 min after incubation (Fig. 2 A), it increases to ∼60 fibrils/μm2 in 90 min (Fig. 2 C). Fibrils that display different growth behavior are shown by colored arrows at the different times. Yellow arrows show a fibril that maintains approximately the same length over time; black arrows show another fibril that maintains a length of ∼125 nm for ∼130 min and then increases its length at a rate of ∼4.3 nm/min. The red arrows (Fig. 2, B and C) show two fibrils stacked on top of one another. In this case, the fibril adsorbed directly on the mica surface (red arrow) grows at a rate of ∼12.2 nm/min and the fibril on top (orange arrow) grows at only 2.1 nm/min. Fibril growth on the surface of another fibril involves adsorption of oligomers on the surface of the lower fibril and this type of interaction appears to be less favorable than the fibril/mica interaction. Consequently, fibril on fibril adsorption is not observed as frequently as fibril on mica adsorption in the AFM images.
The lengths of fibrils were plotted as a function of time to investigate their growth kinetics (Fig. 3 A). Because all curves display plateaus of different time lengths, their growth mechanism appears compatible with sudden growth followed by long pauses, also observed by others (55). Typical growth rates of the fibrils are in the range of 4–4.5 nm/min, although an average value of 6.5 ± 3.8 nm/min is obtained due to the influence of some fibrils with very fast growth rates (see for instance the fibril marked with red arrows in Fig. 2 B). Fluorescence of ThT (Fig. 3 B), a dye known to fluoresce upon binding to amyloid fibrils, suggests that both PG-1 and Aβ1–42 fibrils are produced in solution and not induced by the mica surface.
Figure 3.
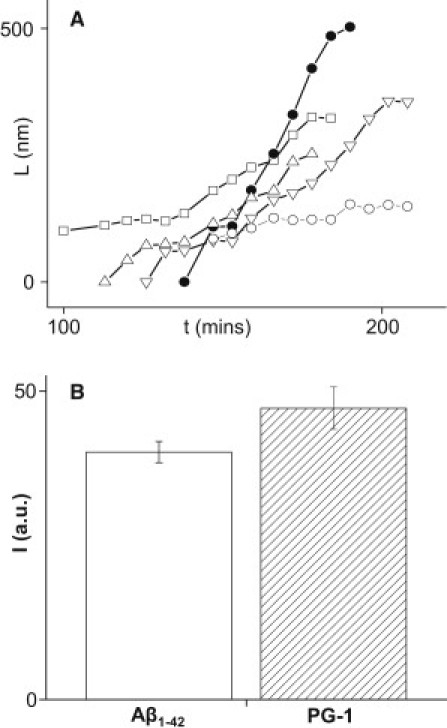
(A) Plot of fibrils length, L, as a function of time, t, for the fibrils marked with arrows in Fig 2. Open squares in this plot correspond to black arrows, solid circles to red arrows, open downward triangles to blue arrows, open upward triangles to green arrows, and open circles to orange arrows. (B) ThT fluorescence intensity, I, specific to amyloid fibrillogenesis shows that PG-1 and Aβ1–42 peptides form amyloid fibrils in solution after 6 h incubation at room temperature. For Aβ1–42, a 10 μM concentration was used, whereas for PG-1 a 200 μg/mL concentration was employed. Higher fluorescence for PG-1 suggests more rapid fiber formation.
When the kinetics of PG-1 fibril growth is compared with Aβ1–42 fibril formation in unseeded conditions at room temperature, a fairly different behavior is observed (Fig. 4). Even after ∼4 h adsorption on mica, only thin Aβ1–42 filaments/protofibrils with heights of ∼2 nm, widths <5 nm, and lengths <100 nm could be observed (Fig. 4 D). At times when PG-1 fibrils are already formed on the mica surface (Fig. 2), only unstable, weakly adsorbed Aβ1–42 filaments/protofibrils and globular structures are seen (Fig. 4, A and B). After a period of 3–3.5 h (Fig. 4 C), a continuous film, consisting of globular oligomers and thin filamentous structures, is observed. Furthermore, the rapid kinetics of PG-1 fibril formation was also examined using staining by ThT (Fig. 3 B). In agreement with AFM data, the higher fluorescence intensity measured for PG-1 suggests faster kinetics of PG-1 fibril formation than for Aβ1–42 under conditions used in this work. It should be noted, however, that morphology and kinetics of Aβ amyloid fibril formation can be highly dependent on preparation conditions such as temperature and seeding; consequently, growth rates as high as 300 nm/min have been measured for Aβ1–40 fibrils seeded for one week and then incubated at 37°C (56).
Figure 4.
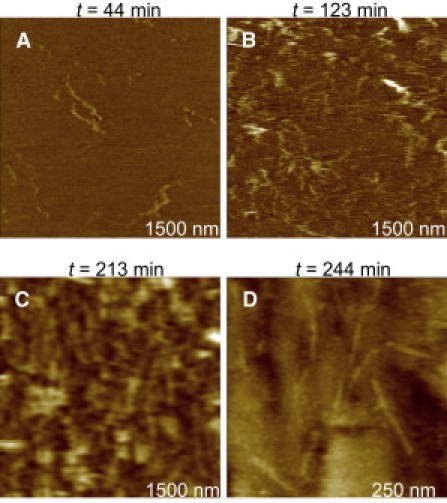
Time sequence of adsorption for a 300 μg/mL solution of Aβ1–42 peptides on mica. Amorphous and weakly adsorbed structures were observed for ∼2 h (A and B). Only after longer times (∼3 h), globular structures and small and thin filaments could be clearly seen (C and D). It should be noticed that images were not acquired in the same spot, but rather in neighboring areas of the same sample.
Absence of PG-1 fibrils on the lipid bilayer
We examined if a biologically relevant surface, e.g., a lipid bilayer would also support PG-1 fibril conformations. For PG-1 peptides added to preformed DOPS/POPE 1:1 (w/w) lipid bilayer, AFM images sometimes showed PG-1 oligomers adsorbed on the bilayer, but no fibril formation (Fig. 5). On the basis of the oligomer height (> 2 nm), we speculate that these oligomers can adopt a multilayered β-sheet structure or interact with the bilayer in different orientations. Considering the heights of some of the particles (up to 20–25 nm), lipid extraction from the membrane and subsequent accumulation around PG-1 oligomers is also possible (57). The final concentration of PG-1 added to the lipid bilayer was ∼100–150 μg/mL, and even prolonged scanning did not show formation of any fibrils.
Figure 5.

AFM images of DOPS/POPE bilayer (A) before and (B) after addition of PG-1 peptide solution. Data were obtained in different regions of the same bilayer and are offline zooms of original 4 μm × 4 μm images. Dark colors denote lower heights in color-coded height scale.
We used MD simulations to examine PG-1 fibril formation on the anionic lipid bilayer. Initially, preformed decameric PG-1 fibrils with antiparallel and parallel β-sheet arrangements were deposited on the surface of DOPS/POPE bilayer (mole ratio 1:2). During the simulations, as expected, the PG-1 fibrils are unstable and break apart into small parts (Fig. 6). For the antiparallel β-sheet, the PG-1 fibril breaks into three parts; trimer, pentamer, and dimer (Fig. 6 A). Similarly, the parallel β-sheet fibril also breaks into three parts; two dimers and hexamer (Fig. 6 D). Discontinuities of the β-sheet network due to breakages of the intermolecular backbone H-bonds in the PG-1 fibrils occur early in the simulations and remain unchanged thereafter (Fig. 6, B and E). In contrast, the intramolecular backbone H-bonds keep intact due to strong disulfide bonds in the β-hairpin (Fig. 6, C and F). Thus, no PG-1 fibril formation on the surface of anionic bilayer is observed, but small PG-1 oligomers might be highly populated. This behavior is in good agreement with the AFM images (Fig. 5).
Figure 6.
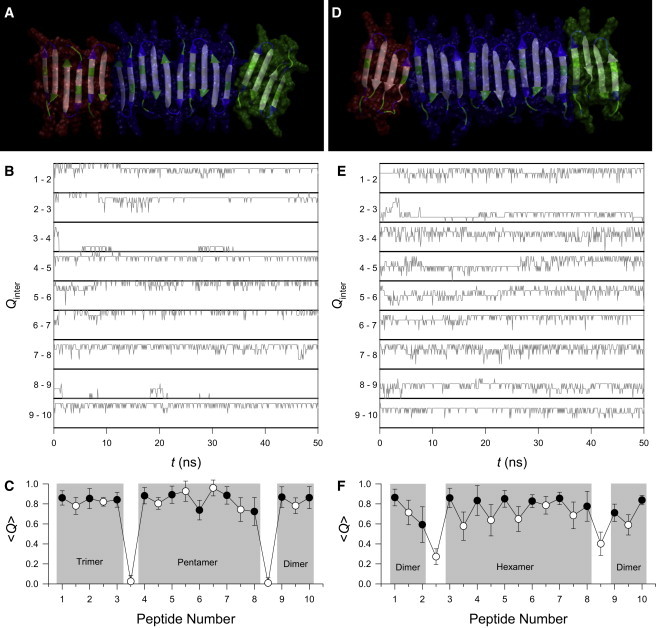
(A) Fibril structure averaged over the simulation in a ribbon representation, (B) fractions of intermolecular backbone H-bonds, Qinter, as a function of time, t, for the neighboring pairs of peptides, and (C) average fractions of intramolecular (solid circles) and intermolecular (open circles) backbone H-bonds, Q, as a function of peptide number for the antiparallel β-sheet fibril of PG-1 on the surface of DOPS/POPE: (1:2) bilayer. (D–F) the same for the parallel β-sheet fibrils of PG-1. In the peptides, hydrophobic residues are shown in white, polar and Gly residues are shown in green, and positively charged residues are shown in blue. Both antiparallel and parallel β-sheet fibrils break into 3 parts; trimer, pentamer, and dimer in the antiparallel, and two dimers and hexamer in the parallel β-sheet fibrils as denoted by different colors of transparent surface for the parts in A and D.
From the MD simulations we observe that these broken pieces, now low molecular mass PG-1 oligomers with further optimized β-sheet, tend to insert into the bilayer. The three-dimensional densities for the positions of the lipid headgroups clearly indicate the roughness of the bilayer surface when embedded with the peptides (Fig. 7). The headgroup density map denotes the average shape of the anionic bilayer surface during the simulation. The roughness of the bilayer surface is clearly related to membrane thinning or disruption. It has been demonstrated that PG-1 monomer (47) and dimer (48) mediate localized membrane thinning, possessing the ability to diffuse into the membrane core, and ultimately clustering to form ion channels (10,13,24). As a whole, the simulations show that the larger 10-mer β-sheet PG-1 fibril is not able to insert into the bilayer, but small PG-1 oligomers can insert into the bilayer. This behavior suggests an explanation for why PG-1 fibrils divide into small pieces leading to channel formation, and why we were unable to observe experimentally fibril formation when PG-1 is in contact with an anionic lipid bilayer.
Figure 7.

Three-dimensional density maps of the lipid headgroup for the anionic DOPS/POPE: (1:2) bilayer. The upper panels are the normal views of the upper bilayer surfaces embedding the (A) antiparallel and (B) parallel β-sheet fibrils of PG-1. The lower panels are the reciprocal normal views of the lower bilayer surfaces without the peptides.
We also simulated the PG-1 fibrils in water. During the simulations, both antiparallel and parallel β-sheet fibrils of PG-1 divided into two separated β-sheets (Fig. S2). However, the morphology of the hexameric and tetrameric β-sheets in water is totally different from those observed in the lipid simulations. While the β-sheets on the bilayer adopt a planar shape, the β-sheets in water are twisted, showing a cross β-structure (or helical structure as mentioned above) commonly observed in amyloid fibrils (58,59). The cross β-sheets may serve as seeds for fibril growth when PG-1 concentration in solution increases. We conclude that PG-1 can produce fibrils in solution, consistent with the ThT staining.
Discussion
We used complementary techniques of AFM, MD simulation, and ThT staining to investigate the amyloidogenic properties of PG-1. Our AFM results show that PG-1 peptides form fibrils on the highly hydrophilic mica surface. The PG-1 fibrils have similar morphologies and dimensions to the well-studied fibrils produced by the amyloidogenic Aβ1–42. In addition, PG-1 fibrils exhibit a helical intrafibrillar structure, which is a typical fibril structure formed by Aβ1–42, Aβ1–40, and other amyloidogenic peptides (27–29), although different periodicity and handedness are occasionally observed in the PG-1 fibrils. These helical structures have been attributed to two protofibrils wrapped around each other, thus forming a helical (28,29) or cross β-structure (58,59).
PG-1 forms amyloid-like fibrils with relatively fast kinetics. Several experimental studies suggested that two or more days of incubation are necessary for Aβ1-42 or Aβ1-40 peptides to form fibrils (27,28,30,36). In our AFM experiments, Aβ1–42 fibrils can be observed after 72 h incubation at 37°C. The dimensions of the thin filamentous structures of Aβ1–42 observed in solution upon 3 h of adsorption are significantly smaller than the PG-1 fibrils, indicating that the growth of Aβ1–42 fibrils is relatively slow. On the other hand, considerably faster kinetics of fibril formation is observed for PG-1, even after taking into account the different molar concentrations used in the experiments (100 μM for PG-1 and 45 μM for Aβ1–42).
Both AFM imaging and MD simulation results show that PG-1 fibril is not produced on the anionic bilayer. Instead, small PG-1 oligomers are observed on the surface of the lipid bilayer, implying PG-1 diffusion into the membrane core. This is in contrast to the results of PG-1 fibrils on the solid mica surface that does not allow PG-1 penetration. Absence of PG-1 fibrils on the fluidic lipid bilayer suggests that PG-1 is evolutionarily optimized for microbial bilayer penetration. This is further supported by works showing that PG-1 binds to LPS and lipid A (the phospholipid core of LPS) moieties, typical components of bacterial cells (4,5). Our simulation results show that preassembled β-sheets of PG-1 fibrils break into several small oligomers, because such large molecules as a whole are not able to dislocate into the membrane core. However, upon size reduction, these small oligomers can significantly induce membrane thinning effects such as those observed in the monomer and dimer PG-1 simulations (47,48). These observations provide important information for PG-1 activity involving cell membrane damage and their ability to form ion channels with loosely attached subunits when they penetrate the cell membrane (13,24). Furthermore, the PG-1 channel morphology with a β-sheet subunit motif is in agreement with Alzheimer's Aβ ion channels (13,32–43), indicating that PG-1 and Aβ share a common cellular mechanism, such as membrane disruption and large pore formation, preceded by toxic ion channel formation, finally leading to cell death.
Further investigation using ThT staining suggests that PG-1 forms fibrils in solution. High levels of fluorescence in solutions incubated with PG-1 peptides under conditions similar to those used in the AFM experiments clearly indicate the PG-1 fibril formations. MD simulation results show that PG-1 β-sheets in water contain a common cross (or helical) β-structure in amyloid fibrils (58,59). Although preassembled planar PG-1 β-sheets in water are divided into two parts, these PG-1 oligomers exhibit more amyloid fibril characteristics than those observed on the lipid bilayer. We speculate that as seeds the small cross β-sheets of PG-1 are capable of developing to protofibrils or mature fibrils in water when PG-1 concentration is increased (45).
PG-1 fibrils on the mica surface are stable and able to grow. We hypothesize that fibril formation on mica occurs by fibrils preformed in solution, which subsequently adsorb on the mica surface and serve as seeds for the fibril growth observed in Fig. 2. Fibrils on mica are likely to be stabilized by favorable electrostatic interactions between the positively charged amino acid residues and the negatively charged mica surface. When fibrils are adsorbed on mica, their configuration in solution may be altered as a result of these interactions. On anionic lipid bilayers, fibril disintegration and bilayer disruption occur faster and these processes predominate over fibril growth.
To conclude, both PG-1 and Aβ form fibrils in solution at room temperature and these fibrils can be adsorbed on the mica surface. In these cases, PG-1 fibrils grow relatively faster than unseeded Aβ fibrils. Thus, our study shows that PG-1 possesses amyloidogenic properties. Furthermore, under proper environments, PG-1 retains its fibril state with morphologies similar to those of amyloids, which are structural hallmarks of amyloid diseases. Moreover, PG-1 abandons the fibril architecture on the lipid bilayer, and forms small oligomers. The presence of small oligomers suggests the peptide's cytolytic mechanism in the cellular membrane. Small PG-1 oligomers diffuse more easily into the membrane core, which is again consistent with the mechanism of amyloid diffusion. These combined results support the conjectures that amyloids are cytotoxic with an antimicrobial activity and that cytotoxic AMPs form fibrils, hence are amyloidogenic. They suggest a possible functional role of amyloids as antibiotics. Interestingly, both are capable of forming channels in the membrane inducing cell toxicity, and in both cases, the channels consist of β-structures, with structural, functional, and biological similarities, suggesting a shared mechanism of action (60–62). Over the last few years, the question of the function of amyloids has been the focus of immense interest in the community (63–67). Although currently there is no direct evidence, the observations that we make here, that PG-1 is like an amyloid and an amyloid behaves like antibacterial peptides such as PG-1, raise the possibility that the Aβ channel may also function in a way similar to that of a cytolytic peptide.
Acknowledgments
This project was funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health (NIH), under contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This research was supported by NIH grant No. R01AG028709 (R.L.). All simulations had been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
Contributor Information
Ruth Nussinov, Email: ruthnu@helix.nih.gov.
Ratnesh Lal, Email: rlal@ucsd.edu.
Supporting Material
References
- 1.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 2.Friedrich C.L., Moyles D., Hancock R.E. Antibacterial action of structurally diverse cationic peptides on gram-positive bacteria. Antimicrob. Agents Chemother. 2000;44:2086–2092. doi: 10.1128/aac.44.8.2086-2092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hancock R.E.W., Sahl H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 4.Yeaman M.R., Yount N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 5.Matsuzaki K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta. 2009;1788:1687–1692. doi: 10.1016/j.bbamem.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Steinberg D.A., Hurst M.A., Fiddes J.C. Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob. Agents Chemother. 1997;41:1738–1742. doi: 10.1128/aac.41.8.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sitaram N., Nagaraj R. Host-defense antimicrobial peptides: importance of structure for activity. Curr. Pharm. Des. 2002;8:727–742. doi: 10.2174/1381612023395358. [DOI] [PubMed] [Google Scholar]
- 8.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 9.Powers J.P.S., Hancock R.E.W. The relationship between peptide structure and antibacterial activity. Peptides. 2003;24:1681–1691. doi: 10.1016/j.peptides.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 10.Sokolov Y., Mirzabekov T., Kagan B.L. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta. 1999;1420:23–29. doi: 10.1016/s0005-2736(99)00086-3. [DOI] [PubMed] [Google Scholar]
- 11.Lam K.L.H., Ishitsuka Y., Lee K.Y. Mechanism of supported membrane disruption by antimicrobial peptide protegrin-1. J. Phys. Chem. B. 2006;110:21282–21286. doi: 10.1021/jp0630065. [DOI] [PubMed] [Google Scholar]
- 12.Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta. 1999;1462:55–70. doi: 10.1016/s0005-2736(99)00200-x. [DOI] [PubMed] [Google Scholar]
- 13.Jang H., Ma B., Nussinov R. Models of toxic β-sheet channels of protegrin-1 suggest a common subunit organization motif shared with toxic alzheimer β-amyloid ion channels. Biophys. J. 2008;95:4631–4642. doi: 10.1529/biophysj.108.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuzaki K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta. 1999;1462:1–10. doi: 10.1016/s0005-2736(99)00197-2. [DOI] [PubMed] [Google Scholar]
- 15.Yang L., Weiss T.M., Huang H.W. Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys. J. 2000;79:2002–2009. doi: 10.1016/S0006-3495(00)76448-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottler L.M., de la Salud Bea R., Marsh E.N. Using fluorous amino acids to probe the effects of changing hydrophobicity on the physical and biological properties of the beta-hairpin antimicrobial peptide protegrin-1. Biochemistry. 2008;47:9243–9250. doi: 10.1021/bi801045n. [DOI] [PubMed] [Google Scholar]
- 17.Kagan B.L., Selsted M.E., Lehrer R.I. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc. Natl. Acad. Sci. USA. 1990;87:210–214. doi: 10.1073/pnas.87.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gössler-Schöfberger R., Hesser G., Jilek A. An orphan dermaseptin from frog skin reversibly assembles to amyloid-like aggregates in a pH-dependent fashion. FEBS J. 2009;276:5849–5859. doi: 10.1111/j.1742-4658.2009.07266.x. [DOI] [PubMed] [Google Scholar]
- 19.Code C., Domanov Y.A., Kinnunen P.K. Activation of phospholipase A2 by temporin B: formation of antimicrobial peptide-enzyme amyloid-type cofibrils. Biochim. Biophys. Acta. 2009;1788:1064–1072. doi: 10.1016/j.bbamem.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Zhao H., Sood R., Kinnunen P.K. Interaction of the antimicrobial peptide pheromone Plantaricin A with model membranes: implications for a novel mechanism of action. Biochim. Biophys. Acta. 2006;1758:1461–1474. doi: 10.1016/j.bbamem.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 21.Zhao H., Tuominen E.K., Kinnunen P.K. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry. 2004;43:10302–10307. doi: 10.1021/bi049002c. [DOI] [PubMed] [Google Scholar]
- 22.Auvynet C., El Amri C., Rosenstein Y. Structural requirements for antimicrobial versus chemoattractant activities for dermaseptin S9. FEBS J. 2008;275:4134–4151. doi: 10.1111/j.1742-4658.2008.06554.x. [DOI] [PubMed] [Google Scholar]
- 23.Yasin B., Lehrer R.I., Wagar E.A. Protegrins: structural requirements for inactivating elementary bodies of Chlamydia trachomatis. Infect. Immun. 1996;64:4863–4866. doi: 10.1128/iai.64.11.4863-4866.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capone R., Mustata M., Lal R. Antimicrobial protegrin-1 forms ion channels: molecular dynamic simulation, atomic force microscopy, and electrical conductance studies. Biophys. J. 2010;98:2644–2652. doi: 10.1016/j.bpj.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 26.Voet D., Voet J.G. Wiley; Hoboken, NJ: 2004. Biochemistry. [Google Scholar]
- 27.Arimon M., Díez-Pérez I., Fernàndez-Busquets X. Fine structure study of Abeta1-42 fibrillogenesis with atomic force microscopy. FASEB J. 2005;19:1344–1346. doi: 10.1096/fj.04-3137fje. [DOI] [PubMed] [Google Scholar]
- 28.Harper J.D., Lieber C.M., Lansbury P.T., Jr. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-β protein. Chem. Biol. 1997;4:951–959. doi: 10.1016/s1074-5521(97)90303-3. [DOI] [PubMed] [Google Scholar]
- 29.Ionescu-Zanetti C., Khurana R., Fink A.L. Monitoring the assembly of Ig light-chain amyloid fibrils by atomic force microscopy. Proc. Natl. Acad. Sci. USA. 1999;96:13175–13179. doi: 10.1073/pnas.96.23.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parbhu A., Lin H., Lal R. Imaging real-time aggregation of amyloid β protein (1-42) by atomic force microscopy. Peptides. 2002;23:1265–1270. doi: 10.1016/s0196-9781(02)00061-x. [DOI] [PubMed] [Google Scholar]
- 31.Miller Y., Ma B., Nussinov R. Polymorphism in Alzheimer Abeta amyloid organization reflects conformational selection in a rugged energy landscape. Chem. Rev. 2010;110:4820–4838. doi: 10.1021/cr900377t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arispe N., Rojas E., Pollard H.B. Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arispe N., Pollard H.B., Rojas E. β-Amyloid Ca(2+)-channel hypothesis for neuronal death in Alzheimer disease. Mol. Cell. Biochem. 1994;140:119–125. doi: 10.1007/BF00926750. [DOI] [PubMed] [Google Scholar]
- 34.Hirakura Y., Lin M.C., Kagan B.L. Alzheimer amyloid abeta1-42 channels: effects of solvent, pH, and Congo Red. J. Neurosci. Res. 1999;57:458–466. [PubMed] [Google Scholar]
- 35.Hirakura Y., Yiu W.W., Kagan B.L. Amyloid peptide channels: blockade by zinc and inhibition by Congo red (amyloid channel block) Amyloid. 2000;7:194–199. doi: 10.3109/13506120009146834. [DOI] [PubMed] [Google Scholar]
- 36.Lin H., Bhatia R., Lal R. Amyloid β protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 37.Quist A., Doudevski I., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang H., Zheng J., Nussinov R. Models of β-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 2007;93:1938–1949. doi: 10.1529/biophysj.107.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jang H., Zheng J., Nussinov R. New structures help the modeling of toxic amyloidbeta ion channels. Trends Biochem. Sci. 2008;33:91–100. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Jang H., Arce F.T., Nussinov R. Misfolded amyloid ion channels present mobile β-sheet subunits in contrast to conventional ion channels. Biophys. J. 2009;97:3029–3037. doi: 10.1016/j.bpj.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang H., Teran Arce F., Nussinov R. Structural convergence among diverse, toxic β-sheet ion channels. J. Phys. Chem. B. 2010;114:9445–9451. doi: 10.1021/jp104073k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jang H., Arce F.T., Lal R. Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer's Disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:6538–6543. doi: 10.1073/pnas.0914251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jang H., Arce F.T., Nussinov R. β-Barrel topology of Alzheimer's β-amyloid ion channels. J. Mol. Biol. 2010;404:917–934. doi: 10.1016/j.jmb.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soscia S.J., Kirby J.E., Moir R.D. The Alzheimer's disease-associated amyloid β-protein is an antimicrobial peptide. PLoS ONE. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang M., Waring A.J., Hong M. Intermolecular packing and alignment in an ordered β-hairpin antimicrobial peptide aggregate from 2D solid-state NMR. J. Am. Chem. Soc. 2005;127:13919–13927. doi: 10.1021/ja0526665. [DOI] [PubMed] [Google Scholar]
- 46.Fahrner R.L., Dieckmann T., Feigon J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996;3:543–550. doi: 10.1016/s1074-5521(96)90145-3. [DOI] [PubMed] [Google Scholar]
- 47.Jang H., Ma B., Nussinov R. Interaction of protegrin-1 with lipid bilayers: membrane thinning effect. Biophys. J. 2006;91:2848–2859. doi: 10.1529/biophysj.106.084046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jang H., Ma B., Nussinov R. Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct. Biol. 2007;7:21. doi: 10.1186/1472-6807-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petrache H.I., Tristram-Nagle S., Nagle J.F. Structure and fluctuations of charged phosphatidylserine bilayers in the absence of salt. Biophys. J. 2004;86:1574–1586. doi: 10.1016/S0006-3495(04)74225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rand R.P., Parsegian V.A. Hydration forces between phospholipid bilayers. Biochim. Biophys. Acta. 1989;988:351–376. [Google Scholar]
- 51.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM:a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 52.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Relini A., Torrassa S., Gliozzi A. Detection of populations of amyloid-like protofibrils with different physical properties. Biophys. J. 2010;98:1277–1284. doi: 10.1016/j.bpj.2009.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vesenka J., Guthold M., Bustamante C. Substrate preparation for reliable imaging of DNA molecules with the scanning force microscope. Ultramicroscopy. 1992;42-44(Pt B):1243–1249. doi: 10.1016/0304-3991(92)90430-r. [DOI] [PubMed] [Google Scholar]
- 55.Kellermayer M.S.Z., Karsai A., Penke B. Stepwise dynamics of epitaxially growing single amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2008;105:141–144. doi: 10.1073/pnas.0704305105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ban T., Hoshino M., Goto Y. Direct observation of Abeta amyloid fibril growth and inhibition. J. Mol. Biol. 2004;344:757–767. doi: 10.1016/j.jmb.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 57.Butterfield S.M., Lashuel H.A. Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 2010;49:5628–5654. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 58.Sipe J.D., Cohen A.S. Review: history of the amyloid fibril. J. Struct. Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 59.Serpell L.C. Alzheimer's amyloid fibrils: structure and assembly. Biochim. Biophys. Acta. 2000;1502:16–30. doi: 10.1016/s0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 60.Kourie J.I., Shorthouse A.A. Properties of cytotoxic peptide-formed ion channels. Am. J. Physiol. Cell Physiol. 2000;278:C1063–C1087. doi: 10.1152/ajpcell.2000.278.6.C1063. [DOI] [PubMed] [Google Scholar]
- 61.Kourie J.I., Henry C.L. Protein aggregation and deposition: implications for ion channel formation and membrane damage. Croat. Med. J. 2001;42:359–374. [PubMed] [Google Scholar]
- 62.Mahalka A.K., Kinnunen P.K. Binding of amphipathic alpha-helical antimicrobial peptides to lipid membranes: lessons from temporins B and L. Biochim. Biophys. Acta. 2009;1788:1600–1609. doi: 10.1016/j.bbamem.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 63.Choi S., Connelly S., Kelly J.W. Chemoselective small molecules that covalently modify one lysine in a non-enzyme protein in plasma. Nat. Chem. Biol. 2010;6:133–139. doi: 10.1038/nchembio.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watt B., van Niel G., Marks M.S. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem. 2009;284:35543–35555. doi: 10.1074/jbc.M109.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fowler D.M., Kelly J.W. Aggregating knowledge about prions and amyloid. Cell. 2009;137:20–22. doi: 10.1016/j.cell.2009.03.030. [DOI] [PubMed] [Google Scholar]
- 66.Maji S.K., Perrin M.H., Riek R. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greenwald J., Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18:1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.