

Abstract
The interaction of cells with extracellular matrix, termed cell-matrix adhesions, importantly governs multiple cellular phenomena. Knowledge of the functional dynamics of cell-matrix adhesion could provide critical clues for understanding biological phenomena. We developed surface plasmon resonance imaging ellipsometry (SPRIE) to provide high contrast images of the cell-matrix interface in unlabeled living cells. To improve the contrast and sensitivity, the null-type imaging ellipsometry technique was integrated with an attenuated total reflection coupler. We verified that the imaged area of SPRIE was indeed a cell-matrix adhesion area by confocal microscopy imaging. Using SPRIE, we demonstrated that three different cell types exhibit distinct features of adhesion. SPRIE was applied to diverse biological systems, including during cell division, cell migration, and cell-cell communication. We imaged the cell-matrix anchorage of mitotic cells, providing the first label-free imaging of this interaction to our knowledge. We found that cell-cell communication can alter cell-matrix adhesion, possibly providing direct experimental evidence for cell-cell communication-mediated changes in cell adhesion. We also investigated shear-stress-induced adhesion dynamics in real time. Based on these data, we expect that SPRIE will be a useful methodology for studying the role of cell-matrix adhesion in important biological phenomena.
Introduction
The extracellular matrix (ECM) provides structural support for cells in all tissues. The interaction between cells and the ECM, termed cell-matrix adhesion, governs various cellular functions including differentiation, proliferation, migration, and gene expression (1–3). Furthermore, cell-matrix adhesion signals provisionally incite pathological processes such as inflammation, tumor growth, and metastasis (4,5). Therefore, elucidating the structure and functional dynamics of cell-matrix adhesions provides critical clues for understanding cellular phenomena.
The optical microscopy techniques used to investigate cell-matrix adhesion have evolved over several decades. The development of tools, including interference reflection microscopy (IRM) (6), total internal reflection fluorescence (TIRF) (7–9), surface plasmon resonance microscopy (SPRM) (10,11), and neutron reflectometry (12), has provided useful information about the cell-matrix interface. Because IRM uses the principle that the interfaces of cells can be differentially illuminated by visible light, depth-dependent information can be provided (13). However, the thickness of the regions of interest can influence the interpretation of the cell-matrix adhesion (14). For TIRF imaging, fluorescently labeled molecules on the cell membrane are excited by an evanescent wave. Thus, high molecular selectivity is permitted with a higher z resolution (≈100 nm).
Surface plasmons (SPs) are electromagnetic waves bound to a metal-dielectric interface that decay exponentially in a direction perpendicular to the sample surface (15) so that the penetration depth of SPs is only a few hundred nanometers. Kretschmann's (16) attenuated total reflection (ATR) geometry is used for the purpose of resonance coupling between light and SPs. In an ATR coupler, a metal film that is several tens of nanometers thick is deposited at the bottom of a prism. The dispersion relation varies sensitively with changing optical properties in the vicinity of the bottom surface of the ATR coupler. This characteristic allows an SPR reflectometer to act as a highly sensitive probe for analyzing protein-protein interactions, protein-DNA interactions, and interactions between other biomaterials (17). Relatively recently, SPRM has been developed to study cell-matrix adhesion. In SPRM, cells are attached to the surface of the prism after proper surface treatment. To collect data, the angle of incidence is adjusted to match the dispersion relation for the case in which cells are not attached. Therefore, the images show gray scale values at the cell-matrix adhesion area, whereas the other undisturbed regions exhibit minimum intensity values. However, the low lateral resolution and contrast obtained in the acquired images still need to be improved (10,11).
Here, we developed surface plasmon resonance imaging ellipsometry (SPRIE) to selectively image cell-matrix adhesion dynamics with improved spatial resolution. The improved resolution is attributable to the integration of an attenuated total reflection coupler for surface sensitivity and null-type imaging ellipsometry for contrast, in the same system. Although the coupling efficiency of the specific ATR coupler is not optimized, the null-type ellipsometry technique was successively applied to acquire an optimal contrast ratio of the cell-matrix adhesion images. In SPRIE, the reflected wave fields from the undisturbed regions, consisting of elliptically polarized light, can be extinguished by adjusting the polarizing components in the ellipsometer (18). These improvements allow for the use of short wavelengths from the light source without impacting the image contrast ratio, as compared with conventional SPRM (19). We applied SPRIE to characterize the adhesion properties of different cell types and then investigated changes in cell dynamics in diverse biological systems. An observation of the cell-substrate attachment of rounding mitotic cells was provided by SPRIE imaging. Cell-cell communication was also visualized by SPRIE. In addition, we report real-time cell adhesion dynamics as influenced by shear stress, based on an analysis of SPRIE images.
Materials and Methods
Surface plasmon resonance imaging ellipsometry system
A schematic diagram of the SPRIE system is depicted in Fig. 1 A. The optical arrangement of SPRIE consists of a light source (L; 50 mW, λ = 532 nm), polarizer (P), compensator (C), ATR coupler (PR), objective lens (O; 20×, SLWD, Nikon), analyzer (A), and CCD camera (D; MC681SPD, Texas Instruments, Dallas, TX). The field of view is ∼170 μm × 240 μm at an incidence angle of 60°. The prism of the ATR coupler was prepared by evaporating a 2-nm chromium thin film on a glass prism (SF10, n = 1.737) and then evaporating a 30-nm gold thin film. A custom-designed incubation chamber was fabricated with polyetheretherketone (PEEK) and was used as the ATR coupler. The inlet and outlet ports were connected with Teflon tubing (1522, Upchurch Scientific, Oak Harbor, WA), and the prism was mechanically clamped to the incubation chamber. A silicone elastomer gasket was inserted between the prism and the chamber as shown in Fig. 1 D. The internal volume of the fluidic chamber was ∼40 μL (6 mm × 13 mm × 0.5 mm). A programmable syringe pump (PHD 22/2000, Harvard Apparatus) and continuous flow tubing segments (DC1 61-0270, Harvard Apparatus, Holliston, MA) were used to circulate the cell incubation media during time course experiments. By changing the flow rate, the influence of shear stress on adhesion dynamics could be observed.
Figure 1.
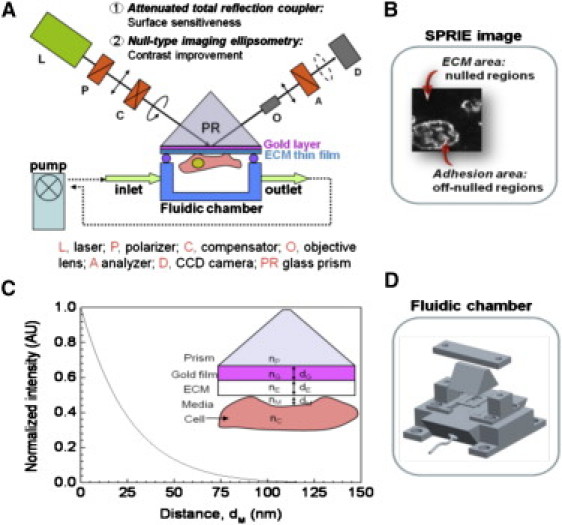
(A) Schematic of the surface plasmon resonance imaging ellipsometry (SPRIE) system. (B) Image of the cell-matrix interface by SPRIE. The bright area indicates the cell-matrix adhesion area obtained by off-nulling. The dark area indicates the cell-free matrix area obtained by nulling. (C) Normalized intensity as a function of distance between the cell membrane and a gold film when nulled at a cell-free medium region. ATR coupler configuration: prism (nP = 1.7367), gold (nG = 0.467 – i 2.4075, dG = 30 nm), ECM (nE = 1.45, dE = 20 nm), media (nM = 1.333), cell (nC = 1.36), wavelength = 532 nm, angle of incidence = 63°. All parameters are indicated in Table S1. (D) Schematic of fluidic chamber integrated in the SPRIE system.
ECM coating on the prisms of the ATR coupler
A self-assembled monolayer on a gold surface was created by incubating 1 mM 1-dodecanethiol for 24 h at room temperature (20). To prepare a fibronectin layer on the gold surface of the prism, 1 mg/mL human fibronectin (Sigma-Aldrich, St. Louis, MO) was diluted to 25 μg/mL with phosphate buffered saline (PBS) and incubated with a previously prepared alkyl-terminated prism surface. The solution was incubated for 2 h at 37°C. The fibronectin-conjugated prism surface was washed with PBS immediately before use.
SPRIE imaging procedure and cell preparation
Human umbilical vein endothelial cells (HUVEC) from Lonza (Walkersville, MD) were maintained in a Clonetics EGM-2 BulletKit (Lonza) that included endothelial cell basal medium-2, penicillin (100 units/mL), streptomycin (100 μg/mL) and the following growth supplements: 0.04% hydrocortisone, 0.4% hFGF-b, 0.1% VEGF, 0.1% R3-IGF-1, 0.1% ascorbic acid, 0.1% heparin, 2% fetal bovine serum (FBS), 0.1% hEGF, and 0.1% gentamicin A. Human coronary artery smooth muscle cells (CASMCs) from Lonza were maintained in a Clonetics SmGM-2 BulletKit (Lonza) containing smooth muscle cell basal medium, penicillin (100 units/mL), streptomycin (100 μg/mL), and the following growth supplements: 0.1% hEGF, 0.1% insulin, 0.2% hFGF-b, 5% FBS, and 0.1% gentamicin A, as described in the manual. A rat vascular smooth muscle cell line (A10) purchased from ATCC (Rockville, MD) was maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco, Gaithersburg, MD) and was supplemented with penicillin (100 units/mL), streptomycin (100 μg/mL), and 10% FBS. All cells were incubated in a humidified 5% CO2 balanced incubator at 37°C. Cells were seeded on fibronectin at the cell density of 1 × 104 per 1.54 cm2 for each SPRIE experiment and were incubated at 37°C in the appropriate media. The cell culture medium was maintained at 37°C and 5% CO2, and the cell culture medium was continuously circulated (0.01 mL/min) over the cells using a syringe pump (Harvard Apparatus).
Immunocytochemical analysis using a confocal laser scanning microscope
After SPRIE imaging, the cells were fixed in 4% paraformaldehyde for 10 min. The cells were then permeabilized in PBS containing 0.3% Triton X-100 and blocked with 10% FBS for 10 min. PBS containing 1% bovine serum albumin was used in each step. Mouse anti-paxillin (1:400; BD Sciences, San Diego, CA) and rabbit anti-tensin (1:200; Santa Cruz Biotechnology, Santa Cruz,CA) antibodies were used for immunocytochemistry. The nuclei were counter-stained by Hoechst 33342 (1:1000; Molecular Probes, Eugene, OR). Appropriate fluorescently tagged secondary antibodies were used to detect the primary antibodies (Invitrogen, Carlsbad, CA). Fluorescent images were acquired with a confocal laser scanning microscope (CLSM, FV 1000; Olympus, Tokyo, Japan).
Image analysis for statistical tests
Image-Pro Plus 6.2 (Media Cybernetics, Bethesda, MD) was used to analyze the data. To determine statistical significance, the results were analyzed using one-way ANOVA and a Bonferroni test. Each p-value is shown in the figures and text.
Results
Development of SPRIE: label-free and real-time visualization of cell adhesion dynamics
To enhance the lateral resolution and image contrast, we applied null-type imaging ellipsometry to the surface plasmon resonance measurement scheme. In regards to the optical arrangement, the major difference between SPRM and SPRIE is the integration of a compensator and an analyzer, as shown in Fig. 1 A. Because the optimal contrast ratio is achieved only when the surface plasmon wave and the measurement light are coupled resonantly, the thickness of the gold film has to be adjusted to fulfill surface plasmon resonance condition. However, the sample-to-sample variation of the refractive index, the thickness of the gold film and the subsequent surface treatment of the ATR coupler make it difficult to couple the resonances. These difficulties could result in reduced coupling efficiency and a decrease in the contrast ratio of the acquired images in SPRM. To overcome these practical limitations, a null-type scheme is adapted in SPRIE, meaning that the polarization state of the light emitted from the source is elliptically polarized after passing consecutively through the polarizer and the compensator. The azimuthal angles of the polarizer and the compensator were adjusted so that the polarization state of the reflected light was linearly polarized. The light is extinguished after passing through the analyzer, whose transmission axis was rotated such that it was perpendicular to the polarization axis of the incident light. Therefore, the medium-contacting region in which cells were not adhered was nulled out during the experiment so that this region exhibited virtually zero intensity values. The cell-matrix adhesion area was off-null so that this region exhibited a grayscale readout in the acquired images, as shown in Fig. 1 B, resulting in a 1-μm spatial resolution (19).
To investigate the contrast mechanism in SPRIE system, the relative intensity was calculated as a function of distance between the cell membrane and the gold surface. First, we applied the Jones matrix formula to the SPRIE system with the ATR coupler. The output electric field and intensity of the SPRIE system are expressed as:
| (1) |
where P, C, and A are the azimuthal angles of the polarizer, compensator and analyzer. RP and Rs are the total reflection coefficients for p- and s-polarized light from the ATR coupler. The ATR coupler was modeled as a multilayered structure with a prism, gold film, ECM, media and cells, as shown in the inset of Fig. 1 C. The refractive index of the cell was assumed to have a constant value, and the cell adhesion property was modeled as the distance between the cell membrane and the ECM surface (dM). The scattering matrix method was used to obtain RP and Rs (18). In calculating the intensity, P, C, and A were set to fulfill the nulling condition of the cell-free region. All parameters used for simulation are shown in Table S1 of the Supporting Material. The relative intensity was defined as:
| (2) |
where I is the intensity, and dM varies between 0 nm and 150 nm. The intensity I(0) is obtained when dM is zero, which means that cells are adhered. The intensity I(∞) is obtained from the cell-free region. Fig. 1 C shows the calculated result of Eq. 2. The normalized intensity decreases exponentially with increasing dM, which agrees well with the evanescent characteristics of the surface plasmon wave (SPW). When dM is >150 nm, which corresponds to the penetration depth of the SPW, the normalized intensity converges to zero. To investigate the real-time adhesion dynamics over long time periods (greater than several days), the fluidic chamber was attached to the SPRIE system, as shown in Fig. 1 D.
Distinct cell-matrix adhesion properties depending on the cell type as imaged by SPRIE
To determine the applicability of SPRIE to investigate cell-matrix adhesion, we characterized the adhesion properties of three cell types, human endothelial cells (HUVECs) and two types of smooth muscle cells (CASMCs and A10) from different species (human and rat). Fig. 2 shows representative SPRIE images (left), in which the white regions indicate cell-matrix adhesion, their corresponding topographical images of the intensity (middle), and the X-Z surface map of interest (the black line in the topography image, right). In the HUVECs shown in Fig. 2 A, each cell shows a finely and flat structured adhesion pattern with a close z distance to the surface, whereas the CASMCs display a concentrated adhesion area at one end (white arrow in Fig. 2 B). Considering the typical morphological shape of migrating cells, it is reasonable to conjecture that this concentrated adhesion area could be a leading edge. In the X-Z surface map, this region is shown to be the region closest to the surface (indicated by the black arrow). The polarization of z-depth along the line between the leading edge and tailing region was shown as well. This result is also in agreement with a previous study that showed that the cell-fibronectin adhesion region becomes the leading edge by inducing local proteolysis of fibronectin (21). Rat smooth muscle cells (A10, Fig. 2 C) primarily exhibit a fibril-structured adhesion pattern. This feature was also clearly shown in the X-Z surface map. We quantitatively analyzed the proportion of adhesion normalized by the area of a single cell (Fig. 2 D). The adhesion patterns of the three cell types were significantly distinct (p < 0.001, n = 15 for each cell type).
Figure 2.
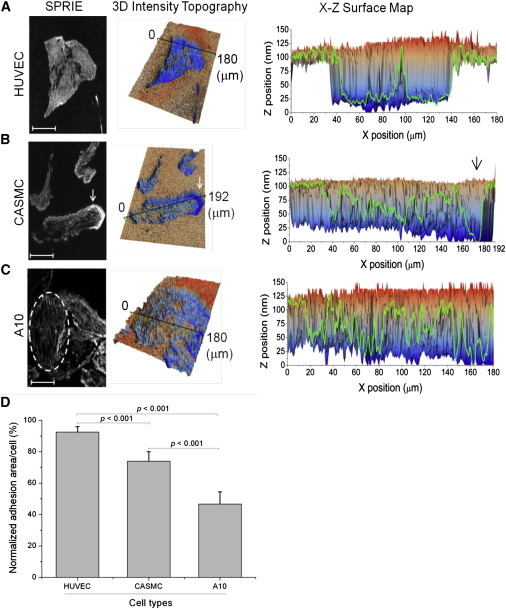
Cell-matrix interfaces imaged by SPRIE. (A–C) Cell-matrix interfaces were imaged for human umbilical vein endothelial cells (HUVECs), human coronary artery smooth muscle cells (CASMCs) and a rat vascular smooth muscle cell line (A10). SPRIE images (left), corresponding three-dimensional intensity topography (middle frame), and X-Z surface map of the black line in topography (right) were compared at identical sites. The leading edge of the migratory cells is indicated by the arrow in B. The fibrillar structure is indicated by a dotted oval in C. Scale bar = 50 μm. (D) Quantification of the adhesion area of three cell types imaged by SPRIE. The adhesion area was quantified at the single cell level after normalization by cell area.
Validation of cell adhesion-selective SPRIE imaging by comparison with CLSM
To test the reliability of the adhesion-selective SPRIE images, we carried out a correlation test between SPRIE and CLSM images showing the distribution of paxillin, a well-known cell-matrix adhesion molecule. After label-free SPRIE imaging, the A10 cells were subjected to immunocytochemistry and CLSM imaging. The two-dimensional correlation of pseudo-red colored SPRIE and CLSM images was tested, resulting in a Pearson's correlation coefficient of 0.46 (Fig. 3 A). The intensity of CLSM highly overlapped with the white regions in the SPRIE images, indicating that the SPRIE intensity can indicate the expression of paxillin. Notably, a dot-like focal adhesion area showed intense overlapping in the merged images (yellow in merged image).
Figure 3.
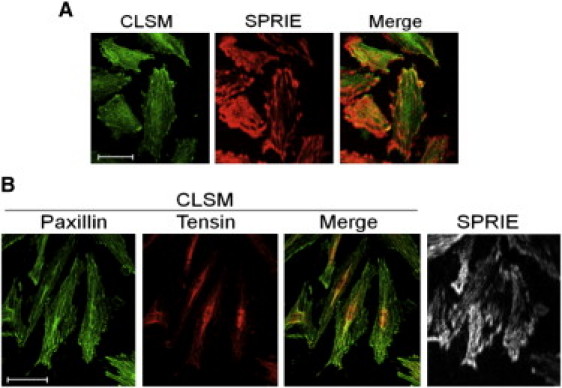
Comparative validation between SPRIE and CLSM. (A) The same cell site was consecutively imaged by SPRIE and CLSM. Cell-matrix adhesions in SPRIE are shown in pseudo-red, and paxillin is shown in green in CLSM. The merged area is shown in yellow. (B) To obtain a functional mapping of adhesion molecules, two types of adhesion molecules were compared using SPRIE and CLSM in the same manner. Green indicates paxillin, and red indicates tensin. In CASMC and A10, the nuclei are stained blue. Scale bar = 50 μm.
To identify the adhesion molecules that contribute to cell-matrix adhesion, we compared the distribution of adhesion molecules in CLSM and the cell-matrix area in SPRIE (Fig. 3 B). Paxillin and tensin, to test the initial and late stage of adhesion molecules, respectively, were subjected to immunocytochemistry. Two-dimensional Pearson's correlation coefficients were analyzed for each pair of paxillin or tensin versus SPRIE (paxillin-SPRIE: 0.56, tensin-SPRIE: 0.38). The results indicate that paxillin contributes more than tensin to the positive signal in SPRIE.
Label-free and real-time imaging of the cell-matrix attachment of mitotic cells
The applicability of SPRIE was verified by analyzing cell adhesion dynamics during division. Because of a lack of appropriate tools, it has been difficult to simultaneously study adhesion dynamics while visualizing cell division. Using the properties of SPRIE with high sensitivity and visualization only in the evanescent field, we imaged the cell-matrix attachment of mitotic cells. Because of a doubling time of 25–30 h, we imaged the cell-matrix adhesion dynamics of HUVECs for at least 2 days and found cell division sites that could be identified by observing one mother cell dividing into two daughter cells. The image gallery in Fig. 4 and Movie S1 show that the mother cell starts to release from the matrix (02:00–06:20) and that the focal anchorage to the matrix then appears (04:05) and is sustained for ≈ 40 min (arrows). The elapsed time is indicated by hour:minute. The elapsed time of the cell-matrix anchorage in our study (47.6 ± 6.8 min, n = 3) is consistent with a previous report stating that the M phase lasts about 1 h in typical mammalian cells. Next, a dot-shaped attachment completely divides and forms two separate cell shapes (04:45–06:20).
Figure 4.
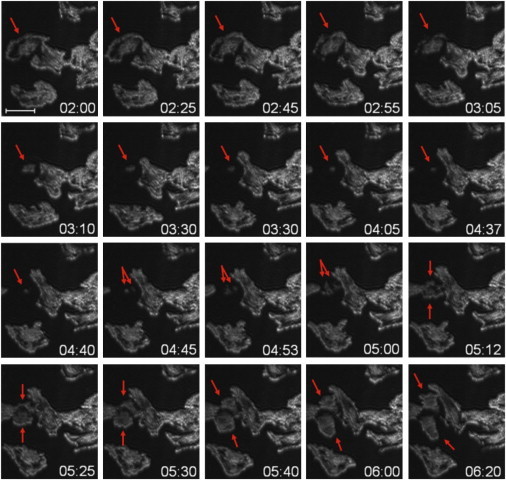
Cell-matrix adhesion dynamics during cell division. The red arrows show one mitotic cell progressing from one mother cell to two daughter cells. The elapsed time is indicated by hour:minute. Scale bar = 50 μm.
Cell-cell communication imaged by SPRIE
Using the improved sensitivity of SPRIE, we examined the cell-cell interactions of endothelial cells (HUVECs) by SPRIE (Fig. 5). We observed cell-cell interactions among confluent cells (Movie S2). At the cell boundaries (shown in Movie S2), cells interact with each other by forming transient fibril-like connections. The SPRIE image shown in Fig. 5 A exhibits fibril-like connections between cells (dashed inserts). Topographical images corresponding to SPRIE clearly show these fibril-like cell-cell interactions between cells (Fig. 5, B and C). The presence of fibril-like connections was demonstrated by a correlative study of SPRIE and CLSM. To confirm that these interactions are cell-cell adhesions, we performed immunocytochemical analyses for vascular endothelial (VE)-cadherin, the principal junctional adhesion molecule, on the cells observed by SPRIE (Fig. 5 D). We observed that the expression of VE-cadherin was higher in the area with the fibril-like shapes shown on the SPRIE image. One possible explanation for this observation is that the signal for VE-cadherin-mediated cell-cell adhesion may be transmitted to cell-matrix adhesion through modulation of the actin cytoskeleton. Therefore, in Fig. 5, A and D, we analyzed histogram intensities of the cell-cell interaction area, indicated by line E in Fig. 5, A and D. As shown in Fig. 5 E, the expression of actin also increased with the expression of VE-cadherin, suggesting that cell-cell communication by VE-cadherin may induce changes in the actin cytoskeleton, leading to a change in cell-matrix adhesion. The histogram in Fig. 5 E shows a partial correlation between SPRIE and confocal intensities, meaning that actin and some adhesion molecules contribute to the generation of SPRIE signals. To determine the contribution of the actin cytoskeleton to generating SPRIE signals in the inclusive footprint region of the cell, we performed a correlation test between Fig. 5, A and D, resulting in a Pearson's correlation coefficient of 0.59.
Figure 5.
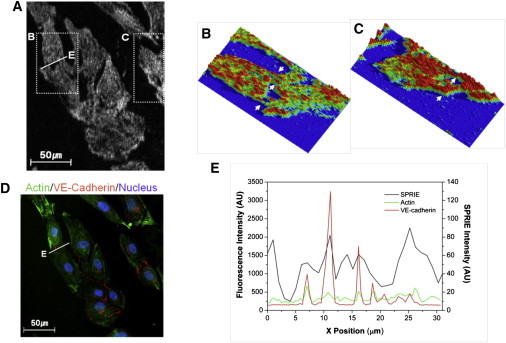
Cell-cell interactions imaged by SPRIE and validated by CLSM. (A) Cell-cell interactions of HUVECs imaged by SPRIE. (B and C) Intensity topography of inserted dotted rectangles in A. The arrows indicate fibril-like cell-cell connections. (D) CLSM image for VE-cadherin (red), actin (green), and nuclei (blue) on the same cells shown in A. (E) Histogram of the fluorescence intensity and SPRIE signal of line E inserted in A and D. The green line indicates actin, and the red line indicates VE-cadherin. The black line indicates SPRIE signal. Note that VE-cadherin and actin overlap, and the length of the peak indicates the thickness of the fibril-like cell-cell connections. Scale bar = 50 μm.
Shear-stress-induced cell adhesion dynamics
Using SPRIE, we examined the real-time modulation of cell adhesion dynamics by shear stress. A shear stress (1.2 Pa), which is physiologically significant in vascular inflammation (22,23), was applied to the SPRIE system for 40 h, and the change in cell-matrix adhesion in the SPRIE images was analyzed as a function of time. Because shear stress is an important factor in endothelial cell dynamics, we used HUVECs for this study. After acquiring SPRIE images once an hour, the centers of individual cells were automatically determined by Image Pro software, and each cell center in the SPRIE images was tracked for analysis. Fig. 6, A and B, show the traction map of cell movement when the cells are exposed to either static or shear stress (1.2 Pa) conditions. In the static conditions, the cells migrate randomly, whereas in the shear stress conditions, the cells migrate in the direction of flow. The velocity of migration by shear stress was analyzed and compared to migration under static conditions in Fig. 6 C. The migration velocity increased significantly with the application of 1.2 Pa of shear stress in one direction. To examine how fast cells could respond to changes in shear stress, we applied the following conditions: 1), static condition (4 h); 2), onset of shear stress; 3), shear stress condition (18 h); 4), cessation of shear stress; and 5) static condition (18 h) sequentially. We observed that some cells started active migration after the onset of shear stress (open circles and triangles). In terms of recovery from shear stress, these migratory cells adapted to static conditions by decreasing their migration distance. We also observed a change in morphology from elongated to round as the cells adapted to static conditions (blue triangles in Fig. 6 D).
Figure 6.
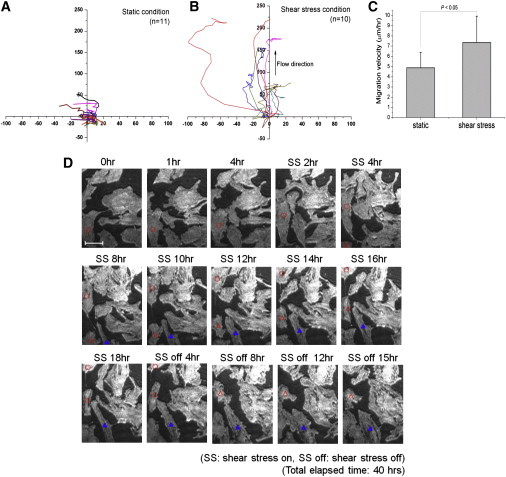
Cell adhesion dynamics modulated by shear stress (1.2 Pa). (A and B) Trajectory maps of cells in both static and shear stress conditions. (C) Migration velocity in both static and shear stress conditions. (D) Cell-matrix adhesion images obtained during modulation of shear stress; static (4 h), shear stress (18 h), static (18 h). The total elapsed time is 40 h. Scale bar = 50 μm.
Discussion
We developed SPRIE to observe cell adhesion dynamics in real time without labeling. The integration of an attenuated total reflection coupler and null-type imaging ellipsometry allowed for selective imaging of cell-matrix adhesion with highly increased contrast. Several adherent cell types were distinctively characterized by SPRIE. Furthermore, the cell-substrate anchorage of rounding mitotic cells was clearly imaged by SPRIE. To our knowledge, this is the first label-free and real-time visualization of cell-substrate anchorage, which may provide a useful platform for identifying detailed dynamic features of cell-adhesion-relevant phenomena. Using SPRIE, we also analyzed the response time of cell adhesion dynamics influenced by shear stress in real time.
Cell adhesion to the matrix occurs through many structures, including focal complexes, focal adhesions, and fibrillar adhesions (24,25). These adhesions are composed of various proteins, including paxillin and vinculin (26). Because these adhesion molecules strongly interact with each other at the interface, cell-matrix adhesion needs to be studied at the interface in a dynamic mode (27). Currently, CLSM is the most popular optical technology for studying adhesion molecules; however, its depth resolution needs to taken into account. The thickness of the cell membrane is much thinner (∼10 nm) than the limit of the z-depth resolution of CLSM, at ∼300 nm. From this point of view, SPRIE can image cell-matrix adhesion at the interface more selectively than CLSM. Using a two-dimensional correlation study between CLSM and SPRIE, we demonstrated that SPRIE can be used to image cell-matrix adhesions. Some regions of the SPRIE images merged with the expression of paxillin, a focal adhesion protein. However, other regions expressing tensin generally correlated with fibrillar adhesion, but did not overlap with the SPRIE image. This result can be explained by the depth difference of focal and fibrillar adhesions. Because these adhesions occur at different depths, there may be some discrepancy between CLSM and SPRIE images. By complementary use of CLSM, we showed that SPRIE is suitable for selective imaging of the cell-matrix interface and can also provide a new combinatorial method for mapping chemical information with label-free imaging. This procedure can be uniquely developed to provide a synergistic method of both label-free long-term observation and chemical identification by labeling.
The visualization of cell-matrix adhesions has been achieved with various optical microscopy techniques. In principle, IRM can provide quantitative information by illuminating various interface depths (13). However, quantitative IRM measurements should be analyzed with caution. Proposing a theory based on microscope interferometry, Gingbell and Todd reported that the cytoplasmic thickness can influence images of the cell-substrate interface in IRM imaging (28). Verschueren also reported that cell-matrix adhesion can be imaged by IRM only in thick cytoplasm regions rather than in thin regions like lamellipodia (14). To study cell-matrix adhesion in thin layers of cytoplasm, TIRF can be used as a complementary method (8). TIRF has become the main method for imaging the cell membrane because of its high contrast and molecular specificity. The first application of TIRF in imaging the cell-substrate interface was reported by Lanni et al. (29). Although they estimated the cell-substrate separation distance, some limitations were also reported, such as a dependency on dye concentration and a variance of the lateral refractive index. The region of target molecules accumulated in the evanescent field can generate increased intensity, regardless of the difference in z depth. In addition, TIRF has several limitations. The fluorescent tags required for TIRF on biomolecules can affect their inherent properties by changing their hydrophobicity (30). The inherent autofluorescence of cell membranes must also be considered when carrying out TIRF imaging because of the possibility of false positive intensity. Most importantly, TIRF is not appropriate for imaging dynamic phenomena over long time periods (on the scale of hours or days) because of photo-bleaching. In contrast, SPRIE allows for label-free and highly selective imaging of cell-matrix interfaces over several days. Additionally, using the principle of imaging ellipsometry, SPRIE provides the relative z-depth information on the cell footprints as shown in Fig. 2.
We demonstrated the applicability of SPRIE by imaging cell-matrix adhesion during cell division, cell-cell interaction and cell migration. Despite the importance of the effects of the cell-matrix adhesions that occur during these phenomena (31–33), simultaneous studies of both processes have been difficult to perform due to a lack of appropriate tools. Using SPRIE, we successfully imaged the cell-matrix anchorage of rounding mitotic cells in real time. To our knowledge, this is the first report of label-free imaging of cell-matrix anchorage during cell division. We also observed lateral cell-cell communication from connecting fibril-like cell extensions using SPRIE live imaging. It is possible that cell-cell interactions could occur at the apical side instead of the basal side of cells. In our SPRIE results (Fig. 5 and Movie S2), the cell-cell connections were dynamically imaged as fibril-like shapes, and these were corroborated by the expression of VE-cadherin, a typical cell-cell adhesion molecule, as shown by CLSM. This observation can be explained by the following: cell-cell contacts induce an interaction between VE-cadherin and the actin cytoskeleton inside the cell, which influences actin re-arrangements on the basal surface, leading to a change in cell-matrix adhesion. Our fluorescence histogram data, which show overlapping regions of VE-cadherin and actin, support this explanation. Our data further support recently published data that demonstrated a basal-apical directional flow-like movement of cadherin adhesion molecules by providing experimental evidence of cell-matrix adhesions (34). In addition, the Pearson's correlation coefficient of 0.59 obtained between the SPRIE imaging (Fig. 5 A) and actin cytoskeleton (Fig. 5 D) indicates that the internal cellular structure of the actin cytoskeleton might partially contribute to the generation of SPRIE. Because the actin cytoskeleton is anchored to each focal adhesion site through the adaptor molecule like vinculin and exerts a force on the matrix. Therefore, the generation of SPRIE signals can be partially affected by cytoskeleton molecules as well as adhesion molecules, which needs further investigation.
Through the shear stress modulation experiments, we studied the response time when shear stress at the apical surface was transmitted to cell-matrix adhesions to the bottom in real-time. Interestingly, the change in cell-matrix adhesion occurred early after the onset of shear stress. After the cessation of shear stress, the migratory movement of the cell-matrix adhesion decreased, and the shape of some cells changed from elongated to round. This change could be considered a recovery process from shear stress. We found that the migratory velocity of cell-matrix adhesions was increased by shear stress, which is consistent with previous reports (22,23). Therefore, SPRIE-based imaging offers a useful methodology to study cell-matrix adhesion under hemodynamic conditions in real-time and in a label-free manner. Integrated fluidic channels also have the potentials to be developed for high-throughput screening system.
The SPRIE intensity is determined not only by the distance between cell membrane and gold surface (dM) but also by the refractive index of the cell. Definitely, the refractive index of the cell is not a constant but a spatial and temporal variable with cell adhesion sites and dynamics. However, in the present monochromatic SPRIE, it is not possible to extract information both dM and details of the refractive index of the cell. That is why the refractive index of the cell was assumed constant and the cell adhesion property was modeled as the distance between cell membrane and gold surface (dM). For further detailed investigation of the spatial and temporal dynamic changes of the intracellular refractive index of cells, a development of spectroscopic SPRIE is in progress. With the further development of spectroscopic SPRIE, we expect get more detailed understanding of intracellular adhesion dynamics of biomolecules including actin cytoskeleton and other adhesion proteins. A disadvantage of SPRIE can be the limitation of available types of matrices. Among various types of matrices, thin layers of matrix under 100 nm of thickness are available by the propagation of surface plasmons. However, this could be resolved by using self-assembled monolayers of matrix components, a technique that allows the absorption of protein in well-ordered form onto the surface (35).
Conclusion
We have developed a SPRIE technique that can selectively and dynamically visualize cell-matrix adhesion interfaces in a label-free manner. Using SPRIE, cell-matrix adhesion patterns for different cell types were observed with 1 μm spatial resolution. SPRIE showed detailed dynamic changes of cell adhesion patterns during cell division and during cell migration, which were modulated by shear stress. Dynamic cell-cell communications were also observed with SPRIE. We expect that this imaging platform will provide a useful method for investigating not only cell adhesion dynamics but also the basic mechanisms of diseases such as cancer metastasis and atherosclerosis.
Acknowledgments
We thank Mihee Park and Jungeun Gil for technical support. We also thank Sujin Kim and Woncheol Jeong for image analysis.
This research was supported by grants from the Development of Nano-Based Convergence Technology for Precision Measurement of the Korea Research Institute of Standards and Science (KRISS) and the Degree Research Center of the Korea Research Council of Fundamental Science and Technology (KRCF), Republic of Korea.
Supporting Material
References
- 1.Danen E.H., Sonnenberg A. Integrins in regulation of tissue development and function. J. Pathol. 2003;200:471–480. doi: 10.1002/path.1416. [DOI] [PubMed] [Google Scholar]
- 2.Streuli C. Extracellular matrix remodeling and cellular differentiation. Curr. Opin. Cell Biol. 1999;11:634–640. doi: 10.1016/s0955-0674(99)00026-5. [DOI] [PubMed] [Google Scholar]
- 3.Bökel C., Brown N.H. Integrins in development: moving on, responding to, and sticking to the extracellular matrix. Dev. Cell. 2002;3:311–321. doi: 10.1016/s1534-5807(02)00265-4. [DOI] [PubMed] [Google Scholar]
- 4.Livant D.L., Brabec R.K., Markwart S. The PHSRN sequence induces extracellular matrix invasion and accelerates wound healing in obese diabetic mice. J. Clin. Invest. 2000;105:1537–1545. doi: 10.1172/JCI8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avraamides C.J., Garmy-Susini B., Varner J.A. Integrins in angiogenesis and lymph angiogenesis. Nat. Rev. Cancer. 2008;8:604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtis A.S. The mechanism of adhesion of cells to glass. a study by interference reflection microscopy. J. Cell Biol. 1964;20:199–215. doi: 10.1083/jcb.20.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zenisek D., Steyer J.A., Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature. 2000;406:849–854. doi: 10.1038/35022500. [DOI] [PubMed] [Google Scholar]
- 8.Axelrod D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol. 1981;89:141–145. doi: 10.1083/jcb.89.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoover D.K., Lee E.J., Yousaf M.N. Total internal reflection fluorescence microscopy of cell adhesion on patterned self-assembled monolayers on gold. Langmuir. 2009;25:2563–2566. doi: 10.1021/la803927k. [DOI] [PubMed] [Google Scholar]
- 10.Giebel K., Bechinger C., Bastmeyer M. Imaging of cell/substrate contacts of living cells with surface plasmon resonance microscopy. Biophys. J. 1999;76:509–516. doi: 10.1016/s0006-3495(99)77219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson A.W., Halter M., Plant A.L. Surface plasmon resonance imaging of cells and surface-associated fibronectin. BMC Cell Biol. 2009;10:16. doi: 10.1186/1471-2121-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith H.L., Hickey J., Majewski J. Mouse fibroblast cell adhesion studied by neutron reflectometry. Biophys. J. 2010;98:793–799. doi: 10.1016/j.bpj.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izzard C.S., Lochner L.R. Formation of cell-to-substrate contacts during fibroblast motility: an interference-reflexion study. J. Cell Sci. 1980;42:81–116. doi: 10.1242/jcs.42.1.81. [DOI] [PubMed] [Google Scholar]
- 14.Verschueren H. Interference reflection microscopy in cell biology: methodology and applications. J. Cell Sci. 1985;75:279–301. doi: 10.1242/jcs.75.1.279. [DOI] [PubMed] [Google Scholar]
- 15.Homola J. Springer-Verlag; Berlin: 2006. Surface Plasmon Resonance Based Sensors. [Google Scholar]
- 16.Kretschmann E. The determination of the optical constants of metals by excitation of surface plasmons. Z. Phys. 1971;241:313–324. [Google Scholar]
- 17.Homola J. Surface plasmon resonance sensors for detection of chemical and biological species. Chem. Rev. 2008;108:462–493. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- 18.Azzam R.M.A., Bashara N.M. North-Holland Publishing; Amsterdam, The Netherlands: 1977. Ellipsometry and Polarized Light. [Google Scholar]
- 19.Brockman J.M., Nelson B.P., Corn R.M. Surface plasmon resonance imaging measurements of ultrathin organic films. Annu. Rev. Phys. Chem. 2000;51:41–63. doi: 10.1146/annurev.physchem.51.1.41. [DOI] [PubMed] [Google Scholar]
- 20.Min H., Park J., Lee T.G. ToF-SIMS study on the cleaning methods of Au surface and their effects on the reproducibility of self-assembled monolayers. Appl. Surf. Sci. 2008;255:1025–1028. [Google Scholar]
- 21.Takino T., Saeki H., Sato H. Inhibition of membrane-type 1 matrix metalloproteinase at cell-matrix adhesions. Cancer Res. 2007;67:11621–11629. doi: 10.1158/0008-5472.CAN-07-5251. [DOI] [PubMed] [Google Scholar]
- 22.Chatzizisis Y.S., Coskun A.U., Stone P.H. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J. Am. Coll. Cardiol. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 23.Urbich C., Dernbach E., Dimmeler S. Shear stress-induced endothelial cell migration involves integrin signaling via the fibronectin receptor subunits alpha(5) and beta(1) Arterioscler. Thromb. Vasc. Biol. 2002;22:69–75. doi: 10.1161/hq0102.101518. [DOI] [PubMed] [Google Scholar]
- 24.Geiger B., Bershadsky A., Pankov R., Yamada K.M. Transmembrane crosstalk between the extracellular matrix–cytoskeleton crosstalk. Nat. Rev. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 25.Berrier A.L., Yamada K.M. Cell-matrix adhesion. J. Cell. Physiol. 2007;213:565–573. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 26.Wehrle-Haller B., Imhof B. The inner lives of focal adhesions. Trends Cell Biol. 2002;12:382–389. doi: 10.1016/s0962-8924(02)02321-8. [DOI] [PubMed] [Google Scholar]
- 27.Katz B.Z., Zamir E., Geiger B. Physical state of the extracellular matrix regulates the structure and molecular composition of cell-matrix adhesions. Mol. Biol. Cell. 2000;11:1047–1060. doi: 10.1091/mbc.11.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gingell D., Todd I. Interference reflection microscopy. A quantitative theory for image interpretation and its application to cell-substratum separation measurement. Biophys. J. 1979;26:507–526. doi: 10.1016/S0006-3495(79)85268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanni F., Waggoner A.S., Taylor D.L. Structural organization of interphase 3T3 fibroblasts studied by total internal reflection fluorescence microscopy. J. Cell Biol. 1985;100:1091–1102. doi: 10.1083/jcb.100.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaiser R.D., London E. Determination of the depth of BODIPY probes in model membranes by parallax analysis of fluorescence quenching. Biochim. Biophys. Acta. 1998;1375:13–22. doi: 10.1016/s0005-2736(98)00127-8. [DOI] [PubMed] [Google Scholar]
- 31.Pugacheva E.N., Roegiers F., Golemis E.A. Interdependence of cell attachment and cell cycle signaling. Curr. Opin. Cell Biol. 2006;18:507–515. doi: 10.1016/j.ceb.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glotzer M. Animal cell cytokinesis. Annu. Rev. Cell Dev. Biol. 2001;17:351–386. doi: 10.1146/annurev.cellbio.17.1.351. [DOI] [PubMed] [Google Scholar]
- 33.Théry M., Bornens M. Cell shape and cell division. Curr. Opin. Cell Biol. 2006;18:648–657. doi: 10.1016/j.ceb.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Kametani Y., Takeichi M. Basal-to-apical cadherin flow at cell junctions. Nat. Cell Biol. 2007;9:92–98. doi: 10.1038/ncb1520. [DOI] [PubMed] [Google Scholar]
- 35.Mrksich M., Whitesides G.M. Using self-assembled monolayers to understand the interactions of man-made surfaces with proteins and cells. Annu. Rev. Biophys. Biomol. Struct. 1996;25:55–78. doi: 10.1146/annurev.bb.25.060196.000415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.