

Abstract
AIM: To identify the differentially over-expressed genes associated with β-catenin accumulation in nuclei of hepatocellular carcinoma (HCC) cells.
METHODS: Differentially expressed genes were identified in radiation-induced B6C3 F1 mouse HCC cells by mRNA differential display, Northern blot and RT-PCR, respectively. Total glutathione-s-transferase (GST) activity was measured by GST activity assay and β-catenin localization was detected with immunostaining in radiation-induced mouse HCC cells and in HepG2 cell lines.
RESULTS: Two up-regulated genes, glutamine synthetase and glutathione-s-transferase M3 (GSTM3), were identified in radiation-induced mouse HCC cells. Influence of β-catenin accumulation in nuclei of HCC cells on up-regulation of GSTM3 mRNA was investigated. The nearby upstream domain of GSTM3 contained the β-catenin/Tcf-Lef consensus binding site sequences [5’-(A/T)(A/T) CAAAG-3’], and the total GST activity ratio was considerably higher in B6C3F1 mouse HCC cells with β-catenin accumulation in nuclei of HCC cells than in those without β-catenin accumulation (0.353 ± 0.117 vs 0.071 ± 0.064, P < 0.001). The TWS119 (a distinct GSK-3β inhibitor)-induced total GST activity was significantly higher in HepG2 cells with β-catenin accumulation than in those without β-catenin accumulation in nuclei of HCC cells. Additionally, the GSTM3 mRNA level was significantly higher at 24 h than at 12 h in TWS119-treated HepG2 cells.
CONCLUSION: β-catenin accumulation increases GST activity in nuclei of HCC cells, and GSTM3 may be a novel target gene of the β-catenin/Tcf-Lef complex.
Keywords: β-catenin accumulation, Differential display analysis, Glutathione-s-transferase M3, Hepatocellular carcinoma, Radiation
INTRODUCTION
Hepatocellular carcinoma (HCC) is a primary cancer of the liver chronically injured by infection, metabolic disease or various drugs[1]. As generally observed in other carcinomas, HCC is attributed to accumulated genetic alterations, including (1) Activation of oncogenes N-ras, H-ras, and K-ras, c-erbA, c-met, RB and c-myc[2-5]; (2) Transcriptional activation of c-jun and nuclear factor kB by hepatitis B virus factors[6]; (3) Repression or mutation of the p53 anti-oncogene[7]; and (4) Accumulation of β-catenin[8]. Although the genetic events responsible for either HCC initiation or progression are not clear, they involve at least three carcinogenesis pathways: the p53, RB and Wnt/β-catenin signaling pathways[1-10].
β-catenin is an essential downstream effector of the canonical Wnt signaling pathway[11,12]. Approximately 20% of HCC cells display β-catenin aberrant activation[9,13]. In the normal steady state, β-catenin is continuously phosphorylated at serine and threonine residues by glycogen synthase kinase 3β (GSK-3β) in a complex with adenomatous polyposis coli (APC)-axin/conductin and is quickly degraded through the ubiquitin/proteasome pathway. In mice, liver-specific deletion of APC induces β-catenin stabilization and increases the number of HCC cells. Although the activation of β-catenin is likely an initiating or contributory factor for HCC, more fundamental information is required for a better understanding of the detailed genetic mechanism underlying HCC associated with β-catenin.
To uncover the detailed genetic mechanisms underlying HCC in the present study, several genes in mouse cancerous liver tissue samples were identified to disclose more of the genes that play a very important role in the regulation of cell proliferation and the development of HCC. We identified several cDNA fragments that were differentially expressed in radiation-induced mouse HCC and compared with those in matched nontumorous liver tissue. Samples using a differential display technique[14]. We determined whether the nearby upstream domain of those genes contain the β-catenin/Tcf-Lef consensus binding site sequences. The influence of β-catenin accumulation in nuclei of HCC cells on activation of protein encoded by the gene containing β-catenin/Tcf-Lef consensus binding site sequences was further investigated.
MATERIALS AND METHODS
Sample preparation
Surgically resected HCC and adjacent nontumorous tissue samples were taken from the livers of 18-mo-old mice irradiated by 3.5 Gy 60Co γ-ray for 1 wk immediately after they were born. B6C3F1 mouse HCC and matched nontumorous liver tissue samples were obtained immediately under the same conditions for measurement of total GST activity[15], isolation of total RNA[16], and immunohistochemical expression of β-catenin and hematoxylin-eosin (HE) staining. Histological analysis of HCC tissue samples from mice was carried out according to the general rules for clinical and pathological study of primary liver cancer. On the other hand, HepG2 cells obtained from China Center for Type Culture Collection were maintained in DMEM supplemented with 10% fetal calf serum (Sigma, Louis, MO), 200 mmol/L L-glutamine and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA) at 37°C in a water-saturated atmosphere containing 5% CO2. Cell cultures were allowed to reach 90% confluence. The cells were then treated with or without 1 μmol/L of 4, 6-disubstituted pyrrolopyrimidine (TWS119, Cayman Chemical, Ann Arbor, MI) and incubated at 37°C for 24 h. Finally, total RNA or cytoplasmic proteins, including total GST proteins, were extracted from surgically resected frozen tissue samples or HepG2 cells by homogenization with a Vibra cell sonicator (Sonics and Materials, Inc., Danbury, CT) under a regularity condition.
mRNA differential display analysis and DNA sequencing
mRNA differential display analysis was performed as previously described[14] with a RNAmap kit A (Genhunter, Nashville, TN) (Table 1). Total RNA (0.4 μg) extracted from radiation-induced mouse HCC and matched nontumorous liver tissue samples was reverse-transcribed with different combinations of arbitrary and anchor primers (Table 1) for initial cDNA synthesis. The thermal cycler parameters were as follows: 1 cycle at 94°C for 4 min, followed by 40 cycles at 94°C for 30 s, at 40°C for 2 min and at 72°C for 30 s. Amplified subpopulations were distributed on a 6% DNA sequencing gel. The bands of interest were cut out from the polyacrylamide gel, and cDNA fragments were re-amplified using the same pair of primers and the same cycle parameters as described above. The re-amplified cDNA fragments were purified from 2% agarose gels and subcloned into pCRII-TOPO vectors using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA). Clones were selected by the same size of bands cut from the polyacrylamide gel as described above, followed by inverse hybridization and DNA sequencing. DNA sequences were compared with those in GenBank by the Blast Service provided by NIH (Bethesda, MD).
Table 1.
Primer sets used in mRNA differential display analysis
| Anchor primers | Arbitrary primers |
| T12MG (10 μmol/L) | AP-10 (2 μmol/L), 5’-TAGCAAGTGC-3’ |
| T12MA (10 μmol/L) | AP-11 (2 μmol/L), 5’-CAGACCGTTC-3’ |
| T12MT (10 μmol/L) | AP-12 (2 μmol/L), 5’-TGCTGACCTG-3’ |
| T12MC (10 μmol/L) | AP-14 (2 μmol/L), 5’-AATGGGCTGA-3’ |
M represents a degenerated mixture of dA, dG and dC. The 16 different primer sets used for PCR amplification were randomly combined from the four arbitrary primers and the four anchor primers.
Northern blot analysis
Total RNA extraction (10 μg) was denatured and electrophoresed in a 1.2% agarose gel containing 0.66 mol/L formaldehyde and then transferred onto a Hybond-nylon membrane (Amersham Biosciences, Buckingham, England). The membranes were UV cross-linked, pre-hybridized and hybridized. The respective cDNA fragments obtained from the differential display reaction were used as a probe for Northern blot analysis. Probes were [α-32P] dCTP (110 TBq/mmol) labeled using the MegaprimeTM DNA labeling kit (Amersham, UK), pre-hybridized to filters at 42°C for 30 min, and then hybridized to the filters overnight at 42°C. The filters were washed twice at 55°C in 1 × SSC, 0.1% SDS for 15 min, and then exposed to X-ray film for 24-72 h at -80°C.
Immune staining
Immunofluorescence and immunocytochemistry analyses of β-catenin localization were performed, respectively, with mouse monoclonal anti-β-catenin antibody diluted at 1:500 for immunofluorescence, and diluted at 1:100 for immunocytochemistry (Sigma, Louis, MO) on 5-μm paraffin-embedded sections and paraformaldehyde-fixed HepG2 cell sections that were differentially resected from 24 radiation-induced B6C3F1 mouse HCC and adjacent nontumorous liver tissue samples[17,18]. The tissue sections were then incubated for 1 h at room temperature with Alexa Fluor 546 goat anti-mouse IgG diluted at 1:1000 (Molecular Probes, Eugene, OR, USA), washed three times with PBS and visualized under a Nikon Eclipse Ti fluorescent microscope (Japan). The HepG2 cell sections were further treated with a Histofine simple stain rat MAX-PO (MULTI) kit (Nichirei, Tokyo, Japan), stained brown with a DAB substrate kit (Nichirei, Tokyo, Japan) and blue with hematoxylin. Negative controls were stained with the omitted primary antibody. Omission of the primary antibody resulted in no staining of the cells.
GST activity assay
Total GST activity was assayed as previously described[15]. Total GST activity with aromatic substrates was determined by monitoring changes in absorbance with a microplate reader (Infinite M200, Tecan, Switzerland). A complete assay mixture without total GST was used as a control. HepG2 cells, HCC cells and matched nontumorous liver tissue cells were broken for 20 s at 4°C, respectively, in 1 mL 0.1 mol/L potassium phosphate buffer (pH 6.5) using a sonicator. Samples were collected into different tubes containing EDTA, centrifuged at 10 000 r/min for 1 h at 4°C, and the supernatant was stored at -20°C. GST activity was assayed with 5 μg protein in duplicate with 1 mmol/L 1-chloro-2, 4-dinitrobenzene and glutathione (Sigma, Louis, MO) and used without further purification in a total volume of 1 mL. Optical density of GST was measured within at least 3 min after incubation at 25°C for 15 min at a wavelength of 340 nm (ε = 9.6 mmol/L per cm). The activity of GST was expressed as a unity of nmol mg per min.
Real-time PCR for detection of glutathione-s-transferase M3 mRNA expression
Total RNA harvested from HepG2 cells was subjected to reverse transcription into cDNA using a Superscript kit (Life Technologies, Gaithersburg, MD) according to its manufacturer’s protocol. Thereafter, 2 μg of cDNA samples was used immediately in measurement of GSTM3 mRNA level by real-time PCR with iQ SYBR Green Supermix (Bio-Rad, Tokyo, Japan), forward primer (5'-GCTCCTGGAGTTCACGGATA-3'), and reverse primer (5'-GCTCCTGGAGTTCACGGATA-3') on a DNA engine Opticon 2 real-time PCR detection system (Bio-Rad, Tokyo, Japan). The thermal cycler parameters were as follows: 1 cycle at 95°C for 3 min, followed by 40 cycles at 95°C for 15 s, at 60°C for 30 s and at 72°C for 30 s[19]. A β-actin control was run simultaneously with the same reaction recipe listed in the instruction manual for the iQ SYBR Green Supermix (Bio-Rad, Tokyo, Japan). All data were normalized to β-actin mRNA levels to account for any variation in RNA concentrations between the samples obtained from three separate experiments.
Statistical analysis
The data are presented as mean ± SE. Statistical analyses were performed among the three groups by a one-way analysis of variance followed by Bonferroni’s test, and between two groups by the unpaired Student’s t-test. P < 0.05 was considered statistically significant.
RESULTS
Identification of differentially expressed genes in mouse HCC and matched nontumorous liver tissue samples
Differential display analysis is a powerful tool for the comparison of differential gene expressions between two or more mRNA populations[14]. Using this method, we compared the mRNA expression patterns of B6C3F1 mouse HCC and matched nontumorous liver tissue samples. Representative differentially displayed autoradiographs of “I and II” cDNAs are shown in Figure 1A. The interesting “I and II” cDNA fragments were successfully recovered from the dried DNA sequencing gel, re-amplified (Figure 1B), purified, and subcloned into the TA cloning site of pCRII-TOPO vector. Its nucleotide sequences were compared with those in GenBank (Figure 1C). Concurrently, the purified products of “I and II” fragments from 2% agarose gels were used as probes for reverse Northern blotting. The Northern blotting patterns of “I and II” fragments differentially expressed in mouse HCC cells are shown in Figure 2A. In comparison with a nucleotide sequence in GenBank, “I and II” nucleotide sequences were identified as the up-regulated gene coding products, such as glutathione-s-transferase M3 (GSTM3) and glutamine synthetase (GLNS) in radiation-induced B6C3 F1 mouse HCC cells.
Figure 1.
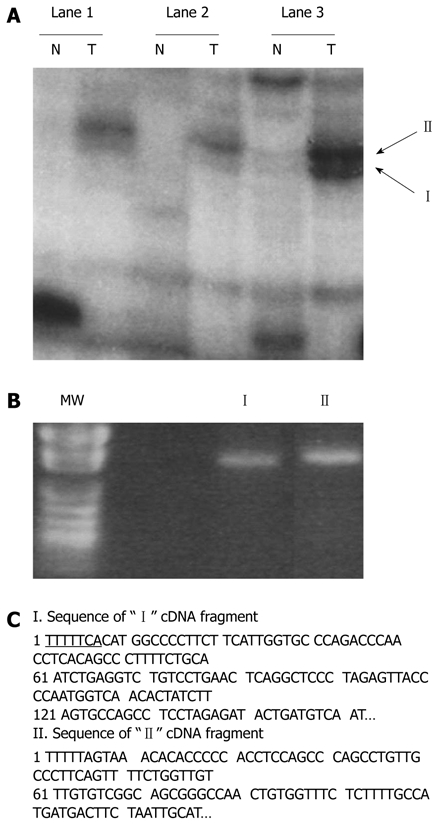
Identification of differentially displayed cDNA fragments from paired hepatocellular carcinoma cells (T) and nontumorous liver tissues (N). A: Differentially displayed PCR products (“I” and “II”) amplified with AP-10 primer and T12MA. Lanes 1-3 denote the three B6C3 F1 mouse samples, respectively; B: Recovered “I” and “II” from the dried DNA sequencing gel reamplified by PCR. MW lane shows the pUC118 DNA fragments cut by HapII (a restriction enzyme) as molecular weight markers; C: Nucleotide sequences of the bands shown in A. Flanking sequences of T12MA primers are underlined. The two insert-containing fragments were sequenced and identified as gene fragments of GLNS and GSTM3 in comparison with those of nucleotide in GenBank.
Figure 2.
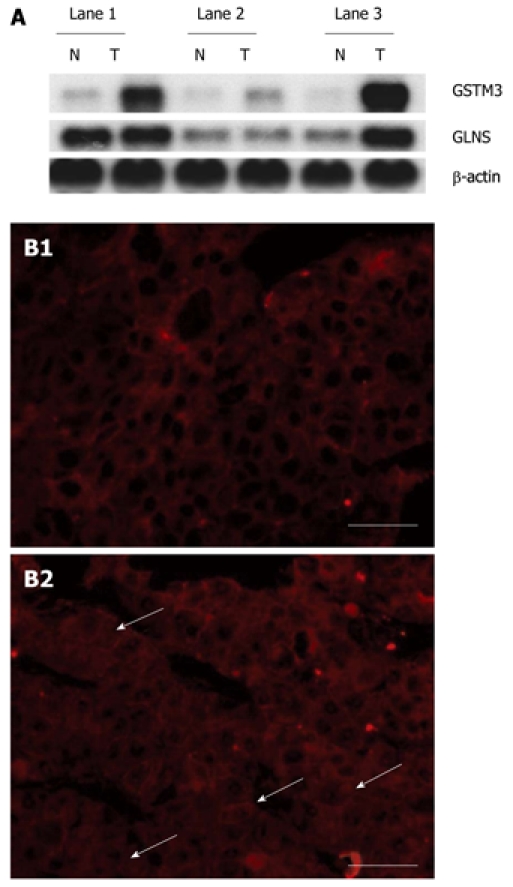
Levels of glutamine synthetase and glutathione-s-transferase M3 mRNA and expression of β-catenin. A: T denotes surgically resected B6C3F1 mouse hepatocellular carcinoma (HCC) tissue samples and N denotes matched nontumorous liver tissue samples; B: Representative immunofluorescence photomicrographs for β-catenin (red photomicrographs) in HCC tissue samples. B1 denotes negative β-catenin staining in nuclei of HCC cells, B2 denotes positive β-catenin staining in nuclei of HCC cells, and white arrows indicate β-catenin detected in nuclei of HCC cells. Bars: 50 μm. GLNS: Glutamine synthetase; GSTM3: Glutathione-s-transferase M3.
Dependence of mouse GSTM3 activity on β-catenin in B6C3F1 mouse HCC
It has been reported that nuclear translocation of β-catenin may represent an early event in liver carcinogenesis[20]. Therefore, we are interested in the relation between β-catenin and the discovered gene described above. Analysis of mRNA expression showed that the expression level of GSTM3 mRNA was significantly higher in cell nuclei of B6C3F1 mouse HCC cells with β-catenin accumulation than in those without β-catenin accumulation (Figure 2B), suggesting that β-catenin can increase the GSTM3 activity. To confirm whether β-catenin increases the GSTM3 activity, the gene near the upstream domain of individual mouse GSTM3 containing the β-catenin/Tcf-Lef consensus binding site sequence [5'-(A/T)(A/T) CAAAG-3'[21]] was detected by searching it in GenBank. On the other hand, whether the increased GSTM3 activity in mouse HCC cells is correlated with β-catenin accumulation in nuclei of HCC cells was also assayed (Figure 3), showing that the nearby upstream domain of mouse GSTM3 contains β-catenin/Tcf-Lef consensus binding site sequences [5'-(A/T)(A/T) CAAAG-3']. Because the GSTM3 activity level was rather variable in normal mouse tissue samples, it was difficult to estimate the β-catenin dependence on GSTM3 activity using the absolute GST activity level. Therefore, we analyzed the increased total GST activity in HCC tissue samples relative to that of normal tissue samples by the ratio of (T-N)/N, where T and N denote the total GST activity in HCC and normal tissue samples, respectively. On the other hand, to see the directly significant relation between total GST activity and β-catenin accumulation in nuclei of HCC cells, the average total GST activity level was also measured in B6C3F1 mouse HCC and matched nontumorous liver tissue samples with or without β-catenin accumulation (Table 2). It can be clearly seen from Table 2 that the total GST activity ratio was considerably higher in B6C3F1 mouse HCC tissue samples with β-catenin accumulation than in those without β-catenin accumulation in nuclei of HCC cells. The averaged GST activity ratio was also significantly higher in HCC tissue samples with β-catenin accumulation than in those without β-catenin accumulation (0.353 ± 0.117 vs 0.071 ± 0.064, P < 0.001), suggesting that the GST activity ratio is significantly different (Table 2).
Figure 3.

Identification of β-catenin/Tcf-Lef consensus binding sites with three β-catenin/Tcf-Lef consensus binding site sequences [5'-(A/T)(A/T) CAAAG-3'] located at the nearby upstream domain of mouse GSTM3 by searching GenBank.
Table 2.
Total glutathione-s-transferase activity in B6C3F1 mice hepatocellular carcinoma cells (T) and matched nontumorous (N) liver tissue samples
| Samples |
Total GST activity (nmol/mg per min) |
Values of GST activity ratio (T-N)/N | |
| T | N | ||
| (+) | 3122 ± 189 | 2447 ± 180 | 0.353 ± 0.117a |
| (n = 13) | (n = 13) | ||
| (-) | 2644 ± 199 | 2523 ± 205 | 0.071 ± 0.064 |
| (n = 11) | (n = 11) | ||
[(T-N)/N] indicates the glutathione-s-transferase (GST) activity values for the samples with and without β-catenin accumulation in hepatocellular carcinoma (HCC) cell nuclei. “(+) or (-)” denotes the samples from B6C3F1 mice with or without β-catenin accumulation in nuclei of HCC cells.
P < 0.001 vs negative β-catenin staining group.
Similarly increased GST activity and β-catenin accumulation in nuclei of HepG2 cells
To further elucidate the above findings, we mimicked the canonical Wnt pathway in cultured HepG2 cells using TWS119 (an inhibitor of GSK-3), which led to phosphorylation, nuclear translocation, and abnormal accumulation of β-catenin in nuclei of HepG2 cells. We treated the cultured HepG2 cells with or without 1 μmol/L TWS119 at 37°C for 24 h, measured the total GST activity in these cells, and then analyzed the relation[15] between GST-GSH protein complex concentration and time (at least 15 min) in “control” and “TWS119” groups using a microplate reader at a wavelength of 340 nm. Concurrently, the β-catenin accumulation in HepG2 cells was detected with immunocytochemical staining with a rabbit polyclonal antibody against β-catenin as previously described[17]. Thereafter, we comprised the linear function of total GST-GSH protein complex concentration between the two groups. It can be clearly seen from Figure 4A that the total GST activity ratio was considerably higher in the “TWS119” group with abnormal nuclear accumulation of β-catenin in HepG2 cells than in the “control” group with low β-catenin nuclear accumulation. Additionally, both the GSTM3 mRNA expression level and the GST activity were significantly higher in HepG2 cells and controls after treatment with 1 μmol/L TWS119 for 24 h (100% ± 5% vs 137% ± 7%, P < 0.05, Figure 4B), supporting that β-catenin nuclear accumulation in nuclei of HCC cells up-regulates the GSTM3 mRNA expression and the GSTM3 activity.
Figure 4.

Total glutathione-s-transferase activity (A) and glutathione-s-transferase M3 mRNA expression (B) in HepG2 cells. aP < 0.05, bP < 0.01 vs TWS119 at 24 h by one-way analysis of variance followed by Bonferroni’s test.
DISCUSSION
The experimental results of this study demonstrate that mRNAs originally identified from gene expression profiles are differentially expressed in mouse HCC cells. The expression levels of GSTM3 and GLNS mRNAs were higher in mouse HCC tissue samples than in matched nontumorous liver tissue samples. However, the detailed genetic mechanism underlying HCC remains unknown.
GSTM3 is a GST Mu-class subunit. Little is known about the role of GSTM3 in metabolism of harmful agents, except for its overlapping substrate specificity to GSTM1[22]. As one of the primary phase II detoxification enzymes, GST can divided into four classes, namely Alpha, Mu, Pi and Theta[23], which protect against the oxidative stress of their products[24]. GST is a potentially important enzyme that regulates the susceptibility to cancer because of its ability to metabolize reactive electrophilic intermediates to usually less reactive and more water soluble glutathione conjugates[25]. It was reported that the GST activity, as an indicator of resistance to chemotherapy, is high in human cancer, because GST increases the formation of drug glutathione (GSH) conjugates[26]. In this study, the total GST activity was assayed to understand why the GSTM3 mRNA expressions are up-regulated in HCC cells. Based upon these observations, the results of this study showing the up-regulated expression of GSTM3 mRNA in B6C3F1 mouse HCC cells suggest that the increased GSTM3 mRNA expression is a significant phenomenon in cellular detoxification, namely enhancing the metastatic potential in HCC cells.
It was reported that β-catenin plays an important role in cell-cell adhesion[27] and in Wnt signaling pathway[28,29]. β-catenin can enter the nuclei of HCC cells by binding the Tcf-Lef family of DNA binding proteins, and regulate the transcription of target genes (for example, c-myc, gastrin, cyclin D1 and PPAR are identified as target genes of the β-catenin/Tcf-Lef complex)[30-33]. In the present study, the GSTM3 mRNA level was higher in B6C3F1 mouse HCC cells with β-catenin accumulation than in those without β-catenin accumulation. To our knowledge, no similar observation has been reported. The total GST activity was much higher in B6C3F1 mouse HCC cells with β-catenin accumulation than in normal tissue samples without β-catenin accumulation (Table 2). The averaged GST activity ratio was significantly higher in HCC cells with β-catenin accumulation than in those without β-catenin accumulation (0.747 ± 0.360 vs 0.071 ± 0.213, P < 0.001), suggesting that the GST activity ratio is significantly different (Table 2). Furthermore, by searching the GenBank, we found that the upstream region of the GSTM3 gene contained three β-catenin/Tcf-Lef consensus binding site sequences in mouse GST polymorphisms. The canonical Wnt pathway in cultured HepG2 cells was further mimicked using TWS119, a GSK-3β inhibitor, which caused abnormal β-catenin accumulation. As a result, TWS119-induced β-catenin accumulation enhanced the GST activity and the GSTM3 mRNA expression in HepG2 cells, suggesting that β-catenin accumulation in nuclei of HCC cells can increase the activity of mouse GSTM3, one of the enzymes responsible for the metabolism of a variety of xenobiotics and carcinogens, and that mouse GSTM3 may be a novel downstream target gene of the β-catenin/Tcf-Lef complex in mouse HCC.
It was reported that GLNS can catalyze the synthesis of glutamine[34], a major energy source of cells (an important ATP source), and is a precursor for the synthesis of nucleotides and numerous amino acids, and up-regulated in a subset of human HCC[35]. In this study, at 3 wk after tumor implantation, the glutamine synthetase activity in rats increased by 34%, which is consistent with the reported findings[36]. However, further study is needed to observe the possible pharmacological action (s) of β-catenin in the up-regulated expression of GLNS mRNA.
In conclusion, GSTM3 and GLNS genes are differentially expressed in mouse HCC cells. The expression level of GSTM3 mRNA and total GST activity are higher in B6C3F1 mouse HCC cells with β-catenin accumulation than in those without β-catenin accumulation, indicating that GSTM3 may be a novel target gene for the β-catenin/Tcf-Lef complex in mouse HCC.
COMMENTS
Background
Hepatocellular carcinoma (HCC) is a primary cancer of the liver. However, the genetic events responsible for HCC initiation and progression are not clear. Since approximately 20% of HCC display β-catenin aberrant activation, Wnt/β-catenin signaling pathways may be involved in HCC occurrence.
Research frontiers
Recent data show that β-catenin may be an initiating or contributory factor for HCC. In this study, the authors demonstrated that glutathione-s-transferase M3 (GSTM3) might be a novel target gene of the β-catenin/Tcf-Lef complex in mouse HCC.
Innovations and breakthroughs
The authors identified two up-regulated genes, glutamine synthetase (GLNS) and GSTM3, in nuclei of radiation-induced mouse HCC cells with β-catenin accumulation. Three β-catenin/Tcf-Lef consensus binding site sequences were observed in mouse glutathione-s-transferase (GST) polymorphisms. GST activity and GSTM3 mRNA levels were induced in cultured HepG2 cells by TWS119 (an inhibitor of GSK-3β). To our knowledge, no similar observation has been reported.
Applications
By demonstrating that GSTM3 may be a novel target gene of the β-catenin/Tcf-Lef complex in mouse HCC, this study may represent a future strategy for therapeutic intervention in patients with HCC.
Terminology
β-catenin plays an important role in cell-cell adhesion and in Wnt signaling pathway, can enter nuclei by binding to the Tcf-Lef family of DNA binding proteins and regulate the transcription of target genes.
Peer review
This paper reports the results of investigations on some differentially over-expressed genes associated with β-catenin accumulation in nuclei of HCC cells, showing that GSTM3 may be a novel target gene of the β-catenin/Tcf-Lef complex in mouse HCC using mRNA differential display, Northern blot analysis, immunostaining and RT-PCR techniques, respectively. It is worthy of publication.
Footnotes
Supported by National Natural Science Foundation of China, 81070887, Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry to Tang HB, and Grants from South-Central University for Nationalities, No. XTZ10001, No. XTZ09001, and No. YZZ09007, China
Peer reviewer: Robert Anthony Fisher, MD, HM Lee Professor of Surgery (tenured), Department of Surgery, Division of Transplant Surgery, Medical College of Virginia Hospitals, Virginia Commonwealth University, MCV Campus, PO Box 980057, Richmond, VA 23298-0057, United States
S- Editor Sun H L- Editor Wang XL E- Editor Ma WH
References
- 1.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 2.Takada S, Koike K. Activated N-ras gene was found in human hepatoma tissue but only in a small fraction of the tumor cells. Oncogene. 1989;4:189–193. [PubMed] [Google Scholar]
- 3.Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM. Cancer gene discovery in hepatocellular carcinoma. J Hepatol. 2010;52:921–929. doi: 10.1016/j.jhep.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calvisi DF, Thorgeirsson SS. Molecular mechanisms of hepatocarcinogenesis in transgenic mouse models of liver cancer. Toxicol Pathol. 2005;33:181–184. doi: 10.1080/01926230590522095. [DOI] [PubMed] [Google Scholar]
- 5.Teufel A, Staib F, Kanzler S, Weinmann A, Schulze-Bergkamen H, Galle PR. Genetics of hepatocellular carcinoma. World J Gastroenterol. 2007;13:2271–2282. doi: 10.3748/wjg.v13.i16.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papa S, Bubici C, Zazzeroni F, Franzoso G. Mechanisms of liver disease: cross-talk between the NF-kappaB and JNK pathways. Biol Chem. 2009;390:965–976. doi: 10.1515/BC.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hosono S, Lee CS, Chou MJ, Yang CS, Shih CH. Molecular analysis of the p53 alleles in primary hepatocellular carcinomas and cell lines. Oncogene. 1991;6:237–243. [PubMed] [Google Scholar]
- 8.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taniguchi K, Roberts LR, Aderca IN, Dong X, Qian C, Murphy LM, Nagorney DM, Burgart LJ, Roche PC, Smith DI, et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene. 2002;21:4863–4871. doi: 10.1038/sj.onc.1205591. [DOI] [PubMed] [Google Scholar]
- 10.Calvisi DF, Factor VM, Loi R, Thorgeirsson SS. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: relationship to phenotype and tumor grade. Cancer Res. 2001;61:2085–2091. [PubMed] [Google Scholar]
- 11.Thompson MD, Monga SP. WNT/beta-catenin signaling in liver health and disease. Hepatology. 2007;45:1298–1305. doi: 10.1002/hep.21651. [DOI] [PubMed] [Google Scholar]
- 12.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 13.Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, Mills AS, Waxman S, Fisher RA, Llovet JM, et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J Clin Invest. 2009;119:465–477. doi: 10.1172/JCI36460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang P, Pardee AB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 15.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 16.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 17.Tang HB, Li YS, Arihiro K, Nakata Y. Activation of the neurokinin-1 receptor by substance P triggers the release of substance P from cultured adult rat dorsal root ganglion neurons. Mol Pain. 2007;3:42. doi: 10.1186/1744-8069-3-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang HB, Li YS, Miyano K, Nakata Y. Phosphorylation of TRPV1 by neurokinin-1 receptor agonist exaggerates the capsaicin-mediated substance P release from cultured rat dorsal root ganglion neurons. Neuropharmacology. 2008;55:1405–1411. doi: 10.1016/j.neuropharm.2008.08.037. [DOI] [PubMed] [Google Scholar]
- 19.Tang HB, Shiba E, Li YS, Morioka N, Zheng TX, Ogata N, Nakata Y. Involvement of voltage-gated sodium channel Na(v)1.8 in the regulation of the release and synthesis of substance P in adult mouse dorsal root ganglion neurons. J Pharmacol Sci. 2008;108:190–197. doi: 10.1254/jphs.08163fp. [DOI] [PubMed] [Google Scholar]
- 20.Calvisi DF, Factor VM, Loi R, Thorgeirsson SS. Activation of beta-catenin during hepatocarcinogenesis in transgenic mouse models: relationship to phenotype and tumor grade. Cancer Res. 2001;61:2085–2091. [PubMed] [Google Scholar]
- 21.Kim WB, Lewis CJ, McCall KD, Malgor R, Kohn AD, Moon RT, Kohn LD. Overexpression of Wnt-1 in thyrocytes enhances cellular growth but suppresses transcription of the thyroperoxidase gene via different signaling mechanisms. J Endocrinol. 2007;193:93–106. doi: 10.1677/JOE-06-0025. [DOI] [PubMed] [Google Scholar]
- 22.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 23.Mannervik B, Awasthi YC, Board PG, Hayes JD, Di Ilio C, Ketterer B, Listowsky I, Morgenstern R, Muramatsu M, Pearson WR. Nomenclature for human glutathione transferases. Biochem J. 1992;282(Pt 1):305–306. doi: 10.1042/bj2820305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao AV, Shaha C. Role of glutathione S-transferases in oxidative stress-induced male germ cell apoptosis. Free Radic Biol Med. 2000;29:1015–1027. doi: 10.1016/s0891-5849(00)00408-1. [DOI] [PubMed] [Google Scholar]
- 25.Chen Q, Luo G, Li B, Samaranayake LP. Expression of p16 and CDK4 in oral premalignant lesions and oral squamous cell carcinomas: a semi-quantitative immunohistochemical study. J Oral Pathol Med. 1999;28:158–164. doi: 10.1111/j.1600-0714.1999.tb02016.x. [DOI] [PubMed] [Google Scholar]
- 26.Abou Ghalia AH, Fouad IM. Glutathione and its metabolizing enzymes in patients with different benign and malignant diseases. Clin Biochem. 2000;33:657–662. doi: 10.1016/s0009-9120(00)00181-8. [DOI] [PubMed] [Google Scholar]
- 27.Jou TS, Stewart DB, Stappert J, Nelson WJ, Marrs JA. Genetic and biochemical dissection of protein linkages in the cadherin-catenin complex. Proc Natl Acad Sci USA. 1995;92:5067–5071. doi: 10.1073/pnas.92.11.5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larabell CA, Torres M, Rowning BA, Yost C, Miller JR, Wu M, Kimelman D, Moon RT. Establishment of the dorso-ventral axis in Xenopus embryos is presaged by early asymmetries in beta-catenin that are modulated by the Wnt signaling pathway. J Cell Biol. 1997;136:1123–1136. doi: 10.1083/jcb.136.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 30.Kolligs FT, Bommer G, Göke B. Wnt/beta-catenin/tcf signaling: a critical pathway in gastrointestinal tumorigenesis. Digestion. 2002;66:131–144. doi: 10.1159/000066755. [DOI] [PubMed] [Google Scholar]
- 31.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 32.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koh TJ, Bulitta CJ, Fleming JV, Dockray GJ, Varro A, Wang TC. Gastrin is a target of the beta-catenin/TCF-4 growth-signaling pathway in a model of intestinal polyposis. J Clin Invest. 2000;106:533–539. doi: 10.1172/JCI9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisenberg D, Gill HS, Pfluegl GM, Rotstein SH. Structure-function relationships of glutamine synthetases. Biochim Biophys Acta. 2000;1477:122–145. doi: 10.1016/s0167-4838(99)00270-8. [DOI] [PubMed] [Google Scholar]
- 35.Osada T, Nagashima I, Tsuno NH, Kitayama J, Nagawa H. Prognostic significance of glutamine synthetase expression in unifocal advanced hepatocellular carcinoma. J Hepatol. 2000;33:247–253. doi: 10.1016/s0168-8278(00)80365-7. [DOI] [PubMed] [Google Scholar]
- 36.Chen MK, Salloum RM, Austgen TR, Bland JB, Bland KI, Copeland EM 3rd, Souba WW. Tumor regulation of hepatic glutamine metabolism. JPEN J Parenter Enteral Nutr. 1991;15:159–164. doi: 10.1177/0148607191015002159. [DOI] [PubMed] [Google Scholar]