

Abstract
PMP1, a regulatory subunit of the yeast plasma membrane H+-ATPase, is a single transmembrane helix protein. Its cytoplasmic C-terminus possesses several positively charged residues and interacts with phosphatidylserine lipids as shown through both 1H- and 2H-NMR experiments. We used all-atom molecular dynamics simulations to obtain atomic-scale data on the effects of membrane interface lipid composition on PMP1 structure and tilt. PMP1 was embedded in two hydrated bilayers, differing in the composition of the interfacial region. The neutral bilayer is composed of POPC (1-palmitoyl-2-oleoyl-3-glycero-phosphatidylcholine) lipids and the negatively charged bilayer is composed of POPC and anionic POPS (1-palmitoyl-2-oleoyl-3-glycero-phosphatidylserine) lipids. Our results were consistent with NMR data obtained previously, such as a lipid sn-2 chain lying on the W28 aromatic ring and in the groove formed on one side of the PMP1 helix. In pure POPC, the transmembrane helix is two residues longer than the initial structure and the helix tilt remains constant at 6 ± 3°. By contrast, in mixed POPC-POPS, the initial helical structure of PMP1 is stable throughout the simulation time even though the C-terminal residues interact strongly with POPS headgroups, leading to a significant increase of the helix tilt within the membrane to 20 ± 5°.
Introduction
Membrane proteins are involved in a wide variety of key cellular processes, such as cell signaling, transport, energy transduction, and cell adhesion. They are embedded within a very complex environment essentially composed of a large variety of proteins and lipids. Lipids differ in headgroups, number of acyl chains, acyl-chain lengths, and bonds. Membranes are composed of a hydrophobic core (acyl-chain region) and a polar interfacial region (headgroup region) due to the arrangement of lipids. Lipids can influence the structure and the function of membrane proteins (1–3) partly through interactions that take place between proteins and lipids especially at the interfacial region of the membrane (4,5).
To understand membrane protein function, it is essential to decipher the interdependency of membrane protein structure and function on lipid composition within the context of the lipid bilayer. Therefore, structural data are needed, but experimental data are difficult to obtain and are sparse. Few high-resolution membrane protein structures, including their lipid components, are available (6–9). The analysis of these structures can provide information about how a membrane protein is likely to interact with lipids in a biological membrane. In addition, model peptides have been extensively used to understand membrane structure and function (10). Data obtained with these models mainly concern the hydrophobic mismatch, the location of tryptophan, and basic residues within the membrane. From the point of view of lipids, some examples describe aliphatic chains tightly bound to the surface of the membrane protein and polar headgroups in specific interactions with the membrane protein within the membrane interface (9).
In this context, molecular dynamics simulation has shown its capacity for studying proteins embedded in bilayers and for providing atomic-scale information (11–13). Various studies illustrate how proteins and lipids interact within the membrane (14–18). Nevertheless, very few studies have investigated the effects of the composition of the interfacial region on transmembrane protein structure and function, at the atomic scale (14,19). Single helix transmembrane proteins can be used as membrane protein models to gain insight into their behavior within the interfacial region. Thus, we studied a single spanning yeast plasma membrane protein, PMP1, embedded within a bilayer. PMP1 copurifies with the major H+-ATPase and enhances its H+-ATPase activity (20,21). PMP1 belongs to a class of single transmembrane domain proteins regulating the activity of membrane cation-transporting P-type ATPases. Phospholamban (22) and sarcolipin (23) are some examples of this class of proteins. The 38-amino-acid sequence of PMP1 is: L-P-G-G-V5-I-L-V-F-I10-L-V-G-L-A15-C-I-A-I-I20-A-T-I-I-Y25-R-K-W-Q-A30-R-Q-R-G-L35-Q-R-F. This sequence comprises two distinct regions: a hydrophobic sequence (from L1 up to I24) followed by a highly positively charged sequence (from Y25 up to F38) located on the cytoplasmic side of the membrane according to the positive-inside rule (24). Structural data on PMP1 have been previously obtained using solution 1H-NMR experiments performed on synthetic fragments of PMP1 in the presence of membrane mimics, i.e., DPC micelles (25,26). Cysteine residue 16 was replaced by a serine to avoid oligomerization effects. Several fragments of different lengths were synthesized (A18–F38, G13–F38, and the longest one F9–F38).
All fragments included the charged C-terminal segment and part of the hydrophobic region. NMR data have shown that PMP1 forms a single transmembrane helix (from the N-terminus up to R33) followed by a five-residue loop, which folds back toward the micelle interior. The conformation of the C-terminal segment results in a specific interfacial distribution of the five basic side chains (see the working model in Beswick et al. (25)). Lipid-PMP1 interactions were then investigated through 2H-NMR experiments performed in pure POPC (1-palmitoyl-2-oleoyl-3-glycero-phosphatidylcholine) bilayers and in mixed POPC-POPS bilayers (27). POPS (1-palmitoyl-2-oleoyl-3-glycero-phosphatidylserine) lipids were added due to the high phosphatidylserine (PS) lipid content (∼34%) of yeast plasma membranes (28). Moreover, POPS lipids represent 64% of the PS lipids present in yeast plasma membranes (29). Experimental results led to the conclusion that the PMP1 fragment specifically segregates eight anionic phospholipids (POPS) when inserted in mixed POPC/POPS bilayers.
However, due to the dynamic properties of the studied systems, we could not obtain, whatever the tested experimental condition, a description at atomic level of the C-terminal structure and of the interactions involving PMP1 and POPC or POPS lipids at the membrane interface. Moreover, neither NMR nor x-ray diffraction could be expected to provide atomic-level resolution for PMP1-lipid interactions within a bilayer. Thus, the only technique remaining is all-atom molecular dynamics simulation. We undertook two molecular dynamics simulations of PMP1 inserted in two different fully hydrated bilayers: a neutral bilayer composed of POPC lipids (named “pure POPC”) and a negatively charged bilayer composed of POPC and POPS lipids (named “mixed POPC-POPS”). The mixed POPC-POPS bilayer was composed of POPC lipids and a shell of eight POPS lipids placed around PMP1 C-terminal segment according to our previous 2H-NMR data (see Materials and Methods in the Supporting Material for more details). We then compared the results obtained from these simulations in both bilayers.
Materials and Methods
See the Supporting Material.
Results
PMP1 inserted in the pure POPC membrane
To investigate the conformational stability of PMP1 during simulation, we measured the conformational drift by calculating the Cα root mean-square deviations (RMSDs) against the initial NMR structure as a function of time (data not shown). After an initial increase, the Cα RMSDs stabilized at ∼3 Å for PMP1 inserted in a pure POPC membrane. To assess how much the helix (from V5 up to R33) and the loop (from G34 up to F38) contribute to these values, we computed Cα RMSD values for the helix and the loop separately (Fig. 1). Helix Cα RMSDs were within 1.6 ± 0.2 Å of the initial structure, indicating a very stable helix throughout simulation. For the loop in pure POPC, Cα RMSDs markedly increase during the first 4 ns and then remain relatively constant at 4.8 ± 0.3 Å.
Figure 1.
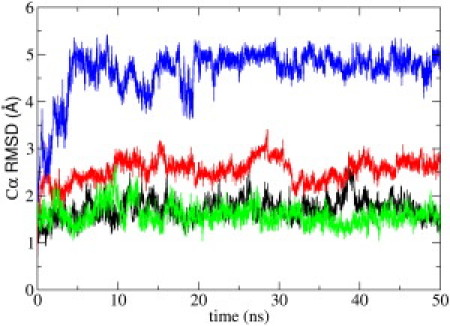
Cα atom root mean-square deviations (RMSDs) from initial NMR structure as a function of simulation time for the PMP1 helix (green) and loop (blue) inserted in pure POPC and for the PMP1 helix (black) and loop (red) in mixed POPC-POPS.
The change in the PMP1 helix tilt, with respect to the normal to the membrane plane as a function of simulation time, is shown in Fig. 2. The helix was first inserted with an angle value of 7°. In pure POPC, the average tilt value is 6 ± 3°. Despite the significant fluctuations affecting the PMP1 helix tilt, we conclude that, in pure POPC, no further tilt was observed.
Figure 2.
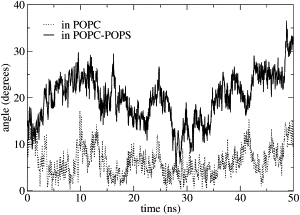
PMP1 helix tilted orientation with respect to the normal to the membrane plane as a function of simulation time. PMP1 inserted in pure POPC (dotted line) and in mixed POPC-POPS (solid line) membranes.
Comparing PMP1 secondary structure with the initial structure throughout the simulation time in pure POPC, shows significant differences. The helix, which initially extended from V5 up to R33, propagates up to L35 after 4 ns of simulation, as shown by the number of COi-NHi+4 contacts involving A30–G34 and R31–L35 (Fig. 3, F and G). Two additional hydrogen bonds are thus formed and this is allowed by the temporary loss of the Q29 (CO)-R33 (NH) contact (Fig. 3 E). Finally, only the last three residues of the sequence were not in a helical conformation. Moreover, analysis of the COi-NHi+3, COi-NHi+4, and COi-NHi+5 contacts demonstrated that the helix is predominantly an α-helix with very rare fluctuations, giving rise to transient local 310 or π-helix turns (data not shown).
Figure 3.
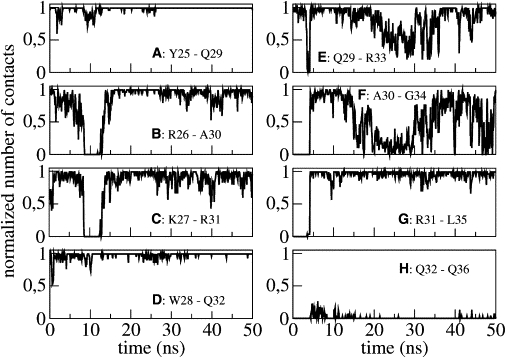
Normalized number of contacts between residue i backbone CO group and residue i+4 backbone NH group (COi-NHi+4) in pure POPC as a function of simulation time. Each panel (from A to H) represents a pair of residues.
Structure of the C-terminal segment within the interface in the pure POPC membrane
An important event takes place after 8 ns of simulation. The PMP1 helix transiently flexes for 4 ns when the R26–A30 and K27–R31 contacts are lost (Fig. 3, B and C) and this break in the helix favors a long-range C-terminal interaction (i.e., a hydrogen bond is formed between the W28 indole NH and the R37 CO backbone; see Fig. 4 B). This interaction is mediated by residue Q32 (Fig. 4 A). Indeed, during the first 8 ns of simulation, the Q32 side chain (NH2) is in contact with the W28 indole NH. Such a contact has been detected by 1H-NMR (26) and could be due to the attractive force between the fractional negative charge of the W28 ring and the fractional positive charge of the Q32 NH2 group. At 8 ns, the helix breaks and the Q32 CO backbone comes into contact with the R37 side chain. As a consequence, W28 and R37 are closer, allowing the W28 indole NH to interact with the R37 CO backbone, by forming a hydrogen bond. When the helix reforms, this interaction is conserved throughout simulation. Another similar event takes place between 15 and 35 ns. Q29–R33 and A30–G34 number of contacts decreases (Fig. 3, E and F), leading to a slight flexing of the helix. This deformation allows the interaction of R31 side chain with R37 CO backbone (Fig. 4 C), which is stable throughout simulation once the helix straightens. These intramolecular interactions lead to a very compact and stable C-terminal segment that is largely accessible to solvent (Fig. 5). Q32 is then found buried within this compact C-terminal segment.
Figure 4.
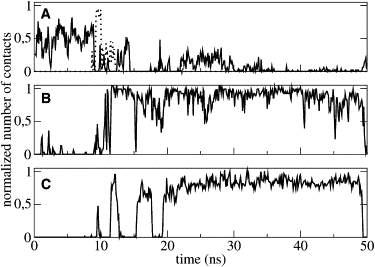
Normalized number of contacts in pure POPC as a function of simulation time. (A) W28 side chain (NH) to Q32 side chain (NH2) (solid line) and Q32 backbone (CO) to R37 side chain (dotted line). (B) W28 side chain (NH) to R37 backbone (CO). (C) R31 side chain to R37 backbone (CO).
Figure 5.
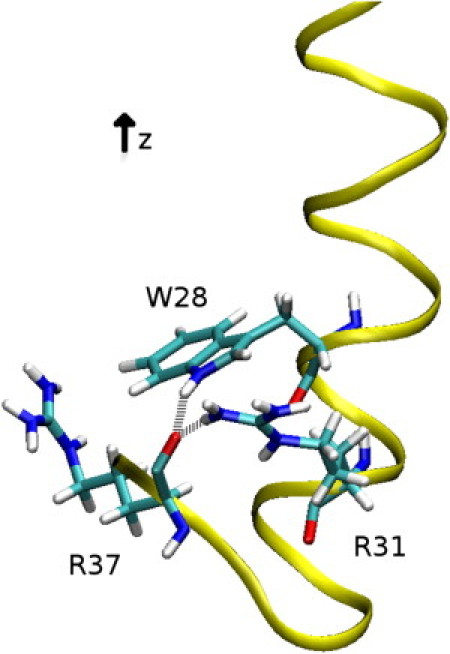
Snapshot from the MD simulation in pure POPC showing PMP1 C-terminal segment. (Yellow) PMP1 ribbon. (Sticks) W28, R31, and R37 residues. (White) Hydrogens; (cyan) carbons; (red) oxygens; (blue) nitrogens. (Gray dotted lines) Hydrogen bonds between W28 indole NH and R37 CO backbone and between R31 side chain (NH2) and R37 CO backbone.
Interaction between the C-terminal segment and the lipid headgroups in the pure POPC membrane
The C-terminal sequence of PMP1—i.e., from Y25 up to F38—is rich in positively charged amino acids (four arginine residues and one lysine residue), polar (three glutamine residues), and aromatic residues (Y25, W28, and F38). The number of contacts between the various side chains and the lipid headgroups was monitored during simulation. All arginine and lysine residues, except for R33, appeared to strongly interact with the phosphate group of POPC lipids. Each residue makes contact with two lipids so that the eight lipids remain in close contact with the C-terminal segment during the whole simulation. As a matter of fact, R33 is largely accessible to solvent. Glutamine and aromatic residues are not in contact with lipids and form intramolecular contacts. The rings of the three aromatic residues were found parallel to the membrane plane and inserted in the region of the lipid glycerol groups. Interestingly, the sn-2 aliphatic chain for one of the POPC lipids, whose phosphate group remains in close contact with the R37 side chain throughout the simulation, is lying on the W28 aromatic ring and in the groove formed on one side of the helix by a series of short side-chain residues (A15, A18, A21, and T22) (Fig. 6). The residence time of the aliphatic chain in the groove is 19 ns.
Figure 6.
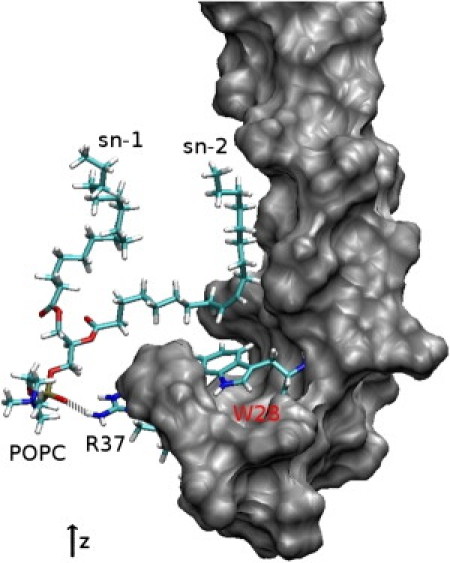
Snapshot from the MD simulation in pure POPC showing the POPC sn-2 aliphatic chain lying on the W28 aromatic ring and in the groove formed on one side of the helix. PMP1 is shown in gray surface representation. The POPC molecule, W28 and R37 side chains are in stick representation. (White) Hydrogens; (cyan) carbons; (red) oxygens; (blue) nitrogens; (yellow) phosphorus. (Gray dotted line) Hydrogen bond between POPC headgroup (PO4−) and R37 side chain (NH2).
PMP1 inserted in the mixed POPC-POPS membrane
Cα RMSDs were calculated against the initial NMR structure as a function of time, to investigate PMP1 stability during simulation (data not shown). After an initial increase, the Cα RMSDs stabilize at 2.5 Å for mixed POPC-POPS. We computed Cα RMSD values for the helix and the loop separately (Fig. 1). Helix Cα RMSDs were within 1.7 ± 0.2 Å of the initial structure, indicating a very stable helix throughout simulation. However, the loop was more flexible, as indicated by Cα RMSDs that slightly increased during simulation, reaching a plateau of 2.6 ± 0.2 Å.
We also studied PMP1 helix-tilted orientations with respect to the normal to the membrane plane as a function of simulation time (Fig. 2). At the start of the simulation, the helix was identical to that in pure POPC, i.e., the initial angle value was 7°. In mixed POPC-POPS, the helix tilt significantly increased to a mean value at ∼20 ± 5°. The tilt distribution was fairly broad, indicating rather large fluctuations. Moreover, the tilt could return to smaller values on a timescale of approximately tens of nanoseconds. The PMP1 secondary structure in mixed POPC-POPS showed no variation compared with the initial structure throughout the simulation time except for the last residue R33. During the first 20 ns, the helix is well defined, and it extends from V5 up to R33, as shown by the number of contacts between the backbone COi and backbone NHi+4 (Fig. 7). After 20 ns, the Q29–R33 contact is lost and the helix is therefore followed by a six-residue C-terminal segment.
Figure 7.
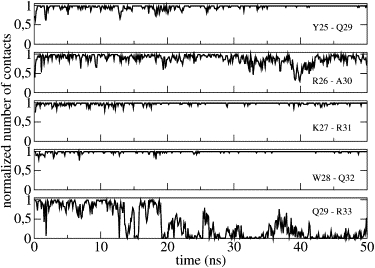
Normalized number of contacts between residue i backbone CO group and residue i+4 backbone NH group (COi-NHi+4) in mixed POPC-POPS as a function of simulation time. Each panel represents a pair of residues.
Structure of the C-terminal segment within the interface in the mixed POPC-POPS membrane
The detailed structural analysis of the C-terminal segment revealed the presence of several intramolecular interactions. A hydrogen bond is formed between L35 backbone (NH) and Q32 backbone (CO), leading to the formation of a β-turn. This takes place immediately and is stable throughout simulation, contributing to the stabilization of the loop (Fig. 8 A). A long-range interaction is mediated by Q32, already implicated in a long-range interaction as described in the case of PMP1 inserted into the pure POPC bilayer. Fig. 8 B shows the number of contacts between the Q32 side chain (NH2) and the W28 side chain (NH). Although this number of contact is small, it is relatively constant because of the presence of a water molecule hydrogen-bonded between both side chains. Moreover, the C-terminal loop is stabilized by the interactions between the Q32 side chain (NH2) and the R37 backbone (CO) (Fig. 8 C and Fig. 9).
Figure 8.
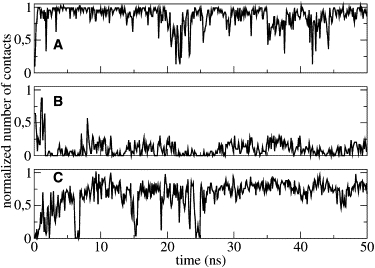
Normalized number of contacts in mixed POPC-POPS as a function of simulation time. (A) Q32 backbone (CO) to L35 backbone (NH). (B) W28 side chain (NH) to Q32 side chain (NH2). (C) Q32 side chain (NH2) to R37 backbone (CO).
Figure 9.
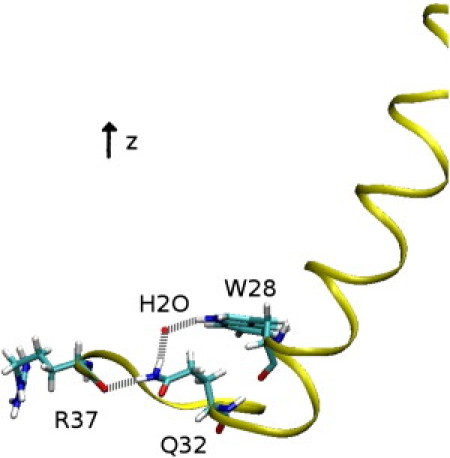
Snapshot from the MD simulation in mixed POPC-POPS showing PMP1 C-terminal segment. PMP1 ribbon is in yellow. W28, Q32, and R37 residues are represented in sticks. (Red sphere) Oxygen of a water molecule. (White) Hydrogens; (cyan) carbons; (red) oxygens; (blue) nitrogens. (Gray dotted lines) Hydrogen bonds between W28 indole NH and the oxygen atom of a water molecule, between the same oxygen atom of the water molecule and Q32 side chain (NH2) and between Q32 side chain (NH2) and R37 backbone (CO).
Interaction between the C-terminal segment and the lipid headgroups in the mixed POPC-POPS membrane
As already mentioned, the C-terminal sequence of PMP1, i.e., from Y25 up to F38, is rich in positively charged (four arginine residues and one lysine residue), polar (three glutamine residues), and aromatic residues (Y25, W28, and F38). The number of contacts between the various residue side chains and the lipid headgroups were monitored during the simulation time. All the arginine and lysine residues, except for R33, appeared to strongly interact with six POPS lipids and one POPC lipid. These interactions involved the phosphate or carboxylate groups of POPS. Residue R33 is largely accessible to solvent. Except for Q36 and F38, all glutamine and aromatic residues form intramolecular contacts and are not involved in contacts with lipids. Q36 and F38 side chains strongly interact with POPS lipids through hydrogen bonds. The Q36 side chain is in contact with the carbonyl group of a POPS aliphatic chain and the F38 C-terminal COO− is in contact with the POPS NH3+ group.
Finally, there were nine lipids (seven POPS and two POPC) in contact with the PMP1 C-terminal segment throughout the simulation time. The aromatic rings of Y25 and W28 were parallel to the membrane plane and inserted in the region of the lipid glycerol groups, whereas the F38 aromatic ring was inserted perpendicular to the membrane plane. Moreover, the POPS molecule, which is not in close contact with the C-terminal segment of the protein during simulation, has its sn-2 aliphatic chain lying on the W28 aromatic ring and in the groove formed on one side of the helix by a series of short side-chain residues (A15, A18, A21, and T22) with a residence time of 42 ns. Lastly, the interactions between the C-terminal residues (Q36, R37, and F38) and POPS lipid headgroups constituted an extremely dense interaction network throughout simulation (Fig. 10).
Figure 10.
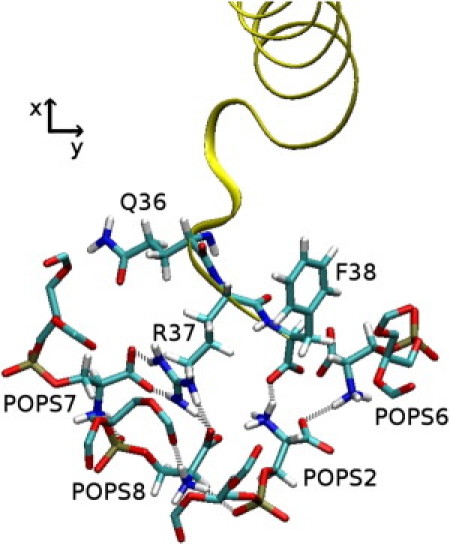
Snapshot from the MD simulation in mixed POPC-POPS showing the hydrogen-bond network between PMP1 C-terminal residues (Q36, R37, and F38) and POPS molecules. (Gray dotted lines) Hydrogen bonds. (Yellow) PMP1 ribbon. (Sticks) Q36, R37, F38 residues, and four POPS headgroups. (White) Hydrogens; (cyan) carbons; (red) oxygens; (blue) nitrogens; (yellow) phosphorus.
Discussion
Molecular dynamics simulation is a powerful method for exploring the atomic details of protein-lipid interactions within a bilayer. We used this technique to obtain detailed structural information on the role of the membrane interface regarding the structure and positioning of a small yeast plasma membrane protein, PMP1. For this purpose, two molecular dynamics simulations in pure POPC and in mixed POPC-POPS bilayers were compared.
Our findings obtained by simulation were in strong agreement with NMR and fluorescence data obtained in a DPC micellar environment. Indeed, in both simulations, one lipid chain, the sn-2 aliphatic chain and not the sn-1 chain, was found lying on the aromatic ring of the tryptophan and in the groove formed on one side of the helix by a series of short side-chain residues (A15, A18, A21, and T22), as previously demonstrated by NMR experiments (30) (Fig. 6). This is of particular interest, as it validates our protocols of simulation and the use of molecular dynamics simulations to obtain structural atomic information on complex biological systems. Moreover, a similar result has been recently described, suggesting that the interactions between lipid acyl chains and membrane proteins within grooves in the protein surface could be the guiding principle for lateral interactions between lipids and membrane proteins (9).
The role of residue Q32, which is involved in long-range intramolecular interactions in both simulations, should be underlined. In pure POPC, Q32 directly participates in the conformational switch that brings two residues (i.e., W28 and R37), far in sequence, closer together, thus allowing them to finally interact (Fig. 4). In mixed POPC-POPS, Q32 side chain (NH2) participates in two interactions: with W28 side chain through a bridging water molecule and with R37 side chain through a hydrogen bond (Figs. 8 and 9). Previous NMR experiments led to the conclusion that residues W28 and Q32 interact and have a concerted influence on the interfacial conformation and lipid-binding specificity of the C-terminal segment of PMP1 (26). Our dynamics clearly show how the W28 and Q32 side chains transiently interact, through an attractive force between the fractional negative charge of the W28 ring and the fractional positive charge of the Q32 NH2 group in pure POPC and through a water bridge in mixed POPC-POPS during simulation. Our dynamics simulation also shows the complementary role of W28 and Q32 in ensuring the appropriate folding of the C-terminal segment.
Other data extracted from both simulations concern the location of the aromatic residues. Aromatic rings are found parallel to the membrane plane and are inserted in the region of the lipid glycerol groups (except for F38 in the POPC-POPS membrane). This finding is consistent with previous experimental data obtained by fluorescence studies showing that the W28 aromatic ring of PMP1 is located below but close to the polar headgroup region (31). Aromatic rings anchor PMP1 within the membrane, as described previously (reviewed by Holt and Killian (10)). The different orientation of the F38 aromatic ring relative to the membrane plane in the POPC-POPS membrane, which is on average perpendicular to the membrane plane, is due to the lipid-protein interactions that take place between the F38 backbone (COO−) and the NH3+ group of a POPS molecule.
Comparison of the simulations of the membrane protein PMP1 in a pure POPC bilayer and in a mixed POPC-POPS bilayer reveals how strongly the lipid composition of the membrane interface influences PMP1 structure, the transmembrane helix tilt, and the interactions of PMP1 with lipids at the membrane interfacial region. Concerning the secondary structure, the helix region—defined by NMR data, i.e., from V5 up to R33—was stable throughout the simulation time, as indicated by weak Cα RMSD values in pure POPC and in mixed POPC-POPS membranes (1.6 and 1.7 Å, respectively) (Fig. 1). By contrast, Cα RMSD values were very different for the C-terminal segment. In pure POPC, RMSD values markedly increase during the first 4 ns and then stabilize at ∼4.8 Å.
These final RMSD values, which are also high, do not reflect the conformational flexibility of the loop, but are a consequence of the significant conformational changes that occur during the first 4 ns of the simulation. In pure POPC, the helix includes part of the initial loop and finally the last three residues of the sequence strongly interact with the helix. Indeed, the PMP1 C-terminal segment is very compact in pure POPC and largely exposed to the solvent. In mixed POPC-POPS, the loop is relatively stable, with a slight increase of RMSD values to 2.6 Å. The weak Cα RMSD values for the loop are consistent with the fact that during simulation, the loop is constrained by several intramolecular interactions (e.g., a turn, long-range interactions) and also strongly interacts with lipids.
The difference in the transmembrane helix tilt observed in pure POPC compared with mixed POPC-POPS membranes (Fig. 2) has to be related to the interactions that take place between the PMP1 C-terminal segment and the lipid headgroups. In POPC, interactions between PMP1 and the lipid headgroups essentially involved arginine and lysine residues (except R33). The structure adopted by PMP1 and its positioning within the membrane allow the long positively charged side chains of these residues to point toward the interfacial region, whereas their backbones are much more deeply inserted into the hydrophobic part of the membrane. This particular positioning of these positively charged side chains was previously described as the snorkel effect (32). Aromatic rings of PMP1 aromatic residues are located within the interfacial region of the membrane. Because no other constraints were imposed on the peptide, the tilt angle is constant and is on average equal to the initial tilt angle value.
This is consistent with results obtained by Kim and Im (33) showing that the helix tilting up to 10° is inherent due to the intrinsic entropic contribution arising from helix precession around the membrane normal. By contrast, in mixed POPC-POPS, PMP1 largely tilts within the membrane. Arginine (except R33) and lysine residues interact with the lipid headgroups, as in pure POPC. In addition, the C-terminal residues Q36 and F38 specifically interact with POPS headgroups, constraining the loop for localization within the membrane interface (Fig. 10). Moreover, the presence of several local structural motifs within the C-terminal segment (a turn, long-range interactions) (Figs. 8 and 9) imposes strong constraints on PMP1 conformation. To satisfy these constraints, the helix tilts within the membrane. This is in agreement with the data by Kim and Im (33) describing that the helix tilt >10° results from specific helix-lipid interactions.
In our case study, the tilt of PMP1 is unfavorable in terms of hydrophobic mismatch (i.e., PMP1 hydrophobic helix length is too short compared to the bilayer hydrophobic thickness, once tilted by 20°). A careful attention to the lipid response shows a lipid adjustment around PMP1 N-terminus with a local membrane thinning. Note that, in mixed POPC-POPS, the helix tilt allows the aromatic rings to be located within the interfacial region of the membrane. This also allows the arginine and lysine backbones to be close to the interfacial region. Thus, the snorkel effect is less pronounced in mixed POPC-POPS than in pure POPC membranes.
Conclusions
Comparison of both MD simulation results showed that the network of intra- and intermolecular interactions within the interfacial region of the membrane differs according to the nature of the lipid headgroups and that this network plays a crucial role in PMP1 structure and tilt within the membrane.
The pure POPC interface induces significant rearrangement in PMP1 structure, if compared with the initial structure derived from NMR data obtained for PMP1 in the presence of DPC micelles. The new structure adopted by PMP1 is driven by the intramolecular interactions that take place at the membrane interface, leading to a very compact C-terminal segment largely exposed to the solvent. The C-terminal segment in this case does not specifically interact with the lipids. By contrast, in mixed POPC-POPS, PMP1 structure is slightly affected and is accommodated within the membrane by tilting. The helix tilt is driven by the specific intermolecular interactions established between the PMP1 C-terminal segment and the POPS lipids.
Supporting Material
References
- 1.Hunte C., Richers S. Lipids and membrane protein structures. Curr. Opin. Struct. Biol. 2008;18:406–411. doi: 10.1016/j.sbi.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Wenz T., Hielscher R., Hunte C. Role of phospholipids in respiratory cytochrome bc1 complex catalysis and supercomplex formation. Biochim. Biophys. Acta. 2009;1787:609–616. doi: 10.1016/j.bbabio.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 3.Lee A.G. The effects of lipids on channel function. J. Biol. 2009;8:86. doi: 10.1186/jbiol178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ernst A.M., Contreras F.X., Wieland F. Determinants of specificity at the protein-lipid interface in membranes. FEBS Lett. 2010;584:1713–1720. doi: 10.1016/j.febslet.2009.12.060. [DOI] [PubMed] [Google Scholar]
- 5.Raja M. The role of phosphatidic acid and cardiolipin in stability of the tetrameric assembly of potassium channel KcsA. J. Membr. Biol. 2010;234:235–240. doi: 10.1007/s00232-010-9251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee A.G. Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 7.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Reichow S.L., Gonen T. Lipid-protein interactions probed by electron crystallography. Curr. Opin. Struct. Biol. 2009;19:560–565. doi: 10.1016/j.sbi.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hite R.K., Li Z., Walz T. Principles of membrane protein interactions with annular lipids deduced from aquaporin-0 2D crystals. EMBO J. 2010;29:1652–1658. doi: 10.1038/emboj.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holt A., Killian J.A. Orientation and dynamics of transmembrane peptides: the power of simple models. Eur. Biophys. J. 2000;39:609–621. doi: 10.1007/s00249-009-0567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ash W.L., Zlomislic M.R., Tieleman D.P. Computer simulations of membrane proteins. Biochim. Biophys. Acta. 2004;1666:158–189. doi: 10.1016/j.bbamem.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 12.Lindahl E., Sansom M.S. Membrane proteins: molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008;18:425–431. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Khalili-Araghi F., Gumbart J., Schulten K. Molecular dynamics simulations of membrane channels and transporters. Curr. Opin. Struct. Biol. 2009;19:128–137. doi: 10.1016/j.sbi.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng M.H., Xu Y., Tang P. Anionic lipid and cholesterol interactions with α4β2 nAChR: insights from MD simulations. J. Phys. Chem. B. 2009;113:6964–6970. doi: 10.1021/jp900714b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khelashvili G., Grossfield A., Weinstein H. Structural and dynamic effects of cholesterol at preferred sites of interaction with rhodopsin identified from microsecond length molecular dynamics simulations. Proteins. 2009;76:403–417. doi: 10.1002/prot.22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pantano D.A., Klein M.L. Characterization of membrane-protein interactions for the leucine transporter from Aquifex aeolicus by molecular dynamics calculations. J. Phys. Chem. B. 2009;113:13715–13722. doi: 10.1021/jp901840u. [DOI] [PubMed] [Google Scholar]
- 17.Lensink M.F., Govaerts C., Ruysschaert J.M. Identification of specific lipid-binding sites in integral membrane proteins. J. Biol. Chem. 2010;285:10519–10526. doi: 10.1074/jbc.M109.068890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deol S.S., Domene C., Sansom M.S. Anionic phospholipid interactions with the potassium channel KcsA: simulation studies. Biophys. J. 2006;90:822–830. doi: 10.1529/biophysj.105.071407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bondar A.N., del Val C., White S.H. Rhomboid protease dynamics and lipid interactions. Structure. 2009;17:395–405. doi: 10.1016/j.str.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navarre C., Ghislain M., Goffeau A. Purification and complete sequence of a small proteolipid associated with the plasma membrane H+-ATPase of Saccharomyces cerevisiae. J. Biol. Chem. 1992;267:6425–6428. [PubMed] [Google Scholar]
- 21.Navarre C., Catty P., Goffeau A. Two distinct genes encode small isoproteolipids affecting plasma membrane H+-ATPase activity of Saccharomyces cerevisiae. J. Biol. Chem. 1994;269:21262–21268. [PubMed] [Google Scholar]
- 22.Tada M. Molecular structure and function of phospholamban in regulating the calcium pump from sarcoplasmic reticulum. Ann. N. Y. Acad. Sci. 1992;671:92–102. doi: 10.1111/j.1749-6632.1992.tb43787.x. discussion 102–103. [DOI] [PubMed] [Google Scholar]
- 23.Wawrzynow A., Theibert J.L., Collins J.H. Sarcolipin, the “proteolipid” of skeletal muscle sarcoplasmic reticulum, is a unique, amphipathic, 31-residue peptide. Arch. Biochem. Biophys. 1992;298:620–623. doi: 10.1016/0003-9861(92)90457-8. [DOI] [PubMed] [Google Scholar]
- 24.van Klompenburg W., Nilsson I., de Kruijff B. Anionic phospholipids are determinants of membrane protein topology. EMBO J. 1997;16:4261–4266. doi: 10.1093/emboj/16.14.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beswick V., Roux M., Neumann J.M. 1H- and 2H-NMR studies of a fragment of PMP1, a regulatory subunit associated with the yeast plasma membrane H+-ATPase. Conformational properties and lipid-peptide interactions. Biochimie. 1998;80:451–459. doi: 10.1016/s0300-9084(00)80012-7. [DOI] [PubMed] [Google Scholar]
- 26.Mousson F., Beswick V., Neumann J.M. Concerted influence of key amino acids on the lipid binding properties of a single-spanning membrane protein: NMR and mutational analysis. Biochemistry. 2001;40:9993–10000. doi: 10.1021/bi010924e. [DOI] [PubMed] [Google Scholar]
- 27.Roux M., Beswick V., Neumann J.M. PMP1 18–38, a yeast plasma membrane protein fragment, binds phosphatidylserine from bilayer mixtures with phosphatidylcholine: a 2H-NMR study. Biophys. J. 2000;79:2624–2631. doi: 10.1016/S0006-3495(00)76501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zinser E., Daum G. Isolation and biochemical characterization of organelles from the yeast, Saccharomyces cerevisiae. Yeast. 1995;11:493–536. doi: 10.1002/yea.320110602. [DOI] [PubMed] [Google Scholar]
- 29.Schneiter R., Brugger B., Kohlwein S.D. Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J. Cell Biol. 1999;146:741–754. doi: 10.1083/jcb.146.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mousson F., Coic Y.M., Neumann J.M. Deciphering the role of individual acyl chains in the interaction network between phosphatidylserines and a single-spanning membrane protein. Biochemistry. 2002;41:13611–13616. doi: 10.1021/bi026274b. [DOI] [PubMed] [Google Scholar]
- 31.Coic Y.M., Vincent M., de Foresta B. Single-spanning membrane protein insertion in membrane mimetic systems: role and localization of aromatic residues. Eur. Biophys. J. 2005;35:27–39. doi: 10.1007/s00249-005-0002-1. [DOI] [PubMed] [Google Scholar]
- 32.Segrest J.P., De Loof H., Anantharamaiah G.M. Amphipathic helix motif: classes and properties. Proteins. 1990;8:103–117. doi: 10.1002/prot.340080202. [DOI] [PubMed] [Google Scholar]
- 33.Kim T., Im W. Revisiting hydrophobic mismatch with free energy simulation studies of transmembrane helix tilt and rotation. Biophys. J. 2010;99:175–183. doi: 10.1016/j.bpj.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.