

Abstract
Low back pain (LBP) is a major cause of disability worldwide that has been linked to intervertebral disc (IVD) degeneration. An improved understanding of the pathogenesis of disc degeneration is now developing, which is leading to the development of a number of possible future therapies targeted at the underlying pathology and regeneration strategies. Although results thus far are promising, the investigation of such therapies in an environment that mimics the mechanical environment of the human disc in vivo is problematic. The development of an in vitro model system that can maintain metabolically active IVD tissue within a loading environment pertaining to that of the human spine is crucial for testing the efficacy of future cell-based and tissue-engineering therapies for IVD degeneration. Here, using our novel loading rig, capable of mimicking the loading environment experienced within the human spine, we have cultured nucleus pulposus tissue explants, applied a daily hydrostatic loading regime for up to 2 weeks and investigated proteoglycan retention, metabolic activity and cellular phenotype. IVD tissue cultured under a loading environment pertaining to the in vivo loading environment maintained metabolic cell activity, proteoglycan content and cellular phenotype. Indeed, all parameters were improved in IVD tissue cultured with load compared to unloaded controls. Such a model is invaluable for investigations assessing the feasibility and efficacy of future therapeutic approaches to inhibiting degeneration or stimulating regeneration of the IVD, where the in vivo loading environment may be crucial to their success or failure.
Keywords: intervertebral disc, loading, culture system, therapeutic testing, tissue culture, low back pain
1. Introduction
Low back pain (LBP) is a major cause of disability, affecting approximately 11 million people within the UK for at least 1 week in each month (Borenstein, 2001). Current therapies are aimed at relieving the symptoms and importantly do not target the underlying cause of LBP, which in a significant proportion of cases has been attributed to intervertebral disc (IVD) degeneration (Luoma et al., 2000), which is associated with decreased matrix synthesis and increased matrix degradation resulting from altered cell function and reduced cell number (Le Maitre et al., 2007b). An improved understanding of the pathogenesis of disc degeneration is now developing, which is leading to the development of a number of possible future therapies which are targeted at the underlying pathology and altered cell biology. These include therapies to inhibit the processes of disc degeneration (Wallach et al., 2003; Le Maitre et al., 2006, 2007a) and regeneration strategies involving cell-based therapies, growth factor treatments and tissue-engineering approaches (Richardson et al., 2007).
Although results from these studies are promising, the investigation of such therapies in an environment that mimics the mechanical environment of the human disc in vivo is problematic. The upright stance of humans and the axial loading of the vertebral column bring with them biomechanical demands upon the lumbar spine, which are unparalleled in the animal kingdom. Although some levels of applied load appear necessary to maintain matrix homeostasis and overall disc health (Holm and Nachemson, 1983), excess load may be involved in the acceleration of disc degeneration (Iatridis et al., 1999). Thus, the loading environment in which prospective therapies are tested could determine their potential success or failure, with the elimination of some therapies which do not work in an unloaded environment but may be well suited to the loaded environment prevailing in vivo. Conversely, those therapies that appear promising in unloaded environments may fail when placed under the loading environment seen in vivo. However, as man is the only animal that has an upright bipedal posture, the ‘load environment’ of human discs is markedly different from those experienced in experimental animals, making their use inappropriate for investigating the effect of load on cell/tissue function or the testing of potential therapies at levels which equate to the human disc loading environment.
As such, there is an urgent need for an in vitro system capable of culturing IVD tissue samples in a loading environment pertaining to the human spine in vivo. The culture of IVD tissue has been hampered by a number of aspects, particularly the high swelling potential of the nucleus pulposus (NP) (Urban and McMullin, 1985), which results in tissue expanding uncontrollably in tissue culture (Urban and McMullin, 1985). Recently the number of studies investigating IVD tissue in in vitro systems has increased, with several investigating the culture of whole organ or explant systems of rabbit (Chiba et al., 1998; Haschtmann et al., 2006a, 2006b), murine (Ariga et al., 2003), rat (Risbud et al., 2003, 2006), bovine (Lee et al., 2006) and ovine (Gantenbein et al., 2006) IVD tissues. Within these culture systems, disc swelling has been restricted in a number of ways, including constriction of the tissue, either by retention of end-plates and adjacent soft tissues (Risbud et al., 2003; Ariga et al., 2003; Gantenbein et al., 2006; Haschtmann et al., 2006b), embedding into gel systems such as alginate (Chiba et al., 1998), application of hyperosmotic solutions (Urban and McMullin, 1985) and/or via the application of external load (Lee et al., 2006; Gantenbein et al., 2006; Haschtmann et al., 2006b; Korecki et al., 2007). Unfortunately, though, many of these still report poor cell viability, loss of proteoglycan from the tissue and altered cellular phenotype and behaviour that often mimics the degenerative phenotype (Haschtmann et al., 2006a).
We previously described a semi-constrained culture system in which human IVD tissue explants could be cultured for up to 3 weeks (the maximum time investigated) without tissue swelling, and in which tissue integrity, cellular phenotype and viability were maintained for the time course investigated (Le Maitre et al., 2004). However, at that time we did not address the problem of whether the system would still function under the load conditions pertaining to that of the human spine. A number of studies have investigated the application of load to explant or organ tissue cultures but these have either been single static loading regimes, or simple diurnal loading patterns which do not represent the loading patterns seen in vivo, i.e. 0.2 MPa or 0.25 MPa static loading (Lee et al., 2006; Korecki et al., 2007) or diurnal loading of 12 h 0.1 MPa and 12 h 0.3 MPa (Korecki et al., 2007) or 8 h 0.2 MPa and 16 h 0.8 MPa (Gantenbein et al., 2006). Here, using our novel loading rig (Le Maitre et al., 2008), which is capable of mimicking the loading environment experienced within the human spine, we have cultured bovine tissue explants in our Perspex ring culture system (Le Maitre et al., 2004) and applied a daily loading regime designed using pressure measurements taken during daily activities in the human spine (Wilke et al., 1999). The aim of this study was to develop an in vitro culture system capable of maintaining IVD tissue without inducing changes in cellular phenotype or altering tissue integrity, in order that such a system could be used for future in vitro testing of novel cell-based or biomaterial therapies under loading conditions experienced in the human spine.
2. Materials and methods
2.1. Tissue samples
Bovine caudal IVDs (age 9–18 months) were excised and NP tissue isolated macroscopically, with the interface zone discarded to ensure no contamination with annulus fibrosus (AF) tissue. Cores of NP tissue (ca. 0.5 cm diameter × 0.6 cm height) were formed and placed into the Perspex ring culture system described previously (Le Maitre et al., 2004).
2.2. Loading apparatus
Our novel loading system, as described previously (Le Maitre et al., 2008) (Figure 1) consists of two parts: a commercial hydraulic biomechanical testing system [bench-mounted HC5 material-testing loading frame (Zwick Testing Machines Ltd)] and an in-house designed stainless steel pressure vessel. Following the insertion of tissue explants, contained in sealed media filled bags, into the pressure vessel, the vessel is filled with water and sealed. Compressive force is then applied via the piston (which is controlled by the hydraulic biomechanical testing system) to the water-filled chamber, which results in the generation of controlled hydrostatic pressure to the tissue samples contained within the pressure vessel. Internal pressure is monitored using a pressure transducer connected to a Hydrotechnik 2042 handheld pressure monitor, and Hydrowin monitoring software enabling live real-time recording of the pressure experienced within the vessel and graphical display readings accurate to ± 1%.
Figure 1.

Photomicrograph of the loading apparatus used in the application of hydrostatic pressure. Tissue explants were cultured in Perspex rings (top right), which for unloaded controls were placed in six-well tissue culture plates and cultured under standard conditions. For application of load, explants in perspex rings were inserted into bags containing medium, the air expelled and the bags sealed. The sealed bags were hung on a carousel (bottom right) to enable free floating in the water-filled pressure vessel (left)
2.3. Design of daily loading regime
Using data derived from studies of intradiscal pressure (Wilke et al., 1999), we designed a daily loading regime consisting of a combination of static and dynamic loading regimes to mimic daily activities. These regimes included two regimes to mimic the load experienced during lying down, i.e. representing the increase in pressure observed during lying down (0.1–0.24 MPa) (Wilke et al., 1999), and a selection of daily activities including walking, sitting/standing and jogging, ordered to mimic a standard daily routine (Table 1). This regime was repeated 24 hourly to generate constant loading.
Table 1.
Daily loading regime, designed using intradiscal pressure measurements
| Step | Load (MPa) | Frequency (Hz) | Duration (h) |
|---|---|---|---|
| Lying 1 | 0.1 | Static | 4 |
| Lying 2 | 0.24 | Static | 4 |
| Walking | 0.53–0.65 | 0.5 | 2 |
| Sitting/standing | 0.5 | Static | 4 |
| Jogging | 0.35–0.95 | 1.0 | 2 |
| Sitting/standing | 0.5 | Static | 4 |
| Walking | 0.53–0.65 | 0.5 | 2 |
| Sitting/standing | 0.5 | Static | 2 |
Pressure measurements from Wilke et al., 1999.
2.4. Application of loading regime
Perspex ring tissue cultures were transferred to sterile Whirl-Pak® bags (Nasco) and 50 ml DMEM/F12 with HEPES medium (Gibco; to maintain pH in the absence of CO2) supplemented with 10% v/v heat-inactivated fetal calf serum (FCS; Gibco), 100 U/ml penicillin (Sigma), 100 μg/ml streptomycin (Sigma), 250 ng/ml amphotericin, 2 mM glutamine (Sigma) and 50 μg/ml ascorbic acid (Sigma) (complete cell culture medium), was added, after which any air was removed and the bags sealed. Unloaded controls were maintained in six-well tissue culture plates in complete cell culture medium in a 37 °C incubator. Bags of Perspex ring tissue cultures for loading were hooked onto the carousel and placed within the pressure vessel (Le Maitre et al., 2008). The pressure vessel was filled with water at 37 °C and sealed. The piston was attached and all air expelled from the system (Figure 1). Hydrostatic loading was then applied for up to 2 weeks, using a repetition of the daily loading regime (Table 1). The medium was changed on samples every 48 h during the 0.1 MPa static loading, to prevent the introduction of an interval in the loading regime. Samples were removed from the loading environment for a maximum of 10 min for each medium change (NB: atmospheric pressure = 0.1 MPa). Tissue samples were removed from loaded and unloaded cultures after 48 h, 1 week and 2 weeks.
2.5. Tissue processing
Following removal from culture, the tissue samples were weighed and at each time point three tissue samples were fixed in 4% paraformaldehyde/PBS overnight, prior to processing to paraffin wax, and three tissue samples were subjected to papain digest to assess GAG content.
2.6. Assessment of glycosaminoglycans
Following culture, triplicate tissue samples were used for quantification of GAG content, using the DMMB assay, and DNA was assayed using the PicoGreen assay. Tissue samples were digested overnight at 60 °C in 1 ml 20 mM sodium phosphate buffer, pH 6.8, containing 1 mM EDTA, 2 mM dithiothreitol and 100 U papain (Sigma). DMMB assay was performed using 25 μl shark chondrotin sulphate (Sigma) standards (62.5, 31.25, 15.625, 7.81, 3.9 and 0 μg/ml) or 5 μl papain-digested tissue sample. Each sample was applied in duplicate in separate wells of the 96-well plate and 200 μl DMMB colour reagent, as described previously (Farndale et al., 1986), was added to each well. Following mixing, absorbance at A525 was read immediately, using a Titertex Multiscan® MC (Labsystems). The concentration of GAGs present within tissue samples was then calculated, using standards. DNA within digested tissue samples was assayed along with calf thymus DNA standards, using the PicoGreen DNA quantification kit (Invitrogen) according to the manufacturer’s instructions.
2.6.1. Statistical analysis
Total GAG per tissue explant was calculated and normalized to DNA content of respective tissue explants. Data were determined as non-parametric, using the Shapiro–Wilke test and Kruskal–Wallis tests, with Mann–Whitney U post hoc tests used to assess the effect of culture on level of GAGs/DNA in tissues.
2.7. Histological examination
Paraffin-embedded samples were subjected to Alcian blue staining, using standard protocols.
2.8. Immunohistochemisty
Paraffin-embedded samples were subjected to immunohistochemistry (IHC) for aggrecan and type I and II collagens, as described previously (Le Maitre et al., 2005). Briefly, 4 μm paraffin sections were dewaxed and rehydrated and endogenous peroxidase blocked using hydrogen peroxide. After washing in distilled water, sections were then treated with a hyaluronidase enzyme antigen retrieval system [0.1% w/v hyaluronidase in TBS (Sigma), 30 min at 37 °C] for aggrecan and type II collagen, or hyaluronidase and trypsin enzyme retrieval system [0.01% hyaluronidase (Sigma), 0.02% trypsin (Sigma) w/v in TBS] for type I collagen. Following washing, non-specific binding sites were blocked at room temperature for 45 min with 20% w/v rabbit serum (Sigma) for aggrecan, type I collagen and type II collagen. Sections were incubated overnight at 4 °C with mouse monoclonal primary antibodies against human aggrecan (1 : 25 dilution; AbCam, Cambridge, UK), collagen type I (1 : 250 dilution; ICN, Basingstoke, UK) and collagen type II (1 : 100 dilution; MP Biomedicals). Negative controls in which mouse IgGs (Dako) replaced the primary antibody (at an equal protein concentration) were used. After washing, sections were incubated in biotinylated rabbit anti-mouse antiserum (1 : 400 dilution, Dako) for 30 min at room temperature. Disclosure of secondary antibody binding was by the streptavidin–biotin complex (Dako) technique with 3,3′-diaminobenzidine tetrahydrochloride solution (Sigma). Sections were counterstained with Mayer’s haematoxylin (Raymond A. Lamb), dehydrated and mounted in XAM (BDH).
2.9. In situ hybridization for polyA mRNA
In situ hybridization for polyA mRNA was performed as an assessment of metabolic cell activity, as described previously (Marles et al., 1991; Walsh et al., 1993).
2.10. Image and statistical analysis
All slides were visualized using a Leica RMDB research microscope and images captured using a digital camera and the Bioquant Nova image analysis system. Total cell counts were performed on uncultured control tissue explants and no significant difference was seen in total cell number between explants (data not shown). Thus, for analysis of immunohistochemistry and polyA mRNA, 200 cells per explant were counted and cell staining recorded as positive or negative and the percentage immunopositive cells calculated. Data were determined as non-parametric, using the Shapiro–Wilke test, and Kruskal–Wallis tests with Mann–Whitney U post hoc tests used to assess the effect of culture on polyA mRNA positive and immunopositive cells during the 2 weeks of culture.
3. Results
3.1. Tissue integrity and proteoglycan content
Tissue integrity was maintained, with no tissue swelling observed throughout the time course investigated in both unloaded and loaded samples. However, within unloaded samples a significant decrease in the proteoglycan content of tissue explants was observed with increasing time in culture (p < 0.05 48 h vs. 2 weeks; Figure 2A). In contrast, an increase was seen in GAG/DNA content in loaded samples between 2 and 7 days, although this was not statistically significant (Figure 2A), with overall proteoglycan content within loaded explants being maintained throughout the time course with no significant decrease in GAG/DNA content observed at any time point compared with 48 h in culture (p > 0.05). No significant differences in proteoglycan content were observed between 2 or 7 days, but significantly less proteoglycan content normalized to DNA was seen in unloaded vs. loaded controls following 2 weeks in culture (p < 0.05; Figure 2A). Improved proteoglycan retention for the duration of the culture period within loaded disc explants compared with unloaded tissue was confirmed with Alcian blue histological staining (Figure 2B–D).
Figure 2.
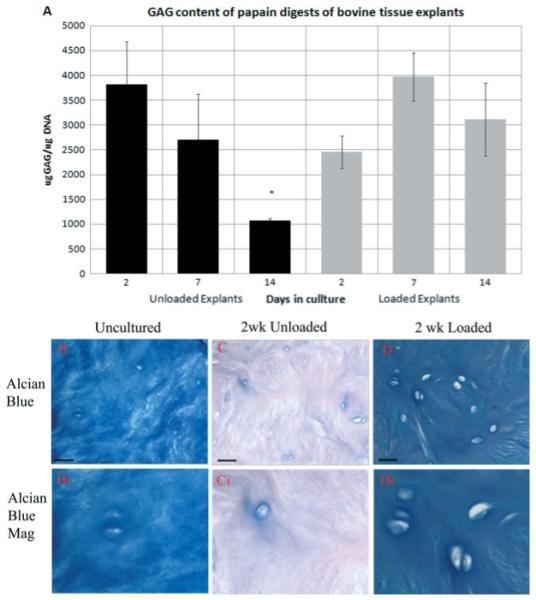
(A) Glycosaminoglycan (GAGs) content of tissue explants during culture with or without load. Error = bars standard error of the mean (SEM). *p < 0.05; n = 3. Photomicrographs of: (B) Alcian blue-stained tissue explants prior to culture; (C) following 2 weeks culture without load; or (D) following 2 weeks in culture with load, together with magnified images. Scale bars = 380 μm
3.2. Metabolic cell activity
Cell metabolic activity was assessed using in situ hybridization for polyA mRNA. Within unloaded tissue samples, a decrease in the number of positively stained cells was observed during the time course investigated, 36% polyA mRNA positive cells in unloaded tissue cultured for 14 days compared to 54% in uncultured tissue (Figures 3, 4). However, samples subjected to the daily loading regime maintained cellular activity for the 2 weeks investigated (66% polyA mRNA-positive cells following culture under load for 14 days compared to 54% in uncultured tissue; Figures 3, 4).
Figure 3.
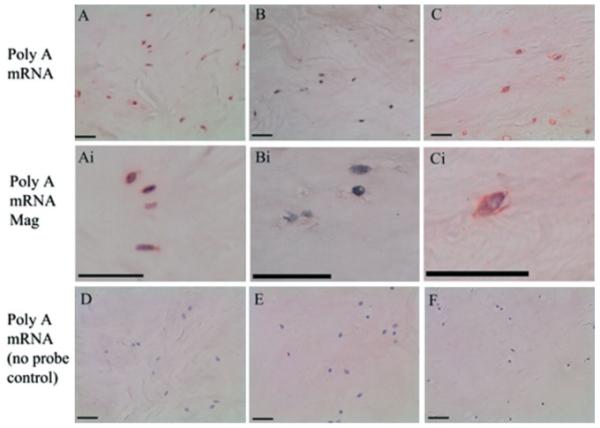
Photomicrographs representative of: (A) in situ hybridization for polyA mRNA within tissue explants prior to culture; and following 2 weeks culture without (B) or with (C) load. Magnified images are also displayed (Ai–Ci). (D–F) Negative control sections for polyA mRNA. Scale bars = 380 μm
Figure 4.

Percentages of cells displaying positive for polyA mRNA and immunopositivity for type II collagen and aggrecan in uncultured tissue samples and those cultured without and with load for 2 weeks. Error bars = SEM; *p < 0.05; n = 3
3.3. Cellular phenotype
Type II collagen and aggrecan immunopositive cells were observed in the uncultured tissue (Figure 5). The number of collagen type II-immunopositive cells was not altered in unloaded tissue after 2 weeks, but in loaded samples a significant increase in the percentage of collagen type II-immunopositive cells was observed (p < 0.05; Figures 4, 5). Following culture for 2 weeks, both unloaded and loaded systems demonstrated a significant increase in the percentage of cells displaying aggrecan immunopositivity (p < 0.05; Figures 4, 5). Type I collagen immunopositivity was not observed in any of the tissue samples examined (Figure 5). No immunopositive staining was observed in IgG controls (Figure 5).
Figure 5.
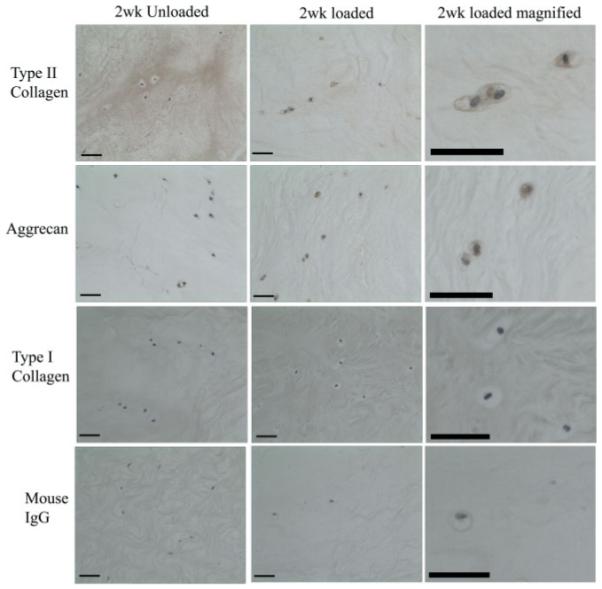
Photomicrographs representative of immunohistochemical staining for type II collagen, aggrecan, type I collagen and IgG control sections in tissue explants following 2 weeks of load or in unloaded controls. Scale bars = 380 μm
4. Discussion
The development of an in vitro model system which can maintain metabolically active IVD tissue within a loading environment pertaining to that of the human spine is crucial for testing the efficacy of future cell-based and tissue-engineering therapies for IVD degeneration, as well as investigating the effects of human loads on IVD cell phenotype/function. This study, for the first time, has successfully maintained IVD tissue in vitro whilst subjecting the living tissue to a load pertaining to that experienced in the human spine. Metabolic cell activity, tissue integrity and cellular phenotype in IVD tissue culture were unaltered for 2 weeks in this loading system. Such a model incorporating this loading regime is invaluable for investigations assessing the feasibility and efficacy of future therapeutic approaches for inhibiting degeneration or repairing the IVD, where the loading environment may be crucial to their success or failure.
We have demonstrated that when these tissue explants were cultured within a loading environment mimicking the complex loads experienced during a daily routine, tissue integrity, proteoglycan retention, cellular phenotype and metabolic activity were maintained to a greater extent than in tissue cultured under standard culture conditions without load. Previous studies have demonstrated poor maintenance of biosynthetic activity and cell viability within unloaded tissue culture systems. Risbud et al. (2003) demonstrated the maintenance of cell viability for 1 week, which was followed by a subsequent loss in cell viability, decreased matrix gene expression and biosynthetic activity after 3 weeks in tissue cultured within a high osmotic environment but without load (Risbud et al., 2003). Such findings are consistent with our data, in that tissue samples cultured without load showed decreased metabolic activity, reduced matrix synthesis and GAGs retention compared to tissue explants maintained under a loaded environment. Our study, together with other data investigating the effect of physiologically normal loading regimes on IVD cells both in vitro and in vivo, demonstrates that load is important in maintaining a normal phenotype. Indeed, within the loaded environment investigated, tissue explants displayed increased matrix protein synthesis, as evidenced by immunohistochemistry over the 2 week culture period. Such results complement several studies that have shown the important role load plays in matrix homeostasis, where the application of ‘normal’ loads has been shown to increase matrix synthesis (Ishihara et al., 1996; Handa et al., 1997; Hutton et al., 2001; Kasra et al., 2003, 2006; Gokorsch et al., 2004; Maclean et al., 2005; Le Maitre et al., 2008)
A number of studies to date have investigated organ culture systems for the in vitro culture of IVD tissue but these have often been hampered by poor cell viability, thought to be caused by poor nutritional diffusion through the end plates or due to the size of the tissue (Lee et al., 2006; Korecki et al., 2007). Some organ culture systems using smaller animal models have been developed and have shown improved cell viability (Risbud et al., 2003, 2006; Haschtmann et al., 2006a, 2006b). Here, we aimed to develop a system that could be transferred to use for human disc tissue samples, and thus favoured the development of an explant culture system. Human samples are generally received as fragments of tissue following surgery, and thus culture of whole IVD organ cultures is not feasible.
Within the current study, we demonstrate the maintenance of metabolic cell activity (as an indication of cell viability) within tissue explants, with no decrease observed over 2 weeks of culture in a loaded environment. One of the key challenges of a successful tissue culture system appears to be the maintenance of cell viability, and studies to date have only succeeded in maintaining viability for periods up to 1 week (Risbud et al., 2006; Gantenbein et al., 2006; Korecki et al., 2007). Within our study, samples cultured without load also showed a decrease in metabolic cell activity during culture; however, those cultured in our apparatus and under a loaded environment demonstrated improved metabolic cell activity. Thus, for the first time we have succeeded in maintaining metabolic cell activity in tissue cultured for up to 2 weeks, which could have been due to improved diffusion through the tissue induced by fluid flow as a result of the application of dynamic loading regimes (Ferguson et al., 2004; Gantenbein et al., 2006; Haschtmann et al., 2006a), or alternatively due to survival signals provided to the cells by the applied loading regime (Holm et al., 1981).
The culture system described here involved the culture of tissue samples in sealed bags for up to 2 weeks, with the medium changed only every 48 h, compared to tissue samples cultured under normal tissue culture conditions (i.e. 5% CO2, 20% O2). Within this ‘bag’ system, gas exchange may differ between the unloaded controls and the loaded samples, due to the sealed culture environment. Notably, however, these factors were not detrimental, but rather appeared to improve cell metabolic activity and matrix protein expression compared to unloaded cultures in standard conditions. Indeed, any resulting hypoxia, due to limited gas exchange, may have improved cell viability, as hypoxic conditions can provide survival signals to NP cells and rescue them from apoptosis (Risbud et al., 2005).
In addition to problems with cell viability, a further limitation of tissue culture systems to date has been the loss of proteoglycans from the tissue and alterations in normal cell phenotype within tissue cultures (Risbud et al., 2003; Haschtmann et al., 2006a, 2006b). Our culture system demonstrated improved proteoglycan retention and cellular phenotype in loaded vs. unloaded samples, with no loss of either proteoglycan content or cell phenotype within loaded tissue samples. Such results suggest that the culture system employed in our loading apparatus, together with the loading regime, is a feasible system for the maintenance of the in vivo disc cell phenotype and tissue integrity.
To date, no studies have attempted to culture IVD tissue under loading regimes which mimic the human spine. Simple static and diurnal loading have been applied in the attempt to improve cell viability, tissue integrity and cellular phenotype of tissue culture systems to varying degrees of success (Ferguson et al., 2004; Lee et al., 2006; Gantenbein et al., 2006; Haschtmann et al., 2006a, 2006b; Korecki et al., 2007). However, such static loading regimes have been reported to induce detrimental effects on IVD cells, in both in vivo and in vitro tests, inducing cell death and abnormal gene expression often associated with matrix destruction (Wang et al., 2007). Similar findings have also been reported for IVD tissue subjected to static load, with a cellular phenotype more akin to that of the degenerate IVD being produced (Lee et al., 2006; Gantenbein et al., 2006; Korecki et al., 2007). Our current study used intradiscal pressure measurements, taken by Wilke et al. (1999) during a series of activities, to derive a daily loading regime which could mimic the loads experienced during a conceivable daily regime. Obviously the regime designed is a simplified pattern, but it provides an important tool for testing future therapies and investigating complex loading patterns on the IVD. The designed loading rig is capable of applying any regime the user chooses, and thus is a vital advance for both feasibility and efficacy testing, but it will also allow investigation of the effects of loading parameters on cells, tissues and tissue-engineered constructs. Although the explant system described here has the advantage that it could be used for human tissue samples from both normal and degenerate patients, its major limitation is that, here, we have only investigated NP tissue. However, the results illustrate an important proof-of-principle concept, in that IVD tissue can be maintained in the system for extended periods under load. As such, our system is ideally suited for testing future therapies in a loading environment pertaining to the human spine and could be expanded to further investigate the maintenance of whole-disc organ cultures.
Of course, in vivo, IVDs are exposed to low levels of glucose, pH and oxygen (Urban et al., 2004; Bibby et al., 2005), which we did not directly address within this study. These are important parameters which have been shown previously to affect cellular metabolism and gene expression (Wang et al., 2001; Horner et al., 2002), and which we aim to investigate directly in future studies using our culture system. Indeed, our culture system allows the humoral environment to be easily manipulated, enabling the complex system seen in vivo within the IVD to be mimicked.
5. Conclusion
This study has described a novel culture system in which NP explant tissue can be cultured in a defined humoral environment for extended periods under load regimes identical to that experienced within the human spine. The further development of this system, including the use of whole-disc explants, will become a vital tool in the efficacy and feasibility testing of future cellular, molecular and tissue-engineering approaches to disc degeneration.
Acknowledgements
The authors wish to thank BackCare and ARC (Ref. No. 18046) for funding this project. The work was undertaken in the Human Tissue Profiling Laboratories of the Tissue Injury and Repair Research Group, who receive core support from the ARC (ICAC Grant No. F0551).
References
- Ariga K, Yonenobu K, Nakase T, et al. Mechanical stress-induced apoptosis of endplate chondrocytes in organ-cultured mouse intervertebral discs: an ex vivo study. Spine. 2003;28:1528–1533. [PubMed] [Google Scholar]
- Bibby SR, Jones DA, Ripley RM, et al. Metabolism of the intervertebral disc: effects of low levels of oxygen, glucose, and pH on rates of energy metabolism of bovine nucleus pulposus cells. Spine. 2005;30:487–496. doi: 10.1097/01.brs.0000154619.38122.47. [DOI] [PubMed] [Google Scholar]
- Borenstein DG. Epidemiology, etiology, diagnostic evaluation, and treatment of low back pain. Curr Opin Rheumatol. 2001;13:128–134. doi: 10.1097/00002281-200103000-00006. [DOI] [PubMed] [Google Scholar]
- Chiba K, Andersson GB, Masuda K, et al. A new culture system to study the metabolism of the intervertebral disc in vitro. Spine. 1998;23:1821–1827. doi: 10.1097/00007632-199809010-00002. [DOI] [PubMed] [Google Scholar]
- Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta. 1986;883:173–177. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]
- Ferguson SJ, Ito K, Nolte LP. Fluid flow and convective transport of solutes within the intervertebral disc. J Biomech. 2004;37:213–221. doi: 10.1016/s0021-9290(03)00250-1. [DOI] [PubMed] [Google Scholar]
- Gantenbein B, Grunhagen T, Lee CR, et al. An in vitro organ culturing system for intervertebral disc explants with vertebral endplates: a feasibility study with ovine caudal discs. Spine. 2006;31:2665–2673. doi: 10.1097/01.brs.0000244620.15386.df. [DOI] [PubMed] [Google Scholar]
- Gokorsch S, Nehring D, Grottke C, et al. Hydrodynamic stimulation and long-term cultivation of nucleus pulposus cells: a new bioreactor system to induce extracellular matrix synthesis by nucleus pulposus cells dependent on intermittent hydrostatic pressure. Int J Artif Organs. 2004;27:962–970. doi: 10.1177/039139880402701109. [DOI] [PubMed] [Google Scholar]
- Handa T, Ishihara H, Ohshima H, et al. Effects of hydrostatic pressure on matrix synthesis and matrix metalloproteinase production in the human lumbar intervertebral disc. Spine. 1997;22:1085–1091. doi: 10.1097/00007632-199705150-00006. [DOI] [PubMed] [Google Scholar]
- Haschtmann D, Stoyanov JV, Ettinger L, et al. Establishment of a novel intervertebral disc/endplate culture model: analysis of an ex vivo in vitro whole-organ rabbit culture system. Spine. 2006a;31:2918–2925. doi: 10.1097/01.brs.0000247954.69438.ae. [DOI] [PubMed] [Google Scholar]
- Haschtmann D, Stoyanov JV, Ferguson SJ. Influence of diurnal hyperosmotic loading on the metabolism and matrix gene expression of a whole-organ intervertebral disc model. J Orthop Res. 2006b;24:1957–1966. doi: 10.1002/jor.20243. [DOI] [PubMed] [Google Scholar]
- Holm S, Maroudas A, Urban JP, et al. Nutrition of the intervertebral disc: solute transport and metabolism. Connect Tissue Res. 1981;8:101–119. doi: 10.3109/03008208109152130. [DOI] [PubMed] [Google Scholar]
- Holm S, Nachemson A. Variations in the nutrition of the canine intervertebral disc induced by motion. Spine. 1983;8:866–874. doi: 10.1097/00007632-198311000-00009. [DOI] [PubMed] [Google Scholar]
- Horner HA, Roberts S, Bielby RC, et al. Cells from different regions of the intervertebral disc: effect of culture system on matrix expression and cell phenotype. Spine. 2002;27:1018–1028. doi: 10.1097/00007632-200205150-00004. [DOI] [PubMed] [Google Scholar]
- Hutton WC, Elmer WA, Bryce LM, et al. Do the intervertebral disc cells respond to different levels of hydrostatic pressure? Clin Biomech (Bristol) 2001;16:728–734. doi: 10.1016/s0268-0033(01)00080-8. [DOI] [PubMed] [Google Scholar]
- Iatridis JC, Mente PL, Stokes IA, et al. Compression-induced changes in intervertebral disc properties in a rat tail model. Spine. 1999;24:996–1002. doi: 10.1097/00007632-199905150-00013. [DOI] [PubMed] [Google Scholar]
- Ishihara H, McNally DS, Urban JP, et al. Effects of hydrostatic pressure on matrix synthesis in different regions of the intervertebral disk. J Appl Physiol. 1996;80:839–846. doi: 10.1152/jappl.1996.80.3.839. [DOI] [PubMed] [Google Scholar]
- Kasra M, Goel V, Martin J, et al. Effect of dynamic hydrostatic pressure on rabbit intervertebral disc cells. J Orthop Res. 2003;21:597–603. doi: 10.1016/S0736-0266(03)00027-5. [DOI] [PubMed] [Google Scholar]
- Kasra M, Merryman WD, Loveless KN, et al. Frequency response of pig intervertebral disc cells subjected to dynamic hydrostatic pressure. J Orthop Res. 2006;24:1967–1973. doi: 10.1002/jor.20253. [DOI] [PubMed] [Google Scholar]
- Korecki CL, Maclean JJ, Iatridis JC. Characterization of an in vitro intervertebral disc organ culture system. Eur Spine J. 2007;16:1029–1037. doi: 10.1007/s00586-007-0327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Maitre CL, Hoyland JA, Freemont AJ. Studies of human intervertebral disc cell function in a constrained in vitro tissue culture system. Spine. 2004;29:1187–1196. doi: 10.1097/00007632-200406010-00006. [DOI] [PubMed] [Google Scholar]
- Le Maitre CL, Frain J, Fotheringham AP, et al. Human cells derived from degenerate intervertebral discs respond differently to those derived from non-degenerate intervertebral discs following application of dynamic hydrostatic pressure. Biorheology. 2008;45:563–575. [PubMed] [Google Scholar]
- Le Maitre CL, Freemont AJ, Hoyland JA. The role of interleukin-1 in the pathogenesis of human intervertebral disc degeneration. Arthritis Res Ther. 2005;7:R732–745. doi: 10.1186/ar1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Maitre CL, Freemont AJ, Hoyland JA. A preliminary in vitro study into the use of IL-1Ra gene therapy for the inhibition of intervertebral disc degeneration. Int J Exp Pathol. 2006;87:17–28. doi: 10.1111/j.0959-9673.2006.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Maitre CL, Hoyland JA, Freemont AJ. Interleukin-1 receptor antagonist delivered directly and by gene therapy inhibits matrix degradation in the intact degenerate human intervertebral disc: an in situ zymographic and gene therapy study. Arthritis Res Ther. 2007a;9:R83. doi: 10.1186/ar2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Maitre CL, Pockert A, Buttle DJ, et al. Matrix synthesis and degradation in human intervertebral disc degeneration. Biochem Soc Trans. 2007b;35:652–655. doi: 10.1042/BST0350652. [DOI] [PubMed] [Google Scholar]
- Lee CR, Iatridis JC, Poveda L, et al. In vitro organ culture of the bovine intervertebral disc: effects of vertebral endplate and potential for mechanobiology studies. Spine. 2006;31:515–522. doi: 10.1097/01.brs.0000201302.59050.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoma K, Riihimaki H, Luukkonen R, et al. Low back pain in relation to lumbar disc degeneration. Spine. 2000;25:487–492. doi: 10.1097/00007632-200002150-00016. [DOI] [PubMed] [Google Scholar]
- Maclean JJ, Lee CR, Alini M, et al. The effects of short-term load duration on anabolic and catabolic gene expression in the rat tail intervertebral disc. J Orthop Res. 2005;23:1120–1127. doi: 10.1016/j.orthres.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Marles PJ, Hoyland JA, Parkinson R, et al. Demonstration of variation in chondrocyte activity in different zones of articular cartilage: an assessment of the value of in situ hybridization. Int J Exp Pathol. 1991;72:171–182. [PMC free article] [PubMed] [Google Scholar]
- Richardson SM, Mobasheri A, Freemont AJ, et al. Intervertebral disc biology, degeneration and novel tissue engineering and regenerative medicine therapies. Histol Histopathol. 2007;22:1033–1041. doi: 10.14670/HH-22.1033. [DOI] [PubMed] [Google Scholar]
- Risbud MV, Di Martino A, Guttapalli A, et al. Toward an optimum system for intervertebral disc organ culture: TGFβ3 enhances nucleus pulposus and annulus fibrosus survival and function through modulation of TGFβR expression and ERK signaling. Spine. 2006;31:884–890. doi: 10.1097/01.brs.0000209335.57767.b5. [DOI] [PubMed] [Google Scholar]
- Risbud MV, Fertala J, Vresilovic EJ, et al. Nucleus pulposus cells upregulate PI3K/Akt and MEK/ERK signaling pathways under hypoxic conditions and resist apoptosis induced by serum withdrawal. Spine. 2005;30:882–889. doi: 10.1097/01.brs.0000159096.11248.6d. [DOI] [PubMed] [Google Scholar]
- Risbud MV, Izzo MW, Adams CS, et al. An organ culture system for the study of the nucleus pulposus: description of the system and evaluation of the cells. Spine. 2003;28:2652–2658. doi: 10.1097/01.BRS.0000099384.58981.C6. [DOI] [PubMed] [Google Scholar]
- Urban JP, McMullin JF. Swelling pressure of the inervertebral disc: influence of proteoglycan and collagen contents. Biorheology. 1985;22:145–157. doi: 10.3233/bir-1985-22205. [DOI] [PubMed] [Google Scholar]
- Urban JP, Smith S, Fairbank JC. Nutrition of the intervertebral disc. Spine. 2004;29:2700–2709. doi: 10.1097/01.brs.0000146499.97948.52. [DOI] [PubMed] [Google Scholar]
- Wallach CJ, Sobajima S, Watanabe Y, et al. Gene transfer of the catabolic inhibitor TIMP-1 increases measured proteoglycans in cells from degenerated human intervertebral discs. Spine. 2003;28:2331–2337. doi: 10.1097/01.BRS.0000085303.67942.94. [DOI] [PubMed] [Google Scholar]
- Walsh L, Freemont AJ, Hoyland JA. The effect of tissue decalcification on mRNA retention within bone for in situ hybridization studies. Int J Exp Pathol. 1993;74:237–241. [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Baer AE, Kraus VB, et al. Intervertebral disc cells exhibit differences in gene expression in alginate and monolayer culture. Spine. 2001;26:1747–1752. doi: 10.1097/00007632-200108150-00003. [DOI] [PubMed] [Google Scholar]
- Wilke HJ, Neef P, Caimi M, et al. New in vivo measurements of pressures in the intervertebral disc in daily life. Spine. 1999;24:755–762. doi: 10.1097/00007632-199904150-00005. [DOI] [PubMed] [Google Scholar]