

Abstract
This article provides an historical and personal perspective on the discovery of genetic causes for hypertrophic cardiomyopathy (HCM). Extraordinary insights of physicians who initially detailed remarkable and varied manifestations of the disorder, collaboration among multidisciplinary teams with skills in clinical diagnostics and molecular genetics, and hard work by scores of trainees, solved the etiologic riddle of HCM, and unexpectedly demonstrated mutations in sarcomere protein genes as the cause of disease. In addition to celebrating 20 years of genetic research in HCM, this article serves as an introductory overview to a thematic review series that will present contemporary advances in the field of hypertrophic heart disease. Through the continued application of advances in genetic methodologies, combined with biochemical and biophysical analyses of the consequences of human mutations, fundamental knowledge about HCM and sarcomere biology has emerged. Expanding research to elucidate the mechanisms by which subtle genetic variation in contractile proteins remodel the human heart remains an exciting opportunity, one with considerable promise to provide new strategies to limit or even prevent HCM pathogenesis.
Keywords: Hypertrophic cardiomyopathy, gene mutations, sarcomere
Late in the 1950’s an enigmatic myocardial disease captured the attention of cardiologists, surgeons, and pathologists. Characterized as asymmetric septal hypertrophy,1 functional aortic stenosis,2, 3 hypertrophic obstructive cardiomyopathy,4 and idiopathic hypertrophic subaortic stenosis,5 early reports described dramatic clinical events associated with the disease: a healthy 14-year old succumbed while being “chased around the playground of his school”; a cyclist collapsed and died “during a thunderstorm without evidence of electric burns or serious injury resulting from the fall”.1 Post mortem examination of the hearts of sudden death victims showed striking cardiomegaly (Fig. 1a), with some heart weights > 500 gm, stunning asymmetry with disproportionate involvement of the interventricular septum, and bizarre histopathology with disordered muscle bundles, myocyte disarray and markedly increased myocardial fibrosis (Fig. 1b).
Figure 1.
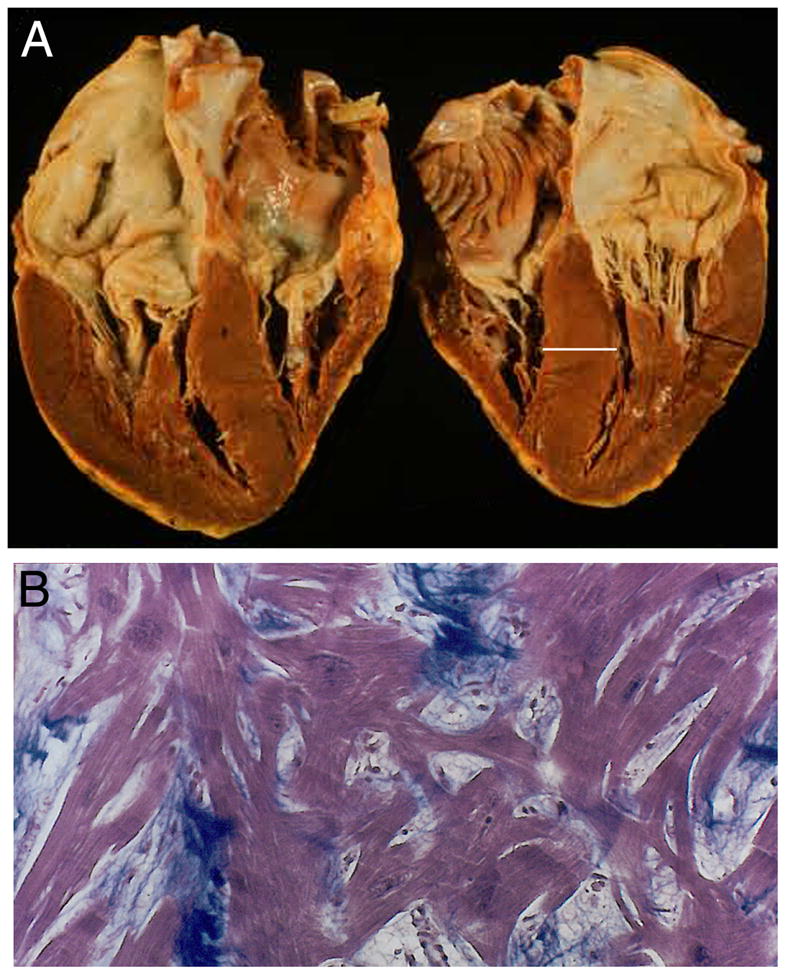
The anatomic and histopathologic findings of HCM. A. Gross anatomy of an HCM heart shows marked hypertrophy with involvement of the interventricular septum, left ventricular free wall, and papillary muscle. Wall thickness (bar) = 20 mm (normal ≤ 12 mm). Left atrial enlargement is also evident. B. Mason trichrome stained ventricular tissue reveals markedly disorganized, enlarged myocytes (magenta) with prominent nuclei. The interstitium (blue) in expanded in HCM due to increased numbers of fibroblasts and extracellular matrix material.
Sudden cardiac death due to unexplained ventricular hypertrophy had in fact been recognized for centuries6 and case reports from 1869 described distinctive cardiac mumurs7 and arrhythmias in patients who at autopsy had muscular obstruction of the left ventricular outflow tract obstruction and distortion of the anterior mitral valve leaflet.8
A plethora of manuscripts expanded upon the anatomic, histopathologic, and hemodynamic features found in affected patients. Authors of these studies appended scores of additional names to the disorder, which was emblematic of the clinical diversity exhibited by affected individuals and the lack of expert consensus on their significance. International task forces were formed, including several NHLBI-sponsored Clinical Bethesda Conferences,9 that established diagnostic criteria, defined the anatomic and hemodynamic underpinnings of obstruction and outflow tract gradients, assessed the natural history of disease, and proposed treatment strategies. The cause(s) of disease was widely debated. Potential etiologies that were put forward included benign tumors of the heart,1 excessive catecholamine activation,10 developmental abnormalities of cardiac neural crest cells,10 and immune processes mediated by the B-12 HLA locus.11 This last hypothesis was found to be predicated on fabricated data and was subsequently retracted.12 Yet despite considerable confusion about the disease, two clinical features were universally recognized. First, left ventricular hypertrophy was idiopathic, and occurred in absence of common triggers such as aortic stenosis or hypertension. Second, cases were clustered in families, with first-degree relatives of patients at highest risk for being affected.
In the mid 1980s, two nascent technologies, human genetics and echocardiography, became sufficiently advanced to apply these to the study of hypertrophic cardiomyopathy (HCM). Until then, genetic researchers had limited biomarkers, for example ABO13 and HLA14 typing that could be used to discover the chromosome location of human disease genes. But in 1980 Botstein and colleagues15 theorized that DNA sequences that were scattered throughout the genome and encoded cleavage sites for restriction enzymes, could theoretically expand this potential. In combination with statistical methodologies to assess the likelihood of co-inheritance of traits while considering genomic recombination events,16 the field of linkage analyses was born. An early success was the mapping of the Huntington disease gene to chromosome 4 by Gusella and colleagues,17 an impressive achievement realized through the analyses of only 12 restriction fragment length polymorphisms (RFLPs) in one large affected kindred. Hundreds of novel RFLPs were soon characterized, and in 1987 the first genetic linkage map of the human genome was reported.18 This resource empowered a simple and elegant experimental design that provided a foothold, the chromosome location, for identification of gene mutations that were responsible for any inherited disease. Four steps were required: accurate ascertainment of affection status in families of sufficient size to define the pattern of inheritance; a renewable source of DNA, usually obtained from EBV-transformed lymphocytes; sequential Southern blot analyses to interrogate potentially hundreds of RFLPs located throughout the genome; statistical analyses that demonstrated significant co-segregation of RFLPs in affected family members.
Despite the feasibility of these steps, analyses were not trivial and success never a guarantee. The early genomic assignments of many RFLPs were erroneous or fairly imprecise, which accounted for a 1985 report of linkage of the cystic fibrosis gene to RFLPs D0CRI-917 and PON,19 rather than to chromosome 7,20 where these markers were eventually shown to reside. More vexing was the problem of inaccurate diagnoses, due to subclinical findings in otherwise healthy individuals or due to prevalent phenocopies of the disease under study. Undisclosed familial structures (e.g., nonpaternity, intrafamilial adoption) posed still more challenges.
Yet the value of knowing the chromosome location of a disease gene was considerable; this information immediately suggested candidates worthy of further study, based on their “guilt” by genomic location. By establishing expression of candidate genes in diseased tissue and identifying sequence variants in affected patients (but not unaffected relatives or controls) that were predicted to deleteriously impact the encoded protein’s structure or function, the definitive disease gene could be identified – even without a prior knowledge of its biologic function or potential mechanistic role in disease pathophysiology. For researchers studying myocardial diseases this strategy allowed triumph over many challenges faced by this discipline - the absence of myocardial cell lines, limited access to human cardiac tissues, and a paucity of robust models of heart disease.
Rapid clinical advances in cardiac imaging during this time also made hypertrophic cardiomyopathy ripe for human genetic analyses. Cardiac catheterization was the initial gold-standard approach for diagnosis,21, 22 but this invasive technique was impractical for defining the affection status of research subjects, in particular those who appeared unaffected based on symptoms and physical examination. Exclusion of subjects without a definitive diagnosis could overcome this issue, but would severely compromise the statistical power of modest sized families. The advent and rapid evolution of echocardiography solved this problem and enabled an accurate, low-risk, and low cost approach for family screening.23–25 The widespread adoption of echocardiography in clinical cardiology revealed the unanticipated finding that unexplained LVH, the diagnostic sine quo non of hypertrophic cardiomyopathy, occurs in 1 of 500 individuals.26
Collaborating Clinicians, Families and Researchers
We approached Bill McKenna to join a collaboration aimed at discovering the genetic basis for hypertrophic cardiomyopathy in 1987. Trained by Goodwin, McKenna had established expertise in the diverse clinical and echocardiographic manifestations of hypertrophic cardiomyopathy.27, 28 We focused on a large family riddled by sudden cardiac death, deemed the “Coaticook curse” by the local community, reported in 1961 by Pare and colleagues,29 as a hereditary cardiovascular dysplasia. Pare’s personal commitment to helping the family engendered such trust that they willingly agreed to participate, despite minimal understanding of genetic research. Over a hundred family members took part in weekend clinics where we performed physical examinations, EKGs, echocardiograms, and obtained blood samples. These studies complemented Pare’s earlier detailed clinical investigations and provided clear evidence for dominant inheritance of the cardiomyopathy and age-related disease expression, information that was critical for designing linkage studies.
In the laboratory scores of Southern blots were performed to assess RFLPs scattered across the genome, until definitive linkage was established with a chromosome 14q marker that yielded a LOD score = 9.3, indicating odds of 2,000,000,000:1 that the disease gene was nearby.30 This finding immediately identified two attractive albeit unexpected candidate genes, MYH6 and MYH7, that encode the α and β myosin heavy chains respectively. Both genes were robustly expressed in the heart, separated by only 3600 bp on chromosome 14,31 and the complete nucleotide sequence of MYH7 was already published.32, 33 Despite these resources, it was technically impractical to directly sequence the 30 kb of both genes in samples from affected individuals. We elected to fine-structure map the locus with restriction enzymes. While this was ultimately a productive decision, this approach initially led to the identification of a polymorphic MYH7 fragment in all samples from affected family members as well as in one unaffected individual. Several vexing possibilities might have accounted for this result - sample mix up, mistaken assignment of clinical status, false familial relationships, or a far more deflating concern, the potential that MYH7 was linked to, but was not the disease gene. As we tackled each possibility, our dilemma was resolved through the confidential disclosure of non-paternity, therein removing the sole piece of data that refuted MYH7 as a disease gene. Divulging this personal secret was truly an heroic contribution that accelerate discovery of the cause of HCM.
Sequence analyses began immediately and revealed a missense variant in exon 13 of MYH7 that substituted a highly conserved arginine residue (403) with glutamine (denoted Arg403Glu) in all affected, but no unaffected family members.34 Concurrent restriction mapping of samples from other families revealed a rearrangement of the chromosome 14q locus characterized by a hybrid MYH6-MYH7 gene as well as non-rearranged MYH6 and MYH7 genes.35 Future studies36 would reveal a missense variant (Arg453Cys) in the non-rearranged MYH7 gene as the definitive cause of disease, a mutation that also arose independently in another HCM family without the locus rearrangement.
The identification of rare non-synonymous variants that segregated with HCM in unrelated families,36–38 which altered residues that were highly conserved during evolution and were absent from hundreds of control samples, provided compelling evidence that MYH7 mutations caused HCM. But analyses of MYH7 in many other HCM families were unrevealing of other mutations39, 40 and new linkage searches began to identify additional disease genes. HCM loci were mapped genes to chromosome 1q341 and 15q242. Identification of the HCM locus on 15q2 location was particularly fortuitous, despite a lack of candidates that were known to reside within that region of the human genome. The syntenic region of the mouse genome was known to encode α tropomyosin,43 one of several thin filament sarcomere proteins that regulates actinomyosin interactions.44 By harnessing information from the mouse gene, we cloned the human homologue, TPM1, mapped this to 15q2 and identified human mutations. Armed with the recognition that 2 HCM genes encoded contractile proteins, the disease gene encoded at 1q3 was soon recognized to be troponin T (TNNT2)42. An enigmatic myocardial disorder was no longer idiopathic, but a disease of the sarcomere.
Multiple research teams solidified the concept that sarcomere mutations caused HCM (Fig. 2). Schwartz and colleagues and our laboratory independently identified another HCM locus on chromosome 11,45 where the thick filament myosin binding protein-C (MYBPC3) was mapped46 and mutations identified.47, 48 Analyses of the myosin essential light chain (MYL3) and regulatory chain (MYL2) genes by Epstein and colleagues,49 troponin I (TNNI3) by Sasazuki and colleagues,50 and cardiac actin (ACTC) by Mogensen, Olson, Fanapazir and colleagues51 soon followed. After these teams reported variants that fulfilled criteria for pathogenic mutations, additional reports of mutations identified by other researchers in HCM families confirmed a definitive role for each of these genes in HCM.
Figure 2.

Definitive and posited HCM genes. A schematic representation of a sarcomere (the unit of contraction that spans two neighboring Z-bands) is shown with the locations of thin and thick filaments in relationship to I, A and H bands. The detailed representation of the A band, highlights definitive HCM genes (black) that encode β myosin heavy chain (MYH7), myosin binding protein C (MYPBC3), troponin T (TNNT2), α tropomyosin (TPMI), myosin regulatory light chain (MYL2), myosin essential light chain (MYL3) and (ACTC). More than 50% of human mutations occur in β myosin heavy chain and myosin binding protein C. Additional genes that have been implicated in HCM (brown) encode Z-disc proteins, troponin C (TNNT1), titin (TNT), α myosin heavy chain (MYH6, not depicted) and phospholamban (PLN, not depicted).
Genes mutations in other sarcomere proteins (α myosin heavy chain,52, 53 titin54 and troponin C55), z disc-associated proteins (actinin,56 ankyrin,57 myozenin 2,58 muscle LIM protein,59 nexilin,60, 61 and telethonin61, 62), and proteins involved in other myocyte functions (phospholamban63 and viniculin64) are also posited to cause HCM (Fig. 2). Among these, only the genes encoding myozenin-258 and actinin56 were identified from unbiased genomic analyses, and novel variants in each were demonstrated to exhibit statistically significant segregation within HCM families. Identification of mutations in additional families will further support the causality of myozenin-2 and actinin in HCM. Whether the variants identified in other posited HCM genes are truly pathogenic remains less certain and warrants further confirmation, especially given the recent recognition that each human genome contains approximately 1000 rare non-synonymous variants, including premature stop signals.65
Genetic Heterogeneity and Founding Mutations in HCM
Over 1000 distinct sarcomere protein gene mutations have been identified to cause HCM, with the vast majority of these found in one or only a few families.66 Moreover haplotype analyses of unrelated patients who share a common HCM mutation typically indicate that these defect arose from independent mutational events.67 Two theories may account for the considerable genetic heterogeneity observed in HCM. Although HCM is not usually thought to have substantial impact on reproductive health, sudden cardiac death in the young and early-onset of significant co-morbidities such as thromboembolism, stroke, and heart failure,68 and increasingly personal choice, promote the gradual loss of HCM mutations from populations. In addition, because mutational events are presumed to occur randomly in the genome, larger genes are more likely to acquire these defects. Protein-encoding sequences for the 8 sarcomere protein genes that definitively cause HCM encompass approximately 23 kb, therein providing an ample target for new mutational events.
Recent genetic studies also indicate that some HCM reflects ancient, founding mutations. In addition to sharing an identical HCM mutation, the extended haplotypes flanking these HCM genes are shared among mutation carriers. Reported in the Netherlands,69 Finland,70 South Africa,71 Italy,72 India,73 Japan,74 and the U.S.,75 founding HCM mutations might be expected in populations that are relatively genetically homogenous due to geographic or social isolationism, that experienced a population bottleneck, or when there is little selective disadvantage from the mutation. That founding mutations almost universally occur in MYBPC3 and typically encode a truncated protein, is remarkable, and implies that these defects have far less deleterious consequences than other HCM mutations. As such, founding mutations provide evidence that distinct clinical outcomes arise from particular HCM mutations. Based upon genetic epidemiologic analyses, the 25-basepair deletion in MYBPC3 found in 4% of individuals with Southeast Indian ancestry, arose approximately 33 ± 23 thousand years ago.73 Maintenance of this genomic defect is best explained by neutral selection throughout human evolution, which would occur if this HCM mutation had minimal impact on survival. Indeed, only with improved human health and increased life expectancy has the medical significance of this MYBPC3 mutation become evident: a 7-fold increased risk for heart failure late in life.
The contrast of genetic heterogeneity and founding mutations in HCM provides a new approach for assessing the relationship of genotype and clinical phenotype. Using an evolutionary perspective, founding mutations are expected to have less adverse impact on cardiac physiology and be associated with better prognosis than other HCM mutations. A corollary of this model is that when a new HCM mutation arises in a genome that carries or lacks a founder mutation, particularly severe consequence may ensue. Comparison of outcomes in patients with one MYBPC3 founding mutation that occurs alone or combination with another HCM defect75 illustrates how clinical course can diverge due to different genotypes. Consideration of these issues is important for clinical application of HCM genotypes and for designing cost-effective gene-based diagnostic platforms.76
Harnessing Mutations to Probe Mechanism
With the possible exception of truncating MYPC3 mutations,77, 78 other pathogenic HCM mutations79 encode the insertion of a single erroneous amino acid into one contractile protein – a change that is unlikely to have egregious effects on stability or prevent incorporation of the mutant peptide into the sarcomere. Moreover because mutations are dominant, HCM sarcomeres contain an admixture of normal and mutant proteins. These factors imply that the consequences of HCM mutations will be significant but subtle, which may account for the slow emergence of clinical disease in mutation carriers.
Early studies of the biochemical and biophysical consequences of HCM mutations capitalized on recombinant technologies that enabled heterologous expression of some sarcomere proteins in bacteria80 and cells.81 Mutant myosin heavy chain fragments were produced and shown to bind ATP normally81, 82, but to have reduced ATPase activity and in vitro actin-sliding motility in comparison to wildtype fragments81. Ex vivo analyses of myosins isolated from skeletal muscles of HCM patients83 similarly showed abnormal actin-myosin interactions. As such, MYH7 mutations appeared to reduce force production by the sarcomere, a result that implied that hypertrophic remodeling was a compensatory response to diminished cardiac performance.
Further insights came from the study of HCM mutations that were engineered into mice.84, 85 Our lab produced mice with 3 different myosin mutations, Arg403Gln,86 Arg719Gln,87 or Arg453Cys.88 Equivalent to disease in human patients, the clinical expression of HCM in mice (ventricular hypertrophy, myocyte disarray and increased myocardial fibrosis) is delayed until early in adulthood.86 Isolation of mutant myosins from young pre-hypertrophic mice allowed assessment of single molecule mechanics (reviewed by Leinwand, Spudich and colleagues89) of myosins carrying HCM mutations. Surprisingly, mutant myosin heavy chains exhibited enhanced contractile properties, including increased force generation, ATP hydrolysis and action-myosin sliding velocities, indicating that HCM mutations produce a gain in function.90 Similar conclusions were reached from analyses of human HCM mutations introduced into troponin T.91 In HCM hearts, the admixture of mutant and wildtype sarcomere proteins might have an additional consequence, of uncoordinated activity.
Based upon these findings, we hypothesized that mutant sarcomeres activate signaling pathways to initiate hypertrophic remodeling. To identify these, myocytes from young pre-hypertrophic mice were studied using biochemical analyses and impairment of calcium homeostasis was found.92 Recent investigations appear to link the early-onset of calcium abnormalities in mutant myocytes with later events in HCM hearts. Abnormal calcium signals can lead to Mef2 activation in HCM hearts through calcium/calmodulin dependent protein kinase II (CaMKII) phosphorylation.93 We observed heterogeneous Mef2 activation in HCM hearts, which was localized in myocytes juxtaposed to foci of necrosis and replacement fibrosis. By studying mice bred to carry homozygous HCM mutations, Mef2 activation was substantially increased and evident shortly before rampant myocyte death that occurs in this model. Together these studies implicate calcium-dependent Mef-2 activation as a signal that defines profoundly stressed and pre-morbid mutant myocytes, and predestines sites of focal myocardial scarring in HCM hearts.
Transcriptional profiling of ventricular tissues94 and isolated cells87 from pre-hypertrophic HCM has also been informative of pathways activated by sarcomere mutations. Expression of Tgfβ (transforming growth factor–β), connective tissue growth factor and periostin are increased in the ventricles of young mutant mice, implying that these potent regulators of fibrosis and collagen deposition contribute to impaired cardiac relaxation and the characteristic histopathology of HCM. Our recent studies indentified the source of these molecules to be non-myocyte cells,87 presumably cardiac fibroblasts, and demonstrated that continual inhibition of Tgfβ, initiated in the pre-hypertrophic phase of disease, reduced the emergence of fibrosis and limited hypertrophy remodeling that emerged in untreated mutant mice.
By combining knowledge of the genetic basis for HCM, with insights gleaned from biochemical, biophysical and transcriptional analyses in HCM models, an integrated understanding of the pathophysiology of disease has begun to emerge. HCM mutations disrupt normal biophysical properties of sarcomere and cause mechanical and calcium-induced biochemical signals that activate gene transcription, including increased Tgfβ expression. Tgfβ may stimulate expression of pro-fibrotic molecules and fibroblast proliferation, which expands the interstitium and may promote development of diastolic dysfunction, a clinical hallmark of HCM. Persistent activation of this pathway is predicted to impose stress on mutant myocytes (evidenced by Mef2 activation), leading to premature death with focal myocardial scarring.
Future Opportunities
The considerable diversity of HCM mutations has provided an unparalleled set of molecular reagents for exploring sarcomere biology. Continued elucidation of the impact of mutations on myocyte contraction, relaxation, and signaling is expected to expand knowledge of cardiac biology in health and disease. Articles that will follow this perspective will explore these concepts in depth.
From a human genetics perspective, there is still more to learn about HCM. Understanding the cause in 25–35% of HCM patients without a pathogenic mutation remains an important challenge. Defining the biochemical, morphologic and functional changes in large genotyped cohorts, from the pre-clinical phase of disease to overt HCM, may provide inform the discovery of molecules and mechanisms that limit clinical expression in young mutation carriers or that account for the emergence of HCM in adolescence and beyond. Developing new cellular models through induced pluripotent stem cells should enable high-throughput investigation of myocyte responses to many more sarcomere gene mutations. Application of whole genome sequencing technologies in patients with atypical clinical presentations may uncover the roles of modifying genetic factors in HCM. Pursuit of these and other research opportunities in HCM95 hold the promise for better ways to diagnose, treat and prevent this extraordinary disease.
Acknowledgments
We thank families, physicians, collaborators, and especially the many past and present trainees who participated in these studies. Their efforts account for discoveries that have advanced understanding of HCM.
Sources of Funding
This work is supported by grants from the NIH and LeDucq Foundation (to J.G.S. and C.E.S) and the Howard Hughes Medical Institute (to C.E.S.).
Footnotes
Disclosures
None.
References
- 1.Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8. doi: 10.1136/hrt.20.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brock R. Functional obstruction of the left ventricle; acquired aortic subvalvar stenosis. Guys Hosp Rep. 1957;106:221–238. [PubMed] [Google Scholar]
- 3.Morrow AG, Braunwald E. Functional aortic stenosis; a malformation characterized by resistance to left ventricular outflow without anatomic obstruction. Circulation. 1959;20:181–189. doi: 10.1161/01.cir.20.2.181. [DOI] [PubMed] [Google Scholar]
- 4.Goodwin JF, Hollman A, Cleland WP, Teare D. Obstructive cardiomyopathy simulating aortic stenosis. Br Heart J. 1960;22:403–414. doi: 10.1136/hrt.22.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braunwald E, Ebert PA. Hemogynamic alterations in idiopathic hypertrophic subaortic stenosis induced by sympathomimetic drugs. Am J Cardiol. 1962;10:489–495. doi: 10.1016/0002-9149(62)90373-9. [DOI] [PubMed] [Google Scholar]
- 6.Coats CJ, Hollman A. Hypertrophic cardiomyopathy: lessons from history. Heart. 2008;94:1258–1263. doi: 10.1136/hrt.2008.153452. [DOI] [PubMed] [Google Scholar]
- 7.Liouville H. Retrecissement cardiaque sous aortique. Gazette Medicale de Paris. 1869;24:161–163. [Google Scholar]
- 8.Hallopeau M. Retrecissement ventriculo-aortique. Gazette Medicale de Paris. 1869;24:683–687. [Google Scholar]
- 9.Maron BJ, Gottdiener JS, Goldstein RE, Epstein SE. Hypertrophic cardiomyopathy: the great masquerader. Clinical conference from the Cardiology Branch of the National Heart, Lung, and Blood Institute, Bethesda, Md. Chest. 1978;74:659–670. doi: 10.1378/chest.74.6.659. [DOI] [PubMed] [Google Scholar]
- 10.Goodwin JF. Prospects and predictions for the cardiomyopathies. Circulation. 1974;50:210–219. doi: 10.1161/01.cir.50.2.210. [DOI] [PubMed] [Google Scholar]
- 11.Darsee JR, Heymsfield SB, Nutter DO. Hypertrophic cardiomyopathy and human leukocyte antigen linkage: differentiation of two forms of hypertrophic cardiomyopathy. N Engl J Med. 1979;300:877–882. doi: 10.1056/NEJM197904193001602. [DOI] [PubMed] [Google Scholar]
- 12.Nutter DO, Heymsfield SB, Glenn JF. Retraction. Darsee JR, Heymsfield SB, Nutter DO. Hypertrophic cardiomyopathy and human leukocyte antigen linkage: differentiation of two forms of hypertrophic cardiomyopathy. N Engl J Med 1979;300:877–82. N Engl J Med. 1983;308:1400. doi: 10.1056/nejm198306093082307. [DOI] [PubMed] [Google Scholar]
- 13.Smith CA. Linkage in blood group genetics. Bibl Haematol. 1965;23:263–267. doi: 10.1159/000384256. [DOI] [PubMed] [Google Scholar]
- 14.Svejgaard A, Platz P, Ryder LP, Nielsen LS, Thomsen M. HL-A and disease associations--a survey. Transplant Rev. 1975;22:3–43. doi: 10.1111/j.1600-065x.1975.tb01550.x. [DOI] [PubMed] [Google Scholar]
- 15.Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 1980;32:314–331. [PMC free article] [PubMed] [Google Scholar]
- 16.Ott J. Estimation of the recombination fraction in human pedigrees: efficient computation of the likelihood for human linkage studies. Am J Hum Genet. 1974;26:588–597. [PMC free article] [PubMed] [Google Scholar]
- 17.Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY, Young AB, Shouldson I, Bonilla E, Martin JB. A polymorphic DNA marker genetically linked to Huntington's disease. Nature. 1983;306(5):234–238. doi: 10.1038/306234a0. [DOI] [PubMed] [Google Scholar]
- 18.Donis-Keller H, Green P, Helms C, Cartinhour S, Weiffenbach B, Stephens K, Keith TP, Bowden DW, Smith DR, Lander ES, Botstein D, Akots G, Rediker KS, Gravius T, Brown VA, Rsing MB, Parker C, Powers JA, Watt DE, Kauffman ER, Bricker A, Phipps P, Muller-Kahle H, Fulton TR, Ng S, Schumm JW, Braman JC, Knowlton RG, Barker DF, Crooks SM, Lincoln SE, Daly MJ, Abrahamson J. A genetic linkage map of the human genome. Cell. 1987;51:319–337. doi: 10.1016/0092-8674(87)90158-9. [DOI] [PubMed] [Google Scholar]
- 19.Tsui LC, Buchwald M, Barker D, Braman JC, Knowlton R, Schumm JW, Eiberg H, Mohr J, Kennedy D, Plavsic N. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science. 1985;230:1054–1057. doi: 10.1126/science.2997931. [DOI] [PubMed] [Google Scholar]
- 20.Buchwald M, Zsiga M, Markiewicz D, Plavsic N, Kennedy D, Zengerling S, Willard HF, Tsipouras P, Schmiegelow K, Schwartz M, Eiberg H, Mohr J, Barker D, Donis-Keller H, Tsue LC. Linkage of cystic fibrosis to the pro alpha 2(I) collagen gene, COL1A2, on chromosome 7. Cytogenet Cell Genet. 1986;41:234–239. doi: 10.1159/000132235. [DOI] [PubMed] [Google Scholar]
- 21.Braunwald E, Lambrew CT, Rockoff SD, Ross J, Jr, Morrow AG. Idiopathic Hypertrophic Subaortic Stenosis. I. A Description of the Disease Based Upon an Analysis of 64 Patients. Circulation. 1964;30(SUPPL 4):3–119. doi: 10.1161/01.cir.29.5s4.iv-3. [DOI] [PubMed] [Google Scholar]
- 22.Wigle ED, Auger P, Marquis Y. Muscular subaortic stenosis. The direct relation between the intraventricular pressure difference and the left ventricular ejection time. Circulation. 1967;36:36–44. doi: 10.1161/01.cir.36.1.36. [DOI] [PubMed] [Google Scholar]
- 23.Popp RL, Harrison DC. Ultrasound in the diagnosis and evaluation of therapy of idiopathic hypertrophic subaortic stenosis. Circulation. 1969;40:905–914. doi: 10.1161/01.cir.40.6.905. [DOI] [PubMed] [Google Scholar]
- 24.Shah PM, Gramiak R, Adelman AG, Wigle ED. Role of echocardiography in diagnostic and hemodynamic assessment of hypertrophic subaortic stenosis. Circulation. 1971;44:891–898. doi: 10.1161/01.cir.44.5.891. [DOI] [PubMed] [Google Scholar]
- 25.Henry WL, Clark CE, Epstein SE. Asymmetric septal hypertrophy. Echocardiographic identification of the pathognomonic anatomic abnormality of IHSS. Circulation. 1973;47:225–233. doi: 10.1161/01.cir.47.2.225. [DOI] [PubMed] [Google Scholar]
- 26.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92(4):785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 27.McKenna WJ, Goodwin JF. The natural history of hypertrophic cardiomyopathy. Curr Probl Cardiol. 1981;6:1–26. doi: 10.1016/0146-2806(81)90015-3. [DOI] [PubMed] [Google Scholar]
- 28.McKenna W, Deanfield J, Faruqui A, England D, Oakley C, Goodwin J. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol. 1981;47:532–538. doi: 10.1016/0002-9149(81)90535-x. [DOI] [PubMed] [Google Scholar]
- 29.Pare JA, Fraser RG, Pirozynski WJ, Shanks JA, Stubington D. Hereditary cardiovascular dysplasia. A form of familial cardiomyopathy. Am J Med. 1961;31:37–62. doi: 10.1016/0002-9343(61)90222-4. [DOI] [PubMed] [Google Scholar]
- 30.Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, Levi T, Donis-Keller H, Seidman JG, Seidman CE. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–1378. doi: 10.1056/NEJM198911163212005. [DOI] [PubMed] [Google Scholar]
- 31.Saez LJ, Gianola KM, McNally EM, Feghali R, Eddy R, Shows TB, Leinwand LA. Human cardiac myosin heavy chain genes and their linkage in the genome. Nucleic Acids Res. 1987;15:5443–5459. doi: 10.1093/nar/15.13.5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liew CC, Sole MJ, Yamauchi-Takihara K, Kellam B, Anderson DH, Lin LP, Liew JC. Complete sequence and organization of the human cardiac beta-myosin heavy chain gene. Nucleic Acids Res. 1990;18:3647–3651. doi: 10.1093/nar/18.12.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M, Bach A, Vosberg HP. The complete sequence of the human beta-myosin heavy chain gene and a comparative analysis of its product. Genomics. 1990;8:194–206. doi: 10.1016/0888-7543(90)90272-v. [DOI] [PubMed] [Google Scholar]
- 34.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 35.Tanigawa G, Jarcho JA, Kass S, Solomon SD, Vosberg HP, Seidman JG, Seidman CE. A molecular basis for familial hypertrophic cardiomyopathy: an alpha/beta cardiac myosin heavy chain hybrid gene. Cell. 1990;62:991–998. doi: 10.1016/0092-8674(90)90273-h. [DOI] [PubMed] [Google Scholar]
- 36.Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326:1108–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 37.Marian AJ, Yu QT, Mares A, Jr, Hill R, Roberts R, Perryman MB. Detection of a new mutation in the beta-myosin heavy chain gene in an individual with hypertrophic cardiomyopathy. J Clin Invest. 1992;90:2156–2165. doi: 10.1172/JCI116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dufour C, Dausse E, Fetler L, Dubourg O, Bouhour JB, Vosberg HP, Guicheney P, Komajda M, Schwartz K. Identification of a mutation near a functional site of the beta cardiac myosin heavy chain gene in a family with hypertrophic cardiomyopathy. J Mol Cell Cardiol. 1994;26:1241–1247. doi: 10.1006/jmcc.1994.1142. [DOI] [PubMed] [Google Scholar]
- 39.Solomon SD, Jarcho JA, McKenna W, Geisterfer-Lowrance A, Germain R, Salerni R, Seidman JG, Seidman CE. Familial hypertrophic cardiomyopathy is a genetically heterogeneous disease. J Clin Invest. 1990;86:993–999. doi: 10.1172/JCI114802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz K, Beckmann J, Dufour C, Faure L, Fougerousse F, Carrier L, Hengstenberg C, Cohen D, Vosberg HP, Sacrez A. Exclusion of cardiac myosin heavy chain and actin gene involvement in hypertrophic cardiomyopathy of several French families. Circ Res. 1992;71:3–8. doi: 10.1161/01.res.71.1.3. [DOI] [PubMed] [Google Scholar]
- 41.Watkins H, MacRae C, Thierfelder L, Chou YH, Frenneaux M, McKenna W, Seidman JG, Seidman CE. A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat Genet. 1993;3:333–337. doi: 10.1038/ng0493-333. [DOI] [PubMed] [Google Scholar]
- 42.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 43.Schleef M, Werner K, Satzger U, Kaupmann K, Jockusch H. Chromosomal localization and genomic cloning of the mouse alpha-tropomyosin gene Tpm-1. Genomics. 1993;17:519–521. doi: 10.1006/geno.1993.1361. [DOI] [PubMed] [Google Scholar]
- 44.Leger JJ, Berson G, Delcaryre C, Klotz C, Schwartz K, Leger J, Stephens M, Swynghedauw B. Heart contractile proteins. Biochimie. 1975;57:1249–1273. doi: 10.1016/s0300-9084(76)80538-x. [DOI] [PubMed] [Google Scholar]
- 45.Carrier L, Hengstenberg C, Beckmann JS, Guicheney P, Dufour C, Bercovici J, Dausse E, Berebbi-Bertrand I, Wisnewsky C, Pulvenis D, et al. Mapping of a novel gene for familial hypertrophic cardiomyopathy to chromosome 11. Nat Genet. 1993;4:311–313. doi: 10.1038/ng0793-311. [DOI] [PubMed] [Google Scholar]
- 46.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP, Fiszman M, Komajda M, Schwartz K. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 48.Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 49.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 50.Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo JA, Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, Matsuzaki M, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16:379–382. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- 51.Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1687–1694. doi: 10.1006/jmcc.2000.1204. [DOI] [PubMed] [Google Scholar]
- 52.Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG, Seidman CE. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105:446–451. doi: 10.1161/hc0402.102990. [DOI] [PubMed] [Google Scholar]
- 53.Carniel E, Taylor MR, Sinagra G, Di Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov D, Graw SL, Feiger J, Zhu XZ, Dao D, Ferguson DA, Bristow MR, Mestroni L. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation. 2005;112:54–59. doi: 10.1161/CIRCULATIONAHA.104.507699. [DOI] [PubMed] [Google Scholar]
- 54.Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun. 1999;262:411–417. doi: 10.1006/bbrc.1999.1221. [DOI] [PubMed] [Google Scholar]
- 55.Hoffmann B, Schmidt-Traub H, Perrot A, Osterziel KJ, Gessner R. First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum Mutat. 2001;17:524. doi: 10.1002/humu.1143. [DOI] [PubMed] [Google Scholar]
- 56.Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, Jormakka M, Lind JM, Semsarian C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J Am Coll Cardiol. 55:1127–1135. doi: 10.1016/j.jacc.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 57.Arimura T, Bos JM, Sato A, Kubo T, Okamoto H, Nishi H, Harada H, Koga Y, Moulik M, Doi YL, Towbin JA, Ackerman MJ, Kimura A. Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:334–342. doi: 10.1016/j.jacc.2008.12.082. [DOI] [PubMed] [Google Scholar]
- 58.Osio A, Tan L, Chen SN, Lombardi R, Nagueh SF, Shete S, Roberts R, Willerson JT, Marian AJ. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2007;100:766–768. doi: 10.1161/01.RES.0000263008.66799.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knoll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, Gunkel S, Kotter S, Babicz K, Sohns M, Miocic S, Didie M, Knoll G, Zimmermann WH, Thelen P, Bickeboller H, Maier LS, Schaper W, Schaper J, Kraft T, Tschope C, Linke WA, Chien KR. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res. 106:695–704. doi: 10.1161/CIRCRESAHA.109.206243. [DOI] [PubMed] [Google Scholar]
- 60.Wang H, Li Z, Wang J, Sun K, Cui Q, Song L, Zou Y, Wang X, Liu X, Hui R, Fan Y. Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am J Hum Genet. 87:687–693. doi: 10.1016/j.ajhg.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bos JM, Poley RN, Ny M, Tester DJ, Xu X, Vatta M, Towbin JA, Gersh BJ, Ommen SR, Ackerman MJ. Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab. 2006;88:78–85. doi: 10.1016/j.ymgme.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi T, Arimura T, Itoh-Satoh M, Ueda K, Hohda S, Inagaki N, Takahashi M, Hori H, Yasunami M, Nishi H, Koga Y, Nakamura H, Matsuzaki M, Choi BY, Bae SW, You CW, Han KH, Park JE, Knoll R, Hoshijima M, Chien KR, Kimura A. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2192–2201. doi: 10.1016/j.jacc.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 63.Landstrom AP, Adekola BA, Bos JM, Ommen SR, Ackerman MJ. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. Am Heart J. 161:165–171. doi: 10.1016/j.ahj.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasile VC, Ommen SR, Edwards WD, Ackerman MJ. A missense mutation in a ubiquitously expressed protein, vinculin, confers susceptibility to hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2006;345:998–1003. doi: 10.1016/j.bbrc.2006.04.151. [DOI] [PubMed] [Google Scholar]
- 65.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010;122:2430–2440. doi: 10.1161/CIRCULATIONAHA.110.978924. discussion 2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watkins H, Thierfelder L, Anan R, Jarcho J, Matsumori A, McKenna W, Seidman JG, Seidman CE. Independent origin of identical beta cardiac myosin heavy-chain mutations in hypertrophic cardiomyopathy. Am J Hum Genet. 1993;53:1180–1185. [PMC free article] [PubMed] [Google Scholar]
- 68.Coats CJ, Elliott PM. Current management of hypertrophic cardiomyopathy. Curr Treat Options Cardiovasc Med. 2008;10:496–504. doi: 10.1007/s11936-008-0042-9. [DOI] [PubMed] [Google Scholar]
- 69.Alders M, Jongbloed R, Deelen W, van den Wijngaard A, Doevendans P, Ten Cate F, Regitz-Zagrosek V, Vosberg HP, van Langen I, Wilde A, Dooijes D, Mannens M. The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J. 2003;24:1848–1853. doi: 10.1016/s0195-668x(03)00466-4. [DOI] [PubMed] [Google Scholar]
- 70.Jaaskelainen P, Miettinen R, Karkkainen P, Toivonen L, Laakso M, Kuusisto J. Genetics of hypertrophic cardiomyopathy in eastern Finland: few founder mutations with benign or intermediary phenotypes. Ann Med. 2004;36:23–32. doi: 10.1080/07853890310017161. [DOI] [PubMed] [Google Scholar]
- 71.Moolman-Smook JC, De Lange WJ, Bruwer EC, Brink PA, Corfield VA. The origins of hypertrophic cardiomyopathy-causing mutations in two South African subpopulations: a unique profile of both independent and founder events. Am J Hum Genet. 1999;65:1308–1320. doi: 10.1086/302623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Girolami F, Olivotto I, Passerini I, Zachara E, Nistri S, Re F, Fantini S, Baldini K, Torricelli F, Cecchi F. A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown) 2006;7:601–607. doi: 10.2459/01.JCM.0000237908.26377.d6. [DOI] [PubMed] [Google Scholar]
- 73.Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, Tharkan JM, Vaideeswar P, Rathinavel A, Narasimhan C, Ayapati DR, Ayub Q, Mehdi SQ, Oppenheimer S, Richards MB, Price AL, Patterson N, Reich D, Singh L, Tyler-Smith C, Thangaraj K. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41:187–191. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kubo T, Kitaoka H, Okawa M, Matsumura Y, Hitomi N, Yamasaki N, Furuno T, Takata J, Nishinaga M, Kimura A, Doi YL. Lifelong left ventricular remodeling of hypertrophic cardiomyopathy caused by a founder frameshift deletion mutation in the cardiac Myosin-binding protein C gene among Japanese. J Am Coll Cardiol. 2005;46:1737–1743. doi: 10.1016/j.jacc.2005.05.087. [DOI] [PubMed] [Google Scholar]
- 75.Saltzman AJ, Mancini-DiNardo D, Li C, Chung WK, Ho CY, Hurst S, Wynn J, Care M, Hamilton RM, Seidman GW, Gorham J, McDonough B, Sparks E, Seidman JG, Seidman CE, Rehm HL. Short communication: the cardiac myosin binding protein C Arg502Trp mutation: a common cause of hypertrophic cardiomyopathy. Circ Res. 106:1549–1552. doi: 10.1161/CIRCRESAHA.109.216291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wordsworth S, Leal J, Blair E, Legood R, Thomson K, Seller A, Taylor J, Watkins H. DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J. 2010;31:926–935. doi: 10.1093/eurheartj/ehq067. [DOI] [PubMed] [Google Scholar]
- 77.van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 78.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 79.Nishi H, Kimura A, Harada H, Koga Y, Adachi K, Matsuyama K, Koyanagi T, Yasunaga S, Imaizumi T, Toshima H, et al. A myosin missense mutation, not a null allele, causes familial hypertrophic cardiomyopathy. Circulation. 1995;91:2911–2915. doi: 10.1161/01.cir.91.12.2911. [DOI] [PubMed] [Google Scholar]
- 80.Eldin P, Le Cunff M, Diederich KW, Jaenicke T, Cornillon B, Mornet D, Vosberg HP, Leger JJ. Expression of human beta-myosin heavy chain fragments in Escherichia coli; localization of actin interfaces on cardiac myosin. J Muscle Res Cell Motil. 1990;11:378–391. doi: 10.1007/BF01739759. [DOI] [PubMed] [Google Scholar]
- 81.Sweeney HL, Straceski AJ, Leinwand LA, Tikunov BA, Faust L. Heterologous expression of a cardiomyopathic myosin that is defective in its actin interaction. J Biol Chem. 1994;269:1603–1605. [PubMed] [Google Scholar]
- 82.Eldin P, Le Cunff M, Mornet D, Leger JJ. The cardiac myosin heavy chain Arg-403-->Gln mutation that causes hypertrophic cardiomyopathy does not affect the actin- or ATP-binding capacities of two size-limited recombinant myosin heavy chain fragments. Biochem J. 1995;306 ( Pt 2):345–351. doi: 10.1042/bj3060345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cuda G, Fananapazir L, Zhu WS, Sellers JR, Epstein ND. Skeletal muscle expression and abnormal function of beta-myosin in hypertrophic cardiomyopathy. J Clin Invest. 1993;91:2861–2865. doi: 10.1172/JCI116530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- 85.Shephard R, Semsarian C. Role of animal models in HCM research. J Cardiovasc Transl Res. 2009;2:471–482. doi: 10.1007/s12265-009-9120-y. [DOI] [PubMed] [Google Scholar]
- 86.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–734. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- 87.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, Seidman JG, Seidman C, Warshaw DM. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007;293:H284–291. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 89.Sivaramakrishnan S, Ashley E, Leinwand L, Spudich JA. Insights into human beta-cardiac myosin function from single molecule and single cell studies. J Cardiovasc Transl Res. 2009;2:426–440. doi: 10.1007/s12265-009-9129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tyska MJ, Hayes E, Giewat M, Seidman CE, Seidman JG, Warshaw DM. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ Res. 2000;86:737–744. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- 91.Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, Moore RL, Robbins J, Leinwand LA. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J Clin Invest. 1999;104:469–481. doi: 10.1172/JCI6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, Giewat M, Seidman CE, Seidman JG. An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Konno T, Chen D, Wang L, Wakimoto H, Teekakirikul P, Nayor M, Kawana M, Eminaga S, Gorham JM, Pandya K, Smithies O, Naya FJ, Olson EN, Seidman JG, Seidman CE. Heterogeneous myocyte enhancer factor-2 (Mef2) activation in myocytes predicts focal scarring in hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:18097–18102. doi: 10.1073/pnas.1012826107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim JB, Porreca GJ, Song L, Greenway SC, Gorham JM, Church GM, Seidman CE, Seidman JG. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science. 2007;316:1481–1484. doi: 10.1126/science.1137325. [DOI] [PubMed] [Google Scholar]
- 95.Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, Anderson ME, Boineau R, Byrne BJ, Cappola TP, Kalluri R, LeWinter MM, Maron MS, Molkentin JD, Ommen SR, Regnier M, Tang WH, Tian R, Konstam MA, Maron BJ, Seidman CE. Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation. 2010;122:1130–1133. doi: 10.1161/CIRCULATIONAHA.110.950089. [DOI] [PMC free article] [PubMed] [Google Scholar]