

Abstract
Intercalated (ITC) amygdala neurons regulate fear expression by controlling impulse traffic between the input (basolateral amygdala; BLA) and output (central nucleus; Ce) stations of the amygdala for conditioned fear responses. Previously, stimulation of the infralimbic (IL) cortex was found to reduce fear expression and the responsiveness of Ce neurons to BLA inputs. These effects were hypothesized to result from the activation of ITC cells projecting to Ce. However, ITC cells inhibit each other, leading to the question of how IL inputs could overcome the inter-ITC inhibition to regulate the responses of Ce neurons to aversive conditioned stimuli (CSs). To investigate this, we first developed a compartmental model of a single ITC cell that could reproduce their bistable electroresponsive properties, as observed experimentally. Next, we generated an ITC network that implemented the experimentally observed short-term synaptic plasticity of inhibitory inter-ITC connections. Model experiments showed that strongly adaptive CS-related BLA inputs elicited persistent responses in ITC cells despite the presence of inhibitory interconnections. The sustained CS-evoked activity of ITC cells resulted from an unusual slowly deinactivating K+ current. Finally, over a wide range of stimulation strengths, brief IL activation caused a marked increase in the firing rate of ITC neurons, leading to a persistent decrease in Ce output, despite inter-ITC inhibition. Simulations revealed that this effect depended on the bistable properties and synaptic heterogeneity of ITC neurons. These results support the notion that IL inputs are in a strategic position to control extinction of conditioned fear via the activation of ITC neurons.
Intercalated (ITC) amygdala neurons occur as multiple densely packed cell clusters distributed along the lateral and medial aspects of the basolateral amygdaloid (BLA) complex. Medial ITC clusters are thought to constitute critical regulators of classically conditioned fear responses (Pare et al. 2004) because they are in a strategic position to control impulse traffic between the sensory input and fear output stations of the amygdala: the BLA and central nucleus (Ce), respectively. Indeed, ITC cells receive glutamatergic afferents from the BLA, and send GABAergic projections to Ce (Pare and Smith 1993a,b; Royer et al. 1999; Jungling et al. 2008). In addition, ITC neurons located dorsally (ITCD) at the BLA–Ce border inhibit more ventral ones (ITCV) (Royer et al. 2000a), thereby allowing for a spatiotemporally differentiated gating of impulse traffic between BLA and Ce (Fig. 1A; Royer et al. 1999).
Figure 1.

(A) Scheme showing connectivity of the amygdala (adapted from Pare et al. 2004 and reprinted with permission from The American Physiological Society © 2004). LA receives thalamic inputs conveying information about the conditioned stimulus (CS) and unconditioned stimulus (US). LA projects to the basal nucleus (BA), and ITC neurons located dorsally (ITCD), which in turn project to ITC cells located more ventrally (ITCV). ITCV cells contribute GABAergic projections to CeM (central medial nucleus). The BA sends excitatory inputs to both ITCD and ITCV cells, and to CeM. IL also projects to both ITCD and ITCV cells. CeM projects to brainstem structures mediating fear responses. (BS) Brainstem; (Glu) glutamate. (B) Structure of the model ITC network with 15 neurons each in ITCD and ITCV clusters. Each ITC neuron inhibits three randomly selected neurons in the same cluster (only one projection per neuron is shown in the figure). Each ITCD neuron also inhibits three randomly selected ITCV neurons (e.g., ITCD2 inhibits ITCV10). For clarity and illustration purpose, the figure only shows partial connectivity, which may not be the actual connectivity used in the model (see Supplementary Table S3). The network has five Ce output neurons that receive excitatory inputs from BA, and inhibitory inputs from ITCV neurons. ITCD and ITCV: Neurons 1–5 are of type A neurons (with facilitating output synapses); neurons 6–10 are of type B (with depressing output synapses); and neurons 11–15 are of type C neurons (with constant synapses). See the Materials and Methods section entitled “Presynaptic release probability” for description of facilitating and depressing ITC synapses.
Much evidence indicates that medial ITC cell clusters participate in the extinction of conditioned fear responses (Royer and Pare 2002; Jungling et al. 2008; Likhtik et al. 2008). It is currently believed (Pare et al. 2004; Quirk and Mueller 2008) that extinguished conditioned stimuli (CS) activate infralimbic (IL) neurons with glutamatergic projections to ITC cells. In turn, ITC cells would reduce conditioned fear responses by generating feedforward inhibition in fear output Ce neurons (Pare et al. 2004). Consistent with this, IL stimulation dramatically reduces the responsiveness of Ce neurons to BLA inputs (Quirk et al. 2003). However, IL axons target all medially located ITC cells clusters (McDonald et al. 1996). Since there are inhibitory connections between (Royer et al. 2000a) as well as within ITC cells clusters (Geracitano et al. 2007), it is not immediately clear how IL inputs could overcome the inter-ITC inhibition.
Unfortunately, it is currently difficult to address this question experimentally, because we lack criteria to identify ITC cells on the basis of their extracellularly recorded activity. Thus, we developed a biophysical conductance-based model of the ITC network (Fig. 1B) to study how inter-ITC inhibitory connections affect their responses to IL inputs. A second objective of our study was to examine how the peculiar electroresponsive properties of ITC cells shape their responsiveness to BLA/IL inputs. Indeed, ITC cells express an unusual voltage-dependent K+ conductance whose slow-deinactivation kinetics allow them to produce prolonged depolarizing plateaus after a transient suprathreshold depolarization (Royer et al. 2000b). The ability of ITC neurons to transform transient excitatory inputs into a prolonged state of increased excitability may have important consequences for how they regulate conditioned fear. In keeping with this idea, during prolonged auditory CSs, BLA principal neurons show rapidly adapting responses (Quirk et al. 1995, 1997; Repa et al. 2001; Herry et al. 2008). Moreover, pairing CSs with brief (300 msec) electrical IL stimuli reduces conditioned freezing in a temporally specific manner (Milad and Quirk 2002; Milad et al. 2004). At present, it is not clear how such transient responses are converted into sustained behavioral output. We therefore tested the hypothesis that the bistable electroresponsive properties of ITC cells allow them to transform transient BLA/IL signals into a more sustained output.
Results
Overview of the model
We developed a biophysical conductance-based model of the medial ITC network (Fig. 1) with dorsal and ventral ITC modules (see Materials and Methods section). We first constructed a single-cell ITC model that could replicate the experimentally observed bistable behavior of ITC cells (Royer et al. 2000b). Short-term facilitation and depression were implemented in the GABAergic ITC–ITC and ITC–Ce connections, and their dynamics were adjusted to reproduce frequency-dependent changes in release probability, as observed experimentally (Geracitano et al. 2007). We first investigated whether the sustained activity seen in a single ITC cell could be maintained in the ITC network with inhibitory connectivity. Next, with experimentally observed changes in BLA firing rates and synaptic strengths, we examined how the ITC network regulated the firing of Ce neurons during fear conditioning and extinction. Finally, the model was used to study the impact of IL inputs on network activity. Note that in this section we used BLA to collectively refer to the lateral (LA) and basal (BA) nuclei of the amygdala.
Single-cell firing properties
ITC model neuron
The responses of the ITC model neuron to current injections from rest reproduced experimentally observed behaviors (Fig. 2A; Royer et al. 2000b; Marowsky et al. 2005; Geracitano et al. 2007). In response to low-amplitude depolarizing current injections (100 pA), there was a delay to the first spike and the firing frequency gradually increased with time due to the inactivation of the ISD current (dashed curve). Such a frequency increase with time was also observed in experimental data (see Fig. 2B in Marowsky et al. 2005). With higher current injections (200 pA), the delay to first spike was reduced and the neuron fired tonically at a higher rate. Also, the ISD current inactivated faster compared with low-amplitude injected currents.
Figure 2.

Firing properties of the ITC neuron model. (A) Voltage responses of ITC neuron model to two 1-sec current injections from rest (top: 200 pA; bottom: 100 pA). Evolution of the inactivation variable h of the ISD current is shown in dash. (B) Instantaneous firing frequency of ITC model to three different levels of current injections (100, 200, and 300 pA) with (gSD = 0.06 mS/cm2) and without ISD current (gSD = 0 mS/cm2).
The impact of ISD on the repetitive firing behavior of the ITC model neuron is shown in Figure 2B, which plots instantaneous firing frequency evoked by depolarizing current injections with (solid lines) and without (dashed lines) the ISD current. In the presence of ISD and with low-amplitude depolarizing current injections (100 pA, bottom), the firing frequency gradually increased. However, when the ISD current was removed from the model, the firing frequency started at a higher rate and slowed down to about 50 Hz due to the activation of the IsAHP current. Thus, the inclusion of the ISD current changed regular frequency adaptation to reverse frequency adaptation. When the applied current increased, the firing frequency with ISD current no longer increased monotonically with time, but showed a triphasic profile: after an initial transient increase, it decreased for about 150 msec, and then increased slightly thereafter. This indicates that with high-current injections, the IsAHP current activated rapidly, which overcame the effect of the ISD current inactivation in the first 200 msec. Without the ISD current, the frequency curve also showed a similar trend, with a larger second-phase adaptation decrease due to the loss of the opposing influence from the ISD current inactivation (Fig. 2B).
A characteristic feature of ITC cells is that due to inactivation of the ISD current, following spike trains, they generate after-depolarizations (ADPs) that increase in amplitude with the amount of depolarizing current (Royer et al. 2000b). To test whether our model could reproduce this behavior, transient suprathreshold current injections of various amplitudes were applied from a constant pre-pulse membrane potential (Fig. 3A). With low-current injections (Fig. 3A1), the ADP had a low amplitude, and a characteristic “hump and sag” developed with increased current reaching the spike threshold (Fig. 3A2). With an even larger current, the ADP generated a burst of spikes (Fig. 3A3, reflects Fig. 2A in Royer et al. 2000b). Time-dependent variations in the activation (m) and inactivation (h) gating variables of the ISD current are shown below each voltage trace in Figure 3A. As the current injection increased, the variable h was inactivated to a lower level, i.e., the conductance was more efficiently reduced, thereby causing a larger ADP.
Figure 3.

Bistable properties of the ITC neuron model due to the ISD current. (A) Dependence of the ADP on the amplitude of current injection. Transient current injections (500 msec) were applied at the same pre-pulse voltage Vm −60 mV) with different intensities (left: 30 pA; middle: 50 pA; right: 60 pA). The pre-pulse voltage Vm was controlled by a baseline current injection, and m & h are the activation and inactivation gating variables of the ISD current, respectively. (B) Dependence of the ADP on the duration of current injections. Current injections were applied at the same pre-pulse voltage Vm (−60 mV) and had the same amplitude. The duration of the current injection was 200, 300, and 400 msec in the left, middle, and right panels, respectively. (C) Dependence of the ADP on the pre-pulse voltage Vm. Current injections (500 msec) adjusted to elicit approximately the same number of spikes were applied at different Vm as indicated. (D) Comparison of the entire model (gSD = 0.06 mS/cm2) with the partial model without ISD current (gSD = 0 mS/cm2) in the presence of TTX (gNa = 0 mS/cm2). The model neuron was set to the same pre-pulse potential in D1 and D2 by applying less depolarizing current in the no ISD condition.
Experimentally, the ADP amplitude was reported to increase with the duration of suprathreshold depolarizing current pulses (Royer et al. 2000b). To test whether our model could reproduce this behavior, current injections of constant intensity but variable durations were applied from the same prepulse Vm (Fig. 3B). In keeping with experimental observations, the ADP amplitude increased with current pulse duration, eventually leading to spike generation (Fig. 3B3). Examination of ISD gating variables shows that longer current injections inactivated the ISD current more effectively, giving rise to larger ADPs.
Last, we examined the effect of the prepulse Vm on the ADP amplitude. Depolarizing current pulses adjusted to elicit that approximately the same number of spikes were delivered at different prepulse voltages (Fig. 3C). When Vm was low (−75 mV), little ADP was produced (Fig. 3C3). An ADP was seen when Vm was depolarized to −66 mV (Fig. 3C2). With a more depolarized Vm (−59 mV), the ADP resulted in tonic firing after the current pulse (Fig. 3C1), consistent with experimental observations (reflects Fig. 2B,C in Royer et al. 2000b). This was because ISD reached its maximal activation at around −60 mV, as found experimentally (Royer et al. 2000b; compares the activation levels of the m variable before current pulses in Fig. 3C). In the sustained firing case (Fig. 3C1), the ISD current remained inactivated for a long time, each spike renewing its inactivation.
To test that the ADP was due to the ISD current, voltage responses to depolarizing inputs were compared under TTX condition (gNa = 0) for the whole (Fig. 3D2) and partial model without ISD current (gSD = 0) (Fig. 3D1). So that the two conditions could be compared, the model neuron was adjusted to the same pre-pulse potential using a smaller baseline current in the no ISD condition. When ISD was removed from the model, the ADP was completely abolished (only AHP remained) (Fig. 3D1), demonstrating that ISD was necessary for ADP generation.
Ce model neurons
The responses of the Ce model neurons to current injections from rest reproduced the behaviors of regular spiking (RS), late firing (LF), and low-threshold bursting (LTB) Ce cells observed experimentally (Dumont et al. 2002; De Armentia and Sah 2004). As shown in Figure 4A, with low-amplitude depolarizing current injections, the RS cell fired only one spike, whereas current pulses of higher amplitude evoked repetitive firing with modest spike frequency adaptation. Also characteristic of RS Ce neurons, negative current injection elicited a hyperpolarizing response with a slowly developing depolarizing sag, produced by the activation of IH current. The response profiles of the LF Ce cell model to current injections are shown in Figure 4B. For a 200-pA depolarizing pulse, a long delay (200 msec) was seen for the first action potential, and the spike interval was also large. With a higher current injection (300 pA), the initial delay was reduced and the neuron fired repetitively, consistent with experimental data (see Fig. 2B in De Armentia and Sah 2004). The firing properties of the LTB model neuron to a series of current injections are shown in Figure 4, C and D. Consistent with the experimental data (Dumont et al. 2002), the LTB model cell generated spike doublets or bursts in response to depolarizing current pulses applied negative to −70 mV (Fig. 4C, top two traces) and at the break of hyperpolarizing current pulses (Fig. 4D, bottom). In contrast, depolarizing current pluses applied from more positive membrane potentials elicited trains of action potentials that displayed variable degrees of frequency adaptation (Fig. 4D, top two traces).
Figure 4.

Firing properties of the Ce neuron models. (A) Voltage responses of the regular spiking model cell to three 600-msec current injections from rest (top: 300 pA; middle: 100 pA; bottom: −100 pA). (B) Voltage responses of the late-firing model cell to three 600-msec current injections from rest (top: 300 pA; middle: 200 pA; bottom: −100 pA). (C) Voltage responses of the LTB neuron model to three 600-msec current injections from −80 mV (top: 200 pA; middle: 100 pA; bottom: −100 pA). (D) Voltage responses of the LTB neuron model to three 600-msec current injections from rest (−68 mV; top: 200 pA; middle: 100 pA; bottom: −100 pA).
Persistent network activity
As mentioned above, the slow deinactivation of ISD allows ITC neurons to generate sustained depolarizations in response to transient suprathreshold current injections (Fig. 3). However, it remains unclear whether transient inputs would have the same effect in a realistic network where ITC cells are interconnected by inhibitory synapses. For instance, LA cells are known to fire transiently at CS onset (Quirk et al. 1995, 1997), yet conditioned fear responses last for the entire duration of the CS. Also, brief IL stimulation (300 msec) was reported to effectively reduce fear expression in response to much longer CSs (Milad and Quirk 2002; Milad et al. 2004). Could ISD allow ITC cells to transform the transient signals arising from the BLA or IL into a more persistent output? To test this, we injected a brief current pulse of 300 pA for 300 msec (red bars in Fig. 5) into all ITC cells, mimicking the transient BLA/IL signal, while more realistic adaptive BLA inputs were turned off in this model experiment. To quantify the persistent activity, the spontaneous rate was measured during the 1 sec preceding the current pulses, while the steady-state firing rate was estimated 1 sec after the start of current injection (from 1 to 2 sec). Also, to test the robustness of the persistent network state, we simulated both a baseline inhibitory network (all ITC-ITC synaptic weights set to 2, i.e., 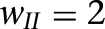 ) and a strong inhibitory network (
) and a strong inhibitory network (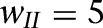 ).
).
Figure 5.

Persistent network activity elicited by transient excitatory inputs. (A,B) Voltage responses of ITCD1, ITCD10, ITCV2, and ITCV11 for the control (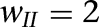 ; A) and strong inter-ITC inhibition (
; A) and strong inter-ITC inhibition (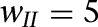 ; B) cases. Evolution of the inactivation variable (h) of the ISD current is shown in green. The red bars indicate the duration of the transient input. (C) Difference between the steady-state firing rate and spontaneous rate of individual ITCD and ITCV neurons. (D) Instantaneous firing frequency of ITCD and ITCV neurons as a population. Error bars indicate SEM and bin width is 100 msec; same in Figure 6.
; B) cases. Evolution of the inactivation variable (h) of the ISD current is shown in green. The red bars indicate the duration of the transient input. (C) Difference between the steady-state firing rate and spontaneous rate of individual ITCD and ITCV neurons. (D) Instantaneous firing frequency of ITCD and ITCV neurons as a population. Error bars indicate SEM and bin width is 100 msec; same in Figure 6.
Figure 5, A and B, shows the voltage responses of four representative cells from the ITCD (ITCD1, ITCD10) and ITCV (ITCV2, ITCV11) clusters for the control and strong inter-ITC inhibition cases, respectively. Prior to current injection, the four ITC neurons had no or little spiking activity. In both cases, current injections produced high-frequency discharges in all ITC cells. However, after the current pulse, ITC cells continued to fire at much higher rates than the spontaneous rates (Fig. 5A,B). Insights into the mechanisms underlying these phenomena can be obtained by considering time-dependent fluctuations of the ISD inactivation variable (green lines superimposed on the voltage responses of Fig. 5A,B). This reveals that ISD inactivated rapidly during the current injection and remained inactivated thereafter (<0.5). Thus, inactivation of ISD enabled both ITCD and ITCV cells to fire continuously beyond the current injection, which in turn kept renewing the inactivation of the ISD current. In keeping with the idea that ISD plays a critical role in generating persistent activity, accelerating the deinactivation kinetics of ISD abolished the persistent activity of ITC cells (Supplemental Fig. S6).
Expectedly, the impact of ISD inactivation on persistent activity varied depending on the strength of the inhibitory connections between the ITC cells. Comparing the control (Fig. 5A) and strong inter-ITC inhibition (Fig. 5B) cases revealed that increasing inhibition strength reduced sustained activity in all four ITC cells, but only to a moderate degree. This can also be seen in Figure 5C, which plots the difference between the steady-state firing rate and spontaneous firing rate for each ITC neuron. The sustained activity of the majority of ITC cells (76.7%) was reduced by stronger inter-ITC inhibition. A small proportion of cells (16.7%) remained unchanged and only two cells (6.7%) had increased activity with larger inhibitory weight. The variability was due to different number and type of inhibitory inputs received by each individual ITC cell (see Supplemental Table S3). If the reduction of firing rate in presynaptic neurons balanced or overcame the effect of increased synaptic weight, the persistent activity of post-synaptic neurons may remain unchanged or increase. This shows that increased inter-ITC coupling strength could differentially modulate the firing rates of individual ITC neurons.
The sustained activity of ITC neurons as a population can be seen by examining the instantaneous firing rates averaged on all ITCD and ITCV cells (Fig. 5D). In the control case, ITC neurons fired spontaneously at low rates to background inputs prior to current injection (ITCD: 2.7 ± 0.7 Hz, ITCV: 1.7 ± 0.7 Hz; mean ± SE). After the current pulse, the average firing rate of ITCD increased more than fourfold (steady-state rate: 11.4 ± 1.5 Hz; t-test P < 0.001), and that of ITCV increased nearly fourfold (steady-state rate: 6.7 ± 1.4 Hz; t-test P < 0.005). When the inter-ITC weight increased 2.5-fold (strong inhibition case), the spontaneous rates remained almost unchanged (ITCD: 2.5 ± 0.8 Hz, ITCV: 1.6 ± 0.7 Hz, t-test P > 0.1). Although reduced compared with the control case, the steady-state frequencies of ITCD and ITCV were still significantly higher than the corresponding spontaneous firing rates (ITCD: 9.1 ± 1.3 Hz, t-test P < 0.001; ITCV: 4.9 ± 1.2 Hz, t-test P < 0.05). Thus, sustained ITC network activity can be generated by transient excitatory inputs, even in the presence of strong inter-ITC inhibition. Note that in both cases, ITCV cells exhibited less persistent activity than ITCD cells (the ratio of steady-state rate to spontaneous rate was lower) because of the unidirectional inhibitory connections from ITCD to ITCV neurons.
Network behaviors during fear and extinction states
Prior to studying the impact of IL inputs on ITC cells, it is important to characterize the model's behavior in response to inputs from LA and BA. Indeed, unit recordings have revealed that after fear conditioning, the responses of LA and BA neurons to CS presentations consist of an early phase of strongly increased firing frequency that quickly decays to levels slightly above baseline for the remainder of the stimulus (Quirk et al. 1995; Herry et al. 2008). It is of great interest to examine how the ITC network processes such adaptive signals to regulate the firing of Ce neurons during fear conditioning and extinction. Toward this end, we modeled three separate network states (habituation, following fear conditioning, and after extinction training) with experimentally observed LA/BA firing rates and synaptic plasticity. Previous experimental work has revealed that expression of fear is a result of potentiated LA responses to the CS (Quirk et al. 1995) relayed to Ce via BA (Pitkanen 2000) and ITC cells (Pare et al. 2004; Pape and Pare 2010), while extinction is accompanied by diminished LA responses (Quirk et al. 1995). A recent experimental study (Herry et al. 2008) reported that BA contains three subpopulations of neurons whose CS responsiveness is differentially altered during fear conditioning and extinction training: “fear cells” that acquire CS responses as a result of fear conditioning, but lose them following extinction training; “extinction cells” that only become CS responsive following extinction training; and “extinction-resistant cells” that acquire CS responses during conditioning, but remain CS responsive after extinction training. Accordingly, the fear state was modeled with elevated firing rate of LA, BA fear (BAF), and extinction-resistant (BAER) neurons, while the extinction state was modeled with diminished LA and BAF inputs, plus increased firing rate of BA extinction (BAE) neurons (see Supplemental Materials section for details). Different synaptic weights were also given for certain synapses in the three network states (Table 1; see Materials and Methods section for details). As indicated in the Materials and Methods section, we considered two possible network architectures with all BA inputs projecting to ITC cells in the first architecture and only extinction inputs projecting to ITC cells in the second architecture. In both architectures, the extinction neurons did not project to Ce because the activation profile of extinction cells was opposite to the expression of fear (Herry et al. 2008). Instead, both fear and extinction-resistant inputs projected to Ce.
Table 1.
Synaptic weights used in the ITC network model during the three network states

Below, we describe the performance of each of the architectures. In these model experiments, we used several terms of firing frequency defined as follows. When a 2-sec CS input was present, the steady-state firing frequency was measured during the last 1 sec of the CS, while the estimate of the CS-induced frequency was based on the entire 2-sec CS period. The instantaneous firing frequency was calculated by dividing the simulation time into 100-msec bins, and the spontaneous firing rate was measured during the 1-sec period prior to CS onset.
Architecture 1
The instantaneous frequency of LA, BAF, BAER, and BAE inputs during CS presentation in three network states is shown in Figure 6A, while that of ITCD, ITCV, and Ce neurons is shown in Figure 6B. The change in CS-induced neuronal firing rate across behavioral states is summarized in Table 2. The steady-state (SS) frequency is also given for Ce. Frequency is given as mean ± SEM. At rest with random background inputs, the average spontaneous firing rates of ITCD, ITCV, and Ce cells were 2.7 ± 0.7 Hz, 1.7 ± 0.7 Hz, and 2.2 ± 0.4 Hz, respectively (the Ce rate was based on Duvarci et al. 2011). In habituation, the LA and BA inputs were delivered at low frequency (LA: 3.0 ± 0.1 Hz, BAF: 1.8 ± 0.1 Hz, BAER: 1.55 ± 0.05 Hz, and BAE: 1.6 ± 0.2 Hz; mean ± SE, average over the CS). The firing pattern of BAF and BAER inputs was similar to that of LA inputs, which showed strong frequency adaptation (Fig. 6A). Due to extra LA inputs and the unidirectional inhibition from ITCD to ITCV, the ITCD neurons fired at a higher frequency than ITCV neurons (14.3 ± 2.2 Hz vs. 4.2 ± 1.4 Hz, t-test P < 0.001, average over the CS). The Ce showed elevated firing in the first 100 msec of CS, which adapted quickly to baseline level (Fig. 6B). Though the average CS-evoked Ce firing rate (3.4 ± 0.6 Hz, t-test P > 0.1) was slightly higher than the average spontaneous rate (2.2 ± 0.4 Hz), the steady-state frequency was slightly lower (1.6 ± 0.9 Hz, t-test P > 0.1). The early Ce response to CS simulated the animal's natural reaction to a neutral stimulus.
Figure 6.

Network behaviors during habituation, fear, and extinction states without IL inputs. (A) Instantaneous firing frequency of LA and BA inputs. (B) Instantaneous firing frequency of ITCD, ITCV, and Ce neurons for architecture 1. (C) Instantaneous firing frequency of ITCD, ITCV, and Ce neurons for architecture 2. The black arrows indicate CS onset. (D) Average CS-evoked firing frequencies of ITCD, ITCV, and Ce neurons for architectures 1 and 2. The dotted lines indicate the spontaneous firing rates.
Table 2.
Spontaneous and CS-induced firing rates (Hz) of ITC and Ce neurons in three network states for architecture 1 (A1) and architecture 2 (A2)

In the fear state, the CS-induced firing frequencies of LA, BAF, and BAER were greatly increased (LA: 8.4 ± 0.4 Hz, BAF: 4.9 ± 0.3 Hz, BAER: 4.2 ± 0.3 Hz, t-test P < 0.001), while that of BAE remained the same (1.7 ± 0.2 Hz, t-test P > 0.1) (Fig. 6A). With potentiation of the LA-ITCD synapses (Table 1), the CS-induced firing rate of ITCD cells greatly increased from 14.3 ± 2.2 Hz to 35.2 ± 4.1 Hz (t-test P < 0.001), while that of ITCV cells slightly decreased from 4.2 ± 1.4 Hz to 3.9 ± 1.4 Hz (t-test P > 0.1). This was because the enhanced inhibition from ITCD overcame the elevated drive from BAF and BAER inputs to ITCV cells. With increase of the BAF and BAER firing rate combined with disinhibition from ITCV cells, the CS-evoked discharge frequency of the Ce output greatly increased from 3.4 ± 0.6 Hz to 9.7 ± 0.8 Hz (t-test P < 0.001) (Fig. 6B), signaling a high “fear” state. The initial frequency in the first bin could even reach above 30 Hz (Fig. 6B) due to high frequency of BA inputs in the first 100 msec. The average steady-state frequency (8.4 ± 0.8 Hz) was also significantly higher than the average spontaneous firing rate (2.2 ± 0.4 Hz, t-test P < 0.001), indicating a persistent state of fear during the entire CS presentation. It should be noted that the 8–10 Hz firing rate of Ce in the fear state is consistent with experimental data (unpublished observations from the Pare lab). To compare the amount of spike frequency adaptation in LA/BA inputs and ITC neurons, we computed the quantity Fadap, proposed by Wang (1998): 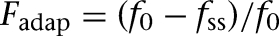 , where f0 is the initial firing rate (frequency in the first bin) and fss is the steady-state firing rate (frequency during the last 1 sec of CS). This revealed that adaptation was much higher in LA/BA inputs (LA: 80.8%, BAF: 84%, BAER: 79.3%) than in ITC cells (ITCD: 51.0%, ITCV: 21.4%). Thus, the strongly adapting signals from BLA neurons can be transformed into a more-sustained, less-adaptive output by ITC cells. The Ce output also exhibited less frequency adaptation compared with BLA inputs in the fear state (75.3%).
, where f0 is the initial firing rate (frequency in the first bin) and fss is the steady-state firing rate (frequency during the last 1 sec of CS). This revealed that adaptation was much higher in LA/BA inputs (LA: 80.8%, BAF: 84%, BAER: 79.3%) than in ITC cells (ITCD: 51.0%, ITCV: 21.4%). Thus, the strongly adapting signals from BLA neurons can be transformed into a more-sustained, less-adaptive output by ITC cells. The Ce output also exhibited less frequency adaptation compared with BLA inputs in the fear state (75.3%).
In the extinction state, the CS-induced firing rates of LA and BAF inputs returned back to the habituation level (LA: 3.0 ± 0.1 Hz, BAF: 1.7 ± 0.1 Hz, t-test P > 0.1), while that of BAER inputs remained as high as the fear state (4.1 ± 0.2 Hz, t-test P > 0.1) (Fig. 6A). Also, the BAE inputs switched on with adaptation (3.6 ± 0.4 Hz, t-test P < 0.002, average over the CS). Due to decrease in LA firing rate with partial depotentiation of the LA–ITCD synapses (Table 1), the drive from LA to ITCD was largely reduced. At the same time, the excitation from BA inputs to ITCD increased as the BA–ITC synaptic strength was potentiated (decrease in BAF firing rate was compensated by increase in BAE firing rate). Since the decrease in LA drive overwhelmed the increase in BA excitation, the CS-evoked firing rate of ITCD was reduced from 35.2 ± 4.1 Hz (in fear state) to 22.7 ± 2.7 Hz (t-test P < 0.02) (Fig. 6B). The reduction of inhibition from ITCD plus potentiation of BA inputs led to a large increase in the CS-evoked ITCV firing rate (from 3.9 ± 1.4 Hz to 13.0 ± 2.1 Hz, t-test P < 0.002) (Fig. 6B), imposing a large inhibition on Ce. On the other hand, the excitatory drive from BA to Ce decreased with extinguished BAF inputs (Fig. 6A). As a result of increased inhibition with decreased excitation, the CS-evoked discharge rate of Ce neurons went below the average spontaneous rate (0.8 ± 0.4 Hz, t-test P < 0.05) (Fig. 6B). In this state, the percentages of adaption in LA/BA inputs (LA: 85%, BAF: 88%, BAER: 78.6%, BAE: 86.3%) were also much higher than those in ITC cells (ITCD: 54.9%, ITCV: 56.6%). Model experiments also revealed that changing all ITC synapses to the depressing type impaired extinction, while using only the facilitating or constant types had little effect on network activity (Supplemental Fig. S7).
Architecture 2
In architecture 2, the BAF and BAER inputs did not project to ITC. For ease of comparison, we denote architecture 1 as “A1” and architecture 2 as “A2”. The instantaneous firing rate of ITCD, ITCV, and Ce neurons is shown in Figure 6C, and the average frequency over the entire CS for A1 and A2 is compared in Figure 6D. The change in CS-induced neuronal firing rate across behavioral states is also summarized in Table 2. In habituation, removal of BAF and BAER inputs to ITC only decreased the CS-induced firing rate of ITCD by 15.4% (A1: 14.3 ± 2.2 Hz, A2: 12.1 ± 1.9 Hz, t-test P > 0.1) (Fig. 6D) since LA inputs still projected to ITCD and were the dominant source of ITCD excitation. In contrast, the CS-induced ITCV firing rate was reduced by 38.1% (A1: 4.2 ± 1.4 Hz, A2: 2.6 ± 1.1 Hz, t-test P > 0.1) (Fig. 6D). The CS-evoked firing rate of Ce was elevated to 5.1 ± 0.5 Hz (A1: 3.4 ± 0.6 Hz, t-test P < 0.1) (Fig. 6D) and showed a high response in the first 100 msec (Fig. 6C). It should be noted that this firing rate of Ce (5.1 ± 0.5 Hz) was significantly higher than the average spontaneous rate (2.2 ± 0.4 Hz, t-test P < 0.002) (Fig. 6D), implying that the animal expresses fear in habituation, which is not true. Similarly, in the fear state, removing BAF and BAER inputs to ITC had a much larger impact on ITCV (76.9% decrease, A1: 3.9 ± 1.4 Hz, A2: 0.9 ± 0.5 Hz, t-test P < 0.05, CS-evoked rate) than on ITCD (7.7% decrease, A1: 35.2 ± 4.1 Hz, A2: 32.5 ± 3.7 Hz, t-test P > 0.1, CS-evoked rate) (Fig. 6D). As a result, the CS-induced Ce firing rate was increased by 51.6% (A1: 9.7 ± 0.8 Hz, A2: 14.7 ± 0.8 Hz, t-test P < 0.005) (Fig. 6D). Thus, less BA inputs to ITC would facilitate expression of fear. In the extinction state, loss of BAF and BAER inputs to ITC decreased the CS-induced ITCD firing rate by 24.7% (A1: 22.7 ± 2.7 Hz, A2: 17.1 ± 2.3 Hz, t-test P > 0.1) and the ITCV firing rate by 58.5% (A1: 13.0 ± 2.1 Hz, A2: 5.4 ± 1.6 Hz, t-test P < 0.01) (Fig. 6D). Such a large decrease in ITCV firing rate resulted in a near sixfold increase in the CS-evoked Ce firing rate (A1: 0.8 ± 0.4 Hz, A2: 4.6 ± 0.7 Hz, t-test P < 0.002) (Fig. 6D). Extinction in architecture 2 was thus not complete because this level of firing rate (4.6 ± 0.7 Hz) was also significantly higher than the average spontaneous rate (2.2 ± 0.4 Hz, t-test P < 0.02). Taken together, the firing rate of ITCV cells was largely decreased, while that of Ce cells was significantly increased in architecture 2. Since such high Ce firing rates were not observed in experiments (Duvarci et al. 2011), we reasoned that the architecture 1 is more realistic.
Impact of IL inputs in network conditions reproducing a high fear state
Brief IL stimulation after tone onset (100–400 msec) has been reported to effectively reduce fear in rats that had not been extinguished (Milad and Quirk 2002), but had less or little effect if given either 1 sec before or 1 sec after tone onset (Milad et al. 2004). It is not clear how such a transient IL input can reduce fear effectively and why it is most effective if delivered shortly after tone onset. To investigate this, brief IL stimulation (300 msec) was modeled by Poisson-distributed spike trains (20 Hz) delivered to ITC neurons 0.1 sec after tone onset (Fig. 7B), 1 sec before tone onset (Fig. 7C), and 1 sec after tone onset (Fig. 7D) during the high fear state (the IL–ITC synaptic weight was set to 1). For comparison, the baseline network activity in the high fear state (without IL stimulation) is shown in Figure 7A. In each figure, we plotted a 3-sec neuronal activity for two representative ITCD cells (ITCD1, ITCD10), two representative ITCV cells (ITCV2, ITCV11), and two Ce cells (one LF and one LTB). Since we inferred that architecture 1 was closer to the realism from previous simulation, we used architecture 1 for this IL experiment.
Figure 7.

Impact of brief IL stimulation (300 msec) on network activity in the high fear state. Voltage responses of ITCD1, ITCD10, ITCV2, ITCV11, and two Ce neurons (one LF type and one LTB type) for the following four situations: (A) High fear state without IL inputs; (B) IL inputs given at 0.1 sec after tone onset; (C) IL inputs given at 1 sec before tone onset; (D) IL inputs given 1 sec after tone onset. The red arrows indicate CS onset and the red bars indicate the presence of IL inputs. (E) Percentage change in the mean firing frequency of ITCD, ITCV, and Ce neurons for three different timings of IL stimulation relative to the fear case without IL inputs. (F) Percentage change in the mean firing frequency of ITCD, ITCV, and Ce neurons for all facilitating, all depressing, and all constant-type synapse cases relative to the control case with heterogeneous synapses. IL stimulation is given at 0.1 sec after tone onset.
IL stimulation caused a large increase in the firing rates of both ITCD and ITCV neurons, which completely eliminated Ce firing during the 300 msec IL presentation (indicated by the red bars, Fig. 7B–D). It should be noted that this reduction in CS-related Ce responses to BLA inputs is consistent with earlier experimental observations (Quirk et al. 2003). However, the effect of IL was not restricted to the 300-msec presentation, as it could facilitate ITC firing beyond the stimulation period due to the persistent inactivation of ISD. Indeed, neuron ITCV2 fired only three action potentials during the 3-sec period in the fear state (Fig. 7A), but fired continuously after IL stimulation (Fig. 7B–D). Such facilitating effect was also evident in neuron ITCV11. Compared with ITCV2 and ITCV11, the facilitating effect of IL on the two ITCD cells (ITCD1 and ITCD10) was much smaller, because these cells already discharged at relatively high frequency in the absence of IL inputs, inactivating the ISD current effectively. As a result, IL stimulation caused little further inactivation of ISD. Thus, the brief IL input modulated individual ITC firing differentially, depending on their firing rates. In the high fear state, ITCV neurons fired at rates much lower than ITCD neurons (3.9 ± 1.4 Hz vs. 35.2 ± 4.1 Hz, t-test P < 0.001, CS-evoked rate) due to the potentiation of LA inputs and the unidirectional inhibitory connections from ITCD to ITCV cells. Hence, the facilitating effect of IL stimulation on ITCV neurons was greater than that on ITCD cells. Indeed, in the case of IL + 0.1 sec, IL stimulation caused a 43.2% increase in the steady-state firing frequency of ITCV neurons (from 3.7 ± 1.4 Hz to 5.3 ± 1.6 Hz, t-test P > 0.1), compared with only a 1.2% increase in ITCD cells (from 32.3 ± 4.1 Hz to 32.7 ± 4.1 Hz, t-test P > 0.1). Such an increase in ITCV activity decreased the Ce steady-state firing rate from 8.4 ± 0.8 Hz to 4.0 ± 1.2 Hz (t-test P < 0.02; Fig. 7, cf. A and B).
To compare the IL effectiveness in three cases of IL timing, we denote the effect during the 300-msec IL presentation as “direct effect” and the effect after IL stimulation as “facilitating effect”. As shown in Figure 7, B–D, the direct effect was always stronger than the facilitating effect. Although IL inputs delivered 1 sec before tone onset could still enhance the CS responses of ITCV neurons due to the slow deinactivation kinetics of the ISD current, the direct effect was lost during the CS presentation so the facilitating effect was also reduced (Fig. 7, cf. B and C). Indeed, the average steady-state frequency of ITCV in the IL + 0.1 case was higher than that in the IL − 1 sec case (5.3 ± 1.6 Hz vs. 4.8 ± 1.5 Hz, t-test P > 0.1). If IL stimulation was given 1 sec after tone onset, its impact was much reduced because it could not inhibit the Ce firing during the first 1 sec of CS where high frequency of Ce spikes were present (Fig. 7D). Figure 7E plots the percentage change in the average firing frequency of ITCD, ITCV, and Ce neurons during the 2-sec CS presentation for the three IL stimulation cases (Fig. 7B–D) relative to the fear case (Fig. 7A). The average Ce firing rate was reduced by 51.6% for the IL + 0.1 sec case (from 9.7 ± 0.8 Hz to 4.7 ± 0.7 Hz, t-test P < 0.005), compared with 43.3% for the IL − 1 sec case (from 9.7 ± 0.8 Hz to 5.5 ± 0.8 Hz, t-test P < 0.01), and 35.1% for the IL + 1 sec case (from 9.7 ± 0.8 Hz to 6.3 ± 0.8 Hz, t-test P < 0.02). This is consistent with the experimental finding that IL stimulation given 0.1 sec after CS onset is most effective in reducing fear expression, followed by IL − 1 sec and then IL + 1 sec (Milad et al. 2004). It can be predicted that if IL stimulation is given at tone onset, fear can be further reduced, since IL inputs can eliminate the Ce burst firing in the first 100 msec. When the strength of IL stimulation increased, little further reduction was seen in the Ce firing frequency (for the case of IL + 0.1 sec; Supplemental Fig. S8), suggesting that low intensity of IL stimulation was as effective as high intensity of IL stimulation (Milad et al. 2004). It should also be noted that the LTB Ce cell fired at a higher rate than the LF Ce cell during CS presentation in the fear state (11 Hz vs. 7 Hz) (Fig. 7A) and was more resistant to IL-induced inhibition (Fig. 7B–D), probably due to partial deinactivation the ICaT current by hyperpolarization.
Setting all inter-ITC synapses to one particular type had little effect in the fear state due to the low firing rate of ITCV neurons (Supplemental Fig. S7). In contrast, when IL inputs were introduced (0.1 sec after CS onset), clear differences were seen as a function of inter-ITC synapse types (Fig. 7F). When all ITC synapses were of the facilitating or constant types (including the ITC–Ce synapses), both ITCD and ITCV firing rates (CS-evoked) decreased as inter-ITC inhibitory efficiency increased. The percentage of decrease was similar for the two cases (ITCD: ∼4%, ITCV: ∼13%) (Fig. 7F). Since ITC cells projecting to Ce only form synapses of the facilitating and constant types (Geracitano et al. 2007), changing them all to the facilitating or constant types had little effect on their efficiency (see Supplemental Fig. S4, the release probability of facilitating synapses at high rates was about 0.8, close to that of the constant synapses). Hence, although not statistically significant, the mean Ce firing rates (over the 2-sec CS presentation) increased as the ITCV firing rates diminished under all facilitating or constant-type situations (all facilitating: 21.3%, t-test P > 0.1; all constant: 25.5%, t-test P > 0.1) (Fig. 7F). When all ITC synapses were of the depressing type, the CS-evoked firing rates of both ITCD and ITCV increased as inter-ITC inhibitory efficiency decreased (ITCD: 10.2%, t-test P > 0.1; ITCV: 30.3%, t-test P > 0.1). However, for this case, the efficiency of the ITC–Ce inhibition was much lower compared with the control case (see Supplemental Fig. S4, the release probability of depressing synapses at high rates was much lower than those of facilitating and constant synapses). This decrease in inhibition efficiency at the ITC–Ce synapses was found to overwhelm the opposing effect of increased ITCV firing rate, and so the CS-evoked Ce firing rate was significantly higher than that in the control case (55.3%, from 4.7 ± 0.7 Hz to 7.3 ± 0.6 Hz, t-test P < 0.05) (Fig. 7F). Thus, these modeling experiments suggest that it is advantageous to have facilitating or constant type synapses for the ITC–Ce connection in order to inhibit Ce neurons effectively. Taken together, the results of our simulations indicate that variations in the short-term dynamics of ITC synapses enhance the effectiveness of IL inputs in reducing the expression of conditioned fear as expressed in the firing rate of Ce neurons.
Discussion
The IL cortex is believed to play a critical role in extinction by inhibiting fear output Ce neurons via the activation of ITC cells (Pare et al. 2004; Quirk and Mueller 2008). However, previous physiological studies have revealed that ITC cells inhibit each other (Royer et al. 2000a; Geracitano et al. 2007) raising the following question: Can IL inputs overcome the inter-ITC inhibition to regulate the CS-evoked activity of Ce neurons? Also, it is not clear how a brief IL stimulation (300 msec) can reduce fear expression effectively if given 100 msec after tone onset, not earlier or later (Milad and Quirk 2002; Milad et al. 2004). Because it is currently difficult to test this experimentally, we investigated this problem using a biophysical network model. Our findings indicate that over a wide range of stimulation strengths, brief IL activation can overwhelm inter-ITC inhibition and reduce the activity of fear output Ce neurons. Importantly, both intrinsic properties (i.e., bistability) and variations in the short-term synaptic dynamics of ITC neurons contributed to the effectiveness of IL stimulation. In addition, we observed that ITC neurons could transform the transient CS-related signals arising in the BLA into a persistent pattern of activity. Below, we consider the significance of these observations in light of previous work about the physiology of ITC neurons.
IL control of ITC cells and the regulation of conditioned fear expression
Mounting evidence implicates ITC neurons in the regulation of conditioned fear expression. For instance, ITC lesions (Likhtik et al. 2008) as well as pharmacological inhibition of BLA inputs to ITC cells (Jungling et al. 2008) interfere with the extinction of conditioned fear responses. The inhibition of conditioned fear responses by ITC cells during extinction is thought to depend on their activation by glutamatergic inputs from the infralimbic cortex (Pare et al. 2004; Quirk and Mueller 2008). Several observations support this view. First, IL lesions interfere with the consolidation of extinction (Quirk et al. 2000). Second, IL sends a massive projection to ITC cells (McDonald et al. 1996; Freedman et al. 2000). Third, disinhibition of IL with local picrotoxin infusions leads to increased c-Fos expression in ITC cells (Berretta et al. 2005). Fourth, extinction testing is associated with marked increases in the number of c-Fos-positive IL and ITC cells (Knapska and Maren 2009). Last, electrical stimulation of IL produces a reduction of conditioned fear (Milad and Quirk 2002; Vidal-Gonzalez et al. 2006) and an abolition of BLA-evoked responses in Ce neurons (Quirk et al. 2003). In principle, ITC cells could produce the latter two effects by generating feedforward inhibition in Ce when activated by IL inputs (Pare and Smith 1993a,b; Royer et al. 1999).
However, other observations cast doubt on the validity of this interpretation. Indeed, ITC cells occur as densely packed clusters and there are connections between ITC cells located in the same (Geracitano et al. 2007) as well as different clusters (Royer et al. 2000a). The latter connections are directionally polarized, in the dorsoventral direction (Royer et al. 2000a). A further complication comes from the fact that inter-ITC connections show short-term plasticity (Geracitano et al. 2007). In a recent in vitro study, when presynaptic ITC cells were repeatedly activated with current injection in a range of frequencies, in an approximately equal proportion of cell pairs, transmitter release probability decreased, increased, or remained constant (Geracitano et al. 2007). As a result, it is not immediately clear whether IL inputs can overcome the inter-ITC inhibition and how the short-term plasticity of inter-ITC synapses might alter the impact of IL activity over time.
To examine these issues, we first developed a compartmental model of a single ITC cell that could reproduce their unusual electroresponsive properties, as observed experimentally (Royer et al. 2000b). These included their very high input resistance, repetitive firing behavior, and ability to transform transient suprathreshold depolarizations into prolonged periods of increased intrinsic excitability via the inactivation of a potassium current with slow deinactivation kinetics (ISD). Next, we generated a larger ITC network that implemented the short-term synaptic plasticity of inter-ITC connections described above. The network behavior was then tested during patterns of BLA activation designed to simulate the rapidly adapting profile of activity evoked by CS presentations, as observed experimentally (Quirk et al. 1995; Pare and Collins 2000).
Persistent modulation of Ce output by ITC neurons
A first interesting observation to emerge from these tests was that despite the presence of inhibitory connections between ITC cells, transient excitatory inputs from BLA or IL were transformed by ITC cells into a sustained state of increased activity via the inactivation of ISD. Although the magnitude of this persistent activity was affected by the strength of inter-ITC inhibitory connections, it remained robust for higher inhibitory synaptic weights (2.5-fold change). Therefore, these results suggest that ITC cells express a form of short-term memory, inscribed in their intrinsic properties, allowing for persistent alterations in fear responsiveness following transient sensory signals. Burgos-Robles et al. (2009) recently demonstrated that prelimbic (PL) neurons transform transient amygdala inputs into a sustained output that drives conditioned fear responses and gates the expression of extinction. Therefore, it is possible that both PL and ITC activity contribute to sustaining the expression of conditioned fear. While PL seems to sustain fear by increasing the excitatory drive onto Ce via BA, the present study suggests that ITCD neurons could contribute to this sustenance by increasing their inhibition on ITCV neurons, resulting in disinhibition of Ce. As shown in Figure 8A, during the high fear state, strongly adaptive LA inputs were transformed into a sustained output by ITCD neurons, leading to persistent inhibition on ITCV cells and allowing for sustained Ce firing. Also, ITC neurons can support the expression of extinction via persistent activity in ITCV cells. In the extinction state (Fig. 8B), the LA responses diminished and the LA–ITCD connection depotentiated, while the BA–ITC connection potentiated (Amano et al. 2010). Strongly adaptive BA inputs were then transformed into sustained firing in ITCV cells, resulting in lowered Ce responses.
Figure 8.

Schematic illustrating the patterns of neuronal activity in the amygdala under different network situations. In the high fear state (A), the LA–ITCD connection strength is potentiated. Strongly adaptive LA inputs are transformed into much sustained output by ITCD neurons, leading to persistent inhibition on ITCV cells allowing for sustained Ce firing. In the extinction state (B), the LA inputs diminish and the LA–ITCD synapses depotentiate, while the BA–ITC synapses potentiate. Similarly, strongly adaptive BA inputs are transformed by ITCV neurons into sustained firing, leading to persistent inhibition of Ce output. When IL stimulation is given in the fear state (C), brief IL inputs increase the excitability of both ITCD and ITCV neurons, with a larger impact on ITCV cells, which significantly reduces the Ce firing. Note that the transmission efficiency (release probability) of the ITCV–Ce synapses is increased during extinction or with IL stimulation, enhancing inhibition on Ce output neurons.
Can other mechanisms lead to sustained activity in the ITC network?
Although diverse mechanisms underlying similar transitions from transient-to-sustained activity have been proposed in various brain areas (e.g., Wang 1999b; Egorov et al. 2002; Miller et al. 2003; Winograd et al. 2008), we believe that the slowly deinactivating current is the ionic basis for sustained ITC network activity due to the following reasons: (1) The ADPs displayed in ITC cells were not dependent on intracellular Ca2+ or Na+ concentration (Royer et al. 2000b), suggesting that ADPs were not mediated by a Ca2+-activated nonspecific cationic current (ICAN). (2) A network-based mechanism seems unlikely here because the ADPs were not dependent on GABA release (Royer et al. 2000b). Indeed, the inhibitory interconnections between ITC cells serve to destabilize the persistent activity (Fig. 5). (3) Although ITC neurons express the HCN current (IH), ITC cells rarely display rebound spikes following hyperpolarizing inputs (unpublished observation from the Pare lab). Indeed, to obtain sustained rebound spikes in the ITC cell model, the HCN channel density had to increase from 0.02 to 0.3 mS/cm2 and a large hyperpolarization was needed (−200 pA) (Supplemental Fig. S9). In light of earlier experimental observations, such a high HCN density is unlikely in ITC neurons. Similarly, the inhibitory connections between ITC cells are unlikely to lead to such large hyperpolarizations. Thus, we concluded that sustained ITC network activity is not due to the IH current.
IL stimulation reduces Ce responses to CS in a temporally specific manner
Finally, we examined the impact of a brief IL stimulation (300 msec) on the responsiveness of ITC cells to strongly adaptive CS-related BLA inputs, in the high fear state. IL inputs in a wide range of strengths caused a marked increase in the firing rate of ITC cells, strongly inhibiting the Ce output. Importantly, the impact of IL inputs extended beyond the 300-msec stimulation window because IL-evoked firing caused a persistent inactivation of ISD in ITC neurons (Fig. 7B–D). Also, the model demonstrated that IL stimulation given shortly after tone onset was most effective in reducing Ce firing, in agreement with experimental findings (Milad et al. 2004). This might be due to the fact that this timing most effectively combines the direct impact of IL in inhibiting early Ce spikes and its indirect (after IL is turned off) impact in inhibiting Ce firing subsequently, via the inactivation of ISD. Furthermore, model experiments revealed that variations in the short-term synaptic dynamics within the ITC network facilitates the inhibitory control of Ce output by IL (see the following section), a prediction that could be tested in future experiments. It can also be predicted that ITC neurons contacted by depressing synapses are more easily recruited by IL inputs than those contacted by facilitating or constant-type synapses.
Implications of variations in the short-term dynamics of ITC synapses
As mentioned above, three different types of short-term synaptic plasticity have been observed in inter-ITC connections (Geracitano et al. 2007), but the role of such synaptic heterogeneity is not clear. Our modeling experiments suggest that this synaptic heterogeneity can enhance the inhibitory control of Ce by IL inputs. First, to effectively inhibit Ce output, the ITCV–Ce connections have to be of the facilitating or constant types, as found experimentally (Geracitano et al. 2007). These two types of synapses maintain efficient levels of inhibition in Ce cells. In contrast, for the inter-ITC connections, pure facilitating or constant type synapses decrease the firing rates of both ITCD and ITCV neurons when IL inputs are active, resulting in elevated Ce responses (Fig. 7F). Hence, depressing synapses, in concert with ISD inactivation, allow IL inputs to overcome the inter-ITC inhibition. Taken together, these considerations suggest that the heterogeneous short-term plasticity of the inter-ITC connections enables sufficiently high activity levels in ITCV cells for an efficient control of fear-related Ce outputs when BA and IL neurons are active.
Conclusions
Overall, our results indicate that although the inhibitory connections between ITC cells tend to oppose excitatory influences onto them, both intrinsic and synaptic properties contribute to the inhibitory control of Ce by IL inputs. In particular, IL inputs do overcome the inhibition produced by inter-ITC connections, leading to an overall excitation of ITC cells and a persistent decrease in Ce fear outputs. These results support the notion that IL inputs are in strategic position to control extinction of conditioned fear via the activation of ITC neurons.
Materials and Methods
ITC single cell model
The ITC cell was modeled using the Hodgkin-Huxley formulation (Byrne and Roberts 2004). It had two compartments representing a soma (diameter of 8 µm; length of 8 µm) and a dendrite (diameter of 5 µm; length of 200 µm) (Millhouse 1986; Marowsky et al. 2005). The current balance equations were of the form given in Eqs. 1 and 2,
| 1 |
| 2 |
where Vs and Vd were the somatic and dendritic membrane potentials (mV), 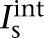 (
(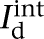 ), and
), and 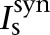 (
(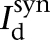 ) were the intrinsic currents and the synaptic currents in the somatic (dendritic) compartments, respectively, Is was the electrode current applied to the soma (or dendrite, Id), Cm was the membrane capacitance, and gc was the coupling conductance between the soma and the dendrite. The values for the specific membrane resistance, membrane capacitance, and cytoplasmic (axial) resistivity were, respectively, Rm = 30 KΩ-cm2, Cm = 1.0 µF/cm2, and Ra = 150 Ω-cm. The leakage reversal potential (EL) was set to −93 mV to match experimental measurements of their resting potential (Vrest, –85 mV; Geracitano et al. 2007; Amano et al. 2010). The resulting input resistance (RIN) was about 600 MΩ when measured from rest, consistent with experimental observations (Royer et al. 2000a; Marowsky et al. 2005; Amano et al. 2010). The ITC model contained several ionic currents including a leakage current IL, a spike-generating sodium current INa, a potassium delayed rectifier IDR, a slow deinactivating current ISD, a voltage-gated persistent muscarinic current IM, a hyperpolarization-activated current IH, a high-voltage activated Ca2+ current ICaL, and a slow Ca2+-dependent afterhyperpolarization current IsAHP. The ionic current for channel i, Ii ,was modeled as
) were the intrinsic currents and the synaptic currents in the somatic (dendritic) compartments, respectively, Is was the electrode current applied to the soma (or dendrite, Id), Cm was the membrane capacitance, and gc was the coupling conductance between the soma and the dendrite. The values for the specific membrane resistance, membrane capacitance, and cytoplasmic (axial) resistivity were, respectively, Rm = 30 KΩ-cm2, Cm = 1.0 µF/cm2, and Ra = 150 Ω-cm. The leakage reversal potential (EL) was set to −93 mV to match experimental measurements of their resting potential (Vrest, –85 mV; Geracitano et al. 2007; Amano et al. 2010). The resulting input resistance (RIN) was about 600 MΩ when measured from rest, consistent with experimental observations (Royer et al. 2000a; Marowsky et al. 2005; Amano et al. 2010). The ITC model contained several ionic currents including a leakage current IL, a spike-generating sodium current INa, a potassium delayed rectifier IDR, a slow deinactivating current ISD, a voltage-gated persistent muscarinic current IM, a hyperpolarization-activated current IH, a high-voltage activated Ca2+ current ICaL, and a slow Ca2+-dependent afterhyperpolarization current IsAHP. The ionic current for channel i, Ii ,was modeled as 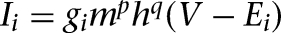 , where gi was its maximal conductance, m its activation variable (with exponent p), h its inactivation variable (with exponent q), and Ei its reversal potential. The kinetic equation for the gating variable x (m or h) had the form
, where gi was its maximal conductance, m its activation variable (with exponent p), h its inactivation variable (with exponent q), and Ei its reversal potential. The kinetic equation for the gating variable x (m or h) had the form
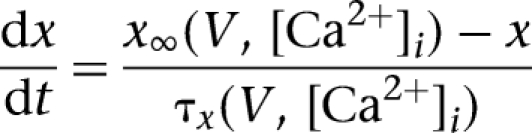 |
3 |
where x∞ was the voltage- or Ca2+-dependent steady state and τx was the voltage- or Ca2+- dependent time constant. The maximal conductance densities for all ionic currents and the expressions of x∞ and τx for each gating variable are listed in Supplemental Tables S1 and S2, respectively. The other reversal potentials were: ENa = 45 mV, EK = −90 mV, ECa = 120 mV, and EH = −43 mV (Huguenard and McCormick 1992).
Ce single-cell models
According to experimental findings (Dumont et al. 2002; De Armentia and Sah 2004), three different types of Ce neurons were modeled: regular spiking (RS), late firing (LF), and low-threshold bursting (LTB). The Ce cell models also contained two compartments: a soma (diameter of 15 µm; length of 15 µm) and a dendrite (diameter of 5 µm; length of 300 µm). The Ce models included a leakage current IL, a sodium current INa, a delayed rectifier IDR, a muscarinic current IM, a hyperpolarization-activated current IH, a high-voltage-activated Ca2+ current ICaL, and a slow Ca2+-dependent afterhyperpolarization current IsAHP. The LF cell contained an additional inactivating transient K+ current IA known to delay the onset of the action potential (Storm 1986), while the LTB cell included an additional low-threshold inactivating calcium current ICaT (Dumont et al. 2002).The passive membrane properties were as follows: Rm = 30 KΩ-cm2, Cm = 1.0 µF/cm2, Ra = 150 Ω-cm, EL = −60 mV for LF cell, and −70 mV for RS and LTB cells.
Current kinetics
The kinetics of INa, IDR, and ICaL were adapted from a prefrontal cortex (PFC) model by Durstewitz et al. (2000), and those for IA, IM, and IsAHP from Warman et al. (1994), who modeled CA1 hippocampal neurons. To adjust spiking threshold, the activation/inactivation functions of the INa current were shifted by −3 mV for ITC cells and 5 mV for Ce neurons. A voltage shift of 20 mV was also made in the inactivation function of the IA current to achieve the desirable delay effect seen in LF Ce cells (Dumont et al. 2002; De Armentia and Sah 2004). The kinetics of the ICaT current were taken from a granule cell model (Inoue and Strowbridge 2008) with the activation and inactivation functions shifted by −15 mV and −10 mV, respectively, to replicate the firing patterns reported by Dumont et al. (2002). The mathematical description of IH was based on a study of the current in rat BLA neurons (Womble and Moises 1993). The detailed kinetics of the slow deinactivating conductance ISD have not yet been characterized in ITC neurons, and we modeled this current as 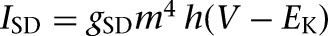 by modifying an “A” current (Huguenard and McCormick 1992) and adjusting the steady-state activation/inactivation with time constant functions to replicate the experimental observations of Royer et al. (2000b). The steady-state functions
by modifying an “A” current (Huguenard and McCormick 1992) and adjusting the steady-state activation/inactivation with time constant functions to replicate the experimental observations of Royer et al. (2000b). The steady-state functions 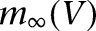 and
and 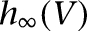 are shown in Supplemental Figure S1A, and the inactivation time constant function
are shown in Supplemental Figure S1A, and the inactivation time constant function 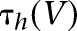 is shown in Supplemental Figure S1B. We note that the half-activation and half-inactivation voltages were about −80 mV and −50 mV, respectively, and ISD reached its maximum at around −60 mV (
is shown in Supplemental Figure S1B. We note that the half-activation and half-inactivation voltages were about −80 mV and −50 mV, respectively, and ISD reached its maximum at around −60 mV (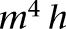 has the largest value), consistent with the observations of Royer et al. (2000b). The time constant of the inactivation variable was 100 msec for V > −30 mV and it increased greatly for V < −50 mV. Hence, ISD activated fully in the subthreshold range (V ≈ −60 mV), inactivated slowly in response to suprathreshold depolarization (V > −40 mV), and deinactivated very slowly (V < −50 mV).
has the largest value), consistent with the observations of Royer et al. (2000b). The time constant of the inactivation variable was 100 msec for V > −30 mV and it increased greatly for V < −50 mV. Hence, ISD activated fully in the subthreshold range (V ≈ −60 mV), inactivated slowly in response to suprathreshold depolarization (V > −40 mV), and deinactivated very slowly (V < −50 mV).
Calcium dynamics
Intracellular calcium was regulated by a simple first-order differential equation of the form (Warman et al. 1994):
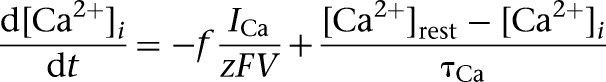 |
4 |
where f is the fraction of the Ca2+ influx (f = 0.024) (Warman et al. 1994), V = wA with w the shell thickness (1 µm), A the cell surface area, z = 2 is the valence of the Ca2+ ion, F is the Faraday constant, and τCa is the Ca2+ removal rate (τCa = 80 msec) (Helmchen et al. 1996). The resting Ca2+ concentration 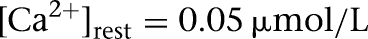 , which was the same as the initial concentration (Durstewitz et al. 2000).
, which was the same as the initial concentration (Durstewitz et al. 2000).
Network structure and inputs
The ITC–Ce network included 15 ITCD and 15 ITCV neurons, plus five Ce output cells (Fig. 1B). Among the five Ce neurons, three were LTB type, since these cells account for the majority of CeM (central medial nucleus) cells in the rat (Dumont et al. 2002), which the model was based on. The remaining two Ce neurons had one RS type and one LF type. Based on experimental findings (Geracitano et al. 2007), the synaptic connections between the ITC neurons were split equally into facilitating, depressing, and constant types (each neuron formed only one type of synapse with all its targets, as seen experimentally) (Geracitano et al. 2007). Each ITC neuron inhibited three other randomly selected neurons within the same cluster. This connectivity ratio (20%) is higher than reported in Geracitano et al. (2007), and was needed to compensate for the fewer number of cells in our reduced network (Bazhenov et al. 2001). Also, based on earlier experimental reports (Royer et al. 1999, 2000a), each ITCD neuron inhibited three randomly selected ITCV neurons (the inhibitory connectivity among ITCD and ITCV cell clusters is shown in Supplemental Table S3).
The network received inputs from LA (lateral amygdala), BA (basal amygdala), and IL (Fig. 1B), which were modeled as realistic spike inputs rather than real neurons. LA inputs projected to ITCD, while IL inputs projected equally to ITCD and ITCV (McDonald et al. 1996). BA inputs also projected to both ITCD and ITCV clusters, but with a lower density to ITCD neurons (Royer et al. 1999, 2000a; Pape and Pare 2010). Based on the findings of Herry et al. (2008), the BA inputs were divided into fear, extinction, and extinction-resistant (ER) groups (Fig. 1B). Since the exact connectivity of each type of BA neurons with ITC and Ce neurons is currently unknown, we considered two possible network architectures. In the first architecture, all three types of BA inputs projected to ITC, while only extinction inputs projected to ITC in the second architecture. In both architectures, the extinction inputs did not project to Ce because the activation profile of extinction cells was opposite to the expression of fear (Herry et al. 2008). Instead, both fear and ER inputs projected to Ce.
We modeled 20 LA, 20 BA, and 20 IL inputs to the network. The relative ratio of each type of BA input was based on experimental data (Herry et al. 2008), which resulted in five fear, five extinction, and 10 ER inputs. Each LA input projected to three randomly selected ITCD neurons, and each BA input projected to two ITCD and 3 ITCV neurons, both again chosen randomly. The projection from IL to ITC appeared to be denser than that from BLA (McDonald et al. 1996). So each IL input projected to five ITCD and five ITCV neurons selected at random. The Ce neurons received excitation from all BA fear and ER inputs, as well as inhibition from ITCV neurons of facilitating and constant type synapses (Pare and Smith 1993b; Royer et al. 1999; Pare et al. 2004; Geracitano et al. 2007). According to experimental observations (Geracitano et al. 2007), ITCV cells forming depressing-type synapses did not project to Ce. The LA and BA inputs were modeled with different degrees of spike frequency adaptation based on previous experimental data (Quirk et al. 1995, 1997; Faber et al. 2001; Herry et al. 2008) and to account for the projection from LA to BA (Fig. 1B; Pitkanen 2000), the firing rate of BA fear inputs was assumed to be dependent on LA inputs due to similar firing patterns across training (Quirk et al. 1995; Herry et al. 2008; see Supplemental Materials and Methods for details). The IL inputs were modeled as Poisson-distributed spike trains with a duration of 300 msec and a mean frequency of 20 Hz (Milad and Quirk 2002). In addition, Poisson-distributed random background inputs were delivered to all ITC and Ce neurons to achieve experimentally observed spontaneous firing rates.
Synaptic currents
In the model, excitatory synaptic transmission was mediated by glutamate acting at AMPA/NMDA receptors, while the inhibitory synapses were mediated by GABA acting at GABAA receptors (Sah et al. 2003). The AMPA/NMDA and GABAA synaptic currents were modeled by dual exponential functions as follows (Li et al. 2009):
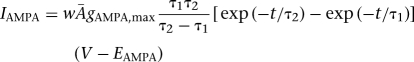 |
5 |
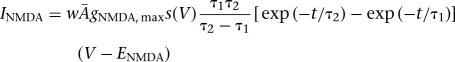 |
6 |
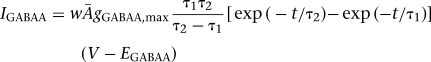 |
7 |
where w was the synaptic weight given in Table 1, 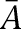 was a normalization constant chosen so that gAMPA, max, gNMDA, max, and gGABAA, max assumed maximum values of the conductances (when w = 1); τ1 and τ2 were the rise and decay time constants, respectively. For AMPA receptor channels, τ1 = 0.5 msec and τ2 = 7 msec for ITC cells and τ1 = 1.8 msec and τ2 = 4.4 msec for Ce neurons (De Armentia and Sah 2003). For NMDA receptor channels, τ1 = 5 msec and τ2 = 125 msec for ITC cells and τ1 = 5 msec and τ2 = 162 msec for Ce neurons (De Armentia and Sah 2003). The voltage-dependent variable s(V), which implements the Mg2+ block of NMDA receptors, was defined as: s(V) = [1 + 0.33 exp(−0.06 V)]−1 (Zador et al. 1990). For the GABAA synaptic current, τ1 = 0.8 msec, τ2 = 13 msec for ITC cells (Geracitano et al. 2007), and τ1 = 0.8 msec, τ2 = 40 msec for Ce neurons (Esmaeili et al. 2009). The maximal conductances were chosen as: gAMPA, max = 1 nS, gNMDA, max = 0.5 nS (Li et al. 2009), and gGABAA, max = 0.6 nS (Geracitano et al. 2007). Synaptic reversal potentials were set as EAMPA = ENMDA = 0 mV, EGABAA = −τ75 mV (Li et al. 2009). The GABAA conductance profile of ITC neurons is shown in Supplemental Figure S3.The synaptic delay was chosen randomly between a minimal and a maximal values with a uniform probability, based on experimental data from the Pare lab (Supplemental Table S4).
was a normalization constant chosen so that gAMPA, max, gNMDA, max, and gGABAA, max assumed maximum values of the conductances (when w = 1); τ1 and τ2 were the rise and decay time constants, respectively. For AMPA receptor channels, τ1 = 0.5 msec and τ2 = 7 msec for ITC cells and τ1 = 1.8 msec and τ2 = 4.4 msec for Ce neurons (De Armentia and Sah 2003). For NMDA receptor channels, τ1 = 5 msec and τ2 = 125 msec for ITC cells and τ1 = 5 msec and τ2 = 162 msec for Ce neurons (De Armentia and Sah 2003). The voltage-dependent variable s(V), which implements the Mg2+ block of NMDA receptors, was defined as: s(V) = [1 + 0.33 exp(−0.06 V)]−1 (Zador et al. 1990). For the GABAA synaptic current, τ1 = 0.8 msec, τ2 = 13 msec for ITC cells (Geracitano et al. 2007), and τ1 = 0.8 msec, τ2 = 40 msec for Ce neurons (Esmaeili et al. 2009). The maximal conductances were chosen as: gAMPA, max = 1 nS, gNMDA, max = 0.5 nS (Li et al. 2009), and gGABAA, max = 0.6 nS (Geracitano et al. 2007). Synaptic reversal potentials were set as EAMPA = ENMDA = 0 mV, EGABAA = −τ75 mV (Li et al. 2009). The GABAA conductance profile of ITC neurons is shown in Supplemental Figure S3.The synaptic delay was chosen randomly between a minimal and a maximal values with a uniform probability, based on experimental data from the Pare lab (Supplemental Table S4).
Presynaptic release probability
As observed experimentally (Geracitano et al. 2007), short-term synaptic plasticity in ITC–ITC connections is heterogeneous (facilitating, depressing, and constant) and depends on presynaptic firing rates. As presynaptic stimulation frequency increases, the release probability of facilitating synapses increases, while that of depressing synapses decreases, and that of constant synapses remains unchanged. Such plasticity is also assumed to exist in the ITCV–Ce connections. In the model, presynaptic release probability was updated according to the following learning rule:
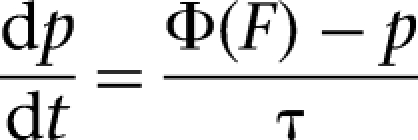 |
8 |
where the time constant τ was selected to be 100 msec (Wang 1999a; a larger time constant of 300 msec yielded similar results), Φ was a function of the presynaptic firing frequency F, determined by nonlinear regression modeling of the experimental data (Geracitano et al. 2007; Supplemental Fig. S4). When a presynaptic spike occurred, the presynaptic frequency was updated as the running average of the current and three previous instantaneous frequencies: 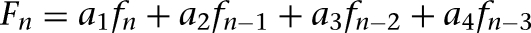 (a1 = 0.4, a3 = 0.2, a3 = 0.2, a4 = 0.1), where
(a1 = 0.4, a3 = 0.2, a3 = 0.2, a4 = 0.1), where 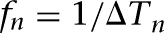 and ΔTn was the interval between the previous and current spikes. Between spikes, F decayed back towards zero with a time constant of 5 sec (a relatively large time constant was chosen to avoid rapid frequency decay between spikes). When a presynaptic spike occurred, a uniform pseudo-random number Pr between 0 and 1 (using the rand function in C++) was generated. If Pr was less than the current release probability p then neurotransmitter release was successful, otherwise it was not. The initial release probability was 0.3 and 0.8 for the facilitating and depressing synapses, respectively, and the release probability of constant synapses was fixed at 0.75 (Geracitano et al. 2007).
and ΔTn was the interval between the previous and current spikes. Between spikes, F decayed back towards zero with a time constant of 5 sec (a relatively large time constant was chosen to avoid rapid frequency decay between spikes). When a presynaptic spike occurred, a uniform pseudo-random number Pr between 0 and 1 (using the rand function in C++) was generated. If Pr was less than the current release probability p then neurotransmitter release was successful, otherwise it was not. The initial release probability was 0.3 and 0.8 for the facilitating and depressing synapses, respectively, and the release probability of constant synapses was fixed at 0.75 (Geracitano et al. 2007).
Synaptic strengths in three network states
We simulated network responses to a 2-sec auditory tone input (CS) during three different network states: habituation, following fear conditioning, and after extinction training. Learning of fear and extinction is accompanied by changes in synaptic strengths in the neural circuitry of the amygdala (Pape and Pare 2010). ITC neurons exhibit NMDA-dependent bidirectional synaptic plasticity (Royer and Pare 2002) and in a recent experimental study, the BA inputs to ITC cells are found to be potentiated in extinction training (approximately threefold; Fig. 4 in Amano et al. 2010). Given the fact that the firing rate of LA neurons is significantly increased after conditioning (Quirk et al. 1995), it is reasonable to assume that the LA–ITCD connection is potentiated by conditioning. Hence, we used a threefold synaptic weight (compared with the habituation state) for the LA–ITCD synapses in the fear state and a threefold synaptic weight for the BA–ITC synapses in the extinction state (Table 1). For the LA–ITCD connection, the potentiated synapses were assumed to be partially depotentiated in the extinction state (Table 1) based on results from a previous LA network model (Li et al. 2009). Note that the potentiation was only imposed on the AMPA components of the synapses (Amano et al. 2010). The BA–Ce, ITC–ITC, and ITC–Ce synaptic weights were assumed to be fixed, but the presynaptic release probability of the ITC–ITC and ITC–Ce synapses was modifiable (Geracitano et al. 2007).
Implementation of model
Model runs were performed on a personal computer using the software package GENESIS (Bower and Beeman 2003) with the Crank-Nicholson integration method, and a time step of 20 µsec. Shorter time steps did not change the results in any significant way. The (background) input onset was 1 sec after the start of the simulation to allow the model cells to stabilize. A simulation of 5 sec of network activity took 15 min of CPU time.
Limitations
Admittedly, our model has some limitations: (1) Given the restricted amount of detailed electrophysiological data about ITC cells, we had to use data on ionic conductance from other brain areas such as hippocampus and cortex. (2) In the absence of more specific information, IL was assumed to project equally to dorsal and ventral clusters. Also, we assumed the same connectivity ratio within the ITC clusters and between the clusters. (3) The network size was designed to be relatively small in order to avoid computational overload. It remains to be determined whether the qualitative conclusions and predictions of the model will hold for a larger network. (4) When dealing with the effectiveness of IL stimulation, the role of IL inputs in facilitating plasticity at BA–ITC synapses (Amano et al. 2010) was not taken into account. (5) Although it was previously reported that the lateral sector of the central nucleus (CeL) projects to CeM (Petrovich and Swanson 1997; Jolkkonen and Pitkanen 1998), CeL was not included in our model. This omission resulted from the paucity of data available to constrain the model. However, as we were completing the present investigation, two studies provided evidence that CeL activity might play an important role in the genesis of conditioned fear responses. In particular, it was shown that pretraining inhibition of CeL with muscimol (Ciocchi et al. 2010) or silencing of a subset of CeL cells expressing PKC-δ (Haubensak et al. 2010) interfered with the acquisition of conditioned fear. Moreover, after fear conditioning, CS presentations inhibited PKC-δ expressing CeL cells (hereafter termed CeL– neurons) (Ciocchi et al. 2010; Haubensak et al. 2010), whereas PKC-δ negative CeL neurons were excited by the CS (hereafter termed CeL+ cells). The existence of inhibitory connections between CeL+ and CeL– neurons implied that when the CS is presented, CeL+ cells inhibit CeL– neurons resulting in the disinhibition of CeM fear output neurons (Ciocchi et al. 2010; Haubensak et al. 2010). Future modeling studies should consider how CeL activity interacts with ITC inputs to regulate the excitability of CeM neurons and fear expression.
Acknowledgments
This research was supported in part by NIMH grants MH087755 to S.S.N. and MH083710 to D.P. We thank Dr. Gregory Quirk for helpful discussions and comments on an earlier draft.
Footnotes
[Supplemental material is available for this article.]
References
- Amano T, Unal CT, Pare D 2010. Synaptic correlates of fear extinction in the amygdala. Nat Neurosci 13: 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Stopfer M, Rabinovich M, Huerta R, Abarbanel HDI, Sejnowski TJ, Laurent G 2001. Model of transient oscillatory synchronization in the locust antennal lobe. Neuron 30: 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta S, Pantazopoulos H, Caldera M, Pantazopoulos P, Pare D 2005. Infralimbic cortex activation increases c-Fos expression in intercalated neurons of the amygdala. Neuroscience 132: 943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower JM, Beeman D 2003. The book of GENESIS: Exploring realistic neural models with the GEneral NEural SImulation System. Internet edition (http://www.genesis-sim.org/GENESIS)
- Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ 2009. Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J Neurosci 29: 8474–8482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JH, Roberts JL 2004. From molecules to networks - An introduction to cellular and molecular neuroscience. Elsevier Academic Press, London, UK [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SBE, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, et al. 2010. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468: 277–282 [DOI] [PubMed] [Google Scholar]
- De Armentia ML, Sah P 2003. Development and subunit composition of synaptic NMDA receptors in the amygdala: NR2B synapses in the adult central amygdala. J Neurosci 23: 6876–6883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Armentia ML, Sah P 2004. Firing properties and connectivity of neurons in the rat lateral central nucleus of the amygdala. J Neurophysiol 92: 1285–1294 [DOI] [PubMed] [Google Scholar]
- Dumont É, Martina M, Samson R, Drolet G, Pare D 2002. Physiological properties of central amygdala neurons: Species differences. Eur J Neurosci 15: 545–552 [DOI] [PubMed] [Google Scholar]
- Durstewitz D, Seamans JK, Sejnowski TJ 2000. Dopamine-mediated stabilization of delay-period activity in a network model of prefrontal cortex. J Neurophysiol 83: 1733–1750 [DOI] [PubMed] [Google Scholar]
- Duvarci S, Popa D, Pare D 2011. Central amygdala activity during fear conditioning. J Neurosci 31: 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA 2002. Graded persistent activity in entorhinal cortex neurons. Nature 420: 173–178 [DOI] [PubMed] [Google Scholar]
- Esmaeili A, Lynch JW, Sah P 2009. GABAA receptors containing gamma1 subunits contribute to inhibitory transmission in the central amygdala. J Neurophysiol 101: 341–349 [DOI] [PubMed] [Google Scholar]
- Faber ESL, Callister RJ, Sah P 2001. Morphological and electrophysiological properties of principal neurons in the rat lateral amygdala in vitro. J Neurophysiol 85: 714–723 [DOI] [PubMed] [Google Scholar]
- Freedman LJ, Insel TR, Smith Y 2000. Subcortical projections of area 25 (subgenual cortex) of the macaque monkey. J Comp Neurol 42: 172–188 [PubMed] [Google Scholar]
- Geracitano R, Kaufmann WA, Szabo G, Ferraguti F, Capogna M 2007. Synaptic heterogeneity between mouse paracapsular intercalated neurons of the amygdala. J Physiol 585: 117–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, et al. 2010. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 468: 270–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B 1996. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J 70: 1069–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A 2008. Switching on and off fear by distinct neuronal circuits. Nature 454: 600–606 [DOI] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA 1992. Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol 68: 1373–1383 [DOI] [PubMed] [Google Scholar]
- Inoue T, Strowbridge BW 2008. Transient activity induces a long-lasting increase in the excitability of olfactory bulb interneurons. J Neurophysiol 99: 187–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolkkonen E, Pitkanen A 1998. Intrinsic connections of the rat amygdaloid complex: Projections originating in the central nucleus. J Comp Neurol 395: 53–72 [DOI] [PubMed] [Google Scholar]
- Jungling K, Seidenbecher T, Sosulina L, Lesting J, Sangha S, Clark SD, Okamura N, Duangdao DM, Xu YL, Reinscheid RK, et al. 2008. Neuropeptide S-mediated control of fear expression and extinction: Role of intercalated GABAergic neurons in the amygdala. Neuron 59: 298–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapska E, Maren S 2009. Reciprocal patterns of c-Fos expression in the medial prefrontal cortex and amygdala after extinction and renewal of conditioned fear. Learn Mem 16: 486–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Nair SS, Quirk GJ 2009. A biologically realistic network model of acquisition and extinction of conditioned fear associations in lateral amygdala neurons. J Neurophysiol 101: 1629–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute A, Fidacaro GA, Pare D 2008. Amygdala intercalated neurons are required for expression of fear extinction. Nature 454: 642–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marowsky A, Yanagawa Y, Obata K, Vogt KE 2005. A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron 48: 1025–1037 [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F, Guo L 1996. Projections of the medial and lateral prefrontal cortices to the amygdala: A Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience 71: 55–75 [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ 2002. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70–74 [DOI] [PubMed] [Google Scholar]
- Milad MR, Vidal-Gonzalez I, Quirk GJ 2004. Electrical stimulation of medial prefrontal cortex reduces conditioned fear in a temporally specific manner. Behav Neurosci 118: 389–394 [DOI] [PubMed] [Google Scholar]
- Miller P, Brody CD, Romo R, Wang X-J 2003. A recurrent network model of somatosensory parametric working memory in the prefrontal cortex. Cerebral Cortex 13: 1208–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millhouse OE 1986. The intercalated cells of the amygdala. J Comp Neurol 247: 246–271 [DOI] [PubMed] [Google Scholar]
- Pape HC, Pare D 2010. Plastic synaptic networks of the Amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev 90: 419–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D, Collins DR 2000. Neuronal correlates of fear in the lateral amygdala: Multiple extracellular recording in conscious cats. J Neurosci 20: 2701–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D, Smith Y 1993a. Distribution of GABA immunoreactivity in the amygdaloid complex of the cat. Neuroscience 57: 1061–1076 [DOI] [PubMed] [Google Scholar]
- Pare D, Smith Y 1993b. The intercalated cell masses project to the central and medial nuclei of the amygdala in cats. Neuroscience 57: 1077–1090 [DOI] [PubMed] [Google Scholar]
- Pare D, Quirk GJ, LeDoux JE 2004. New vistas on amygdala networks in conditioned fear. J Neurophysiol 92: 1–9 [DOI] [PubMed] [Google Scholar]
- Petrovich GD, Swanson LW 1997. Projections from the lateral part of the central amygdalar nucleus to the postulated fear conditioning circuit. Brain Research 763: 247–254 [DOI] [PubMed] [Google Scholar]
- Pitkanen A 2000. Connectivity of the rat amygdaloid complex. In The amygdala: A functional analysis, 2nd ed (ed. Aggleton JP), pp. 31–115 Oxford University Press, Oxford, UK [Google Scholar]
- Quirk GJ, Mueller D 2008. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacol 33: 56–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Repa C, LeDoux JE 1995. Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: Parallel recordings in the freely behaving rat. Neuron 15: 1029–1039 [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Armony JL, LeDoux JE 1997. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron 19: 613–624 [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K 2000. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20: 6225–6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Pare D 2003. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci 23: 8800–8807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE 2001. Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nature Neurosci 4: 724–731 [DOI] [PubMed] [Google Scholar]
- Royer S, Pare D 2002. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 115: 455–462 [DOI] [PubMed] [Google Scholar]
- Royer S, Martina M, Pare D 1999. An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci 19: 10575–10583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Martina M, Pare D 2000a. Polarized synaptic interactions between intercalated neurons of the amygdala. J Neurophysiol 83: 3509–3518 [DOI] [PubMed] [Google Scholar]
- Royer S, Martina M, Pare D 2000b. Bistable behavior of inhibitory neurons controlling impulse traffic through the amygdala: Role of a slowly deinactivating K+ current. J Neurosci 20: 9034–9039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ESL, De Armentia ML, Power J 2003. The amygdaloid complex: Anatomy and physiology. Physiol Rev 83: 803–834 [DOI] [PubMed] [Google Scholar]
- Storm JF 1986. A-current and Ca-dependent transient outward current control the initial repetitive firing in hippocampal neurons. (Abstract). Biophys J 49: 369 [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ 2006. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem 13: 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X-J 1998. Calcium coding and adaptive temporal computation in cortical pyramidal neurons. J Neurophysiol 79: 1549–1566 [DOI] [PubMed] [Google Scholar]
- Wang X-J 1999a. Fast burst firing and short-term synaptic plasticity: A model of neocortical chattering neurons. Neuroscience 89: 347–362 [DOI] [PubMed] [Google Scholar]
- Wang X-J 1999b. Synaptic basis of cortical persistent activity: The importance of NMDA receptors to working memory. J Neurosci 19: 9587–9603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warman EN, Durand DM, Yuen GLF 1994. Reconstruction of hippocampal CA1 pyramidal cell electrophysiology by computer simulation. J Neurophysiol 71: 2033–2045 [DOI] [PubMed] [Google Scholar]
- Winograd M, Destexhe A, Sanchez-Vives MV 2008. Hyperpolarization-activated graded persistent activity in the prefrontal cortex. PNAS 105: 7298–7303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womble MD, Moises HC 1993. Hyperpolarization-activated currents in neurons of the rat basolateral amygdala. J Neurophysiol 70: 2056–2065 [DOI] [PubMed] [Google Scholar]
- Zador A, Koch C, Brown TH 1990. Biophysical model of a Hebbian synapse. Proc Natl Acad Soc 87: 6718–6722 [DOI] [PMC free article] [PubMed] [Google Scholar]