

Summary
Mitogen-activated protein kinases (MAPKs) are key mediators of the T cell receptor (TCR) signals but their roles in T helper (Th) cell differentiation are unclear. Here we showed that the MAPK kinase kinases MEKK2 (encoded by Map3k2) and MEKK3 (encoded by Map3k3), negatively regulated transforming growth factor-β (TGF-β)-mediated Th cell differentiation. Map3k2-/-Map3k3Lck-Cre/- mice showed an abnormal accumulation of regulatory T (Treg) and Th17 cells in the periphery, consistent with Map3k2-/-Map3k3Lck-Cre/- naïve CD4+ T cells' differentiation into Treg and Th17 cells with a higher frequency than wild-type (WT) cells after TGF-β stimulation in vitro. In addition, Map3k2-/-Map3k3Lck-Cre/- mice developed more severe experimental autoimmune encephalomyelitis. Map3k2-/-Map3k3Lck-Cre/- T cells exhibited impaired phosphorylation of the SMAD2 and SMAD3 proteins at their linker regions, which negatively regulated the TGF-β responses in T cells. Thus, the crosstalk between TCR-induced MAPK and the TGF-β signaling pathways is important in regulating Th cell differentiation.
Introduction
Naïve CD4+ T cells must differentiate into distinct helper T (Th) cell lineages in order to mount appropriate immune responses against various classes of pathogens and maintain tolerance to self-tissues. The differentiation of Th cell lineages, which include the Th1, Th2, Th17, and regulatory T (Treg) lineages, depends on signals derived from the T cell receptor (TCR) and the cytokine milieu of the local environment (Murphy and Reiner, 2002; Weaver et al., 2007). Dysregulation of Th cell differentiation has been associated with either immune deficiency or autoimmune disease (Bettelli et al., 2007). TGF-β is a pleiotropic cytokine crucial for many important biological processes including adaptive immunity (Li and Flavell, 2008; Massague, 1998). As a crucial cytokine for T cell mediated immunity, TGF-β induces the differentiation and homeostasis of Foxp3+ Treg cells in the periphery (Chen et al., 2003), mediates the differentiation of Th17 cells in cooperation with IL-6(Bettelli et al., 2006; Veldhoen et al., 2006), and suppresses the differentiation of Th1 and Th2 cell lineages (Gorham et al., 1998; Li and Flavell, 2008). Perturbation of TGF-β signaling specifically in T cells causes severe autoimmunity associated with excessive Th1 cell responses (Li et al., 2006; Marie et al., 2006).
TGF-β triggers a canonical signaling pathway through the TGF-β receptor-mediated phosphorylation of receptor regulated (R)-SMAD proteins; SMAD2 and SMAD3, on conserved serine residues at their carboxyl (C) terminus (Derynck and Zhang, 2003; Massague, 1998). Phosphorylated SMAD2 and SMAD3 subsequently bind to SMAD4 and translocate into the nucleus to mediate the transcription of target genes (Derynck and Zhang, 2003; Massague, 1998). Many of the key components of this canonical pathway have been shown to be essential in mediating the TGF-β dependent Treg and Th17 cell differentiation (Malhotra et al., 2010; Martinez et al., 2010; Martinez et al., 2009; Tone et al., 2008). In addition to the C-terminal phosphorylation, R-SMADs can also be phosphorylated by mitogen-activated protein kinases (MAPKs) at a short region, termed linker region that links the N-terminal MH1 domain (MAD homology domain) and the C-terminal MH2 domain, resulting in the suppression of the TGF-β signaling pathway (Derynck and Zhang, 2003; Kretzschmar et al., 1999; Matsuura et al., 2005; Wrighton et al., 2009). The linker region phosphorylation of R-SMADs can also be mediated by other proline-directed kinases, such as CDKs and GSK3, resulting in either activation or suppression of R-SMAD activity (Alarcon et al., 2009; Guo et al., 2008; Millet et al., 2009; Wrighton et al., 2009). However, the regulation and significance of R-SMAD linker region phosphorylation in Th cell differentiation have not been studied.
MAPKs, which include the extracellular signal regulated kinases (ERK1 and 2), the p38 (α, β, γ and δ), the c-Jun N-terminal protein kinase (JNK1, 2, and 3), and the ERK5 MAPKs, are important intracellular signaling networks employed by all eukaryotic cells to transduce a wide spectrum of extracellular signals triggered by growth factors, cytokines, or stress factors (Dong et al., 2002; Su and Karin, 1996; Weston et al., 2002). These MAPKs are activated by a three-kinase module that include, a MAPK, a MAPK kinase (MAP2K), and MAP2K kinase (MAP3K) (Su and Karin, 1996). Previous studies have established the roles of individual MAPK pathways in T cell development, activation and differentiation (Dong et al., 2002; Su et al., 1994), but the precise molecular mechanisms by which these MAPK modules function in Th cell differentiation remain unclear, nor is much known about their roles in either Treg or Th17 cell differentiation.
Among 19 MAP3Ks in the mammalian genome, MEKK2 and MEKK3 are two highly conserved members of the MEK kinase (MEKK) subgroup of the MAP3K super family. Transient expression of either Map3k2 or Map3k3 in vitro leads to their auto-activation and activates all four major MAPKs: ERK1 and ERK2, p38, JNK, and ERK5 (Cheng et al., 2000; Huang et al., 2004; Yang et al., 2001; Zhang et al., 2006). Previous biochemical and genetic studies show that MEKK2 and MEKK3 regulate either T cell activation or homeostasis, likely through a mechanism involving downstream MAPK activation (Guo et al., 2002; Wang et al., 2009). However, because both MEKK2 and MEKK3 are co-expressed in T cells as well as many other cell types, and share substrate specificity (Cheng et al., 2000; Wang et al., 2009), they may have overlapping function(s) in T cells which cannot be revealed from studying single gene knockout mice.
Here we report that MEKK2 and MEKK3 negatively regulate TGF-β signaling in Th cell differentiation. Mice with MEKK2 and MEKK3 double deficiency in T cells had increased Treg and Th17 cells in the periphery. Map3k2-/-Map3k3Lck-Cre/- (see below) naïve T cells exhibited increased sensitivity to TGF-β stimulation in Treg, Th17 and Th1 cell differentiation due to a defect in ERK1 and ERK2 activation, resulting in hypo-phosphorylation of SMAD2 and SMAD3 at their linker regions. Map3k2-/-Map3k3Lck-Cre/- mice developed more severe EAE disease and had more antigen specific Th17 cells that were preferentially accumulated in the central nervous system. Together, our study uncovers a T cell intrinsic regulatory mechanism for the TGF-β-mediated Th cell differentiation, and demonstrates that T cell sensitivity to TGF-β is dynamically tuned by the TCR-induced activation of MEKK2 and MEKK3 through phosphorylation of SMAD2 and SMAD3 at their linker regions.
Results
Generation of Map3k2-/-Map3k3Lck-Cre/- Mice
To study the roles of MEKK2 and MEKK3 in T cells, we generated MEKK2 and MEKK3 T cell double deficient mice by crossing the Lck-Cre;Map3k3fl/- (referred as Map3k3Lck-Cre/-) mice with the Map3k2-/- mice (Guo et al., 2002; Wang et al., 2009). The resulting Map3k2-/-Map3k3Lck-Cre/- mice have Mekk3 deleted specifically in T cells and Mekk2 deleted globally, resulting in the loss of both MEKK2 and MEKK3 only in the T cell lineage (Supplemental Figure S1A).
Map3k2-/-Map3k3Lck-Cre/- mice were viable with no obvious abnormalities in non-immune tissues (data not shown). The total number of thymocytes in the Map3k2-/-Map3k3Lck-Cre/- mice was similar to that in wild-type (WT) mice, containing all the major subsets of thymocytes suggesting that MEKK2 and MEKK3 are not essential for the thymocyte development (data not shown).
Map3k2-/-Map3k3Lck-Cre/- mice had reduced numbers of CD4+ and CD8+ T cells in the periphery (data not shown), consistent with our recent finding that MEKK3 regulates T cell homeostasis in the periphery (Wang et al., 2009). Despite the reduction of total spleen cell number, the percentages of CD4+ and CD8+ T cells seemed only slightly reduced in the Map3k2-/-Map3k3Lck-Cre/- mice (Figure 1A). However, when compared to WT mice there was a dramatic increase in the frequency of Map3k2-/-Map3k3Lck-Cre/- CD4+ T cells with an effector or memory-like (CD44hiCD62Llo) phenotype (Figure 1A).
Figure 1. Loss of Map3k2 and Map3k3 in T cells Results in Accumulation of Treg and Th17 Cells in vivo.

(A) Splenocytes from wild type (WT) and Map3k2-/-Map3k3Lck-Cre/- (dKO) mice were stained for CD4, CD8, CD44 and CD62L and analyzed by flow cytometry. The percentages of CD4+ and CD8+ T cells are shown at the upper panels. The gated CD4+ T cells were further analyzed for CD44+ and CD62L+ sub-populations (lower panels). Numbers in the profiles indicate the percentages of the gated populations.
(B) Splenocytes were stained for CD4 and Foxp3 to show the percentages (numbers in the gated boxes) of Foxp3+CD4+ T cells (Treg) in the spleen of WT and dKO mice. Data shown are from gated splenic CD4+ cells. FSC, forward scatter.
(C) Summary of splenic Foxp3+CD4+ T cells in WT (n=8), dKO (n=5), Map3k3Lck-Cre/- (3-cKO) (n=6), and Map3k2-/- (2-KO) mice (n=6). Error bars show the standard deviation. **, p<0.01 by two tailed Student's t test.
(D) Ex vivo splenocytes were stimulated with PMA+ionomycin for 6-h then analyzed for IL-17A and IFN-γ expressing CD4+ T cells by flow cytometry. The data shown were gated on CD4+ splenocytes and the percentages of IL-17A and IFN-γ expressing CD4+ T cells from WT and dKO mice are shown in the quadrants.
(E) Summary of splenic Th17+ CD4+ T cells in WT (n=10), dKO (n=8), 3-cKO (n=6), and 2-KO mice (n=6). Error bars show the standard deviation. **, p<0.01 by two tailed Student's t test.
(F, G) Rag1-/- mice were reconstituted with mixed bone marrow cells from B6.SJL (CD45.1+) and dKO (CD45.2+) mice, or from mixed B6.SJL (CD45.1+) and WT (CD45.2+) mice at roughly a 1 to 1 ratio. Total splenocytes in the recipient mice were analyzed 8-week late for CD4+Foxp3+ cellsF), or CD4+IL-17A+ cells (G) as indicated. Data shown are gated on CD4+CD45.1+ (B6.SJL) or CD4+CD45.2+ (WT or dKO) populations. Numbers next to the gated areas show the percentages of the gated population and the results are representative of 6 pairs of mice from two independent experiments.
Increased Treg and Th17 Cells in the Periphery of Map3k2-/-Map3k3Lck-Cre/- Mice
Despite the increased effector or memory-like T cell population in Map3k2-/-Map3k3Lck-Cre/- mice, the percentage of Foxp3+ splenic CD4+ cells (Treg) was two-fold higher than in the WT mice (Figure 1B). However, the absolute number of Foxp3+ cells in the periphery was not altered substantially (Supplemental Figure S1B) because the total splenic CD4+ T cells in Map3k2-/-Map3k3Lck-Cre/- mice was only about half of that in the WT mice (data not shown). The frequency and number of Foxp3+ Treg cells in the thymus of the Map3k2-/-Map3k3Lck-Cre/- mice were not altered (data not shown), indicating that the increased frequency of Treg cells in the periphery of Map3k2-/-Map3k3Lck-Cre/- mice was not due to enhanced thymic production. Deletion of both MEKK2 and MEKK3 was required for the increased Treg cell percentage because neither Map3k2-/- mice nor Map3k3Lck-Cre/- mice manifested this defect (Figure 1C). Furthermore, the Treg cells in the Map3k2-/-Map3k3Lck-Cre/- mice appeared functional and were able to suppress naïve T cell proliferation in vitro with only a slightly reduced efficiency compared to their WT counterparts (Supplemental Figure S1C).
We next examined the effector T cell subsets in the periphery of the Map3k2-/-Map3k3Lck-Cre/- mice. Around 4-5% of the peripheral Map3k2-/-Map3k3Lck-Cre/- CD4+ T cells expressed IL-17A (Th17), whereas less than 0.5% of these cells were found in WT mice (Figure 1D). The total number of Th17 cells was also 3-4 times more in the Map3k2-/-Map3k3Lck-Cre/- mice than in the WT mice (Supplemental Figure S1B). Again, deletion of both MEKK2 and MEKK3 was required for the increased Th17 cells in the periphery (Figure 1E). The percentage of interferon-γ (IFN-γ) producing CD4 T cells (Th1) was marginally increased but the total number of these cells was not increased in the Map3k2-/-Map3k3Lck-Cre/- mice (Figure 1D and data not shown). The IL-4 producing T cells (Th2) were also not markedly changed in the Map3k2-/-Map3k3Lck-Cre/- mice (data not shown). Similar data were obtained from analyzing the lymph node T cells (data not shown). Together, these results suggest that deletion of both Map3k2 and Map3k3 in T cells redirects Th cell differentiation favoring the Treg and Th17 cell lineages.
Because Map3k2-/-Map3k3Lck-Cre/- mice have increased frequency of CD62LloCD44hi T cells in the periphery, it is possible that the increased number of Th17 cells observed in these mice is simply a consequence of more effector cells in the periphery. To examine this possibility, we compared the frequencies of IL17A- and IFN-γ-producing T cells within the effector T cells (CD44hi) between the WT and the Map3k2-/-Map3k3Lck-Cre/- mice. After normalizing to the CD44hi population, the Map3k2-/-Map3k3Lck-Cre/- CD4 T cells still had much higher frequency of Th17 cells than the WT CD4 T cells (8.76% vs. 2.26%), whereas the frequency of IFN-γ+ cells was indeed lower in the same subset of Map3k2-/-Map3k3Lck-Cre/- cells compared to that of the WT T cells (12.1% vs. 21.9%) (Supplemental Figure S1D). Thus, the increased frequency of Th17 cells in the Map3k2-/-Map3k3Lck-Cre/- mice was unlikely caused by the augmented numbers of CD62LloCD44hi T cells in Map3k2-/-Map3k3Lck-Cre/- mice.
To further analyze the effect of MEKK2 and 3 deficiency on T cell differentiation in vivo, we directly compared the gene expression profiles of freshly isolated CD4+CD62LloCD44hi cells between Map3k2-/-Map3k3Lck-Cre/- and WT mice. Using an Illumina Microarray (Supplemental Figure S1E, Table S1) and real time-quantitative RT-PCR (Supplemental Figure S1E), we found that the expression of Th17 cell lineage specific genes (Il17A, Rorc, and Ccr6) was markedly increased in the Map3k2-/-Map3k3Lck-Cre/- T cells, whereas the expression of Th1 cell lineage specific genes (Tbx21, Il12rb1, Ifng, Irf1, and Fasl, etc.) was suppressed. These data together suggest that MEKK2 and 3 may inhibit Th17 cell differentiation while being required for optimal Th1 cell differentiation in the periphery.
MEKK2 and 3 Suppress Treg and Th17 Cell Differentiation Through a Cell Intrinsic Mechanism
To test whether the defects in Treg and Th17 cell differentiation in the Map3k2-/-Map3k3Lck-Cre/- mice were intrinsic to T cells, we reconstituted irradiated Rag1-/- mice with Map3k2-/-Map3k3Lck-Cre/- (CD45.2+) bone marrow cells mixed at a 1:1 ratio with WT congenic (CD45.1+) bone marrow cells. Eight weeks after the reconstitution, we analyzed the phenotypes of donor T cells in the periphery of the chimeric mice. The frequencies of the Map3k2-/-Map3k3Lck-Cre/- Foxp3+ CD4 cells (Figure 1F) and Map3k2-/-Map3k3Lck-Cre/- IL-17A+ CD4 T cells (Figure 1G) in the spleens were consistently higher than their WT counterparts derived from the cotransferred congenic WT bone marrow cells in the same host, resembling the T cell phenotypes found in the parental Map3k2-/-Map3k3Lck-Cre/- mice. We also generated bone marrow chimeric mice by transferring non-mixed control WT bone marrow cells or Map3k2-/-Map3k3Lck-Cre/- bone marrow cells into the Rag1-/- mice. Again, the peripheral Map3k2-/-Map3k3Lck-Cre/- T cells from those chimeric mice also fully recapitulated the phenotypes of T cells from the parental Map3k2-/-Map3k3Lck-Cre/- mice, including the increased activated or memory-like T cells, increased frequencies of Treg, and Th17 cells in the periphery (Supplemental Figure S1F). These data together demonstrate that MEKK2 and 3 suppress Treg and Th17 cell lineage differentiation through a cell intrinsic mechanism.
Map3k2-/-Map3k3Lck-Cre/- Naive T cells Are Hypersensitive to TGF-β-mediated Differentiation
To determine if the Treg and Th17 cell phenotypes in Map3k2-/-Map3k3Lck-Cre/- mice were caused by defects in naïve T cell differentiation, we examined in vitro differentiation of naive Map3k2-/-Map3k3Lck-Cre/- CD4 T cells (CD4+CD62LhiCD44loCD25-). Under the Treg cell differentiation condition, we found that at concentrations of 5 or 1 ng/ml of TGF-β, 82% or 60.8% of Map3k2-/-Map3k3Lck-Cre/- naïve T cells differentiated into the Foxp3+ Treg (iTreg), respectively; whereas 33.2% or 14.2% of WT T cells differentiated into iTreg, respectively, under the same condition (Figure 2A). Interestingly, about 30% of Map3k2-/-Map3k3Lck-Cre/- T cells spontaneously differentiated into iTreg cells without exogenous TGF-β whereas almost no WT iTreg cells were generated under the same condition (Figure 2A). To determine whether the spontaneous induction of Map3k2-/-Map3k3Lck-Cre/- iTreg was caused by the trace amount of TGF-β in the culture medium, a neutralizing antibody against TGF-β was added into the culture, which completely eliminated the iTreg differentiation from the Map3k2-/-Map3k3Lck-Cre/- T cells (Figure 2A).
Figure 2. MEKK2 and MEKK3 Suppress the TGF-β Mediated T Cell Differentiation in vitro.

(A) Naïve CD4+ T cells (CD4+CD62LhiCD44loCD25-) from WT and Map3k2-/-Map3k3Lck-Cre/- (dKO) mice were differentiated into Foxp3+ Treg cells with the indicated concentrations of TGF-β. Induction of the Foxp3+ cells was analyzed five days after differentiation. When indicated, a TGFβ antibody (α-TGFβ) (5 ug/ml) was added throughout the culture.
(B) Naïve dKO CD4+ T cells (CD45.2+) or WT B6.SJL CD4+ T cells (CD45.1+) were differentiated into Treg cells alone or in a mixed culture at a 1:1 ratio without exogenous TGF-β. The frequencies of differentiated Foxp3+ cells from dKO and B6.SJL CD4+ T cells were determined five days later.
(C) Naïve CD4+ T cells from either WT or dKO mice were differentiated into Th17 cells with indicated concentrations of TGF-β and 20 ng/ml IL-6. At day five, the differentiated cells were washed and restimulated with PMA+ionomycin for 4 h and the IL17A+ and Foxp3+ cells were determined by flow cytometry.
(D) Naïve CD4+ T cells from either WT or dKO mice were differentiated into Th1 cells with 10 ng/ml IL-12 plus indicated concentrations of TGF-β. At day five, the IFN-γ+ and IL-17A+ cells were determined by flow cytometry as in panel C.
(A-D) The numbers in the graphs indicate the percentages of the gated populations. Data are representative of either three (A, C, and D) or two (B) independent experiments.
To determine if Map3k2-/-Map3k3Lck-Cre/- T cells were the source of TGF-β, WT and Map3k2-/-Map3k3Lck-Cre/- T cells were co-differentiated under Treg cell condition at 1:1 ratio without exogenous TGF-β. As shown in Figure 2B, only the Map3k2-/-Map3k3Lck-Cre/- T cells but not the co-cultured WT T cells acquired Foxp3 expression indicating that the Map3k2-/-Map3k3Lck-Cre/- T cells did not overproduce TGF-β and the augmented Map3k2-/-Map3k3Lck-Cre/- T cell differentiation into iTreg was not mediated by an autocrine and/or paracrine mechanism. Rather, the Map3k2-/-Map3k3Lck-Cre/- T cells but not the WT T cells were able to respond to trace amount of TGF-β presented in the culture medium. Moreover, the in vitro generated Map3k2-/-Map3k3Lck-Cre/- iTreg cells were as suppressive as the WT iTreg cells differentiated under the same condition (data not shown). Together, these data indicate that Map3k2-/-Map3k3Lck-Cre/- T cells are intrinsically hypersensitive to TGF-β stimulation as measured by iTreg differentiation.
Next, we examined the Th17 cell differentiation from the naive Map3k2-/-Map3k3Lck-Cre/- T cells in vitro with TGF-β and IL-6 (Bettelli et al., 2006; Veldhoen et al., 2006). We found that the Th17 cell differentiation from naïve Map3k2-/-Map3k3Lck-Cre/- T cells was also augmented when compared to WT T cells (Figure 2C). Similar to the iTreg cell differentiation, differentiation of Th17 cells from naive Map3k2-/-Map3k3Lck-Cre/- T cells could be completely blocked by a TGF-β antibody. We found substantial numbers of Map3k2-/-Map3k3Lck-Cre/- T cells (up to 9.7%), but not WT T cells, acquired Foxp3 expression by this Th17 cell differentiation procedure with some cells co-expressing both Foxp3 and IL-17A (Figure 2C). Because strong TGF-β signal could induce Foxp3 expression under Th17 cell conditions (Zhou et al., 2008), this further suggests that the Map3k2-/-Map3k3Lck-Cre/- T cells were much more sensitive to TGF-β stimulation than WT T cells. Under the Th17 cell differentiation procedure, the Map3k2-/-Map3k3Lck-Cre/- T cells also expressed substantially higher amounts of RORγt than WT T cells (Supplemental Figure S2A). Expression of other Th17 cell lineage specific genes, such as Il-17f, Il-23r and Il-21, but not Il-22 or Il-21r, was also elevated in the Map3k2-/-Map3k3Lck-Cre/- T cells (Supplemental Figure S2B). Thus, MEKK2 and MEKK3 negatively regulate the TGF-β response in Th17 cell differentiation.
Because TGF-β signaling is also known to inhibit Th1 cell differentiation, the augmented TGF-β responsiveness in Map3k2-/-Map3k3Lck-Cre/- T cells would predict an enhanced inhibition of Th1 differentiation by TGF-β. Indeed, using the Th1 cell differentiation procedure, we observed a much more sensitive dose response of TGF-β inhibition of IFNγ-producing Th1 cells differentiation from Map3k2-/-Map3k3Lck-Cre/- cells (Figure 2D). Even a low concentration of TGF-β (e.g. 1 ng/ml) that had minimal effect on the WT T cells blocked most of Th1 cell differentiation of the Map3k2-/-Map3k3Lck-Cre/- T cells (Figure 2D). Furthermore, the induction of T-bet, a key transcription factor for Th1 cell lineage differentiation and a direct target of TGF-β (Gorelik et al., 2002), was inhibited more effectively by TGF-β in the Map3k2-/-Map3k3Lck-Cre/- T cells than in the WT T cells (data not shown).
Taken together, these data demonstrate that the Map3k2-/-Map3k3Lck-Cre/- naive CD4+ T cells are hypersensitive to TGF-β mediated induction of Treg and Th17 cell differentiation, and to TGF-β mediated inhibition of Th1 cell differentiation. Consistent with the in vivo data, the augmented TGF-β sensitivity in Map3k2-/-Map3k3Lck-Cre/- T cells in vitro also required deletion of both Map3k2 and Map3k3 because neither Map3k2-/- nor Map3k3Lck-Cre/- naïve T cells had augmented iTreg cell differentiation (Supplemental Figure S2C). Moreover, CD4+CD25- SP thymocytes from the Map3k2-/-Map3k3Lck-Cre/- mice were also hypersensitive to TGF-β stimulation during Th cell differentiation (data not shown). Additionally, we also examined Th2 cell differentiation and found that the Map3k2-/-Map3k3Lck-Cre/- CD4+ T cells appeared to have some reduction in the IL-4 induced Th2 differentiation (Supplemental Figure S2D), which could be due to the increase of basal TGF-β signals in Map3k2-/-Map3k3Lck-Cre/- T cells that is known to inhibit Th2 cell differentiation (Gorelik et al., 2000).
Inhibition of TGF-β Signaling in Map3k2-/-Map3k3Lck-Cre/- Mice Blocks Treg and Th17 cell Differentiation in vivo
The above results thus suggest that the increased Treg and Th17 cell differentiation in Map3k2-/-Map3k3Lck-Cre/- mice could be due to an augmented TGF-β signal in its T cells. To test this possibility, we treated the Map3k2-/-Map3k3Lck-Cre/- mice with a TGF-β receptor kinase inhibitor SB431542 (Inman et al., 2002). The inhibitor showed little effect on the ratios of naïve vs. effector or memory-like T cells in the periphery of either WT mice or the Map3k2-/-Map3k3Lck-Cre/- mice (data not shown). However, ten days after the treatment, the frequencies of Foxp3+ Treg cells in the blood of Map3k2-/-Map3k3Lck-Cre/- mice were reduced to that in the WT mice (Figure 3A-3B). Moreover, the percentages of Foxp3+ Treg cells and Th17 cells in the spleen of Map3k2-/-Map3k3Lck-Cre/- mice were also reduced following the inhibitor treatment (Figure 3C-3D). As a control, the inhibitor had only marginal effect on the frequencies of Treg and Th17 cells in WT mice (Figure 3B, and data not shown). These data suggest that the abnormal accumulation of Treg and Th17 cells in the Map3k2-/-Map3k3Lck-Cre/- mice was due to the elevated TGF-β signals in T cells.
Figure 3. Inhibition of TGF-βSignaling in the Map3k2-/-Map3k3Lck-Cre/- Mice Reduces Treg and Th17 cells in the Periphery.

(A) Map3k2-/-Map3k3Lck-Cre/- (dKO) mice were treated with TGFβRI inhibitor SB431542 or DMSO every other day for ten days. The Foxp3+ CD4+ T cells in the peripheral blood of dKO mice before the treatment (day 0) and 10 days after the treatment (Day 10) were determined. The numbers next to the gated boxes show the percentages of the gated populations. Data are representative of three independent experiments.
(B) Summary of Foxp3+ cells as the percentages of total CD4+ T cells in the peripheral blood of 3-pairs of wild type (WT) and dKO mice before and after treated with SB431542 or DMSO. Each symbol in the graph represents one individual mouse.
(C, D) Splenic CD4 T cells from the dKO mice that were treated with SB431542 or DMSO were analyzed for the frequency of Foxp3+ population (C) or IL17A+ population (D) by flow cytometry. The numbers next to the gated boxes show the percentages of the gated populations. Data are representative of five mice per group from two independent experiments.
MEKK2 and MEKK3 Are Essential for the TCR-Induced Phosphorylation of R-SMAD at the Linker Region
Despite the antagonism between the Th17 and Treg cell lineage differentiation (Zhou et al., 2008), our in vivo and in vitro data indicated that the Map3k2-/-Map3k3Lck-Cre/- T cells were prone to both lineage differentiations, suggesting that MEKK2 and 3 may inhibit the TGF-β pathway, which is shared for both Th17 and iTreg cell lineage differentiation. To confirm that the TGF-β signaling pathway in Map3k2-/-Map3k3Lck-Cre/- T cells was indeed hyperactive, we examined the SMAD transcription activity in the Map3k2-/-Map3k3Lck-Cre/- T cells using CAGA12-Luc, a synthetic SMAD-dependent luciferase reporter (Dennler et al., 1998). As expected, the induction of the reporter activity in Map3k2-/-Map3k3Lck-Cre/- T cells was significantly higher than that in WT T cells (Figure 4A). However, the augmented SMAD reporter activity in the Map3k2-/-Map3k3Lck-Cre/- T cells was not caused by the enhanced SMAD3 phosphorylation at the C-terminal conserved sites at Ser423+425 (Figure 4B), because such phosphorylation was comparable between the Map3k2-/-Map3k3Lck-Cre/- T cells and WT T cells (Figure 4C). Although TGF-β is known to induce a non-canonical pathway through MAPK activation (Derynck and Zhang, 2003), we did not observe substantial induction of ERK1 and 2 or p38 MAPK activation in response to TGF-β stimulation in either WT or Map3k2-/-Map3k3Lck-Cre/- T cells (data not shown).
Figure 4. MEKK2 and MEKK3 Mediate R-SMAD Linker Phosphorylation and Inhibit SMAD Transcriptional Activity.

(A) CAGA12-Luc plasmid was nucleofected into WT or dKO CD4+ T cells. Four hours after nucleofection, cells were stimulated with anti-CD3+anti-CD28 antibodies with or without TGF-β as indicated. Luciferase activity was determined 16 h later and normalized to the activity of non-TGF-β stimulated cells.
(B) An illustration of known phosphorylation sites in SMAD2 and SMAD3. MH1 and MH2: MAD homology domain 1 and 2; linker: the sequence that links MH1 and MH2 domains.
(C) WT or dKO CD4+ T cells were stimulated with 10 ng/ml TGF-β as indicated and the SMAD3 phosphorylation at the C-terminus (Ser423/425) was determined by immunoblotting. The ERK2 level is used as a loading control.
(D) WT or dKO CD4+ T cells were stimulated with anti-CD3+anti-CD28 antibodies (Anti-CD3+28) for the indicated time periods. Phosphorylation of the SMAD2 and SMAD3 linker regions at SMAD2 Ser245+250+255, SMAD3 Ser204, SMAD2 Thr220, and SMAD3 Thr179 was determined by immunoblotting. Total SMAD2 and SMAD3 protein levels were determined by immunoblotting. Actin level is shown as a loading control.
(E) WT and dKO CD4 T cells were stimulated with PMA+ionomycin (PMA+Ion) for the indicated time periods. Phosphorylation of the SMAD2 linker region and total SMAD2 protein was determined by immunoblotting.
(F) SMAD3 expression vector was co-transfected with either empty expression vector (Ctl), or expression vectors for Map3k2 (KK2) or Map3k3 (KK3) into 293T cells. Twenty-four h after transfection, phosphorylation of SMAD3 at the linker region was determined. ERK1 and 2 activation and expression of SMAD3, MEKK2, and MEKK3 in the same transfection was determined by immunoblotting as indicated.
(G) CAGA12-Luc reporter plasmid was co-transfected into 293T cells with either empty expression vector (Ctl), or expression vectors for Map3k2 or Map3k3. Twenty-four h after transfection, cells were stimulated with the indicated concentrations of TGF-β for additional 12 h. The reporter activity was determined with a duel luciferase kit. The reporter activity without TGF-β stimulation was arbitrarily set as value 1 and used for normalization of the relative reporter activity.
(A-G) Data shown are representative of three independent experiments. Error bars in (A) and (G) show standard deviation. **, p<0.01 by two tailed Student's t test.
In addition to the C-terminal phosphorylation, R-SMADs can also be phosphorylated by MAPKs at the linker region (Figure 4B). We thus compared the phosphorylation of SMAD2 and SMAD3 at their linker regions between Map3k2-/-Map3k3Lck-Cre/- T cells and their WT counterparts. T cell activation by anti-CD3 and anti-CD28 or with PMA and ionomycin treatment led to rapid and strong phosphorylation of SMAD2 or SMAD3 at the linker regions in the WT T cells (Figure 4D and 4E). Such phosphorylation was severely impaired in the Map3k2-/-Map3k3Lck-Cre/- T cells, indicating that MEKK2 and MEKK3 are crucial for the TCR-induced phosphorylation of SMAD2 and 3 at their linker regions.
To confirm that MEKK2 or MEKK3 were capable of inducing R-SMAD linker region phosphorylation, and to verify that their activation could suppress R-SMAD transcriptional activity, we co-expressed Map3k2 or Map3k3 with SMAD3 in 293T cells. Transient expression of either MEKK2 or MEKK3 was sufficient to induce SMAD3 phosphorylation (Figure 4F) and SMAD2 (dada not shown) at the linker regions. Furthermore, expression of either Map3k2 or Map3k3 was also able to inhibit the TGF-β induced CAGA12-Luc reporter activity (Figure 4G). Moreover, the transcriptional activity of a mutant SMAD3, EPSM-SMAD3, containing Ser or Thr to Ala or Val mutations in its linker region (Calonge and Massague, 1999) was more resistant than the WT SMAD3 to the MEKK2 or MEKK3 mediated suppression (Supplemental Figure S3), further indicating that MEKK2 and 3 suppress the R-SMAD activity at least in part through the linker region phosphorylation.
ERK1 and ERK2 Mediate MEKK2 and MEKK3 Dependent Phosphorylation of R-SMAD At Their Linker Regions
Because MAPKs are known to phosphorylate R-SMAD at the linker regions, it is possible that MEKK2 and 3 deficiency may result in defective MAPK activation, which in turn leads to the impaired R-SMAD phosphorylation. Indeed, we found that activation of ERK1 and 2 and p38 MAPKs was severely impaired in Map3k2-/-Map3k3Lck-Cre/- T cells as compared to that in WT T cells after TCR stimulation (Figure 5A-5B). Consistent with the reduced activation of ERK1 and 2, induction of ERK1 and 2 target genes after TCR stimulation, such as Egr1 and c-Fos, was greatly diminished in naive Map3k2-/-Map3k3Lck-Cre/- T cells than that in WT T cells (Supplemental Figure S4A). The JNK was marginally activated and appeared not affected in the Map3k2-/-Map3k3Lck-Cre/- T cells (data not shown).
Figure 5. ERK1 and 2 Mediate the TCR-Induced SMAD Linker Region Phosphorylation and Inhibit the TGF-β Induced Treg Differentiation.
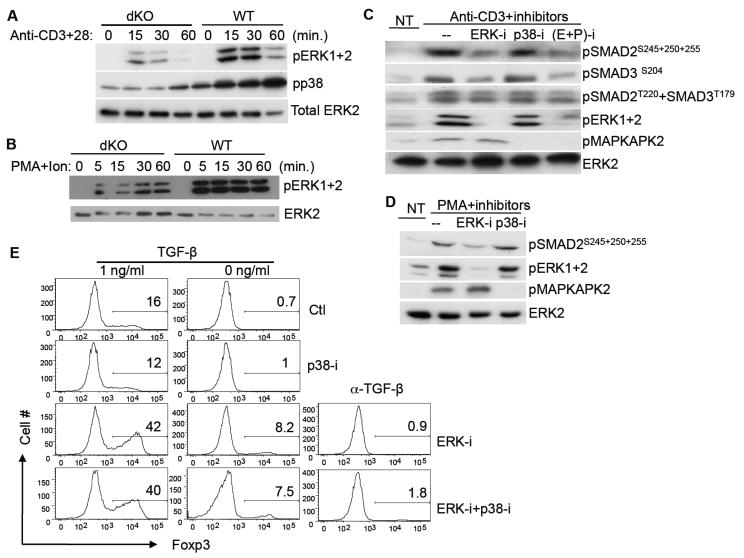
(A, B) WT or dKO CD4+ T cells were stimulated with anti-CD3+anti-CD28 antibodies (Anti-CD3+28) (A) or with PMA+ionomycin (PMA+Ion) (B) for the indicated time periods. Phosphorylation of ERK1 and 2, and p38 was determined by immunoblotting. Total ERK2 protein level was shown as a loading control.
(C, D) WT CD4+ T cells were activated with an anti-CD3 antibody (C) or with PMA treatment (D) in the presence of vehicle DMSO (Ctl), or MEK1/2 inhibitor U0126 (ERK-i), or p38 inhibitor SB203580 (p38-i), or both {(E+P)-i} as indicated. Phosphorylations of SMAD2 or SMAD3 at their linker regions and phosphorylation of ERK1 and 2, and p38 at their activation loop, were determined by immunoblotting. ERK2 protein level was determined as a loading control. NT, non-stimulated cells.
(E) WT naïve CD4 T cells were differentiated into Treg cells with either no TGF-β or 1 ng/ml TGF-β in the presence of no inhibitor (Ctl), or MEK1 inhibitor (ERK-i), or p38 inhibitor (p38-i), or both inhibitors (ERK-i+p38-i), as indicated. Foxp3+ Treg cells were determined five days later by flow cytometry as described in Figure 2.
(A-E) Data presented are representative of three independent experiments.
Because both the ERK1 and 2 and p38 activation was impaired in the Map3k2-/-Map3k3Lck-Cre/- T cells, we then determined whether both of them or either one of them, might mediate the TCR induced phosphorylation of R-SMAD at the linker region. Inhibition of ERK1 and 2 but not p38 MAPK in WT T cells greatly reduced the TCR-induced or PMA-induced phosphorylation of SMAD2 and SMAD3 at their linker regions (Figure 5C-5D). Inhibition of both ERK1 and 2 and p38 did not further reduce the SMAD linker region phosphorylation compared to inhibition of ERK1 and 2, suggesting that there was no synergistic effect between ERK1 and 2 and p38. Because other kinases such as GSK3 and the CDKs are also able to phosphorylate SMAD2 and 3 linker region under certain conditions (Wrighton et al., 2009), to rule out their possible involvement in this process, we treated WT T cells with either GSK3 inhibitor (SB-216763) or CDK inhibitor (flavopiridol), and found that neither of them was able to block the anti-CD3 induced SMAD linker region phosphorylation (Supplemental Figure S4B). Finally, although some cytokines such as IL-2 were able to induce ERK1 and 2 activation in T cells, IL-2 did not induce R-SMAD linker region phosphorylation (Supplemental Figure S4C). Thus, ERK1 and 2 are responsible for the TCR induced, MEKK2 and 3 dependent phosphorylation of SMAD2 and 3 at the linker region.
To further examine the physiological significance of R-SMAD linker region phosphorylation in T cells, we blocked the R-SMAD linker region phosphorylation via inhibition of ERK1 and 2, and determined its effect on Treg differentiation. As shown in Figure 5E, inhibition of ERK1 and 2 activation markedly augmented the TGF-β induced iTreg cell differentiation in vitro, similar to that found in the Map3k2-/-Map3k3Lck-Cre/- T cells. In contrast, inhibition of p38 activation had no effect on iTreg differentiation. In addition, inhibition of both p38 and ERK1 and 2 did not further increase the iTreg differentiation compared to ERK1 and 2 inhibition alone, further confirming that the TCR induced SMAD2 and 3 phosphorylation at the linker region is mediated by ERK1 and 2, which negatively regulates TGF-β signaling during Th cell differentiation.
SMAD3 Linker Region Phosphorylation Mutant Induces More iTreg and Th17 Cells Than WT SMAD3 in WT T cells but Not in Map3k2-/-Map3k3Lck-Cre/- T Cells
To directly test the role of the linker region phosphorylation of R-SMAD during Th cell differentiation, WT naïve T cells were infected with retroviruses that expressed either GFP alone, or GFP plus WT SMAD3, or GFP plus EPSM-SMAD3, which is defective in its linker region phosphorylation (Calonge and Massague, 1999). The infected T cells were then differentiated into either iTreg or Th17 cells in vitro. Expression of WT SMAD3 greatly enhanced both iTreg and Th17 cell differentiation as compared to the expression of the empty vector (Figure 6A). These results confirmed previous findings that SMAD3 is required for iTreg cell differentiation and also suggested that SMAD3 was involved in Th17 cell differentiation. Importantly, we found that EPSM-SMAD3 had much higher activity than the WT SMAD3 in inducing either iTreg (93% vs. 63%, respectively) or Th17 cell differentiation (41% vs. 32%, respectively) (Figure 6A), indicating that phosphorylation of SMAD3 at the linker region is inhibitory to iTreg and Th17 cell differentiation.
Figure 6. MEKK2 and MEKK3 Regulate Treg and Th17 Differentiation Through Mediating R-SMAD Linker Phosphorylation.
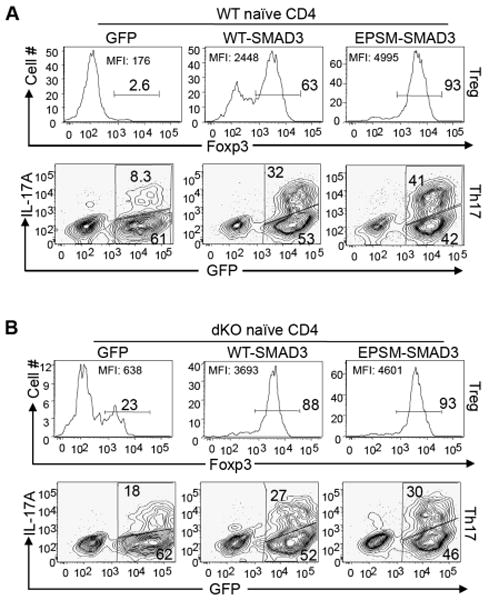
WT (A) or dKO CD4+ T cells (B) were differentiated under the Treg or Th17 conditions, respectively, as described in Figure 2, except that the TGF-β (1 ng/ml) was added at the 24 h time point when the cells were infected with empty retrovirus (GFP), or retroviruses that express WT-SMAD3, or EPSM-SMAD3. The infection was repeated once at 36 h. Five days later, the infected cells were determined by GFP expression and analyzed for Foxp3+ cells directly (upper panels), or analyzed for IL-17A+ cells (lower panels) after re-stimulation with PMA+ionomycin for 4 h. The numbers in the graphs show the percentages of gated populations. Data are representative of two independent experiments.
If MEKK2 and 3 suppress the iTreg and Th17 cell differentiation through the phosphorylation of R-SMAD at their linker regions, we would predict that WT SMAD3 may have comparable activity as the EPSM-SMAD3 in the Map3k2-/-Map3k3Lck-Cre/- T cells because the linker region of WT SMAD3 was hypo-phosphorylated in these cells (Figure 4D). Indeed, the WT SMAD3 induced similar Foxp3 expression as the EPSM-SMAD3 in the Map3k2-/-Map3k3Lck-Cre/- T cells (Figure 6B, upper panels). Consistent with R-SMAD having elevated activity in the Map3k2-/-Map3k3Lck-Cre/- T cells, WT SMAD3 also induced more Foxp3 expressing cells from the Map3k2-/-Map3k3Lck-Cre/- T cells (88%, Figure 6B) than from WT T cell (63%, Figure 6A) under the same condition. Moreover, EPSM-SMAD3 induced similar percentages of Foxp3 expressing cells from both the Map3k2-/-Map3k3Lck-Cre/- T cells and WT T cells (Figure 6A-6B, upper panels). Finally, WT SMAD3 and EPSM-SMAD3 also induced comparable IL-17A expressing cells in the Map3k2-/-Map3k3Lck-Cre/- T cells (Figure 6B, lower panels). Together with the results from the above biochemical study, our data strongly suggest that the TGF-β dependent Th cell differentiation is negatively controlled by a TCR-MEKK2 and 3-ERK1 and 2 MAPK module, which phosphorylates SMAD2 and SMAD3 at their linker regions (Supplemental Figure S5).
Map3k2-/-Map3k3Lck-Cre/- Mice Have Enhanced Th17 cell Response
To examine how MEKK2 and 3 deficiency in T cells may influence effector CD4 T cell differentiation and impact the development of acute inflammatory diseases, we used a well characterized mouse autoimmune disease model, the experimental autoimmune encephalomyelitis (EAE) model. We immunized Map3k2-/-Map3k3Lck-Cre/- mice with MOG35-55 peptide and complete Freund's adjuvant (CFA) and monitored the development of EAE disease. Compared to WT mice, the Map3k2-/-Map3k3Lck-Cre/- mice developed more severe disease after the immunization (Figure 7A) and recovered much slower from the paralysis. Similar results were obtained using the Rag1-/- mice reconstituted with WT or Map3k2-/-Map3k3Lck-Cre/- bone marrows (Figure 7B). Consistent with the more severe disease, the Map3k2-/-Map3k3Lck-Cre/- mice had more Th17 cells in both the central nervous system (CNS) and the spleen compared to the WT mice, whereas the Th1 cells appeared to be comparable (Figure 7C). Treg cells in the CNS were slightly reduced in ratio in the Map3k2-/-Map3k3Lck-Cre/- mice, whereas their absolute numbers were comparable (Figure 7D and data not shown). As in the non-immunized mice, the frequency of Foxp3+ Treg cells in the spleen of Map3k2-/-Map3k3Lck-Cre/- mice remained higher than that in the WT mice (Figure 7D).
Figure 7. Map3k2-/-Map3k3Lck-Cre/- Mice Exhibit More Severe EAE Disease and Accumulate More Th17 cells in the Acute Inflammatory Response.

(A) WT (n=5) and dKO (n=4) mice were immunized with MOG35-55+CFA plus pertussis toxin to induce EAE disease. The immunized mice were observed for the indicated length of time and the mean clinical scores for the severity of EAE were determined.
(B) Sublethally irradiated Rag1-/- mice were reconstituted with either dKO (n=4) or WT (n=5) bone marrow cells. Ten weeks after the reconstitution, EAE disease was induced and analyzed as described in panel A.
(A-B) Data shown are representative of two independent experiments. Error bars show standard error of the mean. **, p<0.01, *, p<0.05, by two tailed Student's t test.
(C, D). EAE disease was induced in WT or dKO mice as described in panel A. Twenty eight days after the immunization, CD4+ T cells were gated from the total leukocytes in the central nervous system (CNS) or spleen (SPL), and further analyzed for frequencies of IL-17A+ and IFN-γ+ cells (C), and Foxp3+ cells (D) by flow cytometry. The numbers in the plots indicate the percentages of the gated populations. Data shown are representative of three independent experiments.
(E, F) On day 8 after MOG+CFA immunization, WT or dKO splenocytes were restimulated with indicated concentrations of MOG peptide in vitro. The production of IL-17A (E) and IFN-γ (F) in culture supernatants was determined three days later by ELISA. Data shown are average values of triplicates. Error bars show standard deviation. The results are representative of four mice of each genotype from two independent experiments. **, p<0.01 by two tailed Student's t test.
To determine whether Map3k2-/-Map3k3Lck-Cre/- mice produced more MOG specific Th17 T cells, we tested the recall response of WT and Map3k2-/-Map3k3Lck-Cre/- T cells after in vivo MOG+CFA immunization. The antigen-induced production of IL-17A by Map3k2-/-Map3k3Lck-Cre/- T cells was significantly elevated over that by the WT T cells (Figure 7E), whereas IFN-γ production by Map3k2-/-Map3k3Lck-Cre/- T cells was also reduced (Figure 7F). These data together indicate that the T cell specific deficiency of Map3k2 and Map3k3 results in augmented antigen-induced Th17 cell differentiation and consequent CNS accumulation, leading to more severe EAE disease.
Discussion
TGF-β is a central regulator of Th cell differentiation, but the underlying molecular mechanism by which the TGF-β signal is regulated during Th cell differentiation is not fully understood. Here we show that MEKK2 and MEKK3 deficiency in T cells resulted in hypersensitivity to the TGF-β signaling during iTreg and Th17 cell differentiation. We demonstrate that MEKK2 and MEKK3 are essential for the TCR-mediated activation of ERK1 and 2 and p38 MAPKs. Further, Map3k2-/-Map3k3Lck-Cre/- mice develop more severe EAE disease and had more antigen specific Th17 cells that preferentially accumulate in the central nervous system, suggesting that the MEKK2 and MEKK3 signaling pathway in T cells plays a crucial regulatory role in inflammation and autoimmunity. This study thus unravels a previously un-identified inhibitory mechanism that dampens the potential of TGF-β response in Th cell differentiation, and reveals a critical interplay between the TCR-induced MAPK activation signal and the cytokine-induced differentiation signals during Th cell differentiation.
MEKK2 and MEKK3 are two highly homologous MAP3Ks. Although previous studies demonstrate that MEKK2 and MEKK3 have non-overlapping function in vivo, our current study indicate that activation of either MEKK2 or MEKK3 alone is sufficient to suppress the TGF-β responses in T cells. In addition, either MEKK2 or MEKK3 is sufficient to activate ERK1 and ERK2 to phosphorylate SMAD2 or SMAD3 at their linker regions, and to suppress R-SMAD-dependent transcriptional activity. These data indicate that MEKK2 and MEKK3 play overlapping roles in regulating the TGF-β response through ERK1 and 2 in Th cell differentiation.
Activation of ERK1 and 2 is mainly mediated by the Raf subgroup of MAP3Ks in T cells and other cell types. Other MAP3Ks such as MEKK1 (Yujiri et al., 1998), MEKK2 (Zhang et al., 2006), MEKK3 (Kim et al., 2007), and Tpl2 (Jager et al.), are also implicated in ERK1 and 2 activation in T cells and other cell types either directly or indirectly. It is conceivable that these various alternative ERK1 and 2 activation pathways could allow cells to use the ubiquitous ERK1 and 2 MAPK module to selectively respond to distinct extracellular stimuli under different conditions. The specific MEKK2 and 3-ERK1 and 2 module in T cells may be one of such examples that is involved in transducing specifically the TCR signals to regulate the TGF-β pathway without affecting the Raf-ERK1 and 2-mediated growth signals during Th cell differentiation. Consistently, activation of ERK1 and 2 by cytokines such as IL-2 in T cells did not induce R-SMAD linker region phosphorylation, Therefore, our work suggest an interesting model that the TGF-β dependent Th cell differentiation is negatively controlled by a TCR-MEKK2 and 3-ERK1 and 2 MAPK module, which phosphorylates SMAD2 and SMAD3 at their linker regions.
The NF-κB pathway has been reported to play a positive role in Treg cell differentiation (Long et al., 2009; Ruan et al., 2009). We previously showed that MEKK3 play a role in NF-κB activation by the TLR ligands and proinflammatory cytokines in fibroblasts (Huang et al., 2004; Yang et al., 2001). However, we did not find obvious augmentation of NF-κB activation in Map3k2-/-Map3k3Lck-Cre/- T cells indicating that the augmented iTreg cell differentiation from naïve Map3k2-/-Map3k3Lck-Cre/- T cells was unlikely due to the altered NF-κB pathway. Although studies from another group reported that MEKK3 was required for NF-κB activation in T cells (Shinohara et al., 2009), we found no obvious defect in the TCR-induced IKKβ activation and IκBα degradation in either Map3k3Lck-Cre/- or Map3k2-/-Map3k3Lck-Cre/- T cells (data not shown). Interestingly, the induction of p65 phosphorylation at Ser534 after TCR stimulation was impaired in Map3k2-/-Map3k3Lck-Cre/- T cells supporting a role of MEKK2 and MEKK3 in NF-κB pathway activation in T cells (data not shown). Nevertheless we believe that this defect is most likely unrelated to the increased Treg cell differentiation of Map3k2-/-Map3k3Lck-Cre/- T cells because defective NF-κB activation should inhibit but not augment the Treg cell differentiation (Long et al., 2009; Ruan et al., 2009).
The peripheral T cells in the Map3k2-/-Map3k3Lck-Cre/- mice appeared to be overtly activated but the cause of this change is unclear. However, this is unlikely the reason why the Map3k2-/-Map3k3Lck-Cre/- T cells are hyper-sensitive to TGF-β signal because naïve peripheral T cells or thymic CD4 SP T cells that have not been previously activated were also hypersensitive to TGF-β signal. Interestingly, although the frequencies of iTreg cells were increased in the Map3k2-/-Map3k3Lck-Cre/- mice and they were able to suppress naïve T cell proliferation in vitro, they failed to suppress the activated T cell phenotype in the Map3k2-/-Map3k3Lck-Cre/- mice. Similarly, the Treg in the Map3k2-/-Map3k3Lck-Cre/- mice failed to suppress the pathogenesis of EAE. In this regard, expansion of iTreg cells has been frequently observed in autoinflammatory conditions such as in the CNS of a mouse EAE model (Korn et al., 2007), or in the inflamed joints of rheumatoid arthritis patients (Ruprecht et al., 2005). One possible cause of this phenomenon could be the over production of IL-6 induced by IL-17A in the Map3k2-/-Map3k3Lck-Cre/- mice, which has been shown to dampen the iTreg cell mediated suppression of naive T cell activation (Pasare and Medzhitov, 2003).
Finally, the augmented T cell activation and the increased Th17 cells in the periphery may predispose the Map3k2-/-Map3k3Lck-Cre/- mice to autoimmune diseases. However, no spontaneous autoimmune syndromes have been observed in Map3k2-/-Map3k3Lck-Cre/- mice up to 15 month old. In contrast, under acute inflammatory condition, after immunization with MOG+CFA, the Map3k2-/-Map3k3Lck-Cre/- mice suffered more severe disease than the WT mice, which is most likely caused by the enhanced production of MOG specific Th17 cells and their CNS accumulation. It is therefore possible that MEKK2 and MEKK3 may positively affect immune tolerance dependent on the inflammatory status, similar to the role of TGF-β in autoimmune diseases. Interestingly, similar to our observation, mice with ERK1 deficiency also had more severe EAE disease after the MOG/CFA immunization (Agrawal et al., 2006). Future studies of the roles of MEKK2 and MEKK3 in immune tolerance and autoimmunity will be rewarding.
Experimental Procedures
Mice
Mekk3 germ line KO mice, Map3k3fl/fl mice, MAP3k3 T cell conditional-deleted mice, and Map3k2-/- mice were described before and were bred with C57BL/6 mice for more than ten generations (Cheng et al., 2000; Guo et al., 2002; Wang et al., 2009). Lck-Cre transgenic mice in C57BL/6 background, C57BL/6 Rag1-/-, WT C57BL/6 mice, and C57BL/6 CD45.1 (B6.SJL) congenic mice were obtained from the Jackson Laboratory. All mice were maintained under pathogen-free conditions. Unless indicated, all mice were used between 10 and 15 weeks old. All animal experimentation was conducted in accordance with institutional guidelines.
Bone Marrow Transplantation
Bone marrow cells were isolated from Map3k2-/-Map3k3Lck-Cre/- mice, WT mice or B6.SJL mice. Five million cells were injected intravenously into Rag1-/- mice, which received a sub-lethal irradiation (400 rad) one day before. Chimeric mice were analyzed six to eight weeks after transplantation.
Chemical Inhibitors
The following inhibitors were used: MEK inhibitor (PD98059, 5 μM), MEK1/2 inhibitor (U0126, 5 μM), p38 inhibitor (SB20190, 5 μM), CDK inhibitor (flavopiridol, 0.3 μM), GSK3α/β inhibitor (SB-216763, 5 μM), TGFβR inhibitor (SB431542). All inhibitors were obtained from Sigma.
Supplementary Material
Acknowledgments
We thank Drs. Richard Flavell, Jordan Pober, Joseph Craft, Martin S Kluger, and Adam Lazorchak for critical reading of the manuscript and for their many insightful suggestions during the course of this work, Drs. Yuan Zhuang and Pumin Zhang for targeting vector construction, and generation of targeted mice, Dr. Min O. Li for sharing the unpublished cDNA array data, Dr. Weiming Ouyang for initial involvement in the characterization of Map3k2-/-Map3k3Lck-Cre/- mice, Dr. Nancy Ruddle and Ms. Cheryl Bergman for help with the EAE study, and Dou Liu, Dr. Adam Lazorchak and other members in Su laboratory for helpful suggestions and technical helps. This work is supported in part by grant AI063348 and HL070225 (NIH) (to BS). XC is a recipient of Gershon and Trudeau Fellowship from Yale University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Referennces
- Agrawal A, Dillon S, Denning TL, Pulendran B. ERK1-/- mice exhibit Th1 cell polarization and increased susceptibility to experimental autoimmune encephalomyelitis. J Immunol. 2006;176:5788–5796. doi: 10.4049/jimmunol.176.10.5788. [DOI] [PubMed] [Google Scholar]
- Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757–769. doi: 10.1016/j.cell.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- Calonge MJ, Massague J. Smad4/DPC4 silencing and hyperactive Ras jointly disrupt transforming growth factor-beta antiproliferative responses in colon cancer cells. J Biol Chem. 1999;274:33637–33643. doi: 10.1074/jbc.274.47.33637. [DOI] [PubMed] [Google Scholar]
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Yang J, Xia Y, Karin M, Su B. Synergistic interaction of MEK kinase 2, c-Jun N-terminal kinase (JNK) kinase 2, and JNK1 results in efficient and specific JNK1 activation. Mol Cell Biol. 2000;20:2334–2342. doi: 10.1128/mcb.20.7.2334-2342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med. 2002;195:1499–1505. doi: 10.1084/jem.20012076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. 2000;165:4773–4777. doi: 10.4049/jimmunol.165.9.4773. [DOI] [PubMed] [Google Scholar]
- Gorham JD, Guler ML, Fenoglio D, Gubler U, Murphy KM. Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J Immunol. 1998;161:1664–1670. [PubMed] [Google Scholar]
- Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF. Axin and GSK3-control Smad3 protein stability and modulate TGF- signaling. Genes Dev. 2008;22:106–120. doi: 10.1101/gad.1590908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Clydesdale G, Cheng J, Kim K, Gan L, McConkey DJ, Ullrich SE, Zhuang Y, Su B. Disruption of Mekk2 in mice reveals an unexpected role for MEKK2 in modulating T-cell receptor signal transduction. Mol Cell Biol. 2002;22:5761–5768. doi: 10.1128/MCB.22.16.5761-5768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Yang J, Lin Y, Walker C, Cheng J, Liu ZG, Su B. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol. 2004;5:98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Jager J, Gremeaux T, Gonzalez T, Bonnafous S, Debard C, Laville M, Vidal H, Tran A, Gual P, Le Marchand-Brustel Y, et al. Tpl2 kinase is upregulated in adipose tissue in obesity and may mediate interleukin-1beta and tumor necrosis factor-{alpha} effects on extracellular signal-regulated kinase activation and lipolysis. Diabetes. 59:61–70. doi: 10.2337/db09-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Duramad O, Qin XF, Su B. MEKK3 is essential for lipopolysaccharide-induced interleukin-6 and granulocyte-macrophage colony-stimulating factor production in macrophages. Immunology. 2007;120:242–250. doi: 10.1111/j.1365-2567.2006.02495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–931. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- Malhotra N, Robertson E, Kang J. SMAD2 is essential for TGF beta-mediated Th17 cell generation. J Biol Chem. 2010;285:29044–29048. doi: 10.1074/jbc.C110.156745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Martinez G, Zhang Z, Reynolds J, Tanaka S, Chung Y, Liu T, Robertson E, Lin X, Feng X, Dong C. Smad2 positively regulates the generation of Th17 cells. J Biol Chem. 2010;285:29039–29043. doi: 10.1074/jbc.C110.155820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Zhang Z, Chung Y, Reynolds JM, Lin X, Jetten AM, Feng XH, Dong C. Smad3 differentially regulates the induction of regulatory and inflammatory T cell differentiation. J Biol Chem. 2009;284:35283–35286. doi: 10.1074/jbc.C109.078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Matsuura I, Wang G, He D, Liu F. Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry. 2005;44:12546–12553. doi: 10.1021/bi050560g. [DOI] [PubMed] [Google Scholar]
- Millet C, Yamashita M, Heller M, Yu LR, Veenstra TD, Zhang YE. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J Biol Chem. 2009;284:19808–19816. doi: 10.1074/jbc.M109.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht C, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, Sallusto F. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med. 2005;201:1793–1803. doi: 10.1084/jem.20050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara H, Yamasaki S, Maeda S, Saito T, Kurosaki T. Regulation of NF-kappaB-dependent T cell activation and development by MEKK3. Int Immunol. 2009;21:393–401. doi: 10.1093/intimm/dxp007. [DOI] [PubMed] [Google Scholar]
- Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Wang X, Chang X, Facchinetti V, Zhuang Y, Su B. MEKK3 is essential for lymphopenia-induced T cell proliferation and survival. J Immunol. 2009;182:3597–3608. doi: 10.4049/jimmunol.0803738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- Weston CR, Lambright DG, Davis RJ. Signal transduction. MAP kinase signaling specificity. Science. 2002;296:2345–2347. doi: 10.1126/science.1073344. [DOI] [PubMed] [Google Scholar]
- Wrighton KH, Lin X, Feng XH. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009;19:8–20. doi: 10.1038/cr.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu Z, Su B. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001;2:620–624. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- Yujiri T, Sather S, Fanger GR, Johnson GL. Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science. 1998;282:1911–1914. doi: 10.1126/science.282.5395.1911. [DOI] [PubMed] [Google Scholar]
- Zhang D, Facchinetti V, Wang X, Huang Q, Qin J, Su B. Identification of MEKK2/3 serine phosphorylation site targeted by the Toll-like receptor and stress pathways. EMBO J. 2006;25:97–107. doi: 10.1038/sj.emboj.7600913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.