

Abstract
The mTOR signaling pathway is dysregulated in ~50% of all human malignancies and is a major cancer drug target. Although rapamycin analogs (rapalogs) have shown clinical efficacy in a subset of cancers, they do not fully exploit the antitumor potential of mTOR targeting. Because the mTOR kinase domain is important for rapamycin-sensitive and -insensitive functions, mTOR catalytic inhibitors have been developed recently as the second generation of anti-mTOR agents. Importantly, they have shown marked improvement of antitumor activity in vivo and in vitro. This review will detail the potential therapeutic value and issues of these novel antineoplastic agents, with emphasis placed on those that have already entered clinical trials.
Keywords: mTOR, PI3K, Kinase inhibitor, Signal transduction, Cancer
mTOR signaling
The mammalian or mechanistic target of rapamycin (mTOR) is an evolutionarily conserved serine/threonine kinase belonging to the phosphoinositide 3-kinase (PI3K)-related kinase family. It lies at the nexus of the regulatory network and acts as a sensor that integrates extracellular and intracellular events, coordinating growth and proliferation. Many cancer-driving molecules such as PI3K, AKT, epidermal growth factor receptor (EGFR), HER2/neu and BCR-ABL stimulate proliferation, growth and survival by activating mTOR kinase [1]. Previous studies from several laboratories, including our own, have found heightened mTOR activity as indicated by an elevated phosphorylation of its downstream substrates in clinical samples of various solid tumors such as colorectal cancer and hematological malignancies such as leukemia [2,3]. This signaling pathway represents one of the major growth and survival pathways that is dysregulated in many human cancers and contributes to cancer pathogenesis and therapy resistance [4]. Therefore, mTOR is a valuable clinically validated target for innovative therapeutic treatment of human cancers. Increasing evidence that many diverse cancers could benefit from mTOR inhibition has fueled the development of small molecule mTOR inhibitors [5]. Like many
The limitation of rapalogs
The historical discovery of rapamycin, the macrolide mTOR inhibitor, illustrates a great example of the modern chemical biology that has illuminated human diseases and therapies [6]. Until recently, we have largely relied on the use of rapalogs to study mTOR function and its anticancer potential. As the first generation mTOR inhibitor, rapalogs have proven effective in a range of preclinical models. However, the clinical success of rapalogs has been limited to a few rare cancers, including mantle cell lymphoma, renal cell carcinoma and endometrial cancer. The overall objective response rates in major solid tumors achieved with rapalogs as single-agent therapy have been, at best, modest [7]. It is increasingly recognized that the mechanism of action of rapamycin as a partial mTOR inhibitor is not sufficient for achieving a broad and robust anticancer effect, at least when these agents are employed in a monotherapy setting [8].
It was reported as early as 1995 that TOR proteins have rapamycin-sensitive and rapamycin-insensitive functions, and that the TOR kinase domain is important for both [9]. This observation revealed, for the first time, that the mTOR kinase domain is a more potent site for signaling inhibition. Subsequent research further supported this notion. It is now understood that mTOR forms at least two functional multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [10]. Rapalogs, which act at a site called the FRB (FKBP-rapamycin-binding) domain, outside the ATP-binding pocket, inhibit the first complex but do not affect the second one, mTORC2, which is also an important driver for cancer cell growth and survival. One of the other proposed shortcomings of rapalogs is that there is a feedback loop between mTORC1 and AKT in certain tumor cells [11] (Figure 1). Treatment with rapalogs in some cases results in elevated AKT activity through insulin receptor substrate 1 (IRS-1), which serves as a mechanism to enhance cell survival when mTORC1 is inhibited [12]. These observations provide an important rationale for PI3K/mTOR or mTORC1/2 dual inhibition, which can prevent the elevated AKT activity. Indeed, recent studies using inhibitors of mTOR kinase achieved higher anticancer effects than rapalogs [13,14].
Figure 1.

Action point of ATP-competitive mTOR inhibitors on PI3K/Akt/mTOR pathway
ATP-competitive mTOR kinase inhibitors
A strong interest now exists for ATP-competitive inhibitors as anticancer agents. The rationale for using ATP-competitive mTOR and/or PI3K kinase inhibitors is mainly 3-fold: i) aberrantly hyperactive signaling by PI3K/AKT/mTOR is a prominent feature of a broad spectrum of human cancers; ii) rapalogs cause activation of AKT through a negative feedback loop; iii) mTORC2 is also involved in cancer cell growth and survival. Not surprisingly, inhibition of mTORC2 and/or PI3K simultaneously with mTORC1 appears to inhibit more robustly the signaling cascades and negating activation of this feedback loop. With multiple efforts under way in academia and industry, a number of small-molecule ATP-competitive mTOR kinase inhibitors have been identified and developed as targeted anticancer agents. Because mTOR and PI3K kinase domains are closely related to each other, some of the compounds, it transpires, also potently inhibit the catalytic activity of PI3K, and are thus called mTOR/PI3K dual inhibitors. The first set of ATP-competitive mTOR kinase inhibitors has already entered early clinical trials (Table 1).
Table 1.
ATP-competitive mTOR inhibitors in clinical trials
| Sponsor | Agent | Stage | Schedule (route) | Condition | |
|---|---|---|---|---|---|
| mTOR/PI3K dual inhibitor | Novartis | NVPBEZ235 | Phase I/II | Oral. Once daily, 28-day cycles. | Advanced solid tumors including advanced breast cancer |
| Phase I | Oral. | Advanced solid tumors (Japanese patients) | |||
| Semafore | SF1126 | Phase I | Intravenous. Twice weekly for 4 weeks. | Advanced or metastatic tumors | |
| GlaxoSmithKline | GSK2126458 | Phase I | Oral. Once/twice daily, or intermittent, 28-day cycles. | Solid tumors or lymphoma | |
| Exelixis | XL765 | Phase I | Oral. Once or twice daily. | Solid tumors | |
| Phase I | Oral. Daily (Combination with erlotinib). | Non-small-cell lung cancer (NSCLC) and other solid tumors | |||
| Phase I | Oral. Daily (Combination with temozolomide). | Malignant glioma | |||
| Phase I/II | Oral. Twice daily (Combination with letrozole). | Breast cancer (hormone-receptor-positive and HER2-negative) | |||
| Novartis | BGT226 | Phase I* | Oral. | Advanced solid tumors (Japanese patients) | |
| Phase I/II | Oral. | Advanced solid malignancies including advanced breast cancer | |||
| Genentech | GDC0980 | Phase I | Oral. Once weekly. | Refractory solid tumors or non-Hodgkin’s lymphoma | |
| Phase I | Oral. Once daily, 3-week on/1-week off [24,25]. | Refractory solid tumors or non-Hodgkin’s lymphoma | |||
| mTORC1/mTORC2 dual inhibitor | OSI | OSI027 | Phase I | Oral. Intermittent, once weekly, or continuous daily. | Advanced solid tumors or lymphoma |
| Intellikine | INK128 | Phase I | Oral. Daily. | Advanced solid malignancies | |
| Phase I | Oral. | Relapsed or refractory multiple myeloma | |||
| AstraZeneca | AZD8055 | Phase I/II | Oral. Twice daily follow a single dose on day 1, or twice daily from day 1, 28-day cycles | Advanced solid tumors | |
| Phase I/II | Oral. Twice daily follow a single dose on day 1 or twice daily from day 1. | Advanced hepatocellular carcinoma (Asian patients) | |||
| Phase I | Oral. Twice daily. | Advanced solid tumors (Japanese patients) | |||
| Phase I | Oral. Twice daily alternate days or continuous twice daily, follow multiple dose on day 1, 28-day cycles. | Advanced solid malignancies or lymphoma | |||
| AstraZeneca | AZD2014 | Phase I | Oral. Twice daily follow a single dose on day 1, or twice daily from day 1. | Advanced solid malignancies |
See also: http://www.clinicaltrials.gov/
Has been completed.
As discussed below, the second-generation mTOR inhibitors bind to the ATP-binding site in the mTOR kinase domain that is required for the functions of both mTOR complexes, resulting in downregulation of mTOR signaling globally. Because PI3K and mTORC2 regulate AKT phosphorylation, these compounds have the advantage of minimizing the feedback activation of AKT. The action point of different ATP-competitive mTOR inhibitors on the PI3K/AKT/mTOR pathway are summarized in Figure 1. The second generation of mTOR inhibitors has opened a new chapter in the 40-plus year odyssey that began with the discovery of rapamycin on the remote Easter Island Rapa Nui.
mTOR/PI3K dual inhibitors
A number of mTOR/PI3K dual inhibitors (TPdIs) have been developed (Table 2). The development of these agents has benefited from previous endeavors with PI3K-selective inhibitors. Through kinase selectivity profiling and cellular assays, it was discovered that several of these PI3K inhibitors are in fact TPdIs, exemplified by PI103 and NVPBEZ235. As a founding member of the TPdI series, PI103 was discovered using HTS followed by a medicinal chemistry campaign [15,16]. Its therapeutic activity against a wide range of human tumor models, including cervical cancer, leukemia and glioma, has been reported providing proof of concept for the therapeutic potential of this pyridofuropyrimidine series [17]. Although PI103 has not been evaluated in humans because of its poor in vivo pharmacokinetics (PK), it is now widely used as a chemical probe for the PI3K/mTOR pathway and as a lead compound for generating other PI3K and mTOR inhibitors. Several ATP-competitive mTOR inhibitors including PI540, PI620, WYE354, WAY600, WYE687 and Ku0063794 were subsequently developed using PI103 as a lead compound [16–18].
Table 2.
mTOR/PI3K dual inhibitors in vitro kinase IC50 (nM)
| PI103 [15,16] | PI540 [16,55] | PI620 [16,55] | NVPBEZ235 [19] | GSK2126458 [22] | SF1126† | NVPBBD130 [41] | PKI402 [45] | |
|---|---|---|---|---|---|---|---|---|
| mTOR | 20/83* | 61 | 231 | 20.7 | 0.180/0.300* | 1060 | n.d./7.7# | 3 |
| PI3Kα | 2/8 | 10 | 7 | 4 | 0.019 | 356 | 72/71 | 2 |
| PI3Kβ | 3/88 | 35 | 63 | 75 | 0.13 | 736 | 2340/2336 | 7 |
| PI3Kγ | 15/150 | 331 | 672 | 5 | 0.06 | 1774 | 382/350 | 16 |
| PI3Kδ | 3/48 | 4 | 8 | 7 | 0.024 | 3225 | 201/201 | 14 |
| DNA-PK | 23/2 | 525 | 147 | n.d. | 0.28 | 357 | n.d. | n.d. |
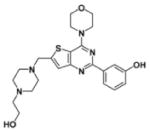
| Chemical structure |  |
 |
 |
 |
 |
 |
 |
 |
| Inventor | Astellas | Piramed | Piramed | Novartis | GlaxoSmithKline | Semafore | Novartis | Wyeth |
n.d., not determined.
IC50 for mTORC1 and mTORC2, respectively.
Values obtained in TSC1 null mouse embryonic fibroblasts using phospho-S6 as output.
Several TPdIs have entered early-stage clinical trials that yielded incomplete but promising results. NVPBEZ235, generated by structure-based design, is an orally bioavailable TPdI. It was reported to inhibit tumor growth in many preclinical models, including prostate, breast, pancreatic, and renal cancers, glioblastoma, multiple myeloma, leukemia and sarcomas, and it enhanced the antitumor activity of several other cancer drugs such as vincristine and doxorubicin [19–21]. NVPBEZ235 potently inhibited several rapamycin-resistant colon cancer cell lines at nanomolar concentrations (our observation), demonstrating its superior antitumor activity NVPBEZ235 has appropriate pharmacological features that allowed it to enter Phase I/II clinical trials in patients with advanced solid tumors, including breast cancer. GSK2126458 is another TPdI with a low picomolar IC50 with excellent antitumor activity. It has a sustained pharmacodynamic (PD) effect at very low circulating drug levels. It is now in a Phase I, open-label, dose-escalation study in subjects with solid tumors or lymphoma [22].
XL765 (structure not disclosed) is the first oral TPdI (mTOR, IC50 = 157 nM; PI3Kα, IC50 = 39 nM) with reported Phase I trial results [23,24]. After the indicated drug administration, XL765 decreased phosphorylation of several components of the PI3K/mTOR pathway including AKT, and reduced the proliferation of tumor tissues. In five out of a total of 19 patients the disease stabilized for at least 3 months, with two having a sustained response for longer than 6 months (one mesothelioma and one colon cancer patient) [25]. GDC0980 (structure not disclosed) is another TDdI (mTOR, IC50 = 17.3 nM; PI3Kα, IC50 = 4.8 nM). GDC0980 demonstrated broad preclinical activity in breast, ovarian, lung and prostate cancer models. It is active against tumor cells bearing mutations in PI3K, PTEN or K-RAS, as well as the wild-type PI3K pathway [26]. In the ongoing Phase I studies, GDC0980 is generally well tolerated with potential signs of antitumor activity and no significant toxicities, while producing PD modulation in surrogate tissues and tumors. Favorable preliminary PK profiles have been observed at the initial dose levels [27].
Finally, SF1126 is a vascular targeted conjugate of the well characterized pan PI3K inhibitor LY294002. LY294002 is also known to inhibit mTOR catalytic activity and thus qualified as a TPdI. It is conjugated to an integrin-targeting peptide, which increases the solubility and enhances its targeting to tumor sites [28]. Whereas SF1126 is not as potent in mTOR and PI3K kinase inhibition as other TPdIs in clinical development, the in vivo activity compares favorably, which is perhaps owing to the unique vascular targeting as well as inhibition of multiple kinase targets. Because SF1126 is targeted to stromal endothelial and tumor cells through RGDS-mediated binding of integrin, this agent exerts anticancer activity based on its effects on the tumor microenvironment (e.g. angiogenesis) and cell-signaling inhibition [28]. SF1126 is in development in multiple Phase I clinical trials as a single agent. The interim results were published recently in patients with solid tumors (2008 ASCO, abstract 14532) [29] and multiple myeloma (2009 ASH, abstract 3879: http://ash.confex.com/ash/2009/webprogram/Paper24232.html). SF1126 is well tolerated with the most common grade 1 adverse events being nausea, vomiting, diarrhea, fever, fatigue, chills and pruritus. Forty-six percent of the dosed patients showed stable disease with a median duration of 13 weeks and a mean duration of ~19 weeks. The Phase I single-agent clinical trials are being expanded to B-cell malignancies such as chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (http://www.semaforepharma.com/semaforeposterkinase.pdf).
mTORC1/mTORC2 dual inhibitors (TORCdIs)
Over the past two years, a new generation of mTOR-specific kinase inhibitors has emerged from screening and drug discovery efforts directed toward the kinase active site of mTOR (Table 3). Because they block the activity of both mTOR complexes they are commonly called mTORC1/mTORC2 dual inhibitors. Among of them, INK128, AZD8055, OSI027 and AZD2014 have already entered clinical trials [30] (Table 1). The pyrazolopyrimidine compounds PP242 and PP30 are selective inhibitors of mTOR kinase [31]. Besides being more effective than rapamycin in achieving cytoreduction and apoptosis in leukemia and multiple myeloma cells, perhaps the most striking thing about PP242 was its effect or lack thereof on the immune system [32,33]. At therapeutic doses in leukemia models PP242 produces much weaker immunosuppression than either rapamycin or PI103, which could translate into a better therapeutic ratio in the clinic [32].
Table 3.
mTORC1/mTORC2 dual inhibitors in vitro kinase IC50 (nM)
| PP242 [31] | PP30 [31] | WAY600 [34] | WYE687 [34] | WYE354 [34] | WYE132 [35] | AZD8055 [37] | Ku0063794 [42] | |
|---|---|---|---|---|---|---|---|---|
| mTOR | 8 | 80 | 9 | 7 | 5 | 0.19 ± 0.07 | 0.8/0.13 | 3/16 |
| PI3Kα | 1960 | 3000 | 1960 | 810 | 1890 | 1179 | 3590 | 8900 |
| PI3Kβ | 2200 | 5800 | n.d. | n.d. | n.d. | >10000 | 18900 | >30000 |
| PI3Kγ | 1270 | 990 | 8450 | 3110 | 7370 | >10000 | >14790 | >30000 |
| PI3Kδ | 102 | 680 | n.d. | n.d. | n.d. | 2380 | 3200 | >5300 |
| DNA-PK | 408 | 339 | n.d. | n.d. | n.d. | n.d. | 1370 | >10000 |
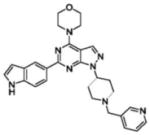
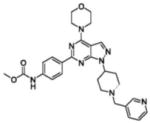
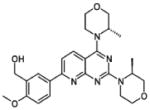
| Chemical structure |  |
 |
 |
 |
 |
 |
 |
 |
| Inventor | Intellikine | Intellikine | Wyeth | Wyeth | Wyeth | Wyeth | AstraZeneca | AstraZeneca |
n.d., not determined.
Another set of TORCdIs is derived from a morpholino pyrazolopyrimidine scaffold that is structurally related to PI103 [34]. Starting from the lead compound WAY001, further derivatization produced WAY600, WYE687 and WYE354. Although these inhibitors exhibit antiproliferative activity against several cancer cell lines and a glioma xenograft model, the poor pharmacological properties prevented their further clinical development. Recently, another compound, WYE132 was reported to show single-agent anticancer activity in tumor models of brain, breast, lung and renal cancer. The data from preclinical pharmacology supported its usefulness as an anticancer agent and provided a strong rationale for clinical development [35].
INK128 (structure not disclosed) is an orally available, potent and selective TORCdI (IC50 = 1 nM). INK128 has demonstrated broad antitumor activity against a range of solid tumor types. Oral administration of INK128 inhibited angiogenesis and tumor growth in multiple preclinical models with a predicted dose exposure relationship. It also exhibited potent inhibition of tumor cell lines resistant to rapamycin and pan-PI3K inhibitors. INK128 was reported to have excellent PK and is currently undergoing Phase I clinical evaluation [36].
AZD8055 is another orally available and ATP-competitive inhibitor of mTOR worth mentioning [37]. AZD8055 is currently in Phase I/II clinical development in patients with advanced solid tumors including advanced hepatocellular carcinoma [38]. Two other TORCdIs from OSI pharmaceuticals (http://www.osip.com/), OSI027 and OXA01, are currently in Phase I or late preclinical testing but their biological profile has not been disclosed. In the in vitro study, OSI027 generates anti-leukemic responses in primitive leukemic progenitors from chronic myelogenous leukemia patients, especially on cells expressing the T315I-BCR-ABL mutation, which is refractory to all BCR/ABL kinase inhibitors currently in clinical use [39]. OSI027 is well tolerated at the doses and schedules tested to date in patients with advanced solid tumors or lymphoma [40].
Other emerging ATP-competitive mTOR inhibitors
There are a number of ATP-competitive inhibitors, including NVPBBD130 (a sister compound of NVPBEZ235) [41], Ku0063794 (a TORCdI derived from PI103) [42,43], WJD008 (a TPdI) [44] and PKI402 (a TPdI) [45], which were all reported to inhibit cap-dependent translation efficiently, and/or to attenuate growth and proliferation of tumor cells. However, the preclinical and clinical therapeutic efficacy and tolerability of such inhibitors has not yet been established.
Potential issues and limitations
ATP-competitive mTOR inhibitors hold great promise for anticancer therapy and are rapidly moving into clinical trials. However, many important issues remain that will determine their ultimate success in the clinic. First, surrogate biomarkers are not yet available to predict what cancer patients will benefit from these inhibitors. Recent studies highlight the emergence of rapamycin-resistant mTOR function in protein synthesis, cell growth, survival and metabolism. Some of these rapamycin-insensitive mTOR functions can be profoundly inhibited by mTOR kinase inhibitors in some but not other cancer cells (e.g. colon cancer cells) [8,46]. Thus, there appear to be genetic determinants that predispose cancer cells to be sensitive or resistant to these anti-mTOR agents. Identification of such factors is likely to be a key to their clinical success.
Solid tumors have significant inter- and intra-tumoral heterogeneity and possess varied genetic abnormalities and treatment responses. Although it is thought tumors ‘addictive’ to the PI3K/mTOR pathway should respond favorably to these inhibitors, it is still unclear if the compounds are similarly efficacious in cancers with distinct genetic lesions, such as PIK3CA, PTEN and K-RAS, in this pathway. Efforts have already been made in this regard, but a clear picture has not emerged so far. It was suggested that breast cancer with HER2 and/or PIK3CA mutations has a favorable prognosis with NVPBEZ235 treatment, but breast cancer with PTEN mutations should be avoided as a single therapy [47]. Another study with PI103 and NVPBEZ235 showed that hyperphosphorylated AKT and K-RAS/B-RAF mutations are correlated with the efficacy and the inefficacy, respectively. By contrast, PIK3CA mutation and PTEN loss did not show any correlation [48]. Obviously, retrospective analysis of genetic biomarkers and clinical efficacy in patients enrolled in Phase I clinical trials with various ATP-competitive mTOR inhibitors is eagerly awaited because this is likely to provide valuable information. Further study of cell lines and primary specimens that are resistant to mTOR inhibitors, especially those with similar molecular pathologies, might lead to biomarkers that can be used to predict efficacy.
Like rapalogs, immunosuppression is a concern for mTOR kinase inhibitors. mTOR is widely known as an immunosuppressive drug target. The immunosuppressive action of rapamycin results from the inhibition of T- and B-cell proliferation through the same mechanisms that blocks cancer cell proliferation [49]. Obviously, chemotherapy-induced impairment of the immune surveillance has an adverse effect and can enhance cancer incidence and metastatic progression. Thus, more-potent suppression of mTOR signaling by ATP-competitive inhibitors is a concern regarding the immunosuppression in a clinical context. It was reported that PI103, one of the TPdIs, promoted immunosuppression and in vivo tumor growth, and increases survival of sorafenib-treated melanoma cells in immunocompetent mice, which contradicts the traditional concept that the anti-mTOR effect on tumor growth and progression outweighs the suppression of the immune system [50]. By contrast, PP242 appears to have weaker immunosuppressive effects than rapamycin and PI103 in assays of adaptive immune function, indicating that anticancer action of PP242 might be dominant over the immunosuppressive action [32]. These results were based on a 1–3 week study, so it remains to be seen what the long-term consequences for PP242 with respect to immunosuppression will be. Currently, because most of the in vivo studies with mTOR kinase inhibitors use xenograft tumor models in immunocompromised mice, certain precautions should be taken for clinical trials. As mTOR kinase-targeted therapies enter the clinical arena it will be important to understand how they can best be used to harness immune responses and eradicate tumor cells. A recent study on leukemia reported that, whereas TORCdIs and TPdIs both have anticancer efficacy, TPdIs cause greater immune suppression. PI3K targeting has been suggested to account for some of the observed immunosuppressive effects [32,51].
As with many other protein kinase inhibitors, some TORCdIs and TPdIs are likely to inhibit multiple kinases at therapeutic concentrations. For example, the IC50 for GSK2126458 toward DNA-PK is at the subnanomolar level (Table 2). Whereas in certain cases pan-kinase blockage can help improve the anticancer efficacy, in other cases it could be problematic because of increased adverse effects. In many published studies, much higher drug concentrations were typically used in cell or animal experiments than that necessary for enzymatic inhibition (μM versus nM). This raises concerns over whether the antitumor activity of some compounds was due to the proposed target inhibition. Much more research is needed to investigate potential off-target effects.
Finally, what are the other adverse effects related to the use of TORCdIs and TPdIs? One issue confronting TPdIs is safety. PI3K has numerous roles in cell survival, differentiation, metabolism and migration, some of which are independent of AKT and mTOR [52,53]. It is notable that metabolic disturbances are likely to arise as a result of inhibiting the PI3Kα isoform [54], which is the key anticancer target in a broad range of tumors for PI3K inhibitors. Because of their intimate role in glucose metabolism, it is expected that hyperglycemia and glucose intolerance will be potential hurdles for TPdIs [55,56]. There is also evidence implicating PI3K in the regulation of hematopoietic stem cells, suggesting that chronic inhibition of the PI3K pathway leads to issues of hematological insufficiency [57]. In XL765 clinical trials, grade 3–4 adverse effects were also observed for elevated hepatic transaminase [24,25]. As for TORCdIs, so far no detailed safety profile is available. The results from treatment with rapalogs might shed some light on the subject. The reported adverse effects of rapalogs are relatively mild, including dyslipidemia, diabetes, myelosuppression, delayed wound healing, infertility, ovarian cysts and mouth ulcers [58,59]. Severe cases of interstitial pneumonia, a rare but potentially lethal adverse effect, have also been reported with sirolimus and everolimus (RAD001) therapy [58,60]. Because TORCdIs generate a more complete suppression of mTOR signaling than rapalogs, global inhibition of mTOR is likely to be accompanied by greater toxicity. In this context, it remains to be seen if mTOR kinase inhibitors can achieve a more favorable balance of efficacy and tolerability. Further study is needed to assess whether blocking multiple points of the PI3K/AKT/mTOR signaling cascade is more effective than blockage at a single node, while providing an acceptable therapeutic window in humans.
Concluding remarks
In summary, recent advances in the development of mTOR catalytic inhibitors not only provide invaluable tools for investigating the increasingly complex mTOR signaling network but also offer considerable new opportunities to exploit fully the therapeutic potential of mTOR targeting in cancer. Studies with various tumor models have shown considerable promises. At present, there are already multiple clinical trials to examine these new agents in a variety of human solid tumors and hematological malignancies. Looking forward, finding predictive biomarkers for human cancers susceptible to these new mTOR inhibitors will be an important future direction, which is likely to determine the clinical success of these agents. Another key future task is to differentiate mechanism-based toxicities from compound-specific ones. This will allow selection of the effective agents with minimal adverse effects. With the help of structural studies, rational design and chemical synthesis, medicinal chemistry efforts are under way to improve the potency, selectivity and pharmacological properties of this class of mTOR inhibitors further. It is conceivable that these commitments will bring the second generation of mTOR inhibitors into clinical practice and will significantly improve cancer patient care in the not-too-distant future.
Acknowledgments
We thank Janice Thomas for reading this manuscript. The work performed in the authors’ laboratory was supported by NIH grant R01CA123391 (XFZ), National Natural Science Foundation of China (30900672) and Shanghai Rising-Star Program (10QA1404300) (YJZ), and the Chinese Ministry of Education 111 Project B08034 (YWD).
Footnotes
Teaser: This article reviews the emergence of ATP-competitive mTOR inhibitors as anticancer drugs, especially those entering clinical development. The potential therapeutic value and limitations of these agents are discussed.
Conflicts of interest
The authors declared no potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tsang CK, et al. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Zhang YJ, et al. mTOR signaling pathway is a target for the treatment of colorectal cancer. Ann Surg Oncol. 2009;16:2617–2628. doi: 10.1245/s10434-009-0555-9. [DOI] [PubMed] [Google Scholar]
- 3.Chapuis N, et al. Perspectives on inhibiting mTOR as a future treatment strategy for hematological malignancies. Leukemia. 2010 doi: 10.1038/leu.2010.170. (in press) [DOI] [PubMed] [Google Scholar]
- 4.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27:2278–2287. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibbons JJ, et al. Mammalian target of rapamycin: discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin Oncol. 2009;36 (Suppl 3):3–17. doi: 10.1053/j.seminoncol.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Don AS, Zheng XF. Recent clinical trials of mTOR-targeted cancer therapies. Rev Recent Clin Trials. 2010 doi: 10.2174/157488711793980147. (in press) [DOI] [PubMed] [Google Scholar]
- 8.Shor B, et al. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831–3837. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- 9.Zheng XF, et al. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell. 1995;82:121–130. doi: 10.1016/0092-8674(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 10.Cybulski N, Hall MN. TOR complex 2: a signaling pathway of its own. Trends Biochem Sci. 2009;34:620–627. doi: 10.1016/j.tibs.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Sun SY, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 12.Wan X, et al. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 13.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldman ME, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. Plos Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Workman P, et al. Drugging the PI3 kinome. Nat Biotechnol. 2006;24:794–796. doi: 10.1038/nbt0706-794. [DOI] [PubMed] [Google Scholar]
- 16.Raynaud FI, et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther. 2009;8:1725–1738. doi: 10.1158/1535-7163.MCT-08-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Workman P, et al. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res. 2010;70:2146–2157. doi: 10.1158/0008-5472.CAN-09-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, et al. mTOR mediated anti-cancer drug discovery. Drug Discov Today: Ther Strat. 2009;6:47–55. doi: 10.1016/j.ddstr.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maira SM, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 20.Chiarini F, et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010 doi: 10.1158/0008-5472.CAN-10-1814. (in press) [DOI] [PubMed] [Google Scholar]
- 21.Manara MC, et al. NVP-BEZ235 as a new therapeutic option for sarcomas. Clin Cancer Res. 2010;16:530–540. doi: 10.1158/1078-0432.CCR-09-0816. [DOI] [PubMed] [Google Scholar]
- 22.Knight SD, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett. 2010;1:39–43. doi: 10.1021/ml900028r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauveur-Michel M, et al. Discovery of novel anticancer therapeutics targeting the PI3K/Akt/mTOR pathway. Future Med Chem. 2009;1:137–155. doi: 10.4155/fmc.09.5. [DOI] [PubMed] [Google Scholar]
- 24.Papadopoulos KP, et al. A Phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of a novel PI3K inhibitor, XL765, administered orally to patients (pts) with advanced solid tumors. J Clin Oncol. 2008;26:3510. [Google Scholar]
- 25.Molckovsky A, Siu LL. First-in-class, first-in-human Phase I results of targeted agents: highlights of the 2008 American society of clinical oncology meeting. J Hematol Oncol. 2008;1:20. doi: 10.1186/1756-8722-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagner AJ, et al. Pharmacokinetics and pharmacodynamic biomarkers for the dual PI3K/mTOR inhibitor GDC-0980: initial Phase I evaluation. Mol Cancer Ther. 2009;8(Suppl 12):B137. [Google Scholar]
- 27.Dolly S, et al. A first-in-human, Phase l study to evaluate the dual PI3K/mTOR inhibitor GDC-0980 administered QD in patients with advanced solid tumors or non-Hodgkin’s lymphoma. J Clin Oncol. 2010;28 (Suppl 15):3079. [Google Scholar]
- 28.Garlich JR, et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008;68:206–215. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 29.Schwertschlag US, et al. Phase 1 pharmacokinetic (PK) and pharmacodynamic (PD) evaluation of SF1126 a vascular targeted pan phosphoinositide 3- kinase (PI3K) inhibitor in patients with solid tumors. J Clin Oncol. 2008;26:14532. [Google Scholar]
- 30.García-Echeverría C. Allosteric and ATP-competitive kinase inhibitors of mTOR for cancer treatment. Bioorg Med Chem Lett. 2010;20:4308–4312. doi: 10.1016/j.bmcl.2010.05.099. [DOI] [PubMed] [Google Scholar]
- 31.Apsel B, et al. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janes MR, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoang B, et al. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood. doi: 10.1182/blood-2010-05-285726. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu K, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69:6232–6240. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- 35.Yu K, et al. Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res. 2010;70:621–631. doi: 10.1158/0008-5472.CAN-09-2340. [DOI] [PubMed] [Google Scholar]
- 36.Jessen K, et al. INK128 is a potent and selective TORC1/2 inhibitor with broad oral anti-tumor activity. Mol Cancer Ther. 2009;8 (Suppl 12):B148. [Google Scholar]
- 37.Chresta CM, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 38.Houghton PJ, et al. Pediatric preclinical testing program (PPTP) stage 1 evaluation of AZD8055, an inhibitor of mTOR kinase. Mol Cancer Ther. 2009;8 (Suppl 12):C57. [Google Scholar]
- 39.Carayol N, et al. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A. 2010;107:12469–12474. doi: 10.1073/pnas.1005114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan DS, et al. First-in-human Phase I study exploring three schedules of OSI-027, a novel small molecule TORC1/TORC2 inhibitor, in patients with advanced solid tumors and lymphoma. J Clin Oncol. 2010;28 (Suppl 15):3006. [Google Scholar]
- 41.Marone R, et al. Targeting melanoma with dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors. Mol Cancer Res. 2009;7:601–613. doi: 10.1158/1541-7786.MCR-08-0366. [DOI] [PubMed] [Google Scholar]
- 42.Malagu K, et al. The discovery and optimisation of pyrido[2,3-d]pyrimidine-2,4-diamines as potent and selective inhibitors of mTOR kinase. Bioorg Med Chem Lett. 2009;19:5950–5953. doi: 10.1016/j.bmcl.2009.08.038. [DOI] [PubMed] [Google Scholar]
- 43.García-Martínez JM, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009;421:29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li T, et al. WJD008, a dual phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin inhibitor, prevents PI3K signaling and inhibits the proliferation of transformed cells with oncogenic PI3K mutant. J Pharmacol Exp Ther. 2010;334:830–838. doi: 10.1124/jpet.110.167940. [DOI] [PubMed] [Google Scholar]
- 45.Mallon R, et al. Antitumor efficacy profile of PKI-402, a dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor. Mol Cancer Ther. 2010;9:976–984. doi: 10.1158/1535-7163.MCT-09-0954. [DOI] [PubMed] [Google Scholar]
- 46.Raynaud FI, et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther. 2009;8:1725–1738. doi: 10.1158/1535-7163.MCT-08-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brachmann SM, et al. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci U S A. 2009;106:22299–22304. doi: 10.1073/pnas.0905152106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dan S, et al. Correlating phosphatidylinositol 3-kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations. Cancer Res. 2010;70:4982–4994. doi: 10.1158/0008-5472.CAN-09-4172. [DOI] [PubMed] [Google Scholar]
- 49.Law BK. Rapamycin: an anti-cancer immunosuppressant? Crit Rev Oncol Hematol. 2005;56:47–60. doi: 10.1016/j.critrevonc.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 50.López-Fauqued M, et al. The dual PI3K/mTOR inhibitor PI-103 promotes immunosuppression, in vivo tumor growth and increases survival of sorafenib-treated melanoma cells. Int J Cancer. 2010;126:1549–1561. doi: 10.1002/ijc.24926. [DOI] [PubMed] [Google Scholar]
- 51.Janes MR, Fruman DA. Targeting TOR dependence in cancer. Oncotarget. 2010;1:69–76. doi: 10.18632/oncotarget.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engelman JA, et al. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 53.Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. 2009;228:253–272. doi: 10.1111/j.1600-065X.2008.00750.x. [DOI] [PubMed] [Google Scholar]
- 54.Foukas LC, et al. Critical role for the p110α phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441:366–370. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 55.Yap TA, et al. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 56.Ihle NT, Powis G. Take your PIK: phosphatidylinositol 3-kinase inhibitors race through the clinic and toward cancer therapy. Mol Cancer Ther. 2009;8:1–9. doi: 10.1158/1535-7163.MCT-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 58.Cravedi P, et al. Sirolimus for calcineurin inhibitors in organ transplantation: contra. Kidney Int. doi: 10.1038/ki.2010.268. (in press) [DOI] [PubMed] [Google Scholar]
- 59.Armstrong AJ, et al. A pharmacodynamic study of rapamycin in men with intermediate- to high-risk localized prostate cancer. Clin Cancer Res. 2010;16:3057–3066. doi: 10.1158/1078-0432.CCR-10-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White DA, et al. Noninfectious pneumonitis after everolimus therapy for advanced renal cell carcinoma. Am J Respir Crit Care Med. 2010;182:396–403. doi: 10.1164/rccm.200911-1720OC. [DOI] [PubMed] [Google Scholar]