

Abstract
Vesicular stomatitis virus (VSV) is an oncolytic virus currently being investigated as a promising tool to treat cancer because of its ability to selectively replicate in cancer cells. To enhance the oncolytic property of the nonpathologic laboratory strain of VSV, we generated a recombinant vector [rVSV(MΔ51)-M3] expressing murine gammaherpesvirus M3, a secreted viral chemokine-binding protein that binds to a broad range of mammalian chemokines with high affinity. As previously reported, when rVSV(MΔ51)-M3 was used in an orthotopic model of hepatocellular carcinoma (HCC) in rats, it suppressed inflammatory cell migration to the virus-infected tumor site, which allowed for enhanced intratumoral virus replication leading to increased tumor necrosis and substantially prolonged survival. These encouraging results led to the development of this vector for clinical translation in patients with HCC. However, a scalable current Good Manufacturing Practice (cGMP)-compliant manufacturing process has not been described for this vector. To produce the quantities of high-titer virus required for clinical trials, a process that is amenable to GMP manufacturing and scale-up was developed. We describe here a large-scale (50-liter) vector production process capable of achieving crude titers on the order of 109 plaque-forming units (PFU)/ml under cGMP. This process was used to generate a master virus seed stock and a clinical lot of the clinical trial agent under cGMP with an infectious viral titer of approximately 2 × 1010 PFU/ml (total yield, 1 × 1013 PFU). The lot has passed all U.S. Food and Drug Administration-mandated release testing and will be used in a phase 1 clinical translational trial in patients with advanced HCC.
Ausubel and colleagues describe a process that is amenable to large-scale GMP manufacturing of vesicular stomatitis virus (VSV) vector. This process was used to generate a master virus seed stock and a clinical lot of the clinical trial agent under cGMP with an infectious viral titer of approximately 2 × 1010 PFU/ml (total yield, 1 × 1013 PFU). This vector lot will be used in a phase 1 clinical translational trial in patients with advanced hepatocellular carcinoma.
Introduction
Oncolytic virotherapy is emerging as a promising strategy to treat cancer because of the unique ability of these replication-competent viruses to selectively replicate only in cancer cells. Early oncolytic viruses shown to be effective against solid tumors in laboratory animals were derived from adenovirus or herpes simplex virus type 1, and hundreds of patients with various types of solid tumors have already been treated in various clinical translational trials (Barker and Berk, 1987; Martuza et al., 1991; Whitley et al., 1993; Mineta et al., 1994, 1995; Bischoff et al., 1996; Heise et al., 1997; Wickham et al., 1997; Hallenbeck et al., 1999; Miyatake et al., 1999; Alemany et al., 2000; Fueyo et al., 2000; Ganly et al., 2000; Khuri et al., 2000; Nemunaitis et al., 2000; Rainov and Ren, 2003; Kasuya et al., 2005). Subsequently, a number of other animal and human viruses are being developed as potential oncolytic agents for cancer treatment, including reovirus, Newcastle disease virus, measles virus, vaccinia virus, influenza virus, and vesicular stomatitis virus (VSV), among others (Kirn et al., 2001). Despite their efficacy in animal models, a major limitation to the successful use of oncolytic viruses to treat cancer in patients is their relatively poor replication efficiency and limitations in spreading within the solid tumor mass in vivo, which typically contains complex stromal structures interspersed with the tumor cells. To overcome these obstacles, more virulent oncolytic viruses that retain tumor specificity and/or oncolytic viruses molecularly engineered to express transgenes that exert bystander effects for enhanced tumor cell killing have been used (Hardcastle et al., 2007; Singleton et al., 2007; Thorne et al., 2007; Gainey et al., 2008; Kuhn et al., 2008; Raykov et al., 2008; Xie et al., 2009; Ziauddin et al., 2010). Another limitation is the neutralizing antiviral antibody responses that are elicited within several days in immune-competent hosts, which shortens the window of opportunity for intratumoral replication. This can be addressed through the use of viruses such as VSV with a short life cycle (8–10 hr), which permits robust intratumoral virus replication before the onset of high-titer neutralizing antibodies (Shinozaki et al., 2005).
VSV is an enveloped, single-strand RNA virus belonging to the family Rhabdoviridae, genus Vesiculovirus, with 16 distinct serotypes, of which 6 can cause animal or human disease (Rose and Whitt, 2001). A high percentage of people living in endemic areas such as the central and southwestern United States and Canada may have been infected. Infections in humans are asymptomatic in most cases or result in a mild febrile illness, oral vesicular lesions, malaise, and pharyngitis (Fields and Hawkins, 1967; Letchworth et al., 1999). Two cases of VSV meningoencephalitis have been reported in children (Quiroz et al., 1988). The envelope G-protein of VSV binds to the surface of most insect and mammalian cell types, accounting for the wide tissue tropism of VSV. VSV replicates in the cytoplasm of infected cells, which die within hours as a result of robust viral mRNA and protein synthesis. Viral replication is inhibited in normal cells because of their responses to type I interferons (IFNs), thereby sparing the cell from cytopathic destruction. In tumor cells, however, viral replication is uninhibited because of cellular defects in the interferon response pathway, resulting in apoptotic cell death (Stojdl et al., 2000a). This selective oncolytic property of VSV makes it a potentially effective agent for selective antitumor treatment (Giedlin et al., 2003). Treatment of human melanoma xenografts with wild-type VSV in nude mice resulted in regression or growth inhibition of the established tumor (Stojdl et al., 2000a). This finding has been postulated to be due to the fact that IFN-responsive antiviral pathways are defective in many tumors including those of human origin, and thus VSV can replicate within these cells regardless of IFN treatment (Stojdl et al., 2000b). Viruses such as VSV and Newcastle disease virus (NDV) activate type I interferon responses in normal cells, which in turn induce protein kinase R (PKR) phosphorylation and inhibition of protein synthesis and viral replication (Kirn et al., 2001; Kasuya et al., 2005). Most tumor cells are defective in type I interferon response to various degrees and PKR remains underphosphorylated, permitting protein synthesis and viral replication to proceed.
To enhance the oncolytic property of the laboratory strain of VSV (Indiana) that is nonpathologic to humans (Sudia et al., 1967), we generated a recombinant vector [rVSV(MΔ51)-M3] expressing the secreted form of the murine gammaherpesvirus M3 protein, a viral chemokine-binding protein that binds to a broad range of chemokines (C, CC, CXC, and CX3C) with high affinity (Parry et al., 2000; van Berkel et al., 2000). When rVSV(MΔ51)-M3 was used in our hepatocellular carcinoma (HCC) animal model, it was shown to block inflammatory cell migration to the tumor site, which allowed for enhanced intratumoral virus replication leading to increased tumor necrosis and substantially prolonged survival (Wu et al., 2008). These encouraging results led us to develop this vector for potential use in treating patients with HCC.
The current manufacturing process has been used under Good Manufacturing Practice (GMP) conditions to produce a clinical lot of rVSV(MΔ51)-M3 with an infectious titer of approximately 2 × 1010 plaque-forming units (PFU)/ml (total yield, 1 × 1013 PFU). The lot is intended for use in phase 1 human clinical trials and has been characterized using the panel of testing described in this paper. The manufacturing methods used for vector production are amenable to further scale-up to provide larger quantities of the vector for future expanded clinical trials.
Materials and Methods
Growth of 293 cells
A vial of 293 cells from a fully characterized 293 master cell bank was thawed and expanded in culture. Cells were seeded at 2.5–4.0 × 104 cells/cm2 in Nunc flasks of increasing size (T-75 and T-500) or 10-layer Cell Factories (Nunc/Thermo Fisher Scientific, Rochester, NY). Cultures were expanded in growth medium consisting of 10% fetal bovine serum (FBS; HyClone, Logan, UT) in Dulbecco's modified Eagle's medium (DMEM; Biowhittaker, East Rutherford, NJ) and passaged every 3–4 days by the addition of trypsin (Irvine Scientific, Santa Ana, CA).
Production of crude rVSV vector
293 cells were seeded in a 10-layer Nunc Cell Factory (Nunc/Thermo Fisher Scientific) at a density of 7.5 × 104 cells/cm2 in DMEM (Biowhittaker) with 10% FBS (HyClone). Approximately 24 hr after seeding, the cell density was determined in one of the culture vessels to calculate the amount of seed virus required for infection at a multiplicity of infection (MOI) of 0.01. Before infection, the DMEM–10% FBS medium was removed and replaced with fresh virus production–serum-free medium (VP-SFM; Invitrogen, Carlsbad, CA) containing the appropriate amount of seed virus. The supernatant containing the rVSV vector was harvested approximately 24 hr after infection.
Clarification of crude virus supernatant
The harvested crude viral supernatant was clarified with a filter train, which consisted of a Milligard LPB (low protein-binding) Opticap XL-4 dual membrane (1.2-μm/0.5-μm) filter in-line with a Milligard LPB Opticap XL-4 single-layer (0.2-μm) filter (Millipore, Billerica, MA), for up to 10 liters of crude viral supernatant. Benzonase (EMD Chemicals, Gibbstown, NJ) was added to the clarified supernatant at a concentration of 50 U/ml to digest host cell nucleic acids. Digestion was carried out at 37°C for 4 hr. After digestion, the supernatant was held at 4°C overnight before purification by column chromatography.
Chromatographic purification
Benzonase-treated supernatant was purified by anion-exchange chromatography, using a Poros HQ-50 (Applied Biosystems/Invitrogen, Foster City, CA) packed column. A 5 × 20 cm column was used to process 10 liters of culture at a linear flow rate of 1.5 cm/min. The column was equilibrated with 150 mM NaCl in 20 mM Tris, pH 7.5, before loading. After loading, the column was washed with 4 column volumes of equilibration buffer until the absorbance at 260 nm returned to baseline. A second wash step was performed with 4 column volumes of 300 mM NaCl in 20 mM Tris, pH 7.5. The elution step was performed with 1.2 M NaCl in 20 mM Tris, pH 7.5. Ultraviolet (UV) absorbance at 260 nm was used to monitor and collect the elution peak, typically between 300 and 370 ml.
Tangential flow ultrafiltration, formulation, and sterile filtration
The column eluate was diluted to 2 liters, using exchange buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA; pH 7.8), and then concentrated to 400 ml by tangential flow filtration (TFF) (GE Healthcare Life Sciences, Little Chalfont, UK). Next, the eluate was diafiltered in continuous mode, using 2.4 liters of exchange buffer for a 6-fold buffer exchange. The diafiltered material was further concentrated from 400 ml to approximately 100 ml, representing an approximately 90-fold volume reduction from the original supernatant. The concentrated bulk was prepared by adding glycerol to the diafiltered material to obtain a final concentration of 10% glycerol. The concentrated bulk was stored at −80°C until pooling of concentrated bulk subbatches for final sterile filtration.
Plaque-forming assay
Six-well plates were seeded with BHK-21 cells in DMEM–10% FBS at 2–4 × 105 cells per well and incubated overnight at 37 ± 2°C with 5 ± 2% CO2. Serial dilutions of the test article were performed in Dulbecco's phosphate-buffered saline with calcium and magnesium The culture medium was aspirated and 1 ml of diluted test article was added to each well in triplicate. The plates were incubated for 30 min at 37 ± 2°C with 5 ± 2% CO2. At the end of the incubation period, inoculum was aspirated and the wells were overlaid with a mixture of equal volumes of 1% agarose and 2 × minimal essential medium (MEM)–15% FBS and incubated overnight at 37 ± 2°C with 5 ± 2% CO2. Plaques were counted after overnight incubation. The virus concentration was determined by taking the average number of plaques per well, at the dilution that gave rise to 5–50 plaques per well, and then multiplying by the corresponding dilution factor. The assay was performed three times to obtain the final titer.
End-labeling assay
Forty microliters of rVSV(ΔM51)-M3 were heat inactivated at 65°C for 30 min. A reaction mixture containing equal volumes of each radiolabeled nucleotide (dCTP, dGTP, dATP, and dTTP) (MP Biomedicals, Solon, OH), Klenow enzyme (New England BioLabs, Ipswich, MA), and 10 × buffer was added to each reaction tube. The original specific activity for each nucleotide was 3000 Ci/mmol and 10 mCi/ml. The contents were mixed by vortexing and the liquid was brought to the bottom of the tube by brief centrifugation at 5000 rpm. The reaction tubes were incubated at 25°C for 30 min. The end-labeling reaction was stopped by adding 10 μl of 0.5 M EDTA and then 0.5 μl of 10% sodium dodecyl sulfate (SDS) and 5 μl of 6 × loading dye were added. The unincorporated radionucleotides were removed with a MicroSpin (G-50) spin column (GE Healthcare Life Sciences). The flowthrough was loaded onto a 3% agarose gel and run for approximately 1.5 hr at 100 V. The dried gel was exposed to a storage phosphor screen (GE Healthcare Life Sciences) overnight in a Kodak film cassette (Carestream Health, Rochester, NY). The exposed storage phosphor screen was scanned with a Storm 860 PhosphorImager (Molecular Dynamics/GE Healthcare Life Sciences) and software.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
A 10% polyacrylamide gel (1.5 mm thick) was prepared and overlaid with a 3.5% stacking gel. A 10-μl aliquot of the rVSV(MD51)-M3 clinical lot was mixed with 10 μl of Laemmli sample buffer (Bio-Rad, Hercules, CA) before being loaded onto the gel. The gel was run at 180 V for approximately 1 hr and then stained with Coomassie Brilliant Blue R250 (Bio-Rad) to visualize the protein bands.
Sequencing of viral genome
Viral RNA was amplified by the strategy of reverse transcription (RT) and polymerase chain reaction (PCR). The rVSV(MΔ51)-M3 genome is 12417 bp long and several RT-PCRs using different primer pairs were performed. The RT-PCR products were then purified by agarose gel separation. Sequencing of the internal fragments was performed on these PCR products, using primers in both directions. Sequencing of the two ends of the viral genome (∼80 bp from the 3′ end and ∼100 bp from the 5′ end) was done in one direction only. All samples were submitted to the DNA Core Sequencing Facility of Mount Sinai School of Medicine (New York, NY). The raw sequence data were analyzed with MacVector software (MacVector, Cary, NC) to align the obtained sequence data with the theoretically expected sequence (based on the VSV-Indiana strain, GenBank accession number J02428.1, and the murine herpesvirus M3, GenBank accession number AF127083).
Neutralization of rVSV
To perform the lot release assays to look for viral or mycoplasma contamination, the rVSV sample needed to be neutralized with antibody against rVSV. The neutralization procedure was performed by incubating the rVSV sample with antiserum to VSV (immune ascites fluid, cat. no. VR-1238AF; American Type Culture Collection [ATCC], Manassas, VA) at a 1:1 ratio for 30 min at room temperature.
Results
Production of master virus seed stock
A vial of the 293 master cell bank (MCB) was expanded until sufficient cells existed for initiation of production of the master virus seed stock (MVSS). The 293 cell line was originally developed as a transformed cell line in 1977 by transfection of primary human embryonic kidney cells with sheared adenovirus type 5 DNA (Graham et al., 1977). Our MCB was frozen at passage 5 and the vector was produced in cells at passage 12–16. 293 cells were seeded at 7.5 × 104 cells/cm2 in 11 × T-175 flasks (Nunc/Thermo Fisher Scientific). Three days later, cells from one of the flasks (approximately 100% confluent) were harvested by trypsinization and the cell density was determined to be 3.2 × 105 cells/cm2 (doubling time, approximately 35–40 hr). The remaining cells (10 × T-175 flasks) were then infected with the VSV seed stock (1.1 × 1010 PFU/ml), using an MOI of 0.01. To initiate the infection, the culture medium was removed from each flask, and the VSV seed stock was diluted in serum-free medium, VP-SFM, and added to each flask. Approximately 24 hr after infection, the supernatant containing the VSV viral vector was harvested and centrifuged (200 × g for 15 min at 4°C) to remove cellular debris. The supernatant was then aliquoted into 240 cryovials (Corning, Corning, NY) at 1 ml each. The titer of the VSV MVSS was determined to be 9.1 × 108 PFU/ml. The assays used to characterize the MVSS are listed in Table 1. A number of assays were performed in vitro using indicator cell lines or in animals, including the in vitro viral assay, in vivo viral assay, and the mycoplasma assay. The viral vector was neutralized as described in Materials and Methods before being tested in these assays. However, despite preincubation with an available neutralizing serum, rVSV breakthrough was observed in the in vitro viral assay (only the portion of the assay performed on MRC-5 cells was free of rVSV breakthrough or evidence of other adventitious agents) and the in vivo viral assay (only the guinea pigs survived the assay). Thus, a conditionally negative result was reported for the in vitro and in vivo viral assays, based on the results of a subset of the cell lines or animals tested in these assays. A more effective neutralizing antibody will need to be identified to address the issue of rVSV breakthrough in these assays before initiation of phase 2 clinical trials.
Table 1.
Lot Release Assays, Specifications, and Results for the Recombinant Vesicular Stomatitis Virus Master Virus Seed Stock
| Assay | Specification | Result |
|---|---|---|
| Sterility testing (USP) | Negative | Negative |
| B&F assay—qualification of sterility testing | Pass | Pass |
| Mycoplasma (PTC) | Negative | Negative |
| In vitro adventitious viruses | Negative | Negative |
| In vivo assay for inapparent viruses | Negative | Negative |
| Hamster antibody production (HAP) test | Negative | Negative |
| Product-enhanced reverse transcriptase (PERT) assay for retrovirus | Negative | Negative |
| PCR-B19 parvovirusa | Negative | Negative |
| PCR-HIV1/2 (human immunodeficiency virus)a | Negative | Negative |
| PCR-HTLV I/II (human lymphotropic virus)a | Negative | Negative |
| PCR-HAV (hepatitis A virus)a | Negative | Negative |
| PCR-HBV (hepatitis B virus)a | Negative | Negative |
| PCR-HCV (hepatitis C virus)a | Negative | Negative |
| PCR-EBV (Epstein-Barr virus)a | Negative | Negative |
| PCR-CMV (cytomegalovirus)a | Negative | Negative |
| PCR-HHV6 (herpesvirus 6)a | Negative | Negative |
| PCR-HHV7 (herpesvirus 7)a | Negative | Negative |
| PCR-HHV8 (herpesvirus 8)a | Negative | Negative |
| Viral genome sequence | Report result | Result reported |
| Infectious titer | Report result | 3.9 × 109 PFU/ml |
B&F, Bacteriostasis and Fungistasis; PTC, Points to Consider; USP, U.S. Pharmacopoeia.
PCR test panel for contaminating viral sequences.
Production of pilot lot
Cell culture and infection
A 10-liter pilot lot was produced to evaluate the manufacturing procedure described in Materials and Methods. In brief, 293 cells were seeded at 7.5 × 104 cells/cm2, in DMEM with 10% FBS in 10 × 10-layer Cell Factories (CFs) and one single-layer Cell Factory. On day 3 (approximately 72 hr postseeding) cells were removed from the single-layer CF and counted (a cell density of 1.44 × 105 cells/cm2 was determined). The culture medium was removed from the 10 × 10-layer CFs and the cells were infected with the rVSV MVSS diluted at an MOI of 0.01, in the serum-free medium VP-SFM supplemented with 4 mM glutamine (Invitrogen). A volume of 900 ml of VP-SFM was used in each 10-layer CF. Approximately 24 hr after infection, cytopathic effect (CPE) was observed and the supernatant was harvested from the infected cells.
Clarification and Benzonase treatment
After the crude supernatant was clarified and treated with Benzonase, it was purified by column chromatography. Approximately 8.8 liters of supernatant was loaded onto an XK 50 column (GE Healthcare Life Sciences) with a 19-cm bed height, packed with Poros HQ-50 resin (column volume, 372 ml). The load ratio was 23.7 ml of supernatant per milliliter of resin and the linear flow rate used for the entire procedure was 1.5 cm/min. After the sample was loaded, the column was equilibrated with 4 column volumes of equilibration buffer and washed with approximately 1500 ml of 300 mM NaCl. The rVSV was eluted in a single step using approximately 550 ml of 1.2 M NaCl (an eluted peak volume of 312 ml was collected). The chromatogram is shown in Fig. 1.
FIG. 1.

Chromatogram of the recombinant vesicular stomatitis virus (rVSV) pilot lot. The culture supernatant was loaded onto the column and eluted with 1.2 M NaCl. Color images available online at www.liebertonline.com/hum.
Tangential flow ultrafiltration, formulation, and sterile filtration
The eluted material was diafiltered and further concentrated to a final volume of 105 ml and formulated by adding 11 ml of glycerol. To evaluate the final filtration step, 8 ml of the formulated bulk was filtered with a Millipore 0.2-μm syringe filter, polyethersulfone (PES) membrane (surface area, 3.9 cm2). Eight 1-ml filtered samples were taken sequentially. Four samples were titered and all of these samples had a similar titer with an average titer of 1.2 × 1010 PFU/ml, indicating minimal loss in infectious titer occurs during the filtration step. Filtration of the remaining 100-ml concentrated bulk was performed with Millipore Steripak-GP-10 filters (0.2-μm PES membrane with a surface area of 100 cm2) that had been prewetted with 25 ml of Tris–NaCl–EDTA (TNE) buffer. The titer of the in-process samples and the final filtered material are summarized in Table 2.
Table 2.
Summary of Product Yield During Pilot Lot Production
| Sample ID | Volume | Infectious titer (PFU/ml) | Total infectious units (PFU) |
|---|---|---|---|
| Clarified supernatant | 9 liters | 2.4 × 108 | 2.2 × 1012 |
| Post-Benzonase | 9 liters | 6.0 × 108 | 5.4 × 1012 |
| Elution peak (1.2M) | 312 ml | 1.7 × 108 | 5.3 × 1011 |
| Post-UF/DF | 105 ml | 5.2 × 109 | 6.0 × 1011 |
| Formulated bulk | 116 ml | 7.2 × 109 | 8.4 × 1011 |
| Filtered (0.2 μm) | 100 ml | 1.2 × 1010 | 1.3 × 1012 |
DF, diafiltration; UF, ultrafiltration.
Production of clinical lot under GMP
Cell culture and infection
The 293 cell line was expanded until sufficient cells existed for initiation of clinical lot production. Expansion of the 293 cell train is depicted in Fig. 2. Production was divided into five subbatches, each having 10 × 10-layer CFs seeded with 293 cells. The manufacturing process for a representative subbatch is described. On day 0, cells were trypsinized, counted with a hemocytometer and trypan blue staining, and plated at 0.75 × 105 cells/cm2 in 11 × 10-layer CFs. On day 3, cells from one of the CFs were removed with trypsin and were counted (8.9 × 109–3.2 × 1010 cells per CF). Monolayers were approximately 85–90% confluent at the time of infection. The cells in the remaining 10 × 10-layer CFs were then infected at an MOI of 0.01, using the rVSV MVSS (3.85 × 109 PFU/ml), by removing the growth medium from each factory and replacing it with 1 liter of VP-SFM containing the appropriate volume of rVSV MVSS. On day 4 (approximately 24 hr after infection), the viral supernatant was harvested. Samples of the crude harvest were collected for quality control (QC) testing (1 × 26-ml and 1 × 2-ml samples for the mycoplasma assay and 2 × 4-ml samples for the in vitro viral assay), as well as for in-process testing and retention samples (40 ml). The same manufacturing process was repeated a total of five times for a total production of 50 liters of rVSV supernatant.
FIG. 2.

Schematic representation of the expansion of the 293 cell train used for the 50-liter rVSV GMP production lot. Cells from the 293 master cell bank were thawed and expanded for the manufacture of VSV. Cells were passaged twice per week with trypsin. An additional 10-layer Cell Factory (CF) was seeded for each week's infection in order to be able to determine the number of cells in each CF (not indicated). Additional flasks (2 × T-175) were also seeded as part of the parallel culture for each week's manufacturing production (not indicated). Color images available online at www.liebertonline.com/hum.
Parallel cell culture
To obtain a sample for in vitro viral adventitious agent testing, a parallel culture of uninfected 293 cells (2 × T-175 flasks) from the same cell train was grown under conditions similar to those used to manufacture the clinical lot of rVSV. Cells were plated at the same density as the cells used for the production of virus (0.75 × 105 cells/cm2) with 28 ml of growth medium per flask. On day 3, the spent medium was removed from the parallel cell culture flasks and 28 ml of VP-SFM without virus was added to each flask. At the time of harvest (day 4), medium from both flasks was collected and saved (conditioned medium). Cells remaining in the flasks were removed with trypsin and then pelleted by centrifugation. The cell pellets were then resuspended in conditioned medium (described previously), aliquoted into multiple samples (4 × 10 ml), and frozen for further QC testing.
Clarification and Benzonase treatment
The harvested crude viral supernatant was clarified with a filter train, which consisted of a Millipore Milligard LPB Opticap XL-4 dual-membrane (1.2-μm/0.5-μm) filter and a second Milligard LPB Opticap XL-4 single-layer (0.2-μm) filter, for up to 10 liters of crude viral supernatant. Benzonase (50 U/ml supernatant) was added to the clarified supernatant to digest the nucleic acids. The digestion was carried out at 37°C for 4 hr. After digestion, the supernatant was held at 4°C overnight until purified by column chromatography.
Column chromatography
The Benzonase-treated supernatant was purified by anion-exchange chromatography, using a Poros HQ-50 packed column. Typically a 5 × 20 cm column was used to process 8–10 liters of culture medium at a linear flow rate of 90 cm/hr. The column was equilibrated with 150 mM NaCl in 20 mM Tris, pH 7.5. After loading, the column was washed with ∼4 column volumes of equilibration buffer until the absorbance returned to baseline. A second wash step was performed with ∼3 column volumes of 300 mM NaCl in 20 mM Tris, pH 7.5. Elution was performed with 1.2 M NaCl in 20 mM Tris, pH 7.5. UV absorbance at 260 nm was used to monitor and collect the elution peak, typically between 300 and 360 ml (Fig. 3).
FIG. 3.

Representative chromatogram of the rVSV GMP lot production. Each subbatch of culture supernatant was loaded onto the column and eluted with 1.2 M NaCl. The chromatogram shown is from one of the five subbatches and is representative of all five. Color images available online at www.liebertonline.com/hum.
Buffer exchange and concentration by ultrafiltration/diafiltration
The column eluate was diluted to 2 liters with exchange buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA; pH 7.8), concentrated by tangential flow filtration (TFF) to 400 ml, and then diafiltered with 2.4 liters of exchange buffer. The product was further concentrated from 400 to 100 ml, which is approximately a 90- to 100-fold concentration compared with the original culture supernatant.
Formulation and sterile filtration
The concentrated product was formulated to 10% glycerol and stored at −80°C until final filtration and pooling of concentrated subbatches. Each subbatch of the rVSV product was sterile filtered with a Steripak-GP10 filter unit (Millipore) that had been prewetted with 30 ml of the product formulation buffer. Filtered subbatches were pooled and subsequently aliquoted. A total of 196 vials were filled, 110 vials with a volume of 0.7 ml and 86 vials with a volume of 5.5 ml.
Production summary
A 50-Cell Factory clinical lot production of an oncolytic rVSV vector was successfully completed. The GMP manufacturing process consisted of five subbatches, with each subbatch being generated from 10 CFs. Each of the five subbatches was processed independently through concentration and formulation and then stored at −80°C until all five subbatches were filtered and pooled. The titration results and volumes of the in-process samples for each of the five subbatches are listed in Table 3. Final product and QC samples were also collected from the filtered, purified bulk. The titers of the purified bulk (after 0.2-μm filtration) and final product (0.7-ml and 5.5-ml vials) are listed in Table 4.
Table 3.
Virus Yield for Clinical Lot In-Process Samples
| Subbatch | Sample ID | Volume | Infectious titer (PFU/ml) | Total infectious units (PFU) |
|---|---|---|---|---|
| 1 | Clarified supernatant | 9 liters | 3.0 × 108 | 2.7 × 1012 |
| Post-Benzonase | 9 liters | 1.5 × 108 | 1.4 × 1012 | |
| Elution peak (1.2M) | 330 ml | 1.6 × 109 | 0.5 × 1012 | |
| Post-UF/DF | 116 ml | 2.4 × 1010 | 2.8 × 1012 | |
| 2 | Clarified supernatant | 9 liters | 3.3 × 108 | 2.7 × 1012 |
| Post-Benzonase | 9 liters | 1.5 × 108 | 1.4 × 1012 | |
| Elution peak (1.2M) | 306 ml | 4.7 × 109 | 1.4 × 1012 | |
| Post-UF/DF | 111 ml | 2.3 × 1010 | 2.6 × 1012 | |
| 3 | Clarified supernatant | 9 liters | 3.8 × 108 | 4.3 × 1012 |
| Post-Benzonase | 9 liters | 2.2 × 108 | 2.0 × 1012 | |
| Elution peak (1.2M) | 342 ml | 4.9 × 109 | 1.7 × 1012 | |
| Post-UF/DF | 117 ml | 2.8 × 1010 | 3.3 × 1012 | |
| 4 | Clarified supernatant | 9 liters | 2.0 × 108 | 1.8 × 1012 |
| Post-Benzonase | 9 liters | ND | ND | |
| Elution peak (1.2M) | 344 ml | ND | ND | |
| Post-UF/DF | 117 ml | 2.3 × 1010 | 2.6 × 1012 | |
| 5 | Clarified supernatant | 9 liters | ND | ND |
| Post-Benzonase | 9 liters | ND | ND | |
| Elution peak (1.2M) | 366 ml | ND | ND | |
| Post-UF/DF | 125 ml | 2.4 × 1010 | 2.7 × 1012 |
DF, diafiltration; ND, not determined; UF, ultrafiltration.
Table 4.
Virus Yield in Purified Bulk and Final Product
| Sample ID | Volume (ml) | Infectious titer (PFU/ml) | Total infectious units (PFU) |
|---|---|---|---|
| Purified bulk | 570 | 1.7 × 1010 | 1.0 × 1013 |
| Final product (0.7-ml vials) | 77 | 1.2 × 1010 | 9.2 × 1011 |
| Final product (5.5-ml vials) | 473 | 1.8 × 1010 | 8.5 × 1012 |
Lot release testing of the GMP lot
The assays performed on the GMP lot as well as the release test results are summarized in Table 5. To address the issue of breakthrough of rVSV vector in the infected indicator cell lines that had been observed during lot release testing of the rVSV MVSS, a parallel 293 cell culture was set up during the cell expansion and infection phase of rVSV production. This culture was used, in addition to the unprocessed bulk from the clinical lot, for the in vitro viral assay. An SDS-polyacrylamide gel of the clinical lot purified vector is shown (Fig. 4). The morphology of the rVSV present in the clinical lot was also examined by transmission electron microscopy (TEM) and shown to be the unique bullet shape expected for rVSV (Fig. 5).
Table 5.
Lot Release Assays and Specifications for the Recombinant Vesicular Stomatitis Virus Clinical Lot
| Assay | Specification | Results |
|---|---|---|
| In vitro viral assay (parallel culture) | Negative | Negative |
| In vitro viral assay (unprocessed bulk) | Negative | Negative |
| Mycoplasma (PTC) | Negative | Negative |
| Residual host DNA | Report result | 64.03 fg/μl |
| Size distribution of residual DNA | <500 bp | Pass |
| Residual Benzonase by ELISA | <100 ng/ml | <1 ng/ml |
| Vector ID (RT-PCR) | Match expected size | Pass |
| LAL—endotoxin | <10 EU/ml | Pass |
| Viral particle counts by TEM | Report result | 1.6 × 1011 particles/ml |
| Sterility assay (USP) | Negative | Negative |
| B&F—Qualification of sterility testing | Pass | Pass |
| Infectious titer | Report result | 1.7 × 1010 PFU/ml |
B&F, Bacteriostasis and Fungistasis; LAL, Limulus amebocyte lysate; PTC, Points to Consider; TEM, transmission electron microscopy; USP, U.S. Pharmacopoeia.
FIG. 4.
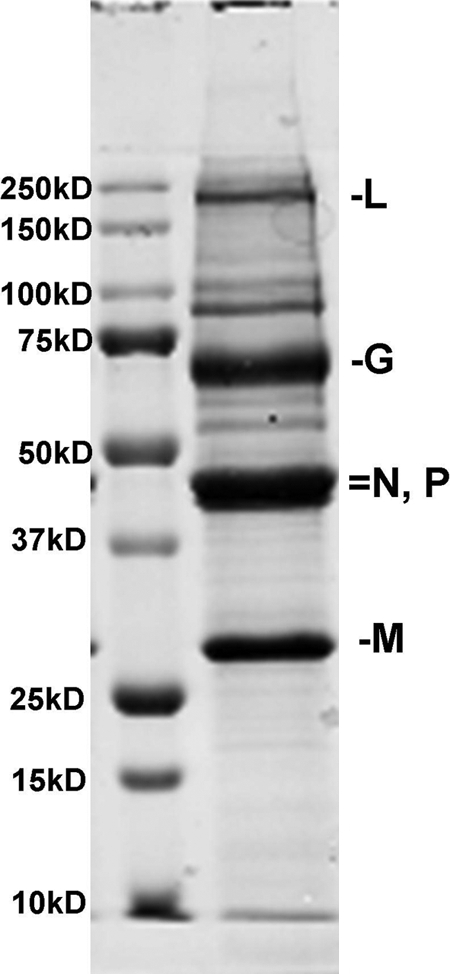
A sodium dodecyl sulfate–polyacrylamide gel of the rVSV(MD51)-M3 clinical lot. Major bands corresponding to the five VSV proteins (L, G, N, P, and M) were present at the expected sizes.
FIG. 5.

Electron micrograph of rVSV particles. An aliquot of the clinical lot was submitted to a contract facility for examination and quantification of viral particles by transmission electron microscopy. A representative image shows the unique bullet shape associated with VSV. The arrowhead indicates a sample rod-shaped structure. Scale bar: 100 nm.
Discussion
A robust and readily scalable manufacturing process has been developed to produce rVSV vector suitable for clinical studies. The crude vector supernatant is generated by infecting adherent 293 cells at a low MOI, then harvesting the excreted rVSV vector when extensive CPE is formed. The purification process involves five steps: (1) a clarification step to remove cellular debris; (2) a nuclease treatment step to degrade host cell DNA; (3) an anion-exchange chromatographic step for purification of the vector; (4) a tangential flow ultrafiltration/diafiltration step for concentration and buffer exchange; and (5) a terminal sterile filtration step. Five independent subbatches representing 10 liters of supernatant each were produced under cGMP conditions with consistent virus yields ranging between 1.8 × 1012 and 2.8 × 1012 PFU per 10-liter subbatch. Although the diafiltration/ultrafiltration and column chromatography steps are easily scalable using existing technology, in order to scale up the infection process for licensed product manufacturing, it will be necessary to investigate the use of bioreactor technology using adherent cells on microbeads or suspension cell cultures to support the production of rVSV at high titers. Ideally, cell expansion would occur in serum-free medium to eliminate the need for a complete media exchange at the time of infection and disposable single-use stirred tank bioreactors which are currently available in the 40–2000 liter range for use in GMP production would be ideal for virus production purposes.
Although manufacturing of the rVSV vector at the current scale was robust and did not present any significant technical challenges, characterization of the rVSV vector, both the MVSS and the clinical lot, did present some difficulties due to the lack of an effective neutralizing antibody. This may prove to be a common issue with replication competent virus products. To test the clinical product for adventitious viruses and inapparent viruses that may be present in the vector lot, the viral vector must be neutralized in order to allow detection of any contaminating viruses in the in vitro and in vivo assays It should also be noted that complete inhibition of the infectivity of the viral vector with existing antibodies might not always be possible. A strategy for the release testing of the viral product should be formulated by assessing the available antibodies for their ability to neutralize virus, identifying indicator cell lines and animal models that may be refractory to viral vector infection, and determining the fold of dilution of the viral vector that will be required to avoid breakthrough or toxicity. If no neutralizing serum is available, effective antiserum may be prepared by immunizing rabbits or other suitable animals with the virus of interest. In addition, the effectiveness of the antiserum may be enhanced by evaluating several indicator cell lines for their sensitivity to the virus infection such that cell lines that are more refractive to the virus infection are selected for the in vitro viral assay.
Because an effective neutralizing antibody is a critical reagent required for release testing and product characterization of a viral product, it is important that it not be overlooked during the process development stage. It is therefore prudent that an effective antiserum to neutralize the viral infectivity or cytotoxicity be identified early during process development and a sufficient quantity of the antiserum be secured. This will avoid unnecessary lengthy delays in the release testing of the product once production is completed. It is also extremely important to keep the regulatory personnel at the Food and Drug Administration (FDA) informed regarding any difficulties encountered during lot release testing and to seek their input and guidance at all key steps in the clinical reagent development process.
Before using this material in phase 1 clinical trials, it will be necessary to submit an Investigational New Drug application to the FDA that contains all of the information regarding production and testing of the clinical grade rVSV, as well as preclinical efficacy, biodistribution, and pharmacological/toxicological testing in laboratory animals, and the draft clinical protocol. This information will also be provided to the Office of Biotechnology Activities at the National Institutes of Health. Pending FDA review, this protocol will be submitted to the Mount Sinai Institutional Review Board and Institutional Biosafety Committee for their review and approval. No patient will be enrolled in the clinical trial until all regulatory approvals are obtained.
Acknowledgments
The authors thank the reviewers at the Food and Drug Administration for critical guidance in devising the plan for lot release testing of the MVSS and clinical lot of rVSV(MΔ51)-M3. This work was supported in part by NIH grant 1R01CA120372.
Author Disclosure Statement
Dr. Savio Woo is a named inventor of a patent application titled “Transgenic Oncolytic Viruses and Uses Thereof.” The pending patent was filed by the Mount Sinai School of Medicine (MSSM). In the event the pending patent is licensed, Dr. Woo would be entitled to a share of any proceeds MSSM receives from the license. The outcome of this research project could affect the ability to license this patent.
References
- Alemany R. Balagué C. Curiel D.T. Replicative adenoviruses for cancer therapy. Nat. Biotechnol. 2000;18:723–727. doi: 10.1038/77283. [DOI] [PubMed] [Google Scholar]
- Barker D.D. Berk A.J. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology. 1987;156:107–121. doi: 10.1016/0042-6822(87)90441-7. [DOI] [PubMed] [Google Scholar]
- Bischoff J.R. Kirn D.H. Williams A., et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- Fields B.N. Hawkins K. Human infection with virus of vesicular stomatitis during an epizootic. N. Engl. J. Med. 1967;277:989–994. doi: 10.1056/NEJM196711092771901. [DOI] [PubMed] [Google Scholar]
- Fueyo J. Gomez-Manzano C. Alemany R., et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- Gainey M.D. Manuse M.J. Parks G.D. A hyperfusogenic F protein enhances the oncolytic potency of a paramyxovirus simian virus 5 P/V mutant without compromising sensitivity to type I interferon. J. Virol. 2008;82:9369–9380. doi: 10.1128/JVI.01054-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganly I. Kirn D. Eckhardt G., et al. A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin. Cancer Res. 2000;6:798–806. [PubMed] [Google Scholar]
- Giedlin M.A. Cook D.N. Dubensky T.W., Jr. Vesicular stomatitis virus: An exciting new therapeutic oncolytic virus candidate for cancer or just another chapter from Fields Virology? Cancer Cell. 2003;4:241–243. doi: 10.1016/s1535-6108(03)00251-4. [DOI] [PubMed] [Google Scholar]
- Graham F.L. Smiley J. Russell W.C., et al. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Hallenbeck P.L. Chang Y.N. Hay C., et al. A novel tumor-specific replication-restricted adenoviral vector for gene therapy of hepatocellular carcinoma. Hum. Gene Ther. 1999;10:1721–1733. doi: 10.1089/10430349950017725. [DOI] [PubMed] [Google Scholar]
- Hardcastle J. Kurozumi K. Chiocca E.A., et al. Oncolytic viruses driven by tumor-specific promoters. Curr. Cancer Drug Targets. 2007;7:181–189. doi: 10.2174/156800907780058880. [DOI] [PubMed] [Google Scholar]
- Heise C. Sampson-Johannes A. Williams A., et al. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat. Med. 1997;3:639–645. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- Kasuya H. Takeda S. Nomoto S., et al. The potential of oncolytic virus therapy for pancreatic cancer. Cancer Gene Ther. 2005;12:725–736. doi: 10.1038/sj.cgt.7700830. [DOI] [PubMed] [Google Scholar]
- Khuri F.R. Nemunaitis J. Ganly I., et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat. Med. 2000;6:879–885. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- Kirn D. Martuza R.L. Zwiebel J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat. Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- Kuhn I. Harden P. Bauzon M., et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS One. 2008;3:e2409. doi: 10.1371/journal.pone.0002409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letchworth G.J. Rodriguez L.L. Del Cbarrera J. Vesicular stomatitis. Vet. J. 1999;157:239–260. doi: 10.1053/tvjl.1998.0303. [DOI] [PubMed] [Google Scholar]
- Martuza R.L. Malick A. Markert J.M., et al. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–856. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- Mineta T. Markert J.M. Takamiya Y., et al. CNS tumor therapy by attenuated herpes simplex viruses. Gene Ther. 1994;1(Suppl. 1):S78. [PubMed] [Google Scholar]
- Mineta T. Rabkin S.D. Yazaki T., et al. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- Miyatake S.I. Tani S. Feigenbaum F., et al. Hepatoma-specific antitumor activity of an albumin enhancer/promoter regulated herpes simplex virus in vivo. Gene Ther. 1999;6:564–572. doi: 10.1038/sj.gt.3300861. [DOI] [PubMed] [Google Scholar]
- Nemunaitis J. Ganly I. Khuri F., et al. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: a phase II trial. Cancer Res. 2000;60:6359–6366. [PubMed] [Google Scholar]
- Parry C.M. Simas J.P. Smith V.P., et al. A broad spectrum secreted chemokine binding protein encoded by a herpesvirus. J. Exp. Med. 2000;191:573–578. doi: 10.1084/jem.191.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz E. Moreno N. Peralta P.H., et al. A human case of encephalitis associated with vesicular stomatitis virus (Indiana serotype) infection. Am. J. Trop. Med. Hyg. 1988;39:312–314. doi: 10.4269/ajtmh.1988.39.312. [DOI] [PubMed] [Google Scholar]
- Rainov N.G. Ren H. Oncolytic viruses for treatment of malignant brain tumours. Acta Neurochir. Suppl. 2003;88:113–123. doi: 10.1007/978-3-7091-6090-9_17. [DOI] [PubMed] [Google Scholar]
- Raykov Z. Grekova S. Leuchs B., et al. Arming parvoviruses with CpG motifs to improve their oncosuppressive capacity. Int. J. Cancer. 2008;122:2880–2884. doi: 10.1002/ijc.23472. [DOI] [PubMed] [Google Scholar]
- Rose J.A. Whitt M.A. Knipe H.P. Howley P.M. Fields Virology. Philadelphia, Lippincott: Williams & Wilkins; 2001. Rhabdoviridae: The viruses and their replication; pp. 1221–1242. [Google Scholar]
- Shinozaki K. Ebert O. Woo S.L. Eradication of advanced hepatocellular carcinoma in rats via repeated hepatic arterial infusions of recombinant VSV. Hepatology. 2005;41:196–203. doi: 10.1002/hep.20536. [DOI] [PubMed] [Google Scholar]
- Singleton D.C. Li D. Bai S.Y., et al. The nitroreductase prodrug SN 28343 enhances the potency of systemically administered armed oncolytic adenovirus ONYX-411NTR nitroreductase-armed oncolytic adenovirus. Cancer Gene Ther. 2007;14:953–967. doi: 10.1038/sj.cgt.7701088. [DOI] [PubMed] [Google Scholar]
- Stojdl D.F. Lichty B. Knowles S., et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000a;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- Stojdl D.F. Abraham N. Knowles S., et al. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol. 2000b;74:9580–9585. doi: 10.1128/jvi.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudia W.D. Fields B.N., et al. Isolation of vesicular stomatitis virus (Indiana strain) and other viruses from mosquitoes in New Mexico 1965. Am. J. Epidemiol. 1967;86:598–602. doi: 10.1093/oxfordjournals.aje.a120769. [DOI] [PubMed] [Google Scholar]
- Thorne S.H. Hwang T.H. O'Gorman W.E., et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J. Clin. Invest. 2007;117:3350–3358. doi: 10.1172/JCI32727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkel V. Barrett J. Tiffany H.L., et al. Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J. Virol. 2000;74:6741–6747. doi: 10.1128/jvi.74.15.6741-6747.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley R.J. Kern E.R. Chatterjee S., et al. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J. Clin. Invest. 1993;91:2837–2843. doi: 10.1172/JCI116527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham T.J. Tzeng E., et al. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. J. Virol. 1997;71:8221–8229. doi: 10.1128/jvi.71.11.8221-8229.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L. Huang T.G. Meseck M., et al. rVSV(MΔ51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum. Gene Ther. 2008;19:635–647. doi: 10.1089/hum.2007.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M. Niu J.H. Chang Y., et al. A novel triple-regulated oncolytic adenovirus carrying PDCD5 gene exerts potent antitumor efficacy on common human leukemic cell lines. Apoptosis. 2009;14:1086–1094. doi: 10.1007/s10495-009-0373-3. [DOI] [PubMed] [Google Scholar]
- Ziauddin M.F. Guo Z.S. O'Malley M.E., et al. TRAIL gene-armed oncolytic poxvirus and oxaliplatin can work synergistically against colorectal cancer. Gene Ther. 2010;17:550–559. doi: 10.1038/gt.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]