

Abstract
Chlamydia trachomatis serovars D-K are sexually-transmitted intracellular bacterial pathogens that replicate in epithelial cells lining the human reproductive tract. It is clear from knockout mice and T cell depletion studies utilizing Chlamydia muridarum that MHC class II and CD4 T cells are critical for clearing bacteria from the murine genital tract. It is not clear how CD4 T cells interact with infected epithelial cells to mediate bacterial clearance in vivo. Previous work using an epithelial tumor cell line showed that a Chlamydia-specific CD4 T cell clone was able to inhibit C. muridarum replication in vitro via induction of epithelial nitric oxide production. We have previously shown that Chlamydia-specific CD4 T cell clones can recognize and be-activated-by infected reproductive tract epithelial cells, and block Chlamydia replication in them. We extend those observations by investigating the mechanism used by a panel of CD4 T cell clones to control Chlamydia replication in epithelial cells. We found that Chlamydia-specific CD4 T cell clones were cytolytic, but that cytolysis was not likely critical for controlling C. muridarum replication. For one CD4 T cell clone induced epithelial nitric oxide production was critical for controlling replication; however, the most potent CD4 T cell clones were dependent on T cell degranulation for replication control with only a minor additional contribution from nitric oxide production. We discuss our data as it relates to existing knockout mouse studies addressing mechanisms of T cell-mediated control of Chlamydia replication, and their implications for intracellular epithelial pathogens in mouse models.
Keywords: Chlamydia, CD4, perforin, epithelial cells, replication, nitric oxide
Introduction
Chlamydia trachomatis serovars D-K are intracellular bacterial pathogens that cause a common sexually transmitted urethritis/cervicitis throughout the world. In the United States 1,210,523 cases were reported to CDC in 2008; the largest number of cases ever reported and an increase of 9.2% from the previous year (1). A similar upward trend in C. trachomatis infections has been previously documented in Canada and thought to reflect an unintended negative consequence of treatment and control efforts on herd immunity (2). In women, untreated Chlamydia infections ascend into the Fallopian tubes causing tubal scarring that leads to chronic pelvic pain, tubal pregnancies and infertility (3).
T cell depletion and knockout mouse studies have clearly shown that MHC class II and CD4 T cells are necessary for primary clearance of Chlamydia muridarum from the murine genital tract (4, 5). C. muridarum is closely related to C. trachomatis serovar D, including gene-for-gene synteny excepting a small region known as the plasticity zone that is associated with species-specific evasion of IFN-γ-induced innate immunity (6, 7). Because rodent and human Chlamydia strains have evolved to evade IFN-γ-induced innate immune defenses in their natural host (8, 9) it is likely that analogous to mice, human clearance of Chlamydia infections requires CD4 T cell-mediated immunity. Because the components of adaptive cellular immunity are highly conserved between mice and humans, it is also likely that mice and humans utilize similar T cell effector mechanisms to clear Chlamydia from the genital tract. While the broad identity of the relevant effector T cell population, CD4 T cells, has been determined using the murine model, the mechanism used by CD4 T cells to clear Chlamydia from the reproductive tract is unknown (10).
CD4 T cell lines protective in adoptive transfer studies have also been shown to control C. muridarum replication in polarized epithelial monolayers (11). The mechanism of control was dependent on IFN-γ and physical interaction between T cells and infected epithelial cells via LFA-1 – ICAM-1. In the presence of IFN-γ, T cell engagement of epithelial cells via LFA-1 was shown to augment epithelial nitric oxide production above that induced by IFN-γ alone, and nitric oxide was shown to be the effector molecule responsible for controlling Chlamydia replication (12). These data identified T cell-induced nitric oxide production as the probable mechanism for clearing Chlamydia from the genital tract. However subsequent studies showed no difference in Chlamydia genital tract clearance kinetics between iNOS-deficient mice and wild type mice (13, 14), though there are important differences in immunopathology (15). In addition, mice deficient in LFA-1, perforin, Fas, FasL, perforin & FasL, p47phox, and TNF receptors all resolve C. muridarum genital tract infections with normal or near normal kinetics, though there are important differences in intensity of shedding (TNF receptor knockout) (reviewed in (4)). Furthermore, mice deficient in IFN-γ clear 99.9% of C. muridarum from the genital tract with near normal kinetics (16, 17). These knockout mouse data argue against direct T cell-mediated killing via perforin and Fas-FasL making a major contribution to bacterial clearance, and argue against indirect mechanisms for CD4 T cell-mediated clearance via IFN-γ/LFA-1 induction of epithelial iNOS (nitric oxide) and IFN-γ/TNF-α induction of epithelial NAPDH oxidase (ROS) defense mechanisms, begging the question of how mice clear Chlamydia genital tract infections.
C. trachomatis replicates predominantly in the reproductive tract epithelium during natural human infections (18, 19) and experimental murine C. muridarum infections in wild type mice (4). Because we have previously shown that Chlamydia-specific CD4 T cell clones can recognize C. muridarum antigens in the context of epithelial MHC class II molecules, and block Chlamydia replication in epithelial cells, it is reasonable to propose that bacterial clearance from the genital tract involves the physical interaction of Chlamydia-specific CD4 T cells with infected epithelial cells. For this report we used our Chlamydia-specific CD4 T cell clones and an upper reproductive tract epithelial cell line to investigate how CD4 T cells control Chlamydia replication in infected epithelial cells. We present the interesting results of those studies here.
Materials and Methods
Mice
4–5 week old female C57BL/6 mice were purchased from Harlan Laboratories (Indianapolis, IN). All mice were housed in Indiana University Purdue University-Indianapolis (IUPUI) specific-pathogen-free facilities (SPF). The IUPUI Institutional Animal Care and Utilization Committee approved all experimental protocols.
Cells, T cell clones and bacteria
Bm1.11 epithelial cells (H-2Kbm1) have been previously (20); C57epi.1 epithelial cells and Chlamydia-specific CD4 T cell clones spl4-10, uvmo-1, uvmo-2 and uvmo-3 were derived from C57BL/6 mice (H-2Kb) and cultured as previously described (21). Mycoplasma-free Chlamydia muridarum (Nigg), previously known as C. trachomatis strain mouse pneumonitis (MoPn) (Nigg) was grown in McCoy cells as previously described (20).
Alloreactive CD8 T cell clone specific for H-2Kbm1 was derived from a C57BL/6 (H-2b) mouse primed with full thickness dorsal tail skin graft from B6.C-H2bm1/ByJ using a protocol previously described (22). The 3 × 6 mm skin grafts were rejected within 2 weeks and the mice were rested for three months. Spleens containing memory T cells were harvested from rested mice, and 10×106 immune-splenocytes plated on Bm1.11 epithelial monolayers (12 well plates) pretreated for 12 h with 1 ml/well cytokine mixture containing murine recombinant IFN-α4 (750 units/ml), IFN-β (750 units/ml), TNF-α (750 ρg/ml), IL-1α (1.5 ηg/ml), IL-6 (1.5 ηg/ml), IL-7 (3 ηg/ml), IL-15 (12 ηg/ml), and human recombinant IL-2 (30 units/ml) overnight. This cytokine milieu reflects that produced by C. muridarum-infected oviduct epithelial cells (20) and bone marrow-derived dendritic cells pulsed with heat-inactivated C. muridarum (23). Final cytokine concentrations in primary wells were 0.3333 that of the overnight pretreatment values; final well volume = 3cc. The resulting polyclonal T cell populations were serially passed and limiting diluted to obtain T cell clones. One T cell clone specific for Bm1.11 epithelial cells, designated CD8bm1, was kept for further experimentation. CD8bm1 is a CD4negativeCD8+ T cell clone with an αβ T cell receptor (TCR) (data not shown).
Epithelial cell infections/Chlamydia replication/titration
C57epi.1 cell monolayers in 48 well plates were untreated or treated with IFN-γ (10 ηg/ml) for 14 h prior to infection. Wells were infected with 3 IFU per cell. After addition of C. muridarum the plates were spun at 1200 rpm (300 × g) for 30 minutes. Mock-infected wells received an equivalent volume of sucrose-phosphate-glutamate acid buffer (SPG buffer) lacking C. muridarum. 4 h post infection the inoculums were removed and 1.5×105 CD4 T cell clone cells were added per well. 32 h later, 36 h post infection, wells were scraped, harvested and stored at −80 C until C. muridarum titers could be determined on McCoy cell monolayers using anti-Chlamydia LPS antibody and FITC-labeled goat anti-mouse IgG (Rockland Immunochemicals, Gilbertsville, PA) as previously described (20). For inhibitor studies, T cells were pretreated for 1 h with 50 nM concanamycin A (CMA) (Sigma, St Louis, MS) or 200 nM phenyl arsine oxide (PAO) (Sigma, St Louis, MS); the inhibitors were added to the epithelial monolayers with the T cells. iNos inhibitors L-N6-(1-Iminoethyl)lysine hydrochloride (NIL) (100 nM) and N-Monomethyal-L-arginine (MLA) (1 mM) (Sigma, St Louis, MO) were added during IFN-γ pretreatment and with subsequent addition of T cell clones to epithelial monolayers. Dose finding studies were done with all inhibitors to identify concentrations that did not cause visible cytopathology to C57epi.1 monolayers (data not shown).
Redirected lysis
Redirected lysis was performed as described by Leo et al. (24). 10,000 P815 cells (ATCC TIB-64), a mastocytoma cell line expressing Fc receptors, were incubated with 10,000 CD4 T cells in the presence of 0.5 μg/ml anti-CD3e (clone145-2c11, NA/LE, BD biosciences, San Jose, CA) in 96 well v-bottom plates spun 1 minute at 300 × g then incubated for 4 h. Killing was quantified using a non-radioactive cytotoxicity assay measuring release of lactate dehydrogenase (LDH) activity in culture supernatant (cyto 96; Promega, Madison, WI) following the manufacturer's protocol. The lysis assays were done using 5% heat-inactivated serum (65 °C for 1 h to inactivate LDH activity present in fetal bovine serum) and phenol red-free RPMI medium. CD4 T cell clones were incubated with inhibitors (50 nM CMA or 200 nM PAO) for 1 h before the assay. In separate experiments evaluating inhibitor stability, the inhibitors were incubated in media at 37°C for 18 h prior to using them in standard 4 h redirected lysis assays.
Microscopy
C57epi.1 cells grown in 8 well chamber slides (Lab-Tek II; Thermo Fisher Scientific,Waltham, MA) were treated with IFN-γ for 14 h then infected with 5 IFU per cell without centrifugation. 18 h post-infection, 1×105 purified (histopaque 1083; Sigma) CD4 T cells were added to the C57epi.1 monolayers in 10 μg/ml tetracycline in presence of inhibitors; either 200 nM PAO, 50 nM of CMA, or 10 μg/ml of antibody specific for CD4 (GK1.5, eBiosciences), Fas (Jo2, NA/LE; BD-biosciences) and FasL (MLF3, functional grade; eBiosciences). T cell clones were incubated with PAO and CMA for 1 h before addition to epithelial cells. After 24 h of co-culture, wells were fixed with 1% paraformaldehyde for 20 minutes and stained with Cyto-quik (Fisher Scientific, Pittsburgh, PA). Photos of representative fields were taken with a Nikan diaphot 200 inverted microscope equipped with a Diagnostic Instruments spot color camera at 20× magnification.
Nitric oxide measurement
C57epi.1 epithelial monolayers in 48 well plates were untreated, or pretreated with 10 ηg/ml IFN-γ for 14 h in the absence or presence of 1 mM MLA or 100 nM NIL, then infected with 3 IFU per cell C. muridarum. Four hours later the inocula were removed and monolayers co-cultured with media or 1.5×105 T cell clone cells, in the presence of iNOS inhibitors where indicated. The T cell to epithelial cell ratio under these conditions was ~1:1. 32 h later, 36 h post infection, culture supernatants were collected and nitric oxide quantified (measured as nitrite) using a commercial Greiss reagent system (Promega, Madison, WI) according to manufacturer's protocol (25).
Statistical Analysis
Summary figures for each experimental investigation are presented as mean and standard deviation (SD) or `pooled' means and with their associated standard error of the mean (SEM). Figure legends indicate the number of independent experiments pooled to generate each figure. Student's two-tailed t test was used to assess significance of experimental data. pvalues < 0.05 were considered statistically significant. For figure 3 Sidak methodology was used to adjust pvalues for multiple comparisons.
Figure 3.
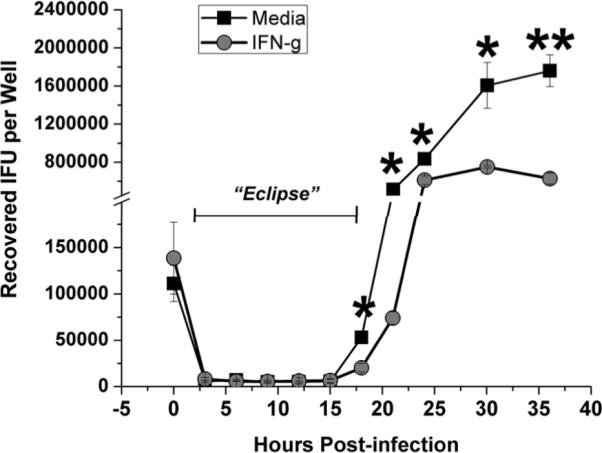
C. muridarum replication time course in C57epi.1 cells. C57epi.1 cells, untreated (squares) or pretreated with 10 ηg per ml IFN-γ for 14 h (circles), were infected with ~1 IFU per cell. Wells were harvested at indicated time points and C. muridarum quantified on McCoy monolayers; mean and SD from one experiment. Note break in Y axis. * = adjusted pvalue <0.05; ** = adjusted pvalue < 0.005.
Results
IFN-γ-independent CD4 T cell clones control C. muridarum replication without nitric oxide production
Igietseme et al previously showed that a Chlamydia-specific CD4 T cell clone designated 2–14.0 controlled C. muridarum replication in an LFA-1- and IFN-γ-dependent fashion through induction of epithelial nitric oxide (12). Blocking nitric oxide production with N-Monomethyl-L-Arginine (MLA) completely negated 2–14.0's ability to control C. muridarum replication in epithelial cells in vitro (26). We have previously shown that the CD4 T cell clones designated uvmo-1, -2, -3 recognized C. muridarum antigens in the context of epithelial MHC class II molecules, and that earliest recognition/activation occurred ~12 h post infection, with maximal T cell activation at 18–21 h post-infection. These three CD4 T cell clones were able to control replication without IFN-γ pretreatment of syngeneic epithelial monolayers. We investigated whether nitric oxide production was critical for CD4 T cell clones uvmo-1, -2, -3 to control C. muridarum replication in epithelial cells in vitro.
C57epi.1 oviduct epithelial cell monolayers, untreated or pretreated with IFN-γ in the absence or presence of nitric oxide synthetase inhibitors MLA or NIL for 14 h, were infected with C. muridarum, and then co-cultured with media or CD4 T cell clones uvmo-1,-2,-3. Supernatants were collected at 36 h post infection, 32 h post T cell co-culture, and analyzed for nitric oxide production measured as nitrite (Figure 1a). IFN-γ significantly induced nitric oxide production in C57.1 epithelial cells, roughly 6-fold with a pvalue < 0.0001. MLA and NIL were effective inhibitors of nitric oxide production as MLA blocked > 90% of IFN-γ-induced nitric oxide; NIL blocked ~60%. Levels of nitric oxide production in the presence of MLA, without or with CD4 T cell co-culture, were not statistically different from nitric oxide produced by infected epithelial cells in media without IFN-γ pretreatment. Uninfected C57epi.1 cells grown in regular culture media for 36 h had basal nitric oxide production of < 1.5 uM. CD4 T cell clones uvmo-1 & uvmo-2 were able to boost nitric oxide produced by infected C57epi.1 epithelial cells that had not been pretreated with IFN-γ. Co-culture of the CD4 T cell clones with IFN-γ-pretreated infected epithelial cells did not boost nitric oxide production above levels seen with IFN-γ pretreatment alone. As seen previously, IFN-γ pretreatment had a modest negative effect on C. muridarum replication in C57epi.1 oviduct epithelial cells (~1.5–2 fold). The modest inhibitory effect of IFN-γ was reversed by blocking nitric oxide production with either MLA or NIL (Fig 1b).
Figure 1.

Infected epithelial cell production of nitric oxide in the absence and presence of CD4 T cell clones; C. muridarum replication in C57epi.1 epithelial cells in the absence and presence of iNOS inhibitors. A) C57epi.1 epithelial cells, untreated (hatched bars) or pretreated with IFN-γ (10 η/ml × 14 h) in the absence (black bars) or presence of iNOS inhibitors MLA (gray bars) or NIL (light gray bars), were infected with C. muridarum (3 IFU per cell). 4 h later the inocula were removed and monolayers co-cultured without and with CD4 T cell clones uvmo-1,-2,-3 for an additional 32 h. Culture supernatants were collected 36 h post infection, 32 h post co-culture, and nitric oxide quantified as nitrite; aggregate means and SEM from two independent experiments. Asterisks indicate comparisons with IFN-γ treatment (black bars) for each of the four conditions (No T cells, uvmo-1,-2,-3). Daggers indicate comparisons with the “no T cell” infected control (hatched bar). ** or†† = pvalue <0.005; ††† or = pvalue <0.0005. B) Effects of iNOS inhibitors on C. muridarum replication in infected C57epi.1 epithelial cells. C57epi.1 cells were untreated (hatched bar) or pretreated with IFN-γ (10 ηg/ml × 14 h) in absence (black bar) or presence of MLA (gray bar) or NIL (light gray bar), and then infected with C. muridarum (3 IFU per cell). Wells were harvested 36 h post infection and C. muridarum quantified on McCoy monolayers. Comparisons were made to the infected control (hatched bar). Aggregate means and SEM from two independent experiments. ††† = pvalue <0.0005; NS = not statistically significant.
Having demonstrated that MLA and NIL were effective inhibitors of nitric oxide production by C57epi.1 epithelial cells in absence or presence of CD4 T cell clones, we tested whether nitric oxide was critical for CD4 T cell clones uvmo-1,-2,-3s ability to control C. muridarum replication in epithelial cells. Darville et al have previously shown detectable IFN-γ levels in genital secretions as soon as 2 days post infection that increase to ~4000 ρg/ml by day 3 and remain elevated at ≥ 1000 ρg/ml through day 10, the last day tested (27). Based on that data it is reasonable to postulate that CD4 T cell interactions with infected epithelial cells occur in a microenvironment that includes IFN-γ. C57epi.1 oviduct epithelial cell monolayers, pretreated with IFN-γ in the absence or presence of nitric oxide synthetase inhibitors MLA & NIL, were infected with C. muridarum. Four hours later the inocula were removed and T cells added at a T cell-to-epithelial cell ratio of 0.75:1 in medium (control), or medium containing nitric oxide synthetase inhibitors MLA or NIL. Thirty two hours later the wells were harvested by scraping, and the C. muridarum yield quantified by titration on McCoy monolayers (Figure 2). Blocking nitric oxide production with MLA (and to a lesser extent NIL) had very modest negative effects on the abilities of uvmo-1 [98% inhibition dropped to 95% → raw IFU data: No T cell control = 3.4×106 IFU recovered per well, + uvmo-1 = 6.4×104 IFU/well, MLA + uvmo-1 = 1.7×105 IFU/well] and uvmo-3 (92% inhibition dropped to 89%), and no effect on the ability of uvmo-2 to control C. muridarum replication in vitro.
Figure 2.
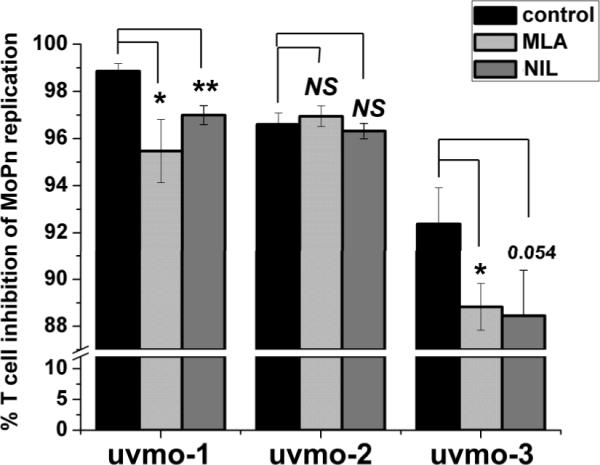
IFN-γ-independent CD4 T cell clones control C. muridarum replication in epithelial cells by an iNOS-independent mechanism. C57epi.1 epithelial cells were pretreated for 14 h with IFN-γ in the absence (black bars) or presence of iNOS inhibitors MLA (light gray bars) or NIL (gray bars), then infected with 3 IFU per cell C. muridarum. Inocula were removed 4 h later and 1.5×105 T cell clone cells added in the absence or presence of MLA or NIL; wells were harvested 36 h post infection and C. muridarum quantified on McCoy monolayers. Aggregate means and SEM from three independent experiments. Comparisons were made to the no inhibitor control (black bar) for each of the three T cell clones. * = pvalue < 0.05; ** = pvalue < 0.005; NS = not statistically significant. Note break in the Y-axis scale necessary to visualize effect.
Role of cytolysis in controlling replication
Replicating Chlamydia species are theoretically vulnerable to T cell-mediated cytolysis during the early stages of infection after infectious elementary bodies (EB) transition to non-infectious replicating reticulate bodies (RB), analogous to the “eclipse phase” in viral replication exploited by CTL to mediate viral clearance (28). Lysis of infected cells during the Chlamydia eclipse phase of replication would release non-infectious reticulate bodies and terminate replication. In figure 3 we show the replication kinetics of C. muridarum in C57epi.1 cells without centrifugation; the protocol used in an upcoming section for investigating the role of CD4 T cell cytolysis in blocking Chlamydia replication. C57epi.1 monolayers, untreated or pretreated with IFN-γ, were infected, then sequentially harvested at 0, 3, 6, 9, 12, 15, 18, 21, 24, 32 and 36 h and IFU quantified on McCoy cell monolayers. In C57epi.1 cells, C. muridarum enters a non-infectious RB replication phase spanning ~3–18 hours post-infection. From ~18 h forward, infectious elementary bodies accumulate with maximal yield at roughly 30 h post infection. Pretreatment with IFN-γ for 14 h caused a modest reduction (~2 fold) of C. muridarum replication in C57epi.1 cells.
The kinetics of C. muridarum replication in C57epi.1 epithelial cells defines a window, roughly 3–18 hours post infection, during which Chlamydia-specific T cells could terminate replication by lysing infected epithelial cells. Our previous work showed that under optimal conditions (pre-treatment of the epithelial monolayer with IFN-γ prior to infection) the earliest time point for recognition by our CD4 clones was ~12 h post infection, and that the maximal recognition/activation time point was 18–21 h post infection. To investigate the role of cytolysis in controlling C. muridarum replication, we first investigated the cytolytic potential of our three Chlamydia-specific CD4 T cell clones, comparing them to a cytolytic alloreactive CD8 T cell clone specific for H-2Kbm1 (designated CD8bm1; see materials and methods).
Redirected lysis assays were performed utilizing an Fc-receptor bearing target cell (P815) and an activating anti-CD3 monoclonal antibody 145-2C11. T cell – target conjugate formation occurs via the anti-CD3 antibodies simultaneously binding to P815 Fc receptors and the T cell receptor CD3 complex. Binding of the CD3 T cell receptor complex triggers T cell-mediated cytolysis. This assay has the advantage of providing a uniform activation signal to the T cells that is independent of their antigen specificity or co-receptor (CD4 or CD8). We also used the redirected lysis assays to determine the potency and stability of the perforin inhibitor concanamycin A (CMA) and the degranulation inhibitor phenylarsine oxide (PAO), used in upcoming experiments.
The perforin-mediated short term cytolytic potential of our Chlamydia-specific CD4 T cell clones was similar to that of an alloreactive CD8 T cell clone CD8bm1. % specific cytolysis at an effector-to-target ratio of 3:1 in a 4 h assay is shown in parenthesis for each clone (Figure 4). Because investigating T cell mechanisms for controlling Chlamydia replication required a 24 h in vitro assay (next section), we did standard 4 h assays with each inhibitor, and also tested the stability of CMA and PAO in culture media overnight (labeled as 24 hour). In standard 4 h killing assays the perforin inhibitor CMA significantly blocked cytolysis by Chlamydia-specific CD4 T cell clones uvmo-2, uvmo-3, and alloreactive clone CD8bm1, with minimal effects on uvmo-1 (Figure 4 light gray bars); in 4 h assays the degranulation inhibitor PAO significantly blocked cytolysis by all clones (Figure 4 black bars). To assess stability of the inhibitors in 24 h assays, working concentrations of the inhibitors were incubated in medium at 37° C for 18 h then used in 4 h redirected lysis assays (18 h incubation + 2 h assay setup + 4 h assay = 24 h) (Figure 4). CMA was stable in medium, retaining its ability to partially inhibit the T cell clones after 18 h in medium at 37°C relative to fresh CMA in the 4 h assays. Conversely PAO lost nearly all its inhibitory activity after 18 h in culture media at 37° C. The data in figure 4 show that CMA is a partial inhibitor based on intrinsic activity; PAO is a potent inhibitor in short term assays but was unstable in culture medium over the course of longer assays. Based on these results CMA and PAO are useful experimental tools for investigating cytolysis, but should be viewed as partial inhibitors in the upcoming 24 h assays used to study epithelial cytolysis and Chlamydia replication.
Figure 4.

Evaluation of the cytolytic potential of Chlamydia-specific CD4 T cell clones using standard 4 h redirected lysis assays, and 24 h inhibitor stability assays in which inhibitors were pre-incubated in media for 18 h at 37°C prior to use with T cell clones in 4 h assays: A) uvmo-1, B) uvmo-2, C) uvmo-3, D) CD8bm1 (H-2Kbm1 alloantigen-specific CD8 T cell clone). 1×104 Chlamydia-specific CD4 T cells or alloreactive CD8bm1 T cells were co-cultured with 1×104 P815 cells in the presence of anti-CD3 antibody 145-2c11 in 4 h assays. T cells were untreated (hatched bars) or pre-incubated with CMA (light gray bars) or PAO (black bars) for 1 h before the assay. Culture supernatants were assayed for release of lactate dehydrogenase activity using a commercial non-radioactive CTL assay to determine % specific lysis. Absolute % specific lysis values for each clone absent inhibitors are shown in parentheses. Aggregate means and SEM from three independent experiments. Comparisons are made to the untreated T cell clone (hatched bars) for each of the two conditions (4 h & 24 h). * = pvalue < 0.05; ** = pvalue < 0.005; *** = pvalue < 0.0005.
CD4 T cell clones lysis infected epithelial cells in delayed fashion
We tested whether the Chlamydia-specific CD4 T cell clones could lyse infected epithelial cells in 4 h killing assays. We have previously shown that maximal recognition/activation for all three CD4 T cell clones occurs 18–21 h post infection using IFN-γ pretreated C57epi.l epithelial cells. C57epi.1 cells were pretreated with IFN-γ for 14 h then infected with C. muridarum. At 18 h post infection the cells were harvested and used as targets in 4 h killing assays in the presence of tetracycline to halt Chlamydia replication. In spite of using an optimal time point post-infection, and earlier demonstration that the CD4 T cells were cytolytic via perforin pathways (Figure 4), none of the CD4 T cell clones showed killing of 18 h infected targets in 4 h 51Chromium release or non-radioactive killing assays (data not shown). While there was no short term killing, we knew that the CD4 T cell clones were able to lyse 18 h infected C57epi.1 cells dramatically within 24 h of co-culture based on direct visualization under phase-contrast microscopy.
To investigate this delayed killing phenomenon we set up C57epi.1 cells in chamber slides, and used direct visualization/photodocumentation as the readout for delayed cytolysis. C57epi.1 monolayers in eight well chamber slides were mock-infected or infected with C. muridarum for 18 h, then tetracycline and T cell clones were added in the absence or presence of inhibitors of cytolysis/apoptosis. In the first set of experiments we used inhibitors of the two classic pathways for T cell-mediated killing; CMA, which blocks perforin-mediated cytolysis by raising the pH in T cell exocytic granules causing perforin degradation (29), and antibodies to Fas and FasL to block activated cell death receptor-ligand interactions (apoptosis) (30, 31) (Figure 5). Photos were taken 24 h later to semi-quantitatively score cytolysis. Addition of tetracycline at 18 h post infection preserved the viability of the infected C57epi.1 monolayer for the duration of the assays (top row). CMA and monoclonal antibodies had little or no toxicity for C57epi.1 monolayers at the concentrations used (top row). None of the clones exhibited visible cytotoxicity toward mock-infected C57epi.1 cells (column A). All of the clones lysed 18 h infected epithelial cells within 24 h (column B). CMA partially protected infected C57epi.1 monolayers from uvmo-2 and uvmo-3, but not uvmo-1, consistent with uvmo-1's relative indifference to CMA documented in the redirected lysis assays (Figure 4). Monoclonal antibodies to Fas/FasL had no visible effect on delayed cytolysis.
Figure 5.

Chlamydia-specific CD4 T cell clones lyse infected C57epi.1 epithelial cells in a delayed fashion: Roles of perforin and Fas/FasLigand. C57epi.1 epithelial monolayers in eight well chamber slides were mock-infected and co-cultured without and with 1×105 T cells (column A), infected 18 h with 5 IFU per cell C. muridarum and co-cultured without and with T cells (column B), infected and co-cultured without and with T cells pretreated and exposed to CMA (column C), and infected co-cultured without and with T cells in the presence of monoclonal antibodies specific for Fas and FasLigand (10 μg/ml each) (column D). All wells, mock-infected or infected for 18 h, included addition of 10 μg/ml tetracycline in the co-culture media after 18 h to halt progression of the Chlamydia infection and preserve epithelial cell viability for duration of the assay. 24 h after addition of the T cells, epithelial monolayers were stained with Cytoquick and photographed at 20× magnification. Representative data from two independent experiments.
Next we investigated the role of T cell degranulation and the CD4 co-receptor in delayed killing of infected epithelial monolayers (Figure 6). Phenylarsine oxide is a tyrosine kinase inhibitor that blocks T cell degranulation (32); GK1.5 is a monoclonal antibody specific for CD4 that blocks the interaction between the CD4 co-receptor and MHC class II molecules (33). Again, the CD4 T cell clones did not lyse mock-infected cells (column A), and the inhibitors were not toxic for the C57epi.1 monolayers at the concentrations used (top row). Blocking T cell degranulation with PAO partially protected infected C57epi.1 monolayers from uvmo-2 and uvmo-3, but not uvmo-1. Anti-CD4 monoclonal antibody almost completely protected infected C57epi.1 monolayers from uvmo-2 and uvmo-3 mediated cytolysis, but not uvmo-1, suggesting that the latter's TCR receptor signal is less dependent on the CD4 co-receptor; a conclusion supported by independent experiments measuring these T cell clones IFN-γ responses to infected epithelial cells without and with 10 μg/ml GK1.5 present (data not shown). We also tested whether simultaneously blocking perforin and activated cell death pathways would be more protective than blocking either pathway separately. The CMA plus anti-Fas monoclonal antibody experiments did not show an unambiguous increased protection of the infected epithelial monolayer from T cell-mediated lysis over that seen with CMA alone (data not shown). Whether Fas-FasL made any contribution to the delayed cytolysis phenomenon could not be determined using this semi-quantitative technique.
Figure 6.

Chlamydia-specific CD4 T cell clones lyse infected C57epi.1 epithelial cells in a delayed fashion: Roles of T cell degranulation and the CD4 co-receptor. C57epi.1 epithelial monolayers in eight well chamber slides were mock-infected and co-cultured without and with 1×105 T cells (column A), infected 18 h with 5 IFU per cell C. muridarum and co-cultured without and with T cells (column B), infected and co-cultured without and with T cells pretreated and exposed to PAO (column C), and infected co-cultured without and with T cells in the presence of monoclonal antibody GK1.5 specific for CD4 (10 μg/ml) (column D). All wells, mock-infected or infected for 18 h, included addition of 10 μg/ml tetracycline in the co-culture media after 18 h to halt progression of the Chlamydia infection and preserve epithelial cell viability for duration of the assay. 24 h after addition of the T cells, epithelial monolayers were stained with Cytoquick and photographed using an inverted microscope at 20× magnification. Representative data from two independent experiments.
Having shown that CMA (perforin) and PAO (degranulation) partially blocked delayed-cytolysis by the CD4 T cell clones uvmo-2 & uvmo-3, we investigated whether blocking cytolysis would interfere with their control of C. muridarum replication in C57epi.1 cells. We first tested whether CMA and PAO affected C. muridarum replication in C57epi.1 epithelial cells. Untreated C57epi.1 monolayers were infected with C. muridarum in the presence of the working concentration of each inhibitor or its stock vehicle. 36 h later the wells were harvested by scraping and C. muridarum replication quantified on McCoy monolayers (Figure 7). PAO had no effect on C. muridarum replication or T cell activation, while CMA, an H+/ATPase inhibitor, significantly inhibited C. muridarum replication, reducing replication yield roughly ten-fold; very similar to the result seen with another H+/ATPase inhibitor bafilomycin (34). Based on these data, we concluded that interpretation of PAO experiments would be straightforward as this inhibitor had no effect on C. muridarum replication. Unfortunately CMA had a significant negative effect on C. muridarum replication. While C. muridarum replication inhibition by CD4 T cells in the presence of CMA would be normalized to the absolute replication of C. muridarum in the presence of CMA, interpretation of CMA experimental results are less straightforward as Chlamydia replication is needed to generate Chlamydia epitopes required for T cell recognition.
Figure 7.
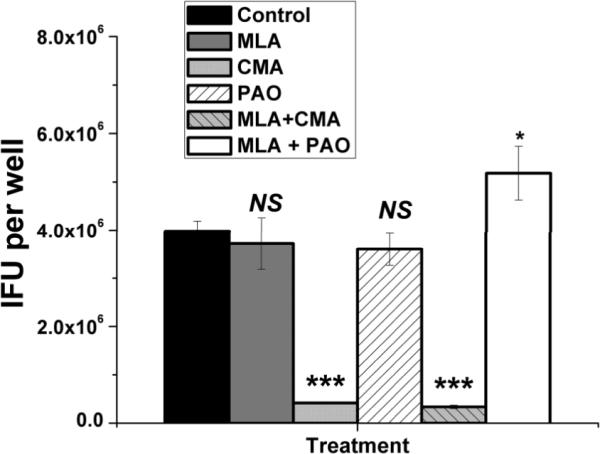
C. muridarum replication in C57epi.1 epithelial cells in the absence and presence of inhibitors of iNOS, perforin, and T cell degranulation. Untreated C57epi.1 cells were infected with C. muridarum at 3 IFU per cell in absence (black bar) or presence of MLA (gray bar) or CMA (light gray bar) or PAO (clear hatched bar) or MLA & CMA (light gray hatched bar) or MLA & PAO (white bar). Wells were harvested 36 h post infection and C. muridarum quantified on McCoy monolayers. Comparisons were made to the infected control (black bar). Means and SD from one experiment. * = pvalue <0.05; *** = pvalue <0.0005.
Cognizant of the above, we tested whether blocking perforin (CMA) or T cell degranulation (PAO), with and without MLA inhibition of epithelial inducible nitric oxide synthetase, would negatively affect the ability of CD4 T cell clones uvmo-2 & uvmo-3 to control C. muridarum replication in C57epi.1 epithelial cells. C57epi.1 monolayers pretreated with IFN-γ, in the presence of MLA where indicated, were infected with C. muridarum. Four hours later the inocula were removed and T cells, pretreated with CMA or PAO where indicated, were added at a T cell to epithelial cell ratio of 0.75:1 in medium (no inhibitor), medium containing MLA (inhibits iNOS), medium containing CMA (inhibits perforin), containing PAO (inhibits degranulation), medium containing MLA & CMA (blocking iNOS & perforin), and medium containing MLA & PAO (blocking iNOS & degranulation). 32 hours later, 36 h post infection, supernatants were collected from each well to measure IFN-γ production; then the wells were harvested by scraping, and the C. muridarum yield quantified by titration on McCoy monolayers. T cell inhibition-of-replication results were normalized to C. muridarum replication in identical parallel C57epi.1 wells exposed to the respected inhibitor(s) without the T cell clones. IFN-γ results, reflecting recognition/activation of each T cell clone by infected epithelial cells, are shown in the top panels of figure 8. For uvmo-2, the presence of CMA caused a 3-fold reduction in IFN-γ production; for uvmo-3, the presence of CMA caused roughly a 10-fold reduction in IFN-γ production, while MLA modestly enhanced (2-fold) and PAO modestly decreased (40% reduction) IFN-γ production for uvmo-3. These results are consistent with the previously documented need for C. muridarum replication for TCR recognition of infected cells by uvmo-2 & uvmo-3 (21), the former apparently requiring less replication than the latter to have its epitope visible in the context of epithelial MHC class II molecules. Consistent with that hypothesis MLA significantly increases replication of C. muridarum under these conditions (figure 1b), and MLA significantly increased the amount of IFN-γ released by uvmo-3 during co-culture with infected epithelial cells (Figure 8 top right panel). CD4 T cell clones uvmo-2 & uvmo-3s inhibition-of-Chlamydia-replication was relatively indifferent to inhibition of epithelial nitric oxide production (as previously shown in figure 2), but both were significantly affected by partial blocking of T cell degranulation with PAO, or partial inhibition of perforin activity with CMA (bottom panels of Figure 8). For both clones, PAO was the most potent single inhibitor, but major reductions, complete reversal for uvmo-3 and 50% reversal for uvmo-2, of T cell-mediated inhibition of Chlamydia replication required inhibition of both nitric oxide production and perforin/degranulation. These results are consistent with dual effector mechanisms, with one pathway dependent on epithelial nitric oxide production and the other on T cell degranulation and likely perforin. Keeping in mind that MLA inhibition of nitric oxide production is >90% and that CMA/PAO inhibition was only partial, complete reversal of uvmo-2 inhibition would be theoretically possible with MLA plus a more stable/potent inhibitor of degranulation or perforin. The alternative explanation for residual inhibition of Chlamydia replication by uvmo-2 in the presence of MLA plus PAO/CMA would be presence of a third anti-Chlamydia mechanism.
Figure 8.

Roles of iNOS, perforin, and T cell degranulation in uvmo-2- (1st column) & uvmo-3-(2nd column) mediated control of Chlamydia replication in C57epi.1 epithelial cells. C57epi.1 epithelial cells were pretreated for 14 h with IFN-γ in the absence (black bars) or presence of iNOS inhibitor MLA, and then infected with C. muridarum at 3 IFU per cell. Inocula were removed 4 h later and infected epithelial cells were co-cultured with 1.5×105 T cell clone cells in the presence of MLA (gray bars), co-cultured with T cells pretreated and exposed to CMA (light gray bars), co-cultured with T cells pretreated and exposed to PAO (clear hatched bars), co-cultured with T cells pretreated and exposed to CMA in the presence of MLA (gray hatched bars), co-cultured with T cells pretreated and exposed to PAO in the presence of MLA (white bars). A sample of supernatant was collected 36 h post infection to quantify IFN-γ by ELISA (top panels); then the wells were harvested and C. muridarum quantified on McCoy monolayers (bottom panels). % T cell inhibition of MoPn replication for each treatment condition (inhibitor) was normalized to C. muridarum replication in identical parallel wells that were not co-cultured with T cells. Aggregate means and SEM from three independent experiments. Comparisons were made to the no inhibitor control (black bars) for each of the T cell clones. * = pvalue < 0.05; ** = pvalue < 0.005; *** = pvalue < 0.0005; NS = not statistically significant.
Other Chlamydia-specific CD4 T cell clones
In this study we initially chose to focus on the most potent Chlamydia-specific CD4 T cell clones identified during an earlier investigation. However, it is reasonable to ask whether all Chlamydia-specific CD4 T cells utilize this dual, apparently redundant, mechanism for controlling replication in epithelial cells. Therefore we investigated the mechanism utilized by a previously described Chlamydia-specific CD4 T cell clone designated spl4-10 that was dependent on IFN-γ pretreatment of C57epi.1 epithelial cells to control C. muridarum replication (21), and compared it to uvmo-2 in the same experiments. C57epi.1 monolayers pretreated with IFN-γ, in the presence of MLA when indicated, were infected with C. muridarum. Four hours later the inocula were removed and T cells, pretreated with PAO where indicated, were added at an effector to target ratio of 0.75:1 in medium (control), medium containing MLA, or medium containing PAO. Thirty two hours later the wells were harvested by scraping and the C. muridarum yield quantified by titration on McCoy monolayers. T cell inhibition-of-replication results were normalized to C. muridarum replication in identical parallel C57epi.1 wells exposed to the respected inhibitor without the T cell clones (no T cell; 0% inhibition). As seen in previous experiments, uvmo-2 inhibition of C. muridarum replication was minimally reversed by MLA, and partially reversed by PAO (Figure 9). In contrast, spl4-10s ability to inhibit Chlamydia replication was completely reversed by either MLA or PAO. These results are consistent with generation of two different Chlamydia-specific CD4 T cell subsets during the adaptive immune response to experimental C. muridarum genital tract infections. One subset, represented by spl4-10 and previously published clone 2–14.0, is absolutely dependent on nitric oxide production for controlling Chlamydia replication; the second subset, represented by uvmo-1,-2,-3, can effectively control Chlamydia replication in epithelial cells in the absence of epithelial production of nitric oxide.
Figure 9.

Role of iNOS and T cell degranulation in IFN-γ-dependent (spl4-10) and -independent (uvmo-2) Chlamydia-specific CD4 T cell clones. C57epi.1 epithelial cells were pretreated for 14 h with IFN-γ in the absence or presence of iNOS inhibitor MLA, and then infected with C. muridarum at 3 IFU per cell. Inocula were removed 4 h later and infected epithelial cells were co-cultured without (white bars; set as 0% inhibition) and with 1.5×105 uvmo-2 T cells (first panel) or spl4-10 T cells (second panel) in the absence of inhibitors (control black bars), in the presence of MLA (gray bars), and with T cells pretreated and exposed to PAO (light gray bars). 36 h post infection the wells were harvested and C. muridarum quantified on McCoy monolayers. % T cell inhibition of MoPn replication for each treatment condition (inhibitor) was normalized to C. muridarum replication in identical parallel wells that were not co-cultured with T cells. Aggregate means and SEM from two independent experiments. Comparisons were made to the no inhibitor control (black bars) for each of the T cell clones. * = pvalue < 0.05; *** = pvalue < 0.0005; NS = not statistically significant.
Discussion
The relevance of the C. muridarum mouse genital tract model to human C. trachomatis genital tract infections is a critical issue for pathogenesis research and vaccine development. Existing data from pathogenesis studies utilizing knockout mice are difficult to reconcile with basic immunologic paradigms. Mice deficient in perforin, Fas, FasL or both perforin & FasL have normal clearance kinetics (35), raising doubts about the role of cytolysis in controlling Chlamydia infections of the genital tract. A strong body of existing research has identified an IFN-γ-dependent mechanism based on induction of epithelial nitric oxide, as a mechanism for clearing Chlamydia from the reproductive tract; yet IFN-γ-deficient mice clear 99.9% of C. muridarum from the genital tract with near normal kinetics (16, 17), and iNOS-deficient mice clear C. muridarum genital tract infections with normal kinetics (13, 14). These perplexing results could be reconciled if there were redundant mechanisms for clearing Chlamydia from the reproductive tract; one dependent on IFN-γ & iNOS and the other independent of IFN-γ & iNOS. Knocking out either mechanism in isolation theoretically would not adversely affect C. muridarum clearance in the mouse model.
Existing knockout mouse data clearly show that MHC class II is critical for clearing C. muridarum from the genital tract (5). We have shown the Chlamydia-specific CD4 T cell clones control C. muridarum replication in oviduct epithelial cells by recognizing Chlamydia antigens in the context of MHC class II molecules, and that IFN-γ was important for up regulating epithelial MHC class II molecules, which correlated with improved T cell recognition, measured as IFN-γ production, and control of C. muridarum replication in epithelial cells (21). Having a panel of Chlamydia-specific CD4 T cell clones allowed us to further investigate mechanisms for controlling C. muridarum replication in epithelial cells in vitro.
One potential mechanism for clearance would be recognition and lysis of infected epithelial cells during the non-infectious RB stage of Chlamydia development. We had previously shown that the earliest CD4 T cell recognition of infected epithelial cells by our clones occurred roughly 12 h post infection, with maximal recognition 18–21 h post infection. In this study we looked at short term perforin-mediated lysis with 18 h infected targets and found no cytolysis in 4 h assays. However there was a delayed lysis phenomenon readily visible via microscopy that occurred 18–24 h post-T cell co-culture with 18 h infected epithelial cells. This visually dramatic lytic event was dependent on T cell degranulation & perforin, but did not occur soon enough in the Chlamydia developmental cycle to lyse infected cells in the RB stage of replication; i.e. optimal epithelial targets at 18 h post infection were not lysed in 4 h assays (18–22 h post infection), and were only visually lysed after 18–24 h of co-culture (36–42 h post infection). C. muridarum replication in oviduct epithelial cells is nearly complete by 24 h post infection and is maximal by ~30 h post infection. Based on the in vitro data presented here and data from perforin/FasL knockout mice (35), the mechanism(s) utilized by CD4 T cells to clear genital tract infections are not likely to be critically dependent on the physical lysis of infected epithelial cells at the RB stage of the Chlamydia life cycle, though we cannot rule out the possibility that replication kinetics in vivo differ from those observed in vitro.
The existing data for controlling Chlamydia replication in epithelial cells supports an IFN-γ & nitric oxide-dependent mechanism, with Chlamydia-specific CD4 T cells up regulating epithelial nitric oxide production to control infection. This iNOS-dependent mechanism did not require direct TCR recognition of Chlamydia antigens presented by epithelial MHC class II molecules; it required recent T cell activation (<48 h) (11) and physical contact between T cells and epithelial cells, with LFA-1:ICAM-1 interactions being critical for boosting epithelial nitric oxide production to Chlamydia-cidal levels (12). This iNOS-dependent mechanism was demonstrable in vivo using systemic MLA treatment and adoptive transfer of 2–14.0 T cells into heterozygous +/nu Balb/c mice (36). However the iNOS-dependent mechanism is not critical in T cell sufficient mice based on normal C. muridarum genital tract clearance kinetics in iNOS knockout mice (13, 14) and wild type mice treated with the iNOS inhibitor MLA (14). In this study we investigated the role of nitric oxide production in controlling C. muridarum replication in oviduct epithelial cells using a small panel of Chlamydia-specific CD4 T cell clones and found that one clone, spl4-10, was dependent on epithelial nitric oxide production for controlling C. muridarum replication similar to the previously reported Chlamydia-specific CD4 T cell clone 2–14.0 (26). However other potent Chlamydia-specific CD4 T cell clones, uvmo-1,-2,-3, were not dependent on epithelial nitric oxide production to control C. muridarum replication. This finding confirms that there are two independent/redundant CD4 T cell-mediated mechanisms for controlling C. muridarum replication in epithelial cells that are mediated by different CD4 T cell subsets, and by extrapolation, two independent/redundant T cell-mediated mechanisms for clearing C. muridarum from the genital tract in the mouse model. iNOS-dependent CD4 T cells apparently have only the IFN-γ & iNOS-dependent pathway as they are completely inhibited by MLA, e.g. spl4-10 (this paper) & 2–14.0 (26), while iNOS-independent T cells, uvmo-1,-2,-3, have both pathways, with PAO having a greater effect than MLA but maximal reversal of T cell-mediated inhibition of C. muridarum replication requiring inhibition of both iNOS (MLA) and T cell degranulation (PAO).
The iNOS-independent mechanism represented by CD4 T cell clones uvmo-1,-2,-3 is dependent on T cell degranulation and likely perforin, though our perforin data based on CMA needs to be interpreted cautiously because of the significant inhibition of C. muridarum replication by this H+/ATPase inhibitor. Though tangential to the focus of the investigations reported here, the finding that two H+/ATPase inhibitors, CMA (this paper) and bafilomycin (34), inhibit replication of C. muridarum in murine epithelial cells identifies a potentially interesting host-pathogen interaction. The iNOS-independent CD4 T cell mechanism utilized by CD4 clones uvmo-1,-2,-3 for limiting C. muridarum replication in vitro is not dependent on the physical lysis of infected cells based on the relative kinetics of C. muridarum replication and timing of CD4 T cell recognition/lysis of infected target cells. The fact that recognition and lysis of infected epithelial cells in vitro occurs after the transition from non-infectious RB to infectious EB suggests that the in vitro iNOS-independent mechanism for controlling C. muridarum replication is a direct attack on EBs; presumably by molecules resident within T cell granules based on a requirement for degranulation, but potentially including T cell-induced epithelial effector molecules other than nitric oxide. Delineation of the specific T cell and possibly epithelial cell anti-Chlamydial effector molecules for the iNOS-independent T cell-mediated mechanism is the focus of ongoing investigations.
In vitro confirmation of two redundant CD4 T cell mechanisms for interrupting Chlamydia replication provides an explanation for otherwise perplexing knockout mouse data. Perforin/degranulation is not critical if the iNOS-dependent mechanism is available; iNOS is not critical if the degranulation-dependent pathway (iNOS-independent) pathway is available. This conclusion is supported by the data showing that heterozygous +/nu Balb/c mice adoptively transferred with a protective iNOS-dependent T cell clone 2–14.0 had reduced clearance of the C. muridarum from the genital tract when mice were treated systemically with MLA (36), but that wild type mice treated with MLA show no delay in resolution of C. muridarum genital tract infections (14).
The presence of two IFN-γ-regulated innate defense mechanisms in mice, p47 GTPases & iNOS, that inhibit Chlamydia replication in mouse epithelial cells but not operative in human reproductive tract epithelium, compromises the mouse model for studying human-relevant Chlamydia immunobiology. Similar concerns can be raised about mice as experimental models for other human intracellular pathogens that replicate in epithelial cells. Human reproductive tract epithelial cells do not express iNOS (7, 9), and iNOS has been shown to inhibit replication of both C. muridarum and human Chlamydia serovars in mouse epithelial cells in vitro (this paper, (12, 37)) and in vivo (36). Second, human epithelial cells do not express IFN-γ regulated murine p47 GTPases that limit replication of human Chlamydia strains, not C. muridarum, in mouse epithelial cells when IFN-γ is present (7, 8, 38, 39). Consistent with susceptibility to these dual IFN-γ-regulated defense mechanisms, IFN-γ knockout (8, 40) and IFN-γ receptor knockout mice (41) are much more permissive for genital tract infections with human C. trachomatis serovars than are wild type mice. Further complicating the mouse model, human reproductive tract epithelial cells have IFN-γ regulated indoleamine dioxygenase (IDO), an innate defense mechanism that is not present in mouse reproductive epithelial cells (7, 9).
One proposed approach to optimizing the mouse model is to knockout murine p47 GTPases and knockin IDO in order to create a mouse that would mirror the IFN-γ-induced innate defenses present in human reproductive tract epithelium (7). Such a mouse would presumably be dependent on human-relevant innate & adaptive immunity to clear infections, and make the mouse model permissive for replication of human Chlamydia serovars. However, the finding of two redundant CD4 T cell-mediated mechanisms for controlling C. muridarum replication makes this approach problematic because a p47 GTPase knockout/IDO knockin mouse would still have an iNOS-dependent T cell-mediated mechanism for controlling replication of rodent or human Chlamydia species that is not operative in epithelium of the human reproductive tract. In light of the findings presented in this report, and the fact that human and rodent Chlamydia serovars are likely relatively indifferent to IFN-γ-induced innate defenses when replicating in their natural host species (human-IDO (6); mouse p47 GTPases (7)), we propose that an optimized mouse model would be a tissue-specific knockout of iNOS in the epithelial cell lineage. Such a mouse, at least when infected with rodent C. muridarum, would be dependent on the iNOS-independent CD4 T cell-mediated mechanism for controlling and clearing Chlamydia. Based on the high degree of conservation of components of T cell adaptive immunity between mice and humans, the iNOS-independent mechanism identified in mouse CD4 T cells is likely to be the critical pathway during resolution of C. trachomatis genital tract infections in humans. A tissue-specific knockout of epithelial iNOS mouse would still be problematic for use with human Chlamydia serovars due to presence of murine p47 GTPases. The existing global iNOS knockout mouse is likely a suboptimal model based on significant dissemination of C. muridarum to the lung and spleen during genital tract infections (13), potentially altering the nature of the adaptive immune response compared to adaptive immune responses seen during typical genital tract infections in wild type mice and humans.
Acknowledgements
None of the authors have any conflicts of interest related to this manuscript.
This research was supported by NIH grant R01AI070514.
References
- 1.CDC, editor. Sexually Transmitted Disease Surveillance, 2008. U.S. Department of Health and Human Services; Atlanta, GA: 2009. [Google Scholar]
- 2.Brunham RC, Pourbohloul B, Mak S, White R, Rekart ML. The Unexpected Impact of a Chlamydia trachomatis Infection Control Program on Susceptibility to Reinfection. J Infect Dis. 2005;192:1836–1844. doi: 10.1086/497341. [DOI] [PubMed] [Google Scholar]
- 3.Haggerty CL, Gottlieb SL, Taylor BD, Low N, Xu F, Ness RB. Risk of sequelae after Chlamydia trachomatis genital infection in women. J Infect Dis. 201(Suppl 2):S134–155. doi: 10.1086/652395. [DOI] [PubMed] [Google Scholar]
- 4.Morrison RP, Caldwell HD. Immunity to murine chlamydial genital infection. Infect Immun. 2002;70:2741–2751. doi: 10.1128/IAI.70.6.2741-2751.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrison RP, Feilzer K, Tumas DB. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect Immun. 1995;63:4661–4668. doi: 10.1128/iai.63.12.4661-4668.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fehlner-Gardiner C, Roshick C, Carlson JH, Hughes S, Belland RJ, Caldwell HD, McClarty G. Molecular basis defining human Chlamydia trachomatis tissue tropism. A possible role for tryptophan synthase. J Biol Chem. 2002;277:26893–26903. doi: 10.1074/jbc.M203937200. [DOI] [PubMed] [Google Scholar]
- 7.Nelson DE, Virok DP, Wood H, Roshick C, Johnson RM, Whitmire WM, Crane DD, Steele-Mortimer O, Kari L, McClarty G, Caldwell HD. Chlamydial IFN-{gamma} immune evasion is linked to host infection tropism. Proc Natl Acad Sci U S A. 2005;102:10658–10663. doi: 10.1073/pnas.0504198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perry LL, Su H, Feilzer K, Messer R, Hughes S, Whitmire W, Caldwell HD. Differential sensitivity of distinct Chlamydia trachomatis isolates to IFN-gamma-mediated inhibition. J Immunol. 1999;162:3541–3548. [PubMed] [Google Scholar]
- 9.Roshick C, Wood H, Caldwell HD, McClarty G. Comparison of gamma interferon-mediated antichlamydial defense mechanisms in human and mouse cells. Infect Immun. 2006;74:225–238. doi: 10.1128/IAI.74.1.225-238.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunham RC, Ladino J. Rey. Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nature reviews. 2005;5:149–161. doi: 10.1038/nri1551. [DOI] [PubMed] [Google Scholar]
- 11.Igietseme JU, Wyrick PB, Goyeau D, Rank RG. An in vitro model for immune control of chlamydial growth in polarized epithelial cells. Infect Immun. 1994;62:3528–3535. doi: 10.1128/iai.62.8.3528-3535.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Igietseme JU, Uriri IM, Hawkins R, Rank RG. Integrin-mediated epithelial-T cell interaction enhances nitric oxide production and increased intracellular inhibition of Chlamydia. J Leukoc Biol. 1996;59:656–662. doi: 10.1002/jlb.59.5.656. [DOI] [PubMed] [Google Scholar]
- 13.Igietseme JU, Perry LL, Ananaba GA, Uriri IM, Ojior OO, Kumar SN, Caldwell HD. Chlamydial infection in inducible nitric oxide synthase knockout mice. Infect Immun. 1998;66:1282–1286. doi: 10.1128/iai.66.4.1282-1286.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramsey KH, Miranpuri GS, Poulsen CE, Marthakis NB, Braune LM, Byrne GI. Inducible nitric oxide synthase does not affect resolution of murine chlamydial genital tract infections or eradication of chlamydiae in primary murine cell culture. Infect Immun. 1998;66:835–838. doi: 10.1128/iai.66.2.835-838.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramsey KH, Miranpuri GS, Sigar IM, Ouellette S, Byrne GI. Chlamydia trachomatis persistence in the female mouse genital tract: inducible nitric oxide synthase and infection outcome. Infect Immun. 2001;69:5131–5137. doi: 10.1128/IAI.69.8.5131-5137.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry LL, Feilzer K, Caldwell HD. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J Immunol. 1997;158:3344–3352. [PubMed] [Google Scholar]
- 17.Cotter TW, Ramsey KH, Miranpuri GS, Poulsen CE, Byrne GI. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect Immun. 1997;65:2145–2152. doi: 10.1128/iai.65.6.2145-2152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swanson J, Eschenbach DA, Alexander ER, Holmes KK. Light and electron microscopic study of Chlamydia trachomatis infection of the uterine cervix. J Infect Dis. 1975;131:678–687. doi: 10.1093/infdis/131.6.678. [DOI] [PubMed] [Google Scholar]
- 19.Kiviat NB, Hanssen P. Wolner, Peterson M, Wasserheit J, Stamm WE, Eschenbach DA, Paavonen J, Lingenfelter J, Bell T, Zabriskie V, et al. Localization of Chlamydia trachomatis infection by direct immunofluorescence and culture in pelvic inflammatory disease. Am J Obstet Gynecol. 1986;154:865–873. doi: 10.1016/0002-9378(86)90473-4. [DOI] [PubMed] [Google Scholar]
- 20.Johnson RM. Murine oviduct epithelial cell cytokine responses to Chlamydia muridarum infection include interleukin-12-p70 secretion. Infection and immunity. 2004;72:3951–3960. doi: 10.1128/IAI.72.7.3951-3960.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jayarapu K, Kerr MS, Katschke A, Johnson RM. Chlamydia muridarum-specific CD4 T-cell clones recognize infected reproductive tract epithelial cells in an interferon-dependent fashion. Infect Immun. 2009;77:4469–4479. doi: 10.1128/IAI.00491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson R, Lancki DW, Fitch FW. Accessory molecules involved in antigen-mediated cytolysis and lymphokine production by cytotoxic T lymphocyte subsets. I. Identification of functions for the T cell surface molecules Ly-6C and Thy-1. Journal of Immunology. 1993;151:2986–2999. [PubMed] [Google Scholar]
- 23.Shaw JH, Grund VR, Durling L, Caldwell HD. Expression of genes encoding Th1 cell-activating cytokines and lymphoid homing chemokines by chlamydia-pulsed dendritic cells correlates with protective immunizing efficacy. Infect Immun. 2001;69:4667–4672. doi: 10.1128/IAI.69.7.4667-4672.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leo O, Sachs DH, Samelson LE, Foo M, Quinones R, Gress R, Bluestone JA. Identification of monoclonal antibodies specific for the T cell receptor complex by Fc receptor-mediated CTL lysis. J Immunol. 1986;137:3874–3880. [PubMed] [Google Scholar]
- 25.Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–2412. [PubMed] [Google Scholar]
- 26.Igietseme JU. The molecular mechanism of T-cell control of Chlamydia in mice: role of nitric oxide. Immunology. 1996;87:1–8. [PMC free article] [PubMed] [Google Scholar]
- 27.Darville T, Neill JMO, Andrews CW, Jr., Nagarajan UM, Stahl L, Ojcius DM. Toll-like receptor-2, but not Toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J Immunol. 2003;171:6187–6197. doi: 10.4049/jimmunol.171.11.6187. [DOI] [PubMed] [Google Scholar]
- 28.Topham DJ, Tripp RA, Doherty PC. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol. 1997;159:5197–5200. [PubMed] [Google Scholar]
- 29.Kataoka T, Shinohara N, Takayama H, Takaku K, Kondo S, Yonehara S, Nagai K. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J Immunol. 1996;156:3678–3686. [PubMed] [Google Scholar]
- 30.Kagi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- 31.Kayagaki N, Yamaguchi N, Nagao F, Matsuo S, Maeda H, Okumura K, Yagita H. Polymorphism of murine Fas ligand that affects the biological activity. Proc Natl Acad Sci U S A. 1997;94:3914–3919. doi: 10.1073/pnas.94.8.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anel A, Richieri GV, Kleinfeld AM. A tyrosine phosphorylation requirement for cytotoxic T lymphocyte degranulation. J Biol Chem. 1994;269:9506–9513. [PubMed] [Google Scholar]
- 33.Wilde DB, Marrack P, Kappler J, Dialynas DP, Fitch FW. Evidence implicating L3T4 in class II MHC antigen reactivity; monoclonal antibody GK1.5 (anti-L3T4a) blocks class II MHC antigen-specific proliferation, release of lymphokines, and binding by cloned murine helper T lymphocyte lines. J Immunol. 1983;131:2178–2183. [PubMed] [Google Scholar]
- 34.Derbigny WA, Hong SC, Kerr MS, Temkit M, Johnson RM. Chlamydia muridarum infection elicits a beta interferon response in murine oviduct epithelial cells dependent on interferon regulatory factor 3 and TRIF. Infect Immun. 2007;75:1280–1290. doi: 10.1128/IAI.01525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perry LL, Feilzer K, Hughes S, Caldwell HD. Clearance of Chlamydia trachomatis from the murine genital mucosa does not require perforin-mediated cytolysis or Fas-mediated apoptosis. Infect Immun. 1999;67:1379–1385. doi: 10.1128/iai.67.3.1379-1385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Igietseme JU. Molecular mechanism of T-cell control of Chlamydia in mice: role of nitric oxide in vivo. Immunology. 1996;88:1–5. [PMC free article] [PubMed] [Google Scholar]
- 37.Igietseme JU, Uriri IM, Chow M, Abe E, Rank RG. Inhibition of intracellular multiplication of human strains of Chlamydia trachomatis by nitric oxide. Biochem Biophys Res Commun. 1997;232:595–601. doi: 10.1006/bbrc.1997.6335. [DOI] [PubMed] [Google Scholar]
- 38.Coers J, Hanley I. Bernstein, Grotsky D, Parvanova I, Howard JC, Taylor GA, Dietrich WF, Starnbach MN. Chlamydia muridarum evades growth restriction by the IFN-gamma-inducible host resistance factor Irgb10. J Immunol. 2008;180:6237–6245. doi: 10.4049/jimmunol.180.9.6237. [DOI] [PubMed] [Google Scholar]
- 39.Al-Zeer MA, Younes H. M. Al, Braun PR, Zerrahn J, Meyer TF. IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS One. 2009;4:e4588. doi: 10.1371/journal.pone.0004588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito JI, Lyons JM. Role of gamma interferon in controlling murine chlamydial genital tract infection. Infect Immun. 1999;67:5518–5521. doi: 10.1128/iai.67.10.5518-5521.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johansson M, Schon K, Ward M, Lycke N. Genital tract infection with Chlamydia trachomatis fails to induce protective immunity in gamma interferon receptor-deficient mice despite a strong local immunoglobulin A response. Infect Immun. 1997;65:1032–1044. doi: 10.1128/iai.65.3.1032-1044.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]