

Abstract
The enantioselective total synthesis of the pyrrolophane natural product streptorubin B is described. Key steps in the concise route include application of a one-pot enantioselective aldol cyclization/Wittig reaction and an anionic oxy-Cope rearrangement to forge the crucial 10-membered ring. Comparisons between CD-spectra of synthetic and natural samples of streptorubin B, coupled with X-ray crystallography, allowed for the determination of the absolute stereochemistry of this natural product for the first time. These studies also provided unambiguous proof of the relative configuration between the butyl sidechain and bis-pyrrole subunit. Additional studies revealed a novel atropstereoselective Paal–Knorr pyrrole condensation and provided fundamental experimental insight into the barrier for atropisomerization of the natural product.
Introduction
The prodigiosin alkaloids have attracted widespread attention from both chemists and biologists due to their unique molecular architectures and the range of potentially useful biological activities they display.1 For example, a prodigiosin-inspired medicinal chemistry program at Gemin X led to the development of a synthetic small-molecule Bcl inhibitor that is currently in Phase I and II clinical trials for the treatment of a variety of cancers.2 From a structural standpoint, streptorubin B (1)3 and metacycloprodigiosin (2)4 are especially notable due to their highly strained pyrrolophane cores, which are formed by oxidative ring-closure from a common precursor, namely, the natural product undecylprodigiosin (3, Figure 1).5 The more recently isolated congener, prodigiosin R1 (5),6 shares a similar structure to metacycloprodigiosin (2) and provides an interesting link between these molecules and the related compound, roseophilin (6),7 while butylcycloheptylprodigiosin (4)8,9 is a constitutional isomer of both 1 and 2. In fact, the structural similarity between streptorubin B (1) and butylcycloheptylprodigiosin (4) led to significant confusion regarding the actual structures of these compounds. In 1975, Gerber and coworkers isolated a red pigment from Streptomyces sp Y-42 and Streptomyces abikoensis, to which they assigned the structure given by 4.8 Likewise, in 1985 the group of Floss assigned the structure 4 to a “pink pigment” they isolated from a strain of Streptomyces coelicolor.9 Subsequent to these publications, in 1991 Weyland and coworkers reported the isolation of a pigment from an actinomycete (strain B 4358), which they showed possessed the pyrrolophane structure given by 1.3 They also went on to state that the assignment put forth earlier by Gerber (and presumably Floss also) for 4 should be revised in favor of 1. Curiously, Gerber herself came to the same conclusion in 1978, although the reasons for this reassignment were not elaborated upon.10 In 2005, Fürstner and coworkers reported the first synthesis of butylcycloheptylprodigiosin (4), and on the basis of comparisons to the data collected by Floss, stated that butylcycloheptylprodigiosin (4) was in fact a natural product.11 A second synthesis of butylcycloheptylprodigiosin (4) by Reeves was reported in 2007, who came to the same conclusion as Fürstner.12 These conclusions are confusing given that Floss reports that his data for the “pink pigment”, 9 to which he assigned the structure 4, “closely matched” that reported by Gerber even though at that time she had already revised her assignment in favor of 1. Furthermore, in 2008, Challis and coworkers reported that Streptomyces coelicolor, the organism that Floss isolated his “pink pigment” from, produced streptorubin B (1) and found no evidence for production of butylcycloheptylprodigiosin (4).5b
Figure 1.

Some prodigiosin alkaloids and roseophilin
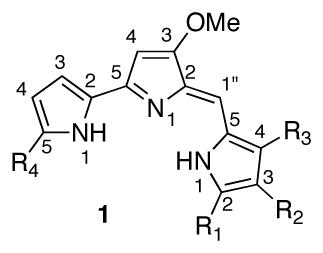
The structure of streptorubin B (1), and its identity as a natural product, is less questionable due to the full complement of NMR spectroscopic analysis conducted Weyland and coworkers in 1991,3 and also by Challis and workers in 2008.5b Key to the structural assignment was the presence of an upfield resonance at −1.55 ppm in the proton NMR spectrum that was attributed to one of the diastereotopic C4′ hydrogens (Weyland numbering), which due to the conformation of the strained 10-membered ring suffers anisotropic shielding by the pyrrole nucleus. Metacycloprodigiosin (2)4 and prodigiosin R1 (5)6 have similar proton resonances. While these two molecules have had their structures confirmed through total synthesis,11,12,13 there have been no reported syntheses of streptorubin B (1), although the groups of Fürstner13c and Chang14 have each prepared the pyrrole core. While the gross structure of streptorubin B (1) is not in doubt, several stereochemical issues remain. Weyland and coworkers noted that streptorubin B (1) possesses an element of planar stereochemistry;3 that is, there are two potential atropdiastereomers of 1 depending on the relative stereochemistry of the butyl sidechain and the bis-pyrrole sidearm (Figure 2). This stereochemical relationship was never established. Additionally, the absolute configuration of streptorubin B (1) was not established. In fact, at the outset of our work, the only compound of this family with known absolute stereochemisty was the related compound, roseophilin (6). The independent synthetic efforts of both the Tius and Boger groups,15 established that roseophilin (6) possessed the absolute stereochemistry indicated in Figure 1. It was against this background that we initiated a synthesis of streptorubin B (1), with the goal of clarifying the structural ambiguities surrounding this fascinating molecule.
Figure 2.

Atropisomerism within streptorubin B (1)
Results and Discussion
Total Synthesis
In 2009, our research group completed the first enantioselective total synthesis of metacycloprodigiosin (2), and its relative, prodigiosin R1 (5).13d At the time of publication we were unable to confirm the absolute configuration of these natural products due to a lack of access to authentic samples. Subsequent to our publication, we were able to confirm the absolute configuration of natural metacycloprodigiosin (2) through comparison of the CD-spectra recorded for our synthetic material and a natural sample isolated by Challis and coworkers.16 The synthetic sample, which was (R), matched the natural sample, thus establishing that metacycloprodigiosin possessed the (R)-configuration for the side chain.
Wishing to also establish the stereochemistry of streptorubin B (1), we sought to apply the same strategy to its synthesis that had proven successful for metacycloprodigiosin (2).13d Our total synthesis of 2 relied upon a ring-closing metathesis (RCM) reaction of diene 7 to produce the requisite 12-membered ring in 69% yield. In order to synthesize streptorubin B (1), we prepared diene 8 by a route analogous to that which we used to prepare 7. In contrast to what we found for 12-membered ring formation, diene 8 failed to undergo the desired ring-closure. Using a variety of metathesis catalysts, the only isolated products were mixtures of dimeric species. Repositioning the alkene did not lead to any improvement. The failure of 8 to undergo efficient ring-closing metathesis is most likely a consequence of severe ring-strain in the 10-membered product, and we therefore devised an alternative strategy.
Our revised synthesis plan for streptorubin B (1) entailed initial disconnection of the bis-pyrrole sidearm of the natural product to generate the pyrrolophane core 11. Simplification of 11 by the Paal–Knorr pyrrole transform, and some functional group interconversions led back to cyclodecanone 12, which contains the full retron for the anionic oxy-Cope rearrangement.17 Thus, the synthesis was simplified to devising a route to gain ready access to the functionalized cyclohexanol precursor 13. To this end, we wished to employ a proline-catalyzed enantioselective desymmetrizing intramolecular aldol reaction on dialdehyde 15,18 followed by an in situ Wittig reaction to form 14, which could be easily converted to cyclohexanol 13.
In 2003, List and coworkers reported the development of a proline-catalyzed enantioselective exo-enol-6-exo-trig aldol reaction as an effective means for desymmetrizing acyclic dialdehydes.18 In a footnote of the manuscript, the authors reported conducting an in situ Horner-Wadsworth-Emmons (HWE) reaction as a means to produce compounds with a chromophore for easy determination of enantiomeric excess by HPLC. We speculated that such a route employing a Wittig reaction rather than the HWE reaction might provide rapid access to the homoallylic alcohol (i.e., 14) we needed for our streptorubin B synthesis. Accordingly, we treated aldehyde 15, prepared in one step19 from commercially available cycloheptene (16), with 10 mol% (S)-proline, and upon consumption of the dialdehyde added ylide 18 to the reaction mixture (Scheme 2). In this way, the homoallylic alcohol 14 was obtained as the major diastereomer in 69% yield as a 98:2 mixture of enantiomers. Oxidation of 14, followed by addition of the vinyl anion 1920 gave the required precursor 13 (97:3 er) to the anionic oxy-Cope rearrangement. Exposure of alcohol 13 to KHMDS and 18-crown-6 produced the desired 10-membered ring 12 in 85% yield. The enantiopurity of 12 was found to be 97:3 er, indicating good levels of stereochemical transfer during the oxy-Cope rearrangement. With 12 in hand, generation of the pyrrole core 11 proceeded smoothly following alkene reduction with concomitant benzyl ether cleavage, oxidation of the liberated alcohol to the aldehyde, and Paal–Knorr pyrrole synthesis (67% yield over 3 steps). Completion of the synthesis involved conducting an acid-promoted condensation between pyrrole 11 and aldehyde 21,21 followed by removal of the Boc-group by basic methanolysis. Analysis of the bright red material thus obtained revealed an approximate 10:1 mixture of two compounds where the major compound did not match the natural product. Reexamination of this NMR sample after ten days, however, revealed that this mixture had transformed almost completely to streptorubin B (1). The synthetic material (as the HCl salt) was spectroscopically (1H NMR, 13C NMR, MS) identical to the natural product.22 Thus, streptorubin B (1) was prepared in nine steps from cycloheptene (16) and in 20% overall yield.
Scheme 2.

Enantioselective total synthesis of streptorubin B (1)
Relative Stereochemistry of Streptorubin B and Key Intermediates
We were intrigued by the observation that the initially formed species of the final condensation step in the synthesis converted into the natural product upon standing. We speculated that the initially formed species was one atropdiastereomer of the natural product, and that over time it isomerized into the other, presumably lower energy atropisomer, which had the same structure as the natural product. Inspection of molecular models of the two possible atropisomers indicated that the lower energy atropisomer should be anti-streptorubin B so as to avoid severe eclipsing interactions present within syn-streptorubin B. Analysis of the two possible atropdiastereomers, anti-1 and syn-1, revealed that anti-1 should display NOESY crosspeaks for HA–HB and HC–HD, and lack them between HA–HC and HB–HD. The isomeric compound syn-1 should display the opposite correlations. The NOESY data obtained, as shown in Figure 4, were fully consistent with the structure assigned as anti-1, where the two sidearms effectively project from opposite faces of the 10-membered ring. An additional NOESY spectrum of the initially formed mixture of condensation between 11 and 21 showed a clear crosspeak corresponding to HB–HD, indicating that the initial adduct is indeed syn-1. From 1H-NMR spectroscopy, the equilibrium ratio between the anti and syn isomers is approximately 10:1 at room temperature, indicating that the anti isomer is thermodynamically favored by approximately 1.4 kcal·mol−1. By monitoring the change in ratio of syn-1 to anti-1 as a function of time at two temperatures we determined the activation barrier for the conversion of syn-1 into anti-1 to be approximately 20.5 kcal·mol−1.23 Thus, on the basis of these NMR spectroscopy studies we established the relative stereochemistry of streptorubin B (1) for the first time. Unequivocal evidence was later obtained through X-ray crystallography (see below).
Figure 4.

NOESY analysis of streptorubin B isomers
The question still remained unanswered, however, as to why we initially observed a mixture of isomers favoring the unnatural atropisomer, syn-streptorubin B, during the final condensation step. We hypothesized that perhaps the pyrrole precursor 11 existed as an atropisomer that favored the unnatural syn configuration, which led to initial generation of syn-streptorubin B following condensation with bis-pyrrole 21 (Scheme 3). This higher energy species then underwent conversion into the more stable anti isomer of the natural product. In fact, Fürstner and coworkers had prepared racemic pyrrole 11 as part of their work on streptorubin B (1) and noted in their manuscript13c that 11 displays an element of planar stereochemistry, but assignment of this stereochemistry and conversion of 11 into the natural product were not reported. They did, however, note that coalescence of pyrrole 11 could not be reached, indicating a barrier to rotation of greater than 17.5 kcal·mol−1. The NOESY experiment conducted in our labs on pyrrole 11 revealed that it exists predominantly as the unnatural syn isomer, syn-11 and that by 1H NMR spectroscopy the ratio of syn-11 to anti-11 is approximately 10:1 when first synthesized. If left to stand over a period of two weeks or longer, the equilibrium ratio appears to be approximately 4:1 in favor of syn-11. Thus, a complete picture of this complex final condensation step has been revealed. When first synthesized the pyrrole 11 exists as a 10:1 mixture favoring the syn atropisomer, which upon condensation with 21 affords a 10:1 mixture of adducts favoring syn-1, which then isomerizes to the more stable compound, anti-1.24
Scheme 3.

Dissection of the atropisomer issue
Further analysis of pyrrole 11 and its immediate precursor cyclodecanone syn-22, led us to speculate that perhaps the syn atropisomer of 11 was formed kinetically during the Paal–Knorr condensation (Scheme 4). If condensation of enamine 23 onto the endocyclic ketone occurred faster than bond rotation to form 24 this might account for the selective (10:1) formation of syn-11 (i.e., in a ratio greater than the equilibrium ratio, which we determined to be 4:1). If this was the case, we hypothesized that diastereomeric cyclodecanone anti-22 would afford anti-11 upon Paal–Knorr pyrrole synthesis via enamine 24. Cyclodecanone anti-22 was synthesized using a modified route to that used to prepare syn-22 (see Supporting Information) and exposed to the conditions for pyrrole synthesis. Analysis of the pyrrole thus obtained, revealed that the major isomer in this case was anti-11, indicating that the cyclodecanone precursors do not converge to the same intermediate upon condensation with ammonium acetate and the intermediate enamines 23 and 24 do not readily interconvert under the reaction conditions. We did observe, however, that over the course of 10 days at room temperature anti-11 equilibrated to a 4:1 mixture favoring syn-11. Furthermore, we showed that condensation of anti-11 with bis-pyrrole 21 afforded streptorubin B (1) directly as the favored atropisomer.
Scheme 4.

Atropselective Paal–Knorr pyrrole synthesis
In hindsight, it was fortunate that the “unnatural” pyrrole isomer, syn-11 could be converted into the natural product due to isomerization of the “unnatural” isomer of streptorubin B (i.e., syn-1) into streptorubin B. Had this final isomerization not occurred, however, completion of the total synthesis would still have been possible by taking advantage of a kinetically controlled Paal–Knorr pyrrole condensation of anti-22 to generate anti-11, and hence the correct atropisomer of streptorubin B (1).
Absolute Configuration of Streptorubin B
As a final goal we wished to establish the absolute stereochemistry of streptorubin B (1), which necessitated that we first establish the absolute stereochemistry of our synthetic material. We had established the configuration of the initial aldol/Wittig reaction product 14 through Mosher ester analysis,25 but were mindful of the fact that the stereochemistry of the butyl sidechain was set through the anionic oxy-Cope rearrangement. While it seemed entirely reasonable to expect the Cope product 12 to possess the stereochemistry indicated in Scheme 2, by way of the minimized chair-like transition state (i.e., 20), we required stronger evidence. Fortunately, the solution to this issue came in the form of X-ray quality crystals of the HCl salt of synthetic streptorubin B (1) itself (Figure 5A). Thus, through anomalous dispersion (Flack parameter of 0.04 for the indicated enantiomer, 0.87 for opposite enantiomer)26 we were able to unambiguously assign the configuration of our synthetic material as (R), as predicted in our synthetic scheme (see Scheme 2). Additionally, this X-ray structure confirmed our NMR structural assignment that streptorubin B (1) exists as the anti atropdiastereomer (Figure 4). Comparisons between the CD spectra of a natural sample and our synthetic streptorubin B were somewhat surprising (Figure 5B). As is shown in Figure 5B, the natural sample (blue) displays the mirror image CD spectrum to synthetic (R)-streptorubin B (green), indicating that natural streptorubin B possesses an (S) configuration for the side chain (Figure 5C).27 This stereochemistry is the opposite of metacycloprodigiosin (2), and is most likely a consequence of the conformation adopted by the linear sidechain within the common biogenetic precursor, undecylprodigiosin (3), when it undergoes enzyme mediated oxidative cyclization to produce either of the two natural products (i.e., streptorubin B or metacycloprodigiosin). We further confirmed this stereochemical assignment through preparation of the (S) enantiomer of streptorubin B (1) by using (R)-proline rather than (S)-proline during the one-pot aldol/Wittig reaction. This material displayed a CD spectrum (Figure 5B, red) identical in shape to that of the natural material.
Figure 5.

A. X-ray structure of synthetic (R)-streptorubin B; B. CD spectra; C. Confirmed absolute and relative stereochemistry of streptorubin B
Conclusions
The absolute and relative stereochemistry of streptorubin B (1) has been established through enantioselective total synthesis, providing definitive proof for the structure of this intriguing compound after decades of speculation. The concise synthesis made use of a novel one-pot enantioselective aldol/Wittig reaction to form a key homoallylic alcohol with high levels of enantiopurity. This powerful reaction, in combination with the anionic oxy-Cope rearrangement proved pivotal for construction of the strained 10-membered ring required for the synthesis. This straightforward procedure should prove useful in the enantioselective synthesis of other substituted 10-membered rings prevalent in many bioactive natural products. The complex question of atropstereoisomerism within streptorubin B (1) and its pyrrole precursor 11 was answered through a combination of NMR analysis and X-ray crystallography, while the absolute stereochemistry of 1 was determined through comparisons of CD spectra. Alongside our established enantioselective routes to the 12-membered pyrrolophane prodigiosins (i.e., metacycloprodigiosin and prodigiosin R1),13d the synthesis outlined herein for the 10-membered isomer (i.e., streptorubin B) will enable ready preparation of these natural products (and analogs) for further biological evaluation.
Supplementary Material
Figure 3.

Failed RCM approach to the streptorubin B core
Scheme 1.

Synthesis plan for streptorubin B (1)
Acknowledgments
This paper is dedicated to Professor David A. Evans. Support for this work was provided by Northwestern University (NU) and the NIH/NIGMS (1R01GM085322). DXH is a 2010 Barry M. Goldwater Scholar, a 2010 ACS SURF recipient, and gratefully acknowledges support from NU in the form of an Undergraduate Research Grant. We thank Professor Gregory Challis (University of Warwick, UK) for a sample of natural streptorubin B, the CD spectrum of natural metacycloprodigiosin, and for helpful discussions. Charlotte Stern (NU) and Dr. Amy Sargeant (NU) are thanked for assistance with X-ray analysis.
Footnotes
Supporting Information Available: Complete ref. for 2b, detailed experimental procedures, spectral data for all compounds, and X-ray structure (CIF file). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of the chemistry and biology of the prodigiosins, see: Fürstner A. Angew Chem Int Ed. 2003;42:3582–3603. doi: 10.1002/anie.200300582.Williamson NR, Fineran PC, Leeper FJ, Salmond GPC. Nature Rev Microbiol. 2006;4:887–899. doi: 10.1038/nrmicro1531.Williamson NR, Fineran PC, Gristwood T, Chawrai SR, Leeper FJ, Salmond GPC. Future Microbiol. 2007;2:605–618. doi: 10.2217/17460913.2.6.605.
- 2.(a) Borthakur G, O’Brien S, Ravandi-Kashani F, Giles F, Schimmer AD, Viallet J, Kantarjian H. Blood. 2006;108:750. [Google Scholar]; (b) Nguyen M, et al. Proc Natl Acad Sci U S A. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laatsch H, Kellner M, Weyland H. J Antibiot. 1991;44:187–191. doi: 10.7164/antibiotics.44.187. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wasserman HH, Rodgers GC, Keith DD. J Am Chem Soc. 1969;91:1263–1264. doi: 10.1021/ja01033a065. [DOI] [PubMed] [Google Scholar]; (b) Wasserman HH, Keith DD, Rodgers GC. Tetrahedron. 1976;32:1855–1861. [Google Scholar]
- 5.Cerdeño AM, Bibb MJ, Challis GL. Chem Biol. 2001;8:817–829. doi: 10.1016/s1074-5521(01)00054-0.Mo S, Sydor PK, Corre C, Alhamadsheh MM, Stanley AE, Haynes SW, Song L, Reynolds KA, Challis GL. Chem Biol. 2008;15:137–148. doi: 10.1016/j.chembiol.2007.11.015.See also, reference 1.
- 6.Kawasaki T, Sakurai F, Hayakawa Y. J Nat Prod. 2008;71:1265–1267. doi: 10.1021/np7007494. [DOI] [PubMed] [Google Scholar]
- 7.Hayakawa Y, Kawakami K, Seto H, Furihata K. Tetrahedron Lett. 1992;33:2701–2704. [Google Scholar]
- 8.(a) Gerber NN. J Antibiot. 1975;28:194–199. doi: 10.7164/antibiotics.28.194. [DOI] [PubMed] [Google Scholar]; (b) Gerber NN, Stahly DP. Appl Microbiol. 1975;30:807–810. doi: 10.1128/am.30.5.807-810.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsao SW, Rudd BAM, He XG, Chang CJ, Floss HG. J Antibiot. 1985;38:128–131. doi: 10.7164/antibiotics.38.128. [DOI] [PubMed] [Google Scholar]
-
10.Gerber NN, McInnes AG, Smith DG, Walter JA, Wright JLC, Vining LC. Can J Chem. 1978;56:1155–1163.In this manuscript, Gerber and coworkers state that “Streptomyces and Streptoverticillium species also produce the related butylcycloheptylprodiginine (1, R1R3 = 1′,7′-cyclo(CH2)6-CH(CH2)3CH3, R2 = R4 = H), which has hitherto been incorrectly identified as a 2,3-cycloalkyl derivative.” In other words, the compound referred to as butylcycloheptylprodiginine is actually streptorubin B.
- 11.Fürstner A, Radkowski K, Peters H. Angew Chem Int Ed. 2005;44:2777–2781. doi: 10.1002/anie.200462215. [DOI] [PubMed] [Google Scholar]
- 12.Reeves JT. Org Lett. 2007;9:1879–1881. doi: 10.1021/ol070341i. [DOI] [PubMed] [Google Scholar]
- 13.For metacycloprodigiosin, see: Wasserman HH, Keith DD, Nadelson J. J Am Chem Soc. 1969;91:1264–1265. doi: 10.1021/ja01033a066.Wasserman HH, Keith DD, Nadelson J. Tetrahedron. 1976;32:1867–1871.Fürstner A, Szillat H, Gabor B, Mynott R. J Am Chem Soc. 1998;120:8305–8314.Clift MD, Thomson RJJ. Am Chem Soc. 2009;131:14579–14583. doi: 10.1021/ja906122g.For prodigiosin R1, see ref. 13d.
- 14.Chang MY, Pai CL, Chen HP. Tetrahedron Lett. 2005;46:7705–7709. [Google Scholar]
- 15.(a) Harrington PE, Tius MA. J Am Chem Soc. 2001;123:8509–8514. doi: 10.1021/ja011242h. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Hong JY. J Am Chem Soc. 2001;123:8515–8519. doi: 10.1021/ja011271s. [DOI] [PubMed] [Google Scholar]
- 16.See Supporting Information for CD spectra of synthetic material prepared as in ref. 12d and the CD spectrum of natural metacycloprodigiosin kindly provided by Prof. Gregory Challis.
- 17.Evans DA, Golob AM. J Am Chem Soc. 1975;97:4765–4766. [Google Scholar]
- 18.Pidathala C, Hoang L, Vignola N, List B. Angew Chem Int Ed. 2003;42:2785–2788. doi: 10.1002/anie.200351266. [DOI] [PubMed] [Google Scholar]
- 19.Hong BC, Tseng HC, Chen SH. Tetrahedron. 2007;63:2840–2850. [Google Scholar]
- 20.Prepared from the corresponding iodide through lithium–halogen exchange, see Supporting Information. The iodide was prepared using the protocol of Huang and Negishi, see: Huang Z, Negishi E. Org Lett. 2006;8:3675–3678. doi: 10.1021/ol061202o.
- 21.Aldrich LN, Dawson ES, Lindsley CW. Org Lett. 2010;12:1048–1051. doi: 10.1021/ol100034p. [DOI] [PubMed] [Google Scholar]
- 22.Key compound data for synthetic 1: IR (neat) 3436, 3371, 3019, 2937, 1764 cm−1; 1H NMR (500 MHz, CDCl3) 12.70 (s, 1H), 12.66 (s, 1H), 12.58 (s, 1H), 7.22 (s, 1H), 7.21 (s, 1H), 7.11 (s, 1H), 6.90 (s, 1H), 6.51 (s, 1H), 6.34 (t, 1H, J = 2.7 Hz), 6.09 (s, 1H), 4.02 (s, 3H), 3.33 (m, 1H), 3.10 (m, 1H), 2.53 (dt, 1H, J = 12.3, 5.4 Hz), 1.96-1.68 (m, 5H), 1.55 (m, 2H), 1.37 (m, 5H), 1.24 (m, 3H), 1.14 (m, 2H), 0.91 (m, 5H), 0.78 (m, 1H), −1.55 (dt, 1H, J = 14.1, 8.8 Hz); 13C NMR (125 MHz, CDCl3) 165.4, 154.4, 150.6, 147.0, 126.7, 125.0, 122.4, 120.3, 116.8, 116.6, 112.5, 111.5, 92.8, 58.7, 38.9, 37.5, 31.7, 30.9, 30.4, 30.0, 29.1, 27.7, 25.4, 22.9, 14.2; HRMS (ESI) Exact mass calcd for C25H33N3O [M+H+], 392.2696. Found 392.2700.
- 23.See Supporting Information for full details regarding measurement and calculation of this activation energy.
- 24.Because the isomers of pyrrole 11 do not interconvert rapidly at room temperature, the Curtin–Hammett principle need not be considered in order to rationalize this outcome. If the barrier for interconversion of the two pyrrole atropisomers were sufficiently low, one might have predicted that anti-1 would be formed preferentially since the rate of condensation of anti-11 with 21 is most likely significantly faster than that of syn-11 with 21. Since this interconversion does not occur rapidly under the reaction conditions, the product distribution reflects the ground state stabilities of the two starting atropisomers. For further discussion on the Curtin–Hammett principle, see: Seeman JI. Chem Rev. 1983;83:83–134.
- 25.Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512–519.Hoye TR, Jeffrey CS, Shao F. Nature Protocols. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354.See Supporting Information for details.
- 26.Flack HD. Acta Crystallogr, Sect A. 1983;39:876–881. [Google Scholar]
- 27.Challis and co-workers came to the same conclusion on the basis of their own studies into the biosynthesis of streptorubin B (Private communication).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.