

Abstract
Glycogen synthase kinase 3 (GSK3) is a constitutively active, proline-directed serine/threonine kinase that plays a part in a number of physiological processes ranging from glycogen metabolism to gene transcription. GSK3 also plays a pivotal and central role in the pathogenesis of both sporadic and familial forms of Alzheimer's disease (AD), an observation that has led us to coin the ‘GSK3 hypothesis of AD’. According to this hypothesis, over-activity of GSK3 accounts for memory impairment, tau hyper-phosphorylation, increased β-amyloid production and local plaque-associated microglial-mediated inflammatory responses; all of which are hallmark characteristics of AD. If our ‘GSK3 hypothesis of AD’ is substantiated and GSK3 is indeed a causal mediator of AD then inhibitors of GSK3 would provide a novel avenue for therapeutic intervention in this devastating disorder.
Keywords: Wnt, insulin, tau, β-amyloid, memory, inflammation
Alzheimer's disease (AD) is a neurodegenerative disorder defined by progressive memory loss and cognitive impairment and at the molecular level by the presence of neurofibrillary tangles (NFTs) and insoluble β-amyloid (Aβ) plaques (Hardy 2006) that are associated with activated microglia (Vehmas et al. 2003). NFTs are composed of hyper-phosphorylated forms of the microtubule-associated protein tau, whereas Aβ is derived from the proteolytic cleavage of β-amyloid precursor protein (APP). Early onset forms of Familial Alzheimer's disease (FAD) typically present before the age of 65 and have been linked to mutations in APP, presenilin-1 (PS-1) and presenilin-2 (PS-2). Mutations in these genes adversely affect APP processing and result in the increased production of insoluble Aβ and its deposition into plaques.
β-Amyloid precursor protein, presenilin and tau are undoubtedly pivotal to understanding the pathogenesis of AD and there are as yet no convincing refutations of the classical amyloid cascade hypothesis of AD, which postulates that, Aβ over-production leads to the pathogenic hyper-phosphorylation of tau resulting in the formation of neurofibrillary tangles (NFTs) and neurodegeneration. However, this hypothesis raises as many questions as it does answers. What regulates the processing of APP in sporadic AD as opposed to those very few families harboring APP or PS mutations? Which species of Aβ (Aβ42, Aβ*56, aggregated fibrillar Aβ or soluble oligomeric Aβ, intracellular Aβ or extracellular Aβ) is neurotoxic and how exactly does Aβ induce tau hyper-phosphorylation and does this lead to tau aggregation, neurotoxicity and loss of neuronal function? These are not idle questions, drug development in the AD field has concentrated very heavily on trying to alter APP processing, but developing alternative approaches is almost certainly going to be necessary for the effective treatment of this devastating and costly condition. We propose that glycogen synthase kinase-3 (GSK3) plays a leading role in the cascade of events that culminate in AD as this kinase is involved in the mechanisms underlying learning and memory, in the hyper-phosphorylation of tau, in the increased production of Aβ from APP and also in local cerebral inflammatory responses.
GSK3 and cell signaling
There are two GSK3 genes from which GSK3α and GSK3β are derived. GSK3β also exists as longer splice variants (Mukai et al. 2002; Schaffer et al. 2003). GSK3α and GSK3β are ubiquitously expressed, constitutively active, proline-directed serine/threonine kinases involved in a variety of cellular processes including glycogen metabolism (Welsh and Proud 1993), gene transcription (Troussard et al. 1999), apoptosis (Turenne and Price 2001) and microtubule stability (Anderton et al. 2001; Brion et al. 2001). Insights from mouse models suggest that the GSK3 isoforms exhibit tissue specific physiological functions. GSK3β knock-out mice die in utero (Hoeflich et al. 2000), whereas GSK3α knock-out mice are viable and display improved glucose tolerance in response to glucose load and elevated hepatic glycogen storage and insulin sensitivity (MacAulay et al. 2007).
GSK3 activity is modulated by insulin and Wnt signaling, both pathways act in a negative regulatory manner. Many, but not all GSK3 substrates require pre-phosphorylation (priming) before phosphorylation by GSK3 can occur, so in both health and disease the activity of the priming kinase might limit GSK3 activity. Insulin signaling leads to the activation of PI3-kinase and subsequently the activation of acutely transforming retrovirus Akt8 in rodent T cell lymphoma (Akt otherwise known as protein kinase B), which in turn phosphorylates free cytoplasmic GSK3β and GSK3α at serine (Ser) residues 9 and 21, respectively (Saltiel and Kahn 2001; Lizcano and Alessi 2002). Regulatory serine phosphorylation results in the generation of an intra-molecular pseudo-substrate, which blocks part of the active site preventing the enzymatic activity of GSK3 towards primed substrates. This in turn leads to the de-phosphorylation of downstream substrates such as glycogen synthase and eukaryotic protein synthesis initiation factor-2B (eIF-2B), which elicits an increase in glycogen and protein synthesis. GSK3 can also be phosphorylated at Ser9/21 by p70 ribosomal S6 kinase-1 (S6K1) and by 90 kDa ribosomal protein S6 kinase (RSK), a downstream mediator of mitogen-activated protein kinase (MAPK) signaling (Doble and Woodgett 2003).
In addition to regulatory Ser phosphorylation, GSK3β and GSK3α activity can be regulated by tyrosine (Tyr) phosphorylation at residues 216 or 279, respectively. Under physiological conditions, GSK3 is phosphorylated at these sites and increases in Tyr phosphorylation augment GSK3 activity. However, regulation of GSK3 at Tyr 216/279 is perhaps less common than regulation at Ser9/21 (Bhat et al. 2000; Bijur and Jope 2001).
Wnt signaling regulates GSK3 activity by physically displacing complexed GSK3 from a number of regulatory binding partners consequently preventing the phosphorylation and degradation of β-catenin. In the absence of Wnt, the signaling pool of β-catenin is maintained at low levels through degradation (Dale 1998; Huelsken and Behrens 2002; Nusse 2005). β-catenin is targeted for ubiquitination by the β-transducin repeat containing protein (βTrCP) and is then degraded by the proteosome. β-catenin is phosphorylated by the serine/threonine kinases casein kinase 1 (CK1) and GSK3β. Phosphorylation of β-catenin occurs in a multi-protein complex (the destruction complex), comprising axin, adenomatous polyposis coli protein and diversin. Upon receipt of a Wnt signal, dishevelled prevents degradation of β-catenin through the recruitment of GSK3 binding protein (GBP)/Frat-1, which displaces GSK3β from the destruction complex. Stabilized β-catenin enters the nucleus and associates with T cell factor/lymphoid enhancer factor transcription factors, leading to the transcription of Wnt target genes, such as cyclin D1, PPARδ and twin. Recently, GSK3α in addition to GSK3β, has been shown to function in the destruction complex and the Wnt signaling pathway, suggesting that this enzyme is equally as important in Wnt biology as is GSK3β (Asuni et al. 2006; Doble et al. 2007).
The role of GSK3 in AD
The evidence that GSK3 plays a central role in AD and that its deregulation accounts for many of the pathological hallmarks of the disease in both sporadic and familial AD cases, has led us to formulate the ‘GSK3 hypothesis of AD.’ Evidence from our group and others suggests that GSK3 is intimately involved in the hyper-phosphorylation of tau, memory impairment, the increased production of Aβ and in inflammatory responses (Fig. 1). GSK3 also reduces acetylcholine synthesis, which is in accordance with the cholinergic deficit present in AD (Hoshi et al. 1996). Moreover, GSK3 is a key mediator of apoptosis (Turenne and Price 2001) and thereby might directly contribute to neuronal loss in AD.
Fig. 1.
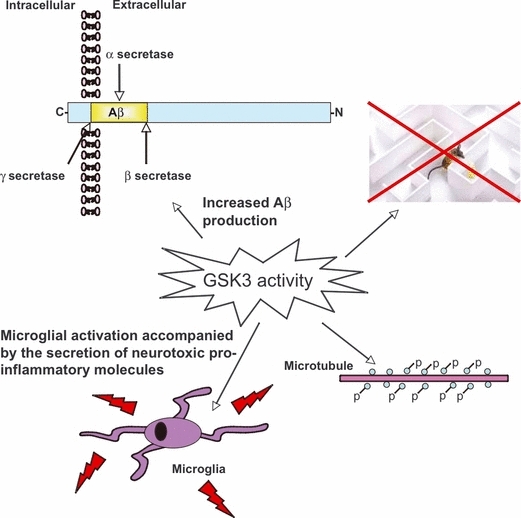
GSK3 and its role in AD. Over-activity of GSK3 caused either by aberrant Wnt or insulin signaling in sporadic AD cases or through familial mutations in PS or APP in FAD, might play an integral role in disease progression. GSK3 mediates the hyper-phosphorylation of tau, the increased production of Aβ from APP (via β and γ secretase-mediated cleavage) and results in impairments in learning and memory and could potentially heighten microglial-mediated inflammatory responses in the local vicinity of Aβ plaques.
Indeed, if GSK3 is central to AD pathogenesis then one would expect evidence for increased activity of this enzyme in AD. However, there is little such evidence, as it is technically difficult, if not impossible, to measure enzymatic activity in post-mortem brain tissue. Nevertheless, indirect evidence does support the role of GSK3 in disease. GSK3 has been shown to co-localize with dystrophic neurites and NFTs (Yamaguchi et al. 1996; Imahori and Uchida 1997; Pei et al. 1997). Active GSK3 appears in neurons with pre-tangle changes (Pei et al. 1999) and there is increased GSK3 activity in the frontal cortex in AD as evidenced by immuno-blotting for GSK3 phosphorylated at Tyr216 (Leroy et al. 2007). Furthermore, GSK3 expression is up-regulated in the hippocampus of AD patients (Blalock et al. 2004) and in post-synaptosomal supernatants derived from AD brain (Pei et al. 1997), although the latter study reports that there is not an increase in GSK3 enzymatic activity. GSK3 expression is also up-regulated in circulating peripheral lymphocytes in both AD and in mild cognitive impairment (Hye et al. 2005). It has recently been reported that a polymorphism in the GSK3 promoter is a risk factor for late onset AD (Mateo et al. 2006), which might account for alterations in GSK3 expression in disease. Collectively, these findings suggest that GSK3 activity might be increased in AD, through changes in its phosphorylation state as well as expression levels, although we acknowledge that direct evidence for this is still limited at present and some studies find no change in GSK3 activity (Pei et al. 1997) or reduced GSK3 activity (Swatton et al. 2004) in AD.
Genetic and epidemiological studies indicate that GSK3 is deregulated in AD through alterations in upstream Wnt and insulin signalling pathway intermediates. The low-density lipoprotein receptor related Protein 6 (LRP6), a co-receptor for Wnt signaling, has recently been identified as a risk gene for late onset AD in apolipoprotein E4-e4 negative individuals (De Ferrari et al. 2007), implicating aberrant Wnt signalling in AD pathology. In addition, an association of AD with diabetes and insulin resistance has been reported (Biessels and Kappelle 2005) and genetic studies find insulin signaling genes to be susceptibility loci for AD (Hamilton et al. 2007; Reiman et al. 2007).
GSK3 and memory
Suppression of Wnt or PI3-kinase signaling impairs long term potentiation (LTP), the best characterized correlate of learning and memory (Sanna et al. 2002; Chen et al. 2006). GSK3 is negatively regulated by both the Wnt and insulin pathways and inhibition of GSK3β follows the induction of LTP in wild-type mice (Hooper et al. 2007; Peineau et al. 2007) suggesting that GSK3β might mediate the effects of insulin and Wnt on LTP. Accordingly, over-expression of GSK3β in mice prevents the induction of LTP (Hooper et al. 2007) and causes a decrease in spatial learning (Hernandez et al. 2002). Inhibitors of GSK3β have also been shown to block long-term depression (LTD) and GSK3β activity is enhanced during LTD (Peineau et al. 2007). Thus, it would appear that GSK3β is critical for the induction of memory formation, switching off LTD and allowing LTP to occur.
Many downstream substrates of GSK3 are involved in synaptic remodeling, which is a vital process required for the proper establishment of connections during memory formation. Adenomatous polyposis coli a synaptic scaffolding protein (Rubinfeld et al. 1996) and collapsin response mediator proteins (CRMPs) are known to influence growth cone dynamics and are hyper-phosphorylated in Aβ plaques (Cole et al. 2004). GSK3 also phosphorylates and inhibits cAMP responsive element-binding protein (Bullock and Habener 1998; Hansen et al. 2004), a universal modulator of memory. Moreover, GSK3 promotes actin and tubulin assembly (Koivisto et al. 2004), processes required for synaptic reorganization during memory formation. We propose, therefore, that GSK3 acts as a gate through which LTP and memory are established. Considering, GSK3 activity is increased in AD, memory failure in this disorder might be attributable to the inhibition of LTP with neuronal loss ensuing as the disease progresses.
GSK3 and tau phosphorylation
Both GSK3β and GSK3α (Hanger et al. 1992; Ishiguro et al. 1992; Lovestone et al. 1994; Cho and Johnson 2003; Asuni et al. 2006) induce the hyper-phosphorylation of tau at both primed and non-primed phosphorylation sites, in vitro and in cell culture models of neurodegeneration, implicating GSK3 as an important tau-kinase possibly involved in the formation of NFTs in vivo. Consistent with this, GSK3β transgenic mice display tau hyper-phosphorylation and neurodegeneration (Lucas et al. 2001) and chronic lithium (GSK3 inhibitor) treatment prevents tau hyper-phosphorylation and NFT formation in double transgenic mice over-expressing GSK3β and tau (harboring a triple mutation associated with frontotemporal dementia and parkinsonism linked to chromosome 17), although reversal of pre-formed tangles was not observed (Engel et al. 2006). Similarly, double-transgenic Drosophila over-expressing tau and shaggy, the Drosophila form of GSK3, exhibit neuronal impairment and tau pathology (Mudher et al. 2004).
Insulin transiently increases tau phosphorylation in human neuroblastoma cells, but this is soon followed by a reduction in tau phosphorylation, which correlates with the activation and subsequent inhibition of GSK3 (Lesort et al. 1999). Dickkopf, a negative regulator of Wnt signaling also promotes tau phosphorylation and neurodegeneration through the activation of GSK3 (Caricasole et al. 2004) and Dickkopf is up-regulated in AD. Furthermore, PS-1 binds both GSK3β and tau, and mutant forms of PS-1, associated with FAD, bind GSK3β more effectively resulting in increased tau phosphorylation (Takashima et al. 1998). PS-1 has been shown to inactivate GSK3 through PI3-kinase/Akt signaling preventing tau phosphorylation and apoptosis. Interestingly, PS-1 FAD mutations inhibit PS-1-dependent PI3-kinase/Akt signaling, facilitating GSK3 activity and thus tau hyper-phosphorylation. This is one potential mechanism through which FAD mutations might lead to accelerated disease progression through secondary aberrations in GSK3 activity (Baki et al. 2004).
GSK3 and Aβ production
GSK3α, but not GSK3β, has been shown to regulate APP cleavage resulting in the increased production of Aβ (Sun et al. 2002; Phiel et al. 2003). Exposure of neurones to Aβ increases GSK3β activity through the inhibition of PI3-kinase signalling and blockade of either GSK3β expression or activity prevents Aβ-induced neurodegeneration (Takashima et al. 1993, 1996; Alvarez et al. 1999). Although heightened GSK3 activity is not the primary cause of disease in this scenario, increased GSK3 activity would serve to augment Aβ production and in turn tau hyper-phosphorylation and neuronal degeneration in both FAD and sporadic cases, in line with the amyloid cascade hypothesis of AD.
Insulin signaling, possibly acting though the suppression of GSK3 activity, has also been shown to exert a beneficial effect on APP and amyloid processing. Insulin increases the expression of the Aβ-protease, insulin degrading enzyme (IDE) (Zhao et al. 2004) and enhances the secretion of sAPPα, which results from the non-amyloidogenic α-secretase-mediated cleavage of APP (Solano et al. 2000). We also note with great interest the finding that in people with AD, insulin given through the intranasal route favorably alters the Aβ40/42 ratio and improves cognition (Reger et al. 2007).
GSK3 and inflammation
As well as being implicated in the core pathogenic events of AD – the increased formation of Aβ, tau hyper-phosphorylation and memory impairment – GSK3 might also play a role in other processes thought to impact on the amyloid cascade. Foremost amongst these are inflammatory processes. There is a growing body of evidence to suggest that inflammation plays an important role in AD. Microglia are known to accumulate around Aβ plaques in AD (Christie et al. 1996; Joshi and Crutcher 1998; Stalder et al. 1999) and there is increased expression of inflammatory mediators in AD brain tissue (McGeer and McGeer 1996). Moreover, Fc-dependent engagement of microglia is thought to be one mechanism through which Aβ immunotherapy operates. This treatment strategy has proven successful in improving cognitive function and reducing Aβ plaque burden in both animal models of AD and in human AD patients. However, phase II clinical trials were aborted as some patients developed symptoms of brain inflammation resembling encephalitis or meningitis following injection with Aβ (Robinson et al. 2003; Schenk et al. 2005). In the periphery, the regulation of GSK3 activity is critical for inflammatory cell differentiation, inflammatory cell migration and the secretion of pro-inflammatory cytokines (Woodgett and Ohashi 2005; Jope et al. 2007; Rodionova et al. 2007). However, little is known about the function of GSK3 in the cerebral inflammatory response, which is predominantly mediated by microglia. If, as we discuss above, GSK3 regulation is aberrant in AD brain, then this might impact upon the cerebral inflammatory response leading to the sustained secretion of neuro-toxic inflammatory mediators by microglia, in turn causing by-stander damage to neighboring neurons and contributing to neurodegenerative processes.
Concluding remarks
There is then substantial data strongly implicating GSK3 in the pathogenesis of AD. GSK3 activity and/or protein levels are increased in afflicted individuals with AD and cell biological, epidemiological and genetic evidence points to an association between AD and pathways that regulate GSK3. We postulate these pathways augment GSK3 activity through secondary changes in both regulatory Ser and Tyr phosphorylation, mediated either directly or through alterations in the insulin and/or Wnt signaling cascades or indeed through other pathways that converge on GSK3.
In various cell culture, invertebrate and mammalian models of AD increasing GSK3 activity leads to the hyper-phosphorylation of tau, increased Aβ generation and deficits in learning and memory accompanied with neurodegeneration. Most importantly inhibiting GSK3 activity reverses some of the pathological effects of over-expression of mutated APP and tau in the best available models of AD (Noble et al. 2005; Rockenstein et al. 2007). Our ‘GSK3 hypothesis of AD’ integrates and extends the well established ‘amyloid cascade hypothesis of AD’ incorporating the known key molecular events and linking these with outcomes such as memory impairment and inflammation. If correct, then this hypothesis strongly implicates GSK3 inhibitors as a novel treatment strategy for AD.
References
- Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, az-Nido J. Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 1999;453:260–264. doi: 10.1016/s0014-5793(99)00685-7. [DOI] [PubMed] [Google Scholar]
- Anderton BH, Betts J, Blackstock WP, et al. Sites of phosphorylation in tau and factors affecting their regulation. Biochem. Soc. Symp. 2001;67:73–80. doi: 10.1042/bss0670073. [DOI] [PubMed] [Google Scholar]
- Asuni AA, Hooper C, Reynolds CH, Lovestone S, Anderton BH, Killick R. GSK3alpha exhibits beta-catenin and tau directed kinase activities that are modulated by Wnt. Eur. J. Neurosci. 2006;24:3387–3392. doi: 10.1111/j.1460-9568.2006.05243.x. [DOI] [PubMed] [Google Scholar]
- Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J. 2004;23:2586–2596. doi: 10.1038/sj.emboj.7600251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci USA. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biessels GJ, Kappelle LJ. Increased risk of Alzheimer's disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem. Soc. Trans. 2005;33:1041–1044. doi: 10.1042/BST0331041. [DOI] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J. Biol. Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. PNAS. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion JP, Anderton BH, Authelet M, Dayanandan R, Leroy K, Lovestone S, Octave JN, Pradier L, Touchet N, Tremp G. Neurofibrillary tangles and tau phosphorylation. Biochem. Soc. Symp. 2001;0:81–88. doi: 10.1042/bss0670081. [DOI] [PubMed] [Google Scholar]
- Bullock BP, Habener JF. Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA-binding affinity, conformation, and increases net charge. Biochemistry. 1998;37:3795–3809. doi: 10.1021/bi970982t. [DOI] [PubMed] [Google Scholar]
- Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, Storto M, Gaviraghi G, Terstappen GC, Nicoletti F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer's brain. J. Neurosci. 2004;24:6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Park CS, Tang SJ. Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J. Biol. Chem. 2006;281:11910–11916. doi: 10.1074/jbc.M511920200. [DOI] [PubMed] [Google Scholar]
- Cho JH, Johnson GV. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J. Biol. Chem. 2003;278:187–193. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- Christie RH, Freeman M, Hyman BT. Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer's disease. Am. J. Pathol. 1996;148:399–403. [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale TC. Signal transduction by the Wnt family of ligands. Biochem. J. 1998;329 (Pt 2):209–223. doi: 10.1042/bj3290209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari GV, Papassotiropoulos A, Biechele T, et al. Common genetic variation within the low-density lipoprotein receptor-related Protein 6 and late-onset Alzheimer's disease. PNAS. 2007;104:9434–9439. doi: 10.1073/pnas.0603523104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Goni-Oliver P, Lucas JJ, Avila J, Hernandez F. Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 2006;99:1445–1455. doi: 10.1111/j.1471-4159.2006.04139.x. [DOI] [PubMed] [Google Scholar]
- Hamilton G, Proitsi P, Jehu L, Morgan A, Williams J, O'Donovan MC, Owen MJ, Powell JF, Lovestone S. Candidate gene association study of insulin signaling genes and Alzheimer's disease: evidence for SOS2, PCK1, and PPARgamma as susceptibility loci. Am. J Med Genet B Neuropsychiatr. Genet. 2007;144:508–516. doi: 10.1002/ajmg.b.30503. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Hansen TO, Rehfeld JF, Nielsen FC. GSK-3beta reduces cAMP-induced cholecystokinin gene expression by inhibiting CREB binding. Neuroreport. 2004;15:841–845. doi: 10.1097/00001756-200404090-00021. [DOI] [PubMed] [Google Scholar]
- Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Hernandez F, Borrell J, Guaza C, Avila J, Lucas JJ. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 2002;83:1529–1533. doi: 10.1046/j.1471-4159.2002.01269.x. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hooper C, Markevich V, Plattner F, et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 2007;25:81–86. doi: 10.1111/j.1460-9568.2006.05245.x. [DOI] [PubMed] [Google Scholar]
- Hoshi M, Takashima A, Noguchi K, Murayama M, Sato M, Kondo S, Saitoh Y, Ishiguro K, Hoshino T, Imahori K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci USA. 1996;93:2719–2723. doi: 10.1073/pnas.93.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsken J, Behrens J. The Wnt signalling pathway. J. Cell Sci. 2002;115:3977–3978. doi: 10.1242/jcs.00089. [DOI] [PubMed] [Google Scholar]
- Hye A, Kerr F, Archer N, Foy C, Poppe M, Brown R, Hamilton G, Powell J, Anderton B, Lovestone S. Glycogen synthase kinase-3 is increased in white cells early in Alzheimer's disease. Neurosci. Lett. 2005;373:1–4. doi: 10.1016/j.neulet.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Imahori K, Uchida T. Physiology and pathology of tau protein kinases in relation to Alzheimer's disease. J Biochem. (Tokyo) 1997;121:179–188. [PubMed] [Google Scholar]
- Ishiguro K, Omori A, Takamatsu M, Sato K, Arioka M, Uchida T, Imahori K. Phosphorylation sites on tau by tau protein kinase I, a bovine derived kinase generating an epitope of paired helical filaments. Neurosci. Lett. 1992;148:202–206. doi: 10.1016/0304-3940(92)90839-y. [DOI] [PubMed] [Google Scholar]
- Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SN, Crutcher KA. Rat microglia exhibit increased density on Alzheimer's plaques in vitro. Exp. Neurol. 1998;149:42–50. doi: 10.1006/exnr.1997.6678. [DOI] [PubMed] [Google Scholar]
- Koivisto L, Hakkinen L, Matsumoto K, McCulloch CA, Yamada KM, Larjava H. Glycogen synthase kinase-3 regulates cytoskeleton and translocation of Rac1 in long cellular extensions of human keratinocytes. Exp. Cell Res. 2004;293:68–80. doi: 10.1016/j.yexcr.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Leroy K, Yilmaz Z, Brion JP. Increased level of active GSK-3beta in Alzheimer's disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007;33:43–55. doi: 10.1111/j.1365-2990.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- Lesort M, Jope RS, Johnson GV. Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J. Neurochem. 1999;72:576–584. doi: 10.1046/j.1471-4159.1999.0720576.x. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Alessi DR. The insulin signalling pathway. Curr. Biol. 2002;12:R236–R238. doi: 10.1016/s0960-9822(02)00777-7. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, Hanger D, Mulot S, Marquardt B, Stabel S. Alzheimer's disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 1994;4:1077–1086. doi: 10.1016/s0960-9822(00)00246-3. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Mateo I, Infante J, Llorca J, Rodriguez E, Berciano J, Combarros O. Association between glycogen synthase kinase-3beta genetic polymorphism and late-onset Alzheimer's disease. Dement. Geriatr. Cogn Disord. 2006;21:228–232. doi: 10.1159/000091044. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Anti-inflammatory drugs in the fight against Alzheimer's disease. Ann. NY Acad. Sci. 1996;777:213–220. doi: 10.1111/j.1749-6632.1996.tb34421.x. [DOI] [PubMed] [Google Scholar]
- Mudher A, Shepherd D, Newman TA, et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- Mukai F, Ishiguro K, Sano Y, Fujita SC. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J. Neurochem. 2002;81:1073–1083. doi: 10.1046/j.1471-4159.2002.00918.x. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. USA. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15:28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Tanaka T, Tung YC, Braak E, Iqbal K, Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, et al. LTP Inhibits LTD in the Hippocampus via Regulation of GSK3beta. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Green PS, et al. Intranasal insulin improves cognition and modulates {beta}-amyloid in early AD. Neurology. 2007 doi: 10.1212/01.WNL.0000265401.62434.36. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Reiman EM, Webster JA, Myers AJ, et al. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron. 2007;54:713–720. doi: 10.1016/j.neuron.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SR, Bishop GM, Munch G. Alzheimer vaccine: amyloid-beta on trial. Bioessays. 2003;25:283–288. doi: 10.1002/bies.10236. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionova E, Conzelmann M, Maraskovsky E, Hess M, Kirsch M, Giese T, Ho AD, Zoller M, Dreger P, Luft T. GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood. 2007;109:1584–1592. doi: 10.1182/blood-2006-06-028951. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J. Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer B, Wiedau-Pazos M, Geschwind DH. Gene structure and alternative splicing of glycogen synthase kinase 3 beta (GSK-3beta) in neural and non-neural tissues. Gene. 2003;302:73–81. doi: 10.1016/s0378-1119(02)01092-2. [DOI] [PubMed] [Google Scholar]
- Schenk DB, Seubert P, Grundman M, Black R. A beta immunotherapy: Lessons learned for potential treatment of Alzheimer's disease. Neurodegener. Dis. 2005;2:255–260. doi: 10.1159/000090365. [DOI] [PubMed] [Google Scholar]
- Solano DC, Sironi MARI, Bonfini CLAU, Solerte SB, Govoni STEF, RACCHI MARC. Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000;14:1015–1022. doi: 10.1096/fasebj.14.7.1015. [DOI] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Sato S, Murayama O, Murayama M, Park J-M, Yamaguchi H, Takashima A. Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci. Lett. 2002;321:61–64. doi: 10.1016/s0304-3940(01)02583-6. [DOI] [PubMed] [Google Scholar]
- Swatton JE, Sellers LA, Faull RLM, Holland A, Iritani S, Bahn S. Increased MAP kinase activity in Alzheimer's and Down syndrome but not in schizophrenia human brain. Eur. J. Neurosci. 2004;19:2711–2719. doi: 10.1111/j.0953-816X.2004.03365.x. [DOI] [PubMed] [Google Scholar]
- Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl Acad. Sci. USA. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima A, Noguchi K, Michel G, Mercken M, Hoshi M, Ishiguro K, Imahori K. Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci. Lett. 1996;203:33–36. doi: 10.1016/0304-3940(95)12257-5. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, et al. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc. Natl. Acad. Sci USA. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troussard AA, Tan C, Yoganathan TN, Dedhar S. Cell-extracellular matrix interactions stimulate the AP-1 transcription factor in an integrin-linked kinase- and glycogen synthase kinase 3-dependent manner. Mol. Cell. Biol. 1999;19:7420–7427. doi: 10.1128/mcb.19.11.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turenne GA, Price BD. Glycogen synthase kinase3 beta phosphorylates serine 33 of p53 and activates p53’s transcriptional activity. BMC Cell Biol. 2001;2:12. doi: 10.1186/1471-2121-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehmas AK, Kawas CH, Stewart WF, Troncoso JC. Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol. Aging. 2003;24:321–331. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Proud CG. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem. J. 1993;294 (Pt 3):625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR, Ohashi PS. GSK3: an in-Toll-erant protein kinase? Nat. Immunol. 2005;6:751–752. doi: 10.1038/ni0805-751. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol. (Berl) 1996;92:232–241. doi: 10.1007/s004010050513. [DOI] [PubMed] [Google Scholar]
- Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, Frautschy SA, Cole GM. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J. Neurosci. 2004;24:11120–11126. doi: 10.1523/JNEUROSCI.2860-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]