

Abstract
Pitx2 is a paired-like homeodomain gene that acts as a key regulator of eye development. Despite its significance, upstream regulation of Pitx2 expression during eye development remains incompletely understood. We use neural crest-specific ablation of Ctnnb1 to demonstrate that canonical Wnt signaling is not required for initial activation of Pitx2 in neural crest. However, canonical Wnt signaling is subsequently required to maintain Pitx2 expression in the neural crest. Eye development in Ctnnb1-null mice appears grossly normal early but significant phenotypes emerge following loss of Pitx2 expression. LEF-1 and β-catenin bind Pitx2 promoter sequences in ocular neural crest, indicating a likely direct effect of canonical Wnt signaling on Pitx2 expression. Combining our data with previous reports, we propose a model wherein a sequential code of retinoic acid followed by canonical Wnt signaling are required for activation and maintenance of Pitx2 expression, respectively. Other key transcription factors in the neural crest, including Foxc1, do not require intact canonical Wnt signaling.
Keywords: Homeodomain transcription factor, Wnt signaling, Pitx2, eye development, anterior segment, neural crest
INTRODUCTION
It is a well-established paradigm that the net expression patterns of important developmental transcriptional regulators result from the integration of multiple inductive signaling inputs, but the underlying molecular details are poorly understood in most systems. The mammalian eye provides an excellent system for further detailed analysis of this model because extensive inductive signaling contributed by multiple major signaling pathways is passed back and forth between the four embryonic primordial contributing to the mature organ. For example, inductive signaling from the neuroectoderm-derived optic vesicle is required for initiation of the lens differentiation program in the overlying surface ectoderm and the newly induced lens placode in turn signals back to the optic vesicle, specifying the position where the presumptive retina will emerge within the neuroectoderm (Chow and Lang, 2001).
Development of the ocular anterior segment, including the cornea, limbus, conjunctiva, eyelids, and ciliary body is particularly complex: it requires the interaction of neuroectoderm, surface ectoderm, and neural crest and mesoderm mesenchyme. The mature cornea, limbus, conjunctiva, eyelids, and ciliary body are chimeric, arising from an integration of neuroectoderm (ciliary body) or surface ectoderm (cornea, limbus, conjunctiva, and eyelids) and mesenchyme. Development of the primordia to these structures commences immediately after transition of the optic vesicle to the optic cup and separation of the lens vesicle from the surface ectoderm, when mesenchyme cells consisting primarily of neural crest begin to migrate between the anterior optic cup/lens vesicle and the overlying surface ectoderm (Cvekl and Tamm, 2004; Gould et al., 2004). Retinoic acid (RA) signaling from the optic cup, lens, and surface ectoderm crest plays a critical role in orchestrating the morphogenesis of the anterior segment at this stage of development by activating expression of key transcription factor genes in the neural crest (Matt et al., 2005; Molotkov et al., 2006; Matt et al., 2008).
A critical downstream target of RA signaling in the ocular neural crest at this stage is the homeobox gene, Pitx2 (Matt et al., 2005; Molotkov et al., 2006; Matt et al., 2008). Global or neural crest-specific loss of Pitx2 in mice results in severe dysmorphogenesis of the eye, including loss of any recognizable anterior segment structures except the lens (Gage et al., 1999; Evans and Gage, 2005). These defects closely phenocopy those found in mice in which RA signaling is disrupted, in part because Pitx2 expression is lost (Matt et al., 2005; Molotkov et al., 2006; Matt et al., 2008). Heterozygous mutations in human PITX2 are one genetic cause of Axenfeld-Rieger Syndrome, which features anterior segment defects and an associated high risk for early-onset glaucoma (Semina et al., 1997; Alward, 2000). An important downstream target of PITX2 is Dkk2, which encodes an extracellular antagonist of canonical Wnt/β-catenin signaling. DKK2 is required to moderate Wnt/β-catenin signaling activity in both the neural crest and overlying surface ectoderm of the anterior segment (Gage et al., 2008). Collectively, these observations implicate PITX2 as an important integration node between these two major signaling pathways during eye development (Gage et al., 2008; Gage and Zacharias, 2009; Kumar and Duester, 2010). In addition, PITX2, β-catenin, and LEF-1 interact synergistically to regulate the Lef1 promoter, further strengthening the connection between PITX2 and canonical Wnt signaling (Vadlamudi et al., 2005).
Pitx2 itself has been implicated as a transcriptional target of canonical Wnt/β-catenin signaling in the developing pituitary gland through sequences located 5’ (Kioussi et al., 2002) and 3’ (Ai et al., 2007) of the gene. Canonical Wnt signaling may also play a role in post-transcriptional stabilization of the Pitx2 mRNA (Briata et al., 2003). Treatment of embryos with LiCl, an agonist of canonical Wnt signaling, stimulates expression of Pitx2 within the periocular mesenchyme (Kioussi et al., 2002). In addition, PITX2 protein levels remain persistently high in the ocular neural crest of Dkk2 deficient mice, providing evidence for an auto-regulatory feedback loop (Gage et al., 2008). Although the precise mechanism underlying this expression change is not known, these data also suggested that a combinatorial code including both RA and canonical Wnt/β-catenin signaling may be required to activate Pitx2 expression in the neural crest.
In this report, we present evidence that canonical Wnt/β-catenin signaling is not required for initial activation of Pitx2, but it is required for maintenance of its continued expression likely via direct binding of Lef-1 and β-catenin to the Pitx2 promoter. Thus, the two signaling pathways do contribute to a code required for normal Pitx2 expression but they act sequentially, rather than in concert. Transient expression of Pitx2 is sufficient to activate a genetic program that is required to anchor the anterior optic cup/lens to the overlying surface ectoderm. Finally, the dependence of Pitx2 expression upon canonical Wnt/β-catenin signaling appears to be unique among a panel of known regulators of early anterior segment development.
RESULTS
Neural crest-specific loss of β-catenin is efficient and does not notably alter early eye development
β-catenin is expressed throughout the tissues of the developing eye, including the neural crest, through the period during which Pitx2 expression is activated (Fig. 1A). This expression pattern was consistent with the possibility that intact responsiveness to canonical Wnt/ β-catenin signaling by the neural crest might be required for activation of Pitx2 in these cells, as well as more generally for normal eye development during this stage.
Figure 1. Loss of β-catenin protein and analysis of early eye morphogenesis in Ctnnb1-ncko mice.

(A & E) Eye sections of e10.5 control and Ctnnb1-ncko littermates were stained for detection of β-catenin by immunofluorescence (green) and imaged with DAPI (blue) to visualized nuclei. Note how the mesoderm-derived endothelial cells of blood vessels (<) stand out in Ctnnb1-ncko mice due to the efficient loss of β-catenin in surrounding neural crest. (B-D) Eye sections of control embryos at the indicated ages were stained by H&E to visualize eye morphogenesis. (F-H) Eye sections of Ctnnb1-ncko littermates of wild type embryos in B-D were stained by H&E to visualize eye morphogenesis. Key: retina (r); lens (l); neural crest (n); blood vessel (<); optic stalk (os); magnification bar = 100 μm.
To test these hypotheses, we generated mice lacking β-catenin in neural crest by mating mice carrying the conditionally null Ctnnb1tm2Kem allele (Brault et al., 2001) with mice transmitting the neural crest-specific Wnt1Cre1 transgene (Danielian et al., 1998). Since β-catenin is the essential downstream effector of the pathway, this genetic approach very efficiently and comprehensively prevents neural crest in these mice from responding to canonical Wnt signaling (Brault et al., 2001). However, the effects of abating β-catenin on eye development were not previously described (Brault et al., 2001). β-catenin is undetectable in neural crest of Ctnnb1-ncko (Wnt1Cre;Ctnnb1tm2Kem/tm2Kem) embryos at e10.5 (Fig. 1E). Eyes of the mutant embryos are grossly normal at this time point (Fig. 1 B vs. F), indicating that intact Wnt signaling in neural crest is not required for their migration into the eye field and may not be required for early stages of eye morphogenesis. In contrast, neural crest in mutants with constitutively activated β-catenin does not efficiently populate the developing eye (data not shown) (Lee et al., 2004). Ctnnb1-ncko eyes begin to exhibit a thick, neuroblastic optic stalk that is different from wild type controls and a modest misorientation of the optic cup towards the ventral side by e11.5 (Fig. 1 C vs. G). These features are more accentuated by e12.5, when the optic stalk remains abnormally thick and contiguous with the ventral diencephalon, which is displaced peripherally, and the optic cup is rotated ventrally (Fig 1 D vs. H). Collectively, these data suggest that the overt requirement for Wnt/β-catenin responsiveness in neural crest begins at approximately e11.5 during mouse eye development.
Canonical Wnt/β-catenin signaling is required for maintenance of PITX2 expression in neural crest
We next assessed whether intact canonical Wnt/β-catenin signaling is required for activation or maintenance of Pitx2 expression in neural crest by immunostaining Ctnnb1-ncko embryos for both β-catenin and PITX2. Activation of the R26R Cre reporter locus (Soriano, 1999) was used to fate map neural crest in the mutant eyes and to confirm comprehensive loss of β-catenin in these cells (data not shown). The overall pattern of PITX2 expression in eyes of Ctnnb1-ncko embryos, including in neural crest lacking β-catenin, is indistinguishable from control littermates at e10.5, although levels of PITX2 expression sometimes appear diminished in individual neural crest cells (Fig. 2 A-D). PITX2 expression is still detectable but dramatically reduced in neural crest of Ctnnb1-ncko mice compared to control littermates at e11.5 (Fig. 2E-H). By e12.5, PITX2 expression is undetectable in neural crest of Ctnnb1-ncko embryos but high-level expression is observed in neural crest surrounding eyes of control littermates (Fig. 2I-L). Mesoderm expression appears to be unaffected in Ctnnb1-ncko eyes at any timepoint examined (Fig. 2).
Figure 2. PITX2 protein expression in Ctnnb1-ncko versus control eyes during early eye development.
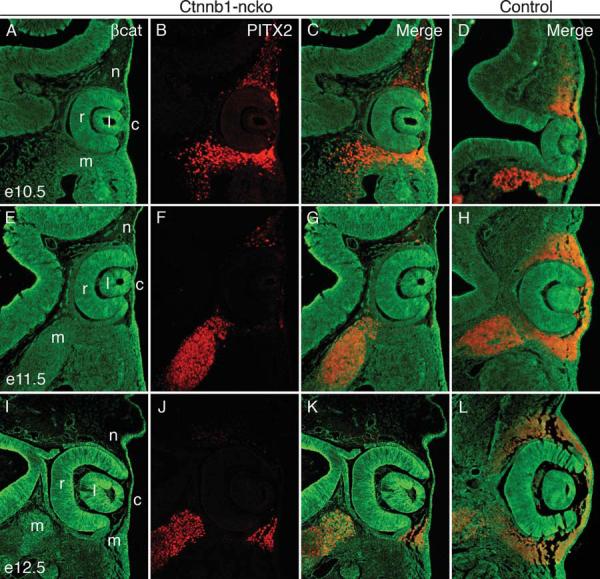
β-catenin (green) and PITX2 (red) protein expression were detected by double immunofluorescence on eye sections taken from Ctnnb1-ncko and control littermates at the indicated time points. Overall PITX2 expression is comparable in the two genotypes at e10.5 but is rapidly reduced (e11.5) and then lost (e12.5) specifically in neural crest cells of Ctnnb1-ncko eyes. Mesoderm expression of PITX2 is unaffected in the mutants. Key: retina (r); lens (l); cornea (c); neural crest (n); mesoderm (m).
We considered the possibility that intact canonical Wnt/β-catenin signaling could be required for survival of neural crest and that the apparent loss of PITX2 expression might result from the death of these cells. In addition to Pitx2flox, mutant animals also carried the R26R reporter allele, allowing us to fate map neural crest following Cre-mediated activation of lacZ (Soriano, 1999). Neural crest expressing lacZ are abundant in Ctnnb1-ncko eyes throughout the relevant developmental stages (Fig. 3A,D). Consistent with this finding, TUNEL assays detected no differences in apoptotic cell death in mutant versus control littermate eyes (Fig. 3B,C,E,F). Altogether, these data indicate that maintenance of PITX2 expression in neural crest during eye development requires intact canonical Wnt/β-catenin signaling.
Figure 3. β-catenin is not required for survival of ocular neural crest.

(A,D) Survival of neural crest mesenchyme in Ctnnb1-ncko eyes at the indicated time points is demonstrated by using immunofluorescence to detect β-galactosidase expressed following Cre-mediated activation of the R26R reporter allele. (B,C,E,F) Apoptotic cells in Ctnnb1-ncko and control littermates at the indicated time points as detected by TUNEL assay. No difference in TUNEL-positive cells is observed between the two genotypes. Key: retina (r); lens (l); cornea (c); neural crest (n); mesoderm (m). Note: The image in panel A of this figure is taken from an adjacent section to the image in Fig. 2A and the image in panel D of this figure is taken from an adjacent section to the image in Fig. 2E.
β-catenin and LEF1 bind Pitx2 promoter sequences in developing periocular mesenchyme
A functional TCF/LEF responsive element is located 200-bp 5’ to the promoter for the Pitx2a and Pitx2b mRNA isoforms (Fig. 4A) (Kioussi et al., 2002). This element is conserved between mice and humans, and is active in cells lines from the anterior pituitary gland (Kioussi et al., 2002). A second functional TCF/LEF responsive element is present within an enhancer located 3’ to the Pitx2 gene and is required for in vivo enhancer activity within Rathke's pouch, the embryonic precursor to the anterior pituitary gland (Ai et al., 2007). However, the potential relevance of these elements to Pitx2 expression in periocular mesenchyme has not been explored.
Figure 4. β-catenin and LEF1 physically associate with Pitx2 promoter sequences.
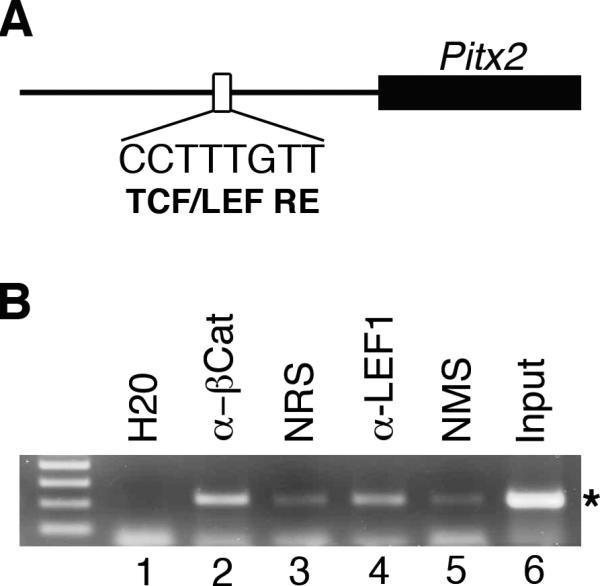
(A) Schematic illustrating location of conserved TCF/LEF responsive element located 200-bp upstream of the 5’ Pitx2 promoter. (Modified from Kioussi et al). (B) ChIP analysis on primary cultures of mesenchyme grown from wild type e12.5 eye primordia with specific antibodies, normal control sera, water and sheared input DNA. Specific PCR product (*) is significantly enriched in samples immunoprecipitated with rabbit anti- β-catenin and mouse anti-LEF1 antibodies as compared with the normal rabbit serum (NRS) and normal mouse serum (NMS), respectively, indicating physical association of β-catenin and LEF1 with the Pitx2 promoter sequences in cells derived from periocular mesenchyme.
To determine whether either of the two previously identified TFC/LEF responsive elements are occupied during eye development, we performed chromatin immunoprecipitation on periocular mesenchyme derived from e12.5 embryos. These assays revealed that Pitx2 promoter sequences containing the TCF/LEF responsive element are significantly enriched over background levels in samples immunoprecipitated with antibodies specific to either β-catenin or LEF1 (Fig. 4B). In contrast, neither sequences spanning the TCF/LEF responsive element located within the 3’ enhancer, nor sequences within a Pitx2 intron that are not associated with a potential TCF/LEF site were enriched by immunoprecipitation with the specific antibody (data not shown). These data indicate that the TCF/LEF responsive element within the 5’ Pitx2 promoter sequences is physically occupied by β-catenin and LEF1 in periocular mesenchyme and suggest that canonical Wnt signaling acts directly through this site to maintain Pitx2 expression in these cells during development.
Transient expression of Pitx2 is sufficient to rescue initiation of anterior segment development
The optic stalk of wild type eyes elongates as development proceeds and the optic cup remains in intimate contact with the overlying surface ectoderm (Fig. 5A). In contrast, global or neural crest-specific Pitx2 knockout mice exhibit severe defects including a thickened and substantially shortened the optic stalk, as well as displacement of the optic cup to a midline position substantially removed from the overlying ocular surface ectoderm (Fig. 5B) (Gage et al., 1999; Kitamura et al., 1999). We have previously hypothesized that displacement of the optic cup in Pitx2 mutant mice results primarily from the foreshortened optic stalk “pulling” the optic cup towards the midline and away from the surface ectoderm (Evans and Gage, 2005). However, optic cups of Ctnnb1-ncko mice are appropriately positioned directly beneath the overlying ocular surface ectoderm, despite the fact that morphogenesis of the optic stalk appears to be disrupted at least as severely in these animals as in Pitx2 mutant mice (Fig. 5C). This raised the possibility that transient expression of Pitx2 in Ctnnb1-ncko mice is sufficient to activate a genetic program required to anchor the optic cup/lens to the overlying ocular surface ectoderm.
Figure 5. Comparison of eye phenotypes in Pitx2 and Ctnnb1 mutant mice.

Transverse sections of eyes from e14.5 embryos with the indicated genotypes were stained with hematoxylin and eosin. Images of each genotype were taken at the same magnification. Key: lens (l); surface ectoderm (e).
We tested this hypothesis by performing temporal knockouts of Pitx2 (Pitx2-tko) in mice carrying our conditional Pitx2flox allele (Gage et al., 1999) and UBC-CreER (Gruber et al., 2007). The Cre-ER fusion protein required to delete Pitx2 is ubiquitously expressed by this transgene but is functionally inactive except in the presence of tamoxifen (Gruber et al., 2007). In animals with the genotype UBC-CreER;Pitx2flox/flox, injection of timed pregnant dams with tamoxifen resulted in complete loss of PITX2 protein within 24 hours (Supplemental Fig. 1). We therefore injected timed pregnant dams from the appropriate matings with tamoxifen at e10.5, which permitted normal activation but resulted in the loss of all PITX2 protein by e11.5, a time course similar to that observed in Ctnnb1-ncko mice.
In contrast to global or neural crest-specific Pitx2 knockout eyes (Fig. 5B and data not shown), the eyes of Pitx2-tko mutant mice are located peripherally, similar to, although not identical to the eyes of wild type and Ctnnb1-ncko mice (Fig. 5D). The corneal mesenchyme of e14.5 Pitx2-tko eyes is significantly thicker than that found in wild type or Ctnnb1-ncko eyes, an early manifestation of the severe corneal defects found in these mice at later time points (data not shown). The Pitx2-tko eyes are connected to the lateral diencephalon by an optic stalk that is thickened and shortened relative to wild type eyes (Fig. 5A vs. 5D), a defect that appears similar to what we have reported previously for global and neural crest-specific Pitx2 knockout eyes (Gage et al., 1999; Evans and Gage, 2005). The observation that Ctnnb1-ncko and Pitx2-tko eyes are located peripherally, despite significant defects in development of the optic stalk, indicates that the elongation of the optic stalk alone cannot determine placement of the optic cup and lens. Rather, these data suggest a model wherein early expression of PITX2 protein at e10.5-11 also activates a program of adhesion that facilitates a stable interaction between the anterior optic cup, the lens, the anterior segment mesenchyme, and the overlying ocular surface ectoderm.
Other regulators of anterior segment development do not depend on canonical Wnt signaling in ocular neural crest
Foxc1, Foxc2, and Tfap2b each encode transcription factors that are essential in the ocular neural crest for normal anterior segment development and function. We analyzed the expression of these genes in Ctnnb1-ncko mice to determine whether their expression in neural crest might require intact canonical Wnt/β-catenin signaling and because the results could provide insight into the extent to which initial patterning of the anterior segment might be disrupted in these mice.
The forkhead transcription factor gene, Foxc1 is required for anterior segment development in mice, and mutations in human FOXC1 are a second genetic cause of Axenfeld-Rieger Syndrome (Mears et al., 1998; Lehmann et al., 2000; Lehmann et al., 2003). Like Pitx2, retinoic acid signaling is also required to activate Foxc1 expression in the ocular neural crest (Matt et al., 2005; Molotkov et al., 2006; Matt et al., 2008). FOXC1 protein is normally expressed in neural crest of the periocular mesenchyme and in the surface ectoderm of the palpebral conjunctiva and palpebral epidermis in the eyelid (Fig. 6A,C) (Kume et al., 2000; Gage et al., 2008). This expression pattern within the anterior segment is unchanged in Ctnnb1-ncko mice at e12.5 and e14.5, suggesting that Foxc1 expression within these structures of the eye is not dependent on canonical Wnt signaling (Fig. 6B,D). However, FOXC1 protein levels are diminished in mesenchyme adjacent to the optic cup and normally fated to become sclera in the Ctnnb1-ncko mutants. However, the sclera is absent in Ctnnb1-ncko eyes. Therefore, its not clear whether this loss of Foxc1 expression is a direct result of inactivating canonical Wnt signaling in these cells or an indirect result of losing scleral specification, which requires Pitx2 expression in neural crest (Evans and Gage, 2005).
Figure 6. Additional transcription factor expression in Ctnnb1-ncko versus control eyes.

Eye sections of control and Ctnnb1-ncko littermates at the indicated developmental stages were immunostained to detect expression of the transcription factors, FOXC1 (A-D), FOXC2 (E-H), or TFAP2B (I-L). For each, expression in neural crest mesenchyme and surface ectoderm was independent of β-catenin function in the neural crest.
Mutations in Foxc2/FOXC2 cause distichiasis (extra eyelashes) in mice and humans, as well as additional anterior segment defects in mice (Smith et al., 2000; Kriederman et al., 2003). In developing wild type eyes, FOXC2 protein is initially expressed in the neural crest surrounding the optic cup, but expression in corneal neural crest is down regulated by e12.5 and beyond (Fig. 6E,G) (Kidson et al., 1999; Gage et al., 2008). FOXC2 is also expressed in surface ectoderm of the palpebral conjunctiva (Fig. 6E,G) (Gage et al., 2008). These expression patterns are conserved in both the neural crest and palpebral conjunctiva of Ctnnb1-ncko mice, suggesting that initial patterning of anterior segment structures is not completely lost in these mutants and Foxc2 does not appear to be regulated by canonical Wnt signaling (Fig. 6F,H).
Tfap2b encodes an AP-2 family transcription factor whose expression from e12.5-14.5 marks neural crest of the corneal, limbal, and conjunctival mesenchyme, as well as surface ectoderm of the presumptive conjunctiva and eyelid (Fig. 6I,K). These expression patterns are retained in Ctnnb1-ncko mice (Fig. 6J,L), providing further evidence that initial patterning of anterior segment structures is not completely lost in these mutants.
DISCUSSION
Inappropriately low or high levels of PITX2 activity lead to anterior segment defects in mice and humans (Semina et al., 1996; Kozlowski and Walter, 2000; Priston et al., 2001; Holmberg et al., 2004), indicating that tight control of Pitx2 in time and space is a prerequisite for normal eye development and function. Similar control of Pitx2 expression is required for normal development of organs such as the eye, pituitary gland, craniofacial structures, and heart but the underlying mechanisms regulating expression in any of these structures are not well defined. In the present study, we extend our understanding of the molecular requirements leading to normal Pitx2 expression during eye development, information that may have relevance in other tissues and organs.
Normal Pitx2 expression in ocular neural crest is the net response to inputs from multiple signaling pathways
Pitx2 is not expressed in neural crest migrating to the eye primordia. Rather, retinoic acid directly activates Pitx2 expression in neural crest after arrival of these cells in the developing eye field (Fig. 7) (Matt et al., 2005; Molotkov et al., 2006; Matt et al., 2008; Kumar and Duester, 2010). The anterior optic cup, lens vesicle, and ocular surface ectoderm are the sources of the retinoic acid, providing an effective means for stimulating Pitx2 activation only within neural crest adjacent to the developing eye. The action of retinoic acid on the neural crest is direct as demonstrated by neural crest specific ablation of the appropriate nuclear receptors (Matt et al., 2005; Matt et al., 2008). Recently, a DR5 RA response element that binds all three RA receptors in the developing eye was identified upstream of Pitx2, supporting the idea that Pitx2 is a direct RA target (Kumar and Duester, 2010). The competence of neural crest to activate Pitx2 in response to retinoic acid appears to be transient and ends within days of the stage when Pitx2 is normally first expressed (See and Clagett-Dame, 2009). Therefore, a second mechanism likely underlies persistent Pitx2 expression within the neural crest, a fact that fits well with the data presented herein.
Figure 7. Model for regulation of Pitx2 expression in the anterior segment by retinoic acid and Wnt signaling.

Phase I: Pitx2 expression is initiated by retinoic acid signaling produced by the anterior optic cup, anterior lens vesicle, and surface ectoderm. Phase II: Pitx2 expression is maintained by canonical Wnt signaling produced by the surface ectoderm. Phase III: Pitx2 expression levels are reduced to lower steady-state levels due to inhibition of canonical Wnt signaling activity by DKK2.
We tested the potential requirements for canonical Wnt signaling activity on Pitx2 expression in ocular neural crest. Unlike retinoic acid signaling, canonical Wnt/β-catenin signaling is not required for activation of Pitx2, as initial expression in Ctnnb1-ncko mice is comparable to control littermates. In contrast, Pitx2 expression rapidly diminishes and is soon lost in ocular neural crest of Ctnnb1-ncko mice. We conclude that intact canonical Wnt/β–catenin signaling is required for maintenance of Pitx2 expression in neural crest but is not sufficient for activation in these cells (Fig. 7). The action of Wnt signaling appears to be direct as a functional TCF/LEF site within the Pitx2 promoter is occupied by LEF-1 and β-catenin in ocular neural crest. Collectively, we propose a model in which a serial code of signaling pathways is required for normal overall Pitx2 expression in ocular neural crest, with retinoic acid required for activation and canonical Wnts subsequently required for maintained expression (Fig. 7). As with retinoic acid, the ocular surface ectoderm and optic cup appear are likely to be the sources of Wnt ligands (Liu et al., 2003).
Modulation of canonical Wnt/β-catenin signaling activity levels also mediates net local PITX2 protein levels over time in the developing and mature eye (Fig. 7) (Gage et al., 2008). Most Axenfeld-Rieger cases linked to PITX2 arise via a haploinsufficiency mechanism, although an apparent gain-of-function mutation has also been reported (Kozlowski and Walter, 2000; Priston et al., 2001). Over expression of PITX2 also results in features of Axenfeld-Rieger in mice, including corneal opacification, corneal hypertrophy, and irido-corneal adhesions (Holmberg et al., 2004). Therefore, the ability to ultimately control and fine-tune PITX2 protein levels through modulation of canonical Wnt signaling activity is likely critical for normal eye development in humans and mice. Additional negative regulators of canonical Wnt signaling are expressed in the developing and mature eye (Chow and Lang, 2001; Chen et al., 2004; Diep et al., 2004; Fjeld et al., 2005). Although their effects on PITX2 expression levels have not been tested, it may be that the net effects of DKK2 and additional negative regulators on canonical Wnt signaling activity provide a key mechanism for providing temporal and spatial control over PITX2 protein levels once the Pitx2 gene is initially activated by retinoic acid.
It appears that complex regulation is a general feature of Pitx2 expression in multiple tissues, as initiation and maintenance of asymmetric Pitx2 expression in the left lateral plate mesoderm is also dependent upon a two-step process (Shiratori et al., 2001). Regulation of expression at multiple steps and, potentially by divergent mechanisms in different lineages as development proceeds, likely provides advantages over more simple forms of regulation. In the eye, RA signaling activates Pitx2 expression within a large pool of neural crest cells that are subsequently fated to generate multiple mature tissues over the course of eye development, including corneal stroma, corneal endothelium, and trabecular meshwork. Local and temporal differences in signaling environment may determine whether Pitx2 expression is maintained, and at what levels - conditions that likely have a profound effect on fate decisions undertaken by individual cells.
Inputs from other signaling pathways, in addition to retinoic acid and Wnts are likely to contribute to the code required to achieve appropriate temporal and spatial Pitx2 expression levels during eye development. Hedgehog appears to be a third component of the signaling code required to maintain Pitx2 expression during eye development. At e12.5, Pitx2 expression is largely equivalent between Ihh-/- eyes and those of control littermates (Dakubo et al., 2008). By e13.5, however, Pitx2 expression is lost from mesenchyme located at the posterior and periphery of Ihh-/- eyes, establishing that hedgehog signaling is required to maintain Pitx2 expression within the subset of periocular mesenchyme fated to contribute the scleral coating (Dakubo et al., 2008). Notably, hedgehog signaling is not sufficient to activate Pitx2 expression indicating that this step is mediated by retinoic acid or another signaling pathway. Therefore, in posterior periocular mesenchyme, loss of hedgehog and canonical Wnt signaling has similar consequences, suggesting that the two pathways may act in parallel, a possibility that should be experimentally tested. However, hedgehog signaling is not required to maintain Pitx2 expression in the anterior segment, indicating that there are clear differences in the requirements for the two pathways, depending upon the cellular position within the eye primorium (Dakubo et al., 2008). This provides further evidence that overall regulation of Pitx2 expression in these cells is accomplished by a multistep mechanism. TGB-β signaling has also been implicated in Pitx2 expression within anterior segment neural crest but the functions of this pathway with respect to early development are unclear (Ittner et al., 2005).
The requirement for intact canonical Wnt signaling activity in ocular neural crest is not shared by several other key transcription factors in anterior segment development. The inclusion of Foxc1 in this group is particularly surprising since it shares many features with Pitx2, including a dependence on retinoic acid signaling for its activation and Indian hedgehog for persistent expression in peripheral and posterior mesenchyme (Matt et al., 2005; Molotkov et al., 2006; Dakubo et al., 2008; Matt et al., 2008). Alterations in Foxc1/FOXC1 gene dose lead to Axenfeld-Rieger Syndrome in humans and cause anterior segment defects in mice. This indicates that achieving appropriate FOXC1 activity levels is a prerequisite for normal eye development and, similar to PITX2, are likely subject to tight temporal and spatial control(s) (Mears et al., 1998; Lehmann et al., 2000; Smith et al., 2000). However, despite the implication of common regulatory mechanisms suggested by these functional similarities, it does not appear that a requirement for canonical Wnt signaling activity for maintenance of Foxc1/FOXC1 expression is among the similarities.
Roles for canonical Wnt signaling in the neural crest during early eye morphogenesis
Canonical Wnt signaling has the capacity to control many functions in neural crest fated to contribute ocular tissues, including induction, migration, cell survival, cell proliferation, and differentiation. Our data suggest that intact canonical Wnt signaling is not required for migration of neural crest to the eye primordia, as these cells populate early Ctnnb1-ncko eyes at a level comparable to control eyes. Similarly, intact canonical Wnt signaling appears to be dispensable for early eye morphogenesis from the neural and surface ectoderm, as the optic cup and lens vesicle are grossly normal in Ctnnb1-ncko mutants at e10.5. Therefore, we conclude that the earliest stages of eye development do not require competence of the neural crest to respond to canonical Wnt signaling.
In contrast, Ctnnb1-ncko eyes begin to exhibit morphological differences from control littermates by e11.5, when deviations in the cornea, orientation of the optic cup, and the optic stalk first become apparent. These phenotypes are enhanced over the next several days and are followed by agenesis of the sclera and eyelids. The initial emergence of these phenotypes correlates strongly with the timing of loss of Pitx2 expression. In addition, these phenotypes encompass many of the phenotypes observed in Pitx2 mutant mice (Gage et al., 1999; Evans and Gage, 2005). Collectively, these data suggest that maintenance of Pitx2 expression is an important early functional target of canonical Wnt signaling activity in the neural crest during eye development.
Development of the cornea, optic cup, and optic stalk are complex processes and must be regulated by many factors and pathways. Therefore, it was somewhat surprising that none of the other key regulators of eye development that we tested were dependent on canonical Wnt signaling for their expression. This was likely due to the fact that we only tested a small number of genes that had previously been identified as important transcriptional regulators of anterior segment development, comparable to Pitx2. We expect that canonical Wnt signaling regulates the expression and function of additional genes in the ocular neural crest that are required for eye development, and that these will be revealed in the future using more open-ended, unbiased approaches. Discovery of these genes and ascertaining their functions are prerequisites for a more complete understanding of eye development.
Pitx2 expression facilitates cohesion between tissues during early anterior segment morphogenesis
The optic cups in global and neural crest-specific Pitx2 mutant eyes are displaced a considerable distance from the surface ectoderm (Gage et al., 1999; Evans and Gage, 2005), whereas eyes of Ctnnb1-ncko mice are located in the wild type position at the periphery of the head, immediately beneath the surface ectoderm. Transient expression of Pitx2 in temporal knockout mice also largely rescues the displaced optic cup and lens phenotypes of global and neural crest-specific Pitx2 mutant mice, permitting them to remain localized beneath the surface ectoderm in association with the presumptive corneal epithelium. Collectively, these data suggest that early Pitx2 expression in the neural crest activates a mechanism for bonding anterior segment elements together, maintaining the position of these elements relative to each other as the head grows rapidly in size during development. This bonding mechanism likely works in concert with the Pitx2-dependent signaling that is required for optic stalk extension (Evans and Gage, 2005).
The molecular mechanism(s) contributing to this process remains to be determined. At an earlier stage of eye development, during formation of the optic cup and lens vesicle, contractile filopodia tether the presumptive lens to the retina via a Cdc42- and IRSp53-dependent mechanism, providing a method for coordinated evagination between the two epithelial layers (Chauhan et al., 2009). This raises the possibility that early expression of Pitx2 may activate similar connections between the anterior optic cup, lens, neural crest, and surface ectoderm during the subsequent phase of eye development. Alternatively, Pitx2 may activate expression of adhesive extracellular matrix molecules, such as integrins or collagens that bond together the various tissue components. Further work will be required to identify the underlying molecular mechanism(s).
MATERIALS AND METHODS
Mouse strains and generation of timed pregnant litters
Generation of alleles for Cre-mediated inactivation of Ctnnb1 (Ctnnb1tm2Kem) (Brault et al., 2001) and Pitx2 (Pitx2flox) (Gage et al., 1999), or Cre-mediated activation of a lacZ reporter (R26Rflox) was described previously (Soriano, 1999). Transgenic mice expressing Cre under the control of the Wnt1 promoter (Wnt1-Cre) were obtained from A. McMahon (Danielian et al., 1998). Transgenic mice expressing a tamoxifen-inducible form of Cre (Cre-ERT2) under the control of the ubiquitously expressed Ubc9 promoter (Ubc-CreERT2) were obtained from The Jackson Laboratories (Bar Harbor, ME) (Gruber et al., 2007; Ruzankina et al., 2007). The Wnt1-Cre; Ctnnb1+/tm2Kem parental line was generated by mating Wnt1-Cre and Ctnnb1tm2Kem mice. The Ubc-CreERT2; Pitx2+/null parental line was generated by mating Ubc-CreERT2 and Pitx2null mice. Mice were maintained as separate breeding colonies at the University of Michigan. All procedures involving mice were approved by the University of Michigan Committee on Use and Care of Animals. All experiments were conducted in accordance with the principles and procedures outlined in the NIH Guidelines for the Care and Use of Experimental Animals.
Wnt1-Cre; Ctnnb1+/tm2Kem mice were mated to Ctnnb1tm2Kem/tm2Kem; R26Rflox/flox mice to generate timed pregnancies for neural crest-specific Ctnnb1 knockout mice, designating the day after mating as embryonic day 0.5. Embryos were collected by caesarian section at the appropriate stages, after euthanasia of the mother. The resulting embryos were genotyped for Cre (Gage et al., 2005), Ctnnb1 (Brault et al., 2001), or R26R (Soriano, 1999) using PCR-based methods. Embryos with a Wnt1-Cre; Ctnnb1tm2Kem/tm2Kem genotype were considered mutant, while Ctnnb1+/tm2Kem or Ctnnb1tm2Kem/tm2Kem embryos lacking the Wnt1-Cre transgene were used as controls.
Ubc-CreERT2; Pitx2+/null mice were mated to Pitx2flox/flox;R26Rflox/flox mice to generate timed pregnancies for temporal-specific Pitx2 knockout mice, designating the day after mating as embryonic day 0.5. A single intraperitoneal injection of tamoxifen (Sigma, T5648-1G) suspended in corn oil at a dose of 100 μg per gram of body weight was administered to the pregnant dam at noon on e10.5. Embryos were collected on either e11.5 or e14.5 by caesarian section after euthanasia of the mother. The resulting embryos were genotyped for Cre (Gage et al., 2005), Pitx2 (Gage et al., 1999), or R26R (Soriano, 1999) using PCR-based methods. Embryos with a Ubc-CreERT2; Pitx2flox/null genotype were considered mutant. Embryos with the genotype Pitx2+/flox were considered wild type controls while embryos with the genotype Pitx2flox/null were considered heterozygous controls.
Embryo processing and histology
All embryos were fixed in 4% paraformaldehyde in PBS, washed, dehydrated, and processed for sectioning in Paraplast Plus (McCormick Scientific; St. Louis, MO). Mounted paraffin sections for morphological analysis were dewaxed, rehydrated, and stained with hematoxylin and eosin.
Immunofluorescence
Paraffin sections were dewaxed and treated for antigen retrieval by boiling for 10 minutes in 10 mM citrate buffer (pH6.0). Sections were treated with Image-iT FX signal enhancer (Molecular Probes; Eugene, OR) for 30 minutes. Immunostaining was performed according to standard protocols. Briefly, sections were incubated with primary antibodies against FoxC1, FoxC2 (Abcam; Cambridge, MA), TFAP2B (Novus Biologicals; Littleton, CO), β-catenin (Santa Cruz Biotechnology; San Diego, CA), β-galactosidase (kind gift from Tom Glaser), and PITX2 (Hjalt et al., 2000). Treatment with the primary antibody was followed with biotinylated species-specific secondary antibodies (Jackson Immuno Research; West Grove, PA). Signals were detected using tyramide signal amplification kits (Molecular Probes; Eugene, OR). Photography was performed on a Nikon Eclipse 800 fluorescence microscope.
TUNEL assay
Terminal dUTP nick end labeling (TUNEL) was performed using an In situ Cell Death Detection kit (Roche) per manufacturer protocol. If followed with an antibody staining, treatment with hydrogen peroxide was omitted.
Primary Cell Isolation and Chromatin Immunoprecipitation Assays
Eye primordia and surrounding mesenchyme microdissected from multiple wild type e12.5 embryos were pooled and cultured until primary mouse fibroblasts (MEF) were obtained as previously described (Crofford et al., 1994). ChIP assays were performed on the MEFs (passages 3 through 8) as previously described (Gummow et al., 2006). Specific immunoprecipitations were performed using a rabbit polyclonal antibody generated against β-catenin (Santa Cruz; Santa Cruz, CA) or a mouse monoclonal antibody generated against LEF1 (Millipore; Temecula, CA). Parallel control immunoprecipitations were performed using normal rabbit serum (Vector; Burlingame, CA) or normal mouse serum (Millipore; Temecula, CA), respectively. Immune complexes were captured using a mix of protein A and protein G beads (Active Motif; Carlsbad, CA) and eluted DNA fragments were purified with a QIAquick Spin Kit (Qiagen; Valencia, CA). Purified DNA was analyzed by PCR using region-specific Pitx2 genomic primers as described below. Water and sheared input DNA were used as negative and positive controls, respectively. Sequences spanning the TCF/LEF site within the 5’ Pitx2 promoter were amplified using the forward and reverse primers 5’-TTA GAA GGG GTT TGA CGC AC-3’ and 5’-CCA CTA GGG CTG AGA AGT GA-3’, respectively. Sequences spanning the TCF’LEF site within the 3’ enhancer were used as described previously (Ai et al., 2007).
Supplementary Material
Supplemental Figure 1: Rapid extinction of PITX2 protein expression following tamoxifen-induced temporal knockout of the Pitx2 gene. (A) Experimental timeline for generation of temporal Pitx2 knockout (Pitx2-tko) mice. Tamoxifen was injected into timed pregnant dams at e10.5 to activate the dormant Cre-ER fusion protein and embryos were harvested for analysis either at e11.5 (B-G) or e14.5 (Fig. 4). (B-G) PITX2 immunofluorescence (red), DAPI nuclear stain (blue) and merge on eyes taken from control (B-D) and e11.5 Pitx2-tko littermates harvested from a timed pregnant dam injected with tamoxifen at e10.5. PITX2 protein is completely lost from Pitx2-tko eyes within 24 hours post-injection.
Supplemental Figure 2: LEF1 and b-catenin do not bind the Pitx2c promoter or 3’ pituitary enhancer in ocular neural crest. (A) Schematic of the Pitx2c promoter and 3’ pituitary enhancer is diagramed. Locations of the primer pairs for amplifying Pitx2c promoter (P2c) and 3’ pituitary enhancer (En) sequences are indicated. (B) ChIP analysis was performed on mouse embryonic fibroblasts cultured from microdissected e12.5 wild type eyes using rabbit anti-β-catenin antibody, normal rabbit serum (NRS), mouse anti-LEF1 antibody, and normal mouse serum (NMS). Precipitated and control samples were amplified by PCR using primers directed against the Pitx2c promoter sequences (P2c) that contain no predicted TCF/LEF sites. (C) ChIP analysis was performed as in (B) except that precipitated and control samples were amplified by PCR using primers directed against the 3’ pituitary enhancer sequences (En) that contain a single TCF/LEF site. Apart from input, no enrichment of PCR product was reproducibly observed in any sample using either primer set.
ACKNOWLEDGMENTS
The authors wish to thank Christopher Momont, David Lingenfelter, and Ilea Swinehart for technical assistance. This work was supported by grants from NEI/NIH (EY014126 (PJG), EY007003 (PJG), and EY013934 (ALZ)).
Funding: NEI/NIH EY014126 (PJG), EY007003 (PJG), EY013934 (ALZ)
REFERENCES
- Ai D, Wang J, Amen M, Lu MF, Amendt BA, Martin JF. Nuclear factor 1 and T-cell factor/LEF recognition elements regulate Pitx2 transcription in pituitary development. Mol Cell Biol. 2007;27:5765–5775. doi: 10.1128/MCB.01848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alward WL. Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol. 2000;130:107–115. doi: 10.1016/s0002-9394(00)00525-0. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Briata P, Ilengo C, Corte G, Moroni C, Rosenfeld MG, Chen CY, Gherzi R. The Wnt/beta-catenin-->Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol Cell. 2003;12:1201–1211. doi: 10.1016/s1097-2765(03)00407-6. [DOI] [PubMed] [Google Scholar]
- Chauhan BK, Disanza A, Choi SY, Faber SC, Lou M, Beggs HE, Scita G, Zheng Y, Lang RA. Cdc42- and IRSp53-dependent contractile filopodia tether presumptive lens and retina to coordinate epithelial invagination. Development. 2009;136:3657–3667. doi: 10.1242/dev.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Stump RJ, Lovicu FJ, McAvoy JW. Expression of Frizzleds and secreted frizzled-related proteins (Sfrps) during mammalian lens development. Int J Dev Biol. 2004;48:867–877. doi: 10.1387/ijdb.041882yc. [DOI] [PubMed] [Google Scholar]
- Chow RL, Lang RA. Early eye development in vertebrates. Annu Rev Cell Dev Biol. 2001;17:255–296. doi: 10.1146/annurev.cellbio.17.1.255. [DOI] [PubMed] [Google Scholar]
- Crofford LJ, Wilder RL, Ristimaki AP, Sano H, Remmers EF, Epps HR, Hla T. Cyclooxygenase-1 and -2 expression in rheumatoid synovial tissues. Effects of interleukin-1 beta, phorbol ester, and corticosteroids. J Clin Invest. 1994;93:1095–1101. doi: 10.1172/JCI117060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Tamm ER. Anterior eye development and ocular mesenchyme: new insights from mouse models and human diseases. Bioessays. 2004;26:374–386. doi: 10.1002/bies.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakubo GD, Mazerolle C, Furimsky M, Yu C, St-Jacques B, McMahon AP, Wallace VA. Indian hedgehog signaling from endothelial cells is required for sclera and retinal pigment epithelium development in the mouse eye. Dev Biol. 2008;320:242–255. doi: 10.1016/j.ydbio.2008.05.528. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Diep DB, Hoen N, Backman M, Machon O, Krauss S. Characterisation of the Wnt antagonists and their response to conditionally activated Wnt signalling in the developing mouse forebrain. Brain Res Dev Brain Res. 2004;153:261–270. doi: 10.1016/j.devbrainres.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Evans AL, Gage PJ. Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum Mol Genet. 2005;14:3347–3359. doi: 10.1093/hmg/ddi365. [DOI] [PubMed] [Google Scholar]
- Fjeld K, Kettunen P, Furmanek T, Kvinnsland IH, Luukko K. Dynamic expression of Wnt signaling-related Dickkopf1, -2, and -3 mRNAs in the developing mouse tooth. Dev Dyn. 2005;233:161–166. doi: 10.1002/dvdy.20285. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Qian M, Wu D, Rosenberg KI. The canonical Wnt signaling antagonist DKK2 is an essential effector of PITX2 function during normal eye development. Dev Biol. 2008;317:310–324. doi: 10.1016/j.ydbio.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage PJ, Rhoades W, Prucka SK, Hjalt TA. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005;46:4200–4208. doi: 10.1167/iovs.05-0691. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643–4651. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Zacharias AL. Signaling “cross-talk” is integrated by transcription factors in the development of the anterior segment in the eye. Dev Dyn. 2009;238:2149–2162. doi: 10.1002/dvdy.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould DB, Smith RS, John SW. Anterior segment development relevant to glaucoma. Int J Dev Biol. 2004;48:1015–1029. doi: 10.1387/ijdb.041865dg. [DOI] [PubMed] [Google Scholar]
- Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci U S A. 2007;104:2301–2306. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gummow BM, Scheys JO, Cancelli VR, Hammer GD. Reciprocal regulation of a glucocorticoid receptor-steroidogenic factor-1 transcription complex on the Dax-1 promoter by glucocorticoids and adrenocorticotropic hormone in the adrenal cortex. Mol Endocrinol. 2006;20:2711–2723. doi: 10.1210/me.2005-0461. [DOI] [PubMed] [Google Scholar]
- Hjalt TA, Semina EV, Amendt BA, Murray JC. The Pitx2 protein in mouse development. Dev Dyn. 2000;218:195–200. doi: 10.1002/(SICI)1097-0177(200005)218:1<195::AID-DVDY17>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Holmberg J, Liu C-Y, Hjalt T. PITX2 gain-of-function in Rieger Syndrome eye model. American Journal of Pathology. 2004;165:1633–1641. doi: 10.1016/S0002-9440(10)63420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Wurdak H, Schwerdtfeger K, Kunz T, Ille F, Leveen P, Hjalt TA, Suter U, Karlsson S, Hafezi F, Born W, Sommer L. Compound developmental eye disorders following inactivation of TGFbeta signaling in neural-crest stem cells. J Biol. 2005;4:11. doi: 10.1186/jbiol29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidson SH, Kume T, Deng K, Winfrey V, Hogan BL. The forkhead/winged-helix gene, Mf1, is necessary for the normal development of the cornea and formation of the anterior chamber in the mouse eye. Dev Biol. 1999;211:306–322. doi: 10.1006/dbio.1999.9314. [DOI] [PubMed] [Google Scholar]
- Kioussi C, Briata P, Baek SH, Rose DW, Hamblet NS, Herman T, Ohgi KA, Lin C, Gleiberman A, Wang J, Brault V, Ruiz-Lozano P, Nguyen HD, Kemler R, Glass CK, Wynshaw-Boris A, Rosenfeld MG. Identification of a Wnt/Dvl/beta-Catenin --> Pitx2 pathway mediating cell-type-specific proliferation during development. Cell. 2002;111:673–685. doi: 10.1016/s0092-8674(02)01084-x. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Miura H, Miyagawa-Tomita S, Yanazawa M, Katoh-Fukui Y, Suzuki R, Ohuchi H, Suehiro A, Motegi Y, Nakahara Y, Kondo S, Yokoyama M. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development. 1999;126:5749–5758. doi: 10.1242/dev.126.24.5749. [DOI] [PubMed] [Google Scholar]
- Kozlowski K, Walter MA. Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Hum Mol Genet. 2000;9:2131–2139. doi: 10.1093/hmg/9.14.2131. [DOI] [PubMed] [Google Scholar]
- Kriederman BM, Myloyde TL, Witte MH, Dagenais SL, Witte CL, Rennels M, Bernas MJ, Lynch MT, Erickson RP, Caulder MS, Miura N, Jackson D, Brooks BP, Glover TW. FOXC2 haploinsufficient mice are a model for human autosomal dominant lymphedema-distichiasis syndrome. Hum Mol Genet. 2003;12:1179–1185. doi: 10.1093/hmg/ddg123. [DOI] [PubMed] [Google Scholar]
- Kumar S, Duester G. Retinoic acid signaling in perioptic mesenchyme represses Wnt signaling via induction of Pitx2 and Dkk2. Dev Biol. 2010;340:67–74. doi: 10.1016/j.ydbio.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume T, Deng K, Hogan BL. Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development. 2000;127:1387–1395. doi: 10.1242/dev.127.7.1387. [DOI] [PubMed] [Google Scholar]
- Lee HY, Kleber M, Hari L, Brault V, Suter U, Taketo MM, Kemler R, Sommer L. Instructive role of Wnt/beta-catenin in sensory fate specification in neural crest stem cells. Science. 2004;303:1020–1023. doi: 10.1126/science.1091611. [DOI] [PubMed] [Google Scholar]
- Lehmann OJ, Ebenezer ND, Jordan T, Fox M, Ocaka L, Payne A, Leroy BP, Clark BJ, Hitchings RA, Povey S, Khaw PT, Bhattacharya SS. Chromosomal duplication involving the forkhead transcription factor gene FOXC1 causes iris hypoplasia and glaucoma. Am J Hum Genet. 2000;67:1129–1135. doi: 10.1016/s0002-9297(07)62943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann OJ, Tuft S, Brice G, Smith R, Blixt A, Bell R, Johansson B, Jordan T, Hitchings RA, Khaw PT, John SW, Carlsson P, Bhattacharya SS. Novel anterior segment phenotypes resulting from forkhead gene alterations: evidence for cross-species conservation of function. Invest Ophthalmol Vis Sci. 2003;44:2627–2633. doi: 10.1167/iovs.02-0609. [DOI] [PubMed] [Google Scholar]
- Liu H, Mohamed O, Dufort D, Wallace VA. Characterization of Wnt signaling components and activation of the Wnt canonical pathway in the murine retina. Dev Dyn. 2003;227:323–334. doi: 10.1002/dvdy.10315. [DOI] [PubMed] [Google Scholar]
- Matt N, Dupe V, Garnier JM, Dennefeld C, Chambon P, Mark M, Ghyselinck NB. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development. 2005;132:4789–4800. doi: 10.1242/dev.02031. [DOI] [PubMed] [Google Scholar]
- Matt N, Ghyselinck NB, Pellerin I, Dupe V. Impairing retinoic acid signalling in the neural crest cells is sufficient to alter entire eye morphogenesis. Dev Biol. 2008;320:140–148. doi: 10.1016/j.ydbio.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, Ritch R, Koop B, Kuo WL, Collins C, Marshall J, Gould DB, Pearce W, Carlsson P, Enerback S, Morissette J, Bhattacharya S, Hogan B, Raymond V, Walter MA. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet. 1998;63:1316–1328. doi: 10.1086/302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov A, Molotkova N, Duester G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development. 2006;133:1901–1910. doi: 10.1242/dev.02328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priston M, Kozlowski K, Gill D, Letwin K, Buys Y, Levin AV, Walter MA, Heon E. Functional analyses of two newly identified PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome. Hum Mol Genet. 2001;10:1631–1638. doi: 10.1093/hmg/10.16.1631. [DOI] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See AW, Clagett-Dame M. The temporal requirement for vitamin A in the developing eye: mechanism of action in optic fissure closure andnew roles for the vitamin in regulating cell proliferation and adhesion in the embryonic retina. Dev Biol. 2009;325:94–105. doi: 10.1016/j.ydbio.2008.09.030. [DOI] [PubMed] [Google Scholar]
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- Semina EV, Reiter RS, Murray JC. Isolation of a new homeobox gene belonging to the Pitx/Riegfamily: expression during lens development and mapping to the aphakia region on mouse chromosome 19. Hum Mol Genet. 1997;6:2109–2116. doi: 10.1093/hmg/6.12.2109. [DOI] [PubMed] [Google Scholar]
- Shiratori H, Sakuma R, Watanabe M, Hashiguchi H, Mochida K, Sakai Y, Nishino J, Saijoh Y, Whitman M, Hamada H. Two-step regulation of left-right asymmetric expression of Pitx2: initiation by nodal signaling and maintenance by Nkx2. Mol Cell. 2001;7:137–149. doi: 10.1016/s1097-2765(01)00162-9. [DOI] [PubMed] [Google Scholar]
- Smith RS, Zabaleta A, Kume T, Savinova OV, Kidson SH, Martin JE, Nishimura DY, Alward WL, Hogan BL, John SW. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum Mol Genet. 2000;9:1021–1032. doi: 10.1093/hmg/9.7.1021. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Vadlamudi U, Espinoza HM, Ganga M, Martin DM, Liu X, Engelhardt JF, Amendt BA. PITX2, beta-catenin and LEF-1 interact to synergistically regulate the LEF-1 promoter. J Cell Sci. 2005;118:1129–1137. doi: 10.1242/jcs.01706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Rapid extinction of PITX2 protein expression following tamoxifen-induced temporal knockout of the Pitx2 gene. (A) Experimental timeline for generation of temporal Pitx2 knockout (Pitx2-tko) mice. Tamoxifen was injected into timed pregnant dams at e10.5 to activate the dormant Cre-ER fusion protein and embryos were harvested for analysis either at e11.5 (B-G) or e14.5 (Fig. 4). (B-G) PITX2 immunofluorescence (red), DAPI nuclear stain (blue) and merge on eyes taken from control (B-D) and e11.5 Pitx2-tko littermates harvested from a timed pregnant dam injected with tamoxifen at e10.5. PITX2 protein is completely lost from Pitx2-tko eyes within 24 hours post-injection.
Supplemental Figure 2: LEF1 and b-catenin do not bind the Pitx2c promoter or 3’ pituitary enhancer in ocular neural crest. (A) Schematic of the Pitx2c promoter and 3’ pituitary enhancer is diagramed. Locations of the primer pairs for amplifying Pitx2c promoter (P2c) and 3’ pituitary enhancer (En) sequences are indicated. (B) ChIP analysis was performed on mouse embryonic fibroblasts cultured from microdissected e12.5 wild type eyes using rabbit anti-β-catenin antibody, normal rabbit serum (NRS), mouse anti-LEF1 antibody, and normal mouse serum (NMS). Precipitated and control samples were amplified by PCR using primers directed against the Pitx2c promoter sequences (P2c) that contain no predicted TCF/LEF sites. (C) ChIP analysis was performed as in (B) except that precipitated and control samples were amplified by PCR using primers directed against the 3’ pituitary enhancer sequences (En) that contain a single TCF/LEF site. Apart from input, no enrichment of PCR product was reproducibly observed in any sample using either primer set.