

Abstract
First recognized as regulators of development in worms and fruitflies, microRNAs are emerging as pivotal modulators of mammalian cardiovascular development and disease. Individual microRNAs modulate the expression of collections of messenger RNA targets that often have related functions, thereby governing complex biological processes. The wide-ranging functions of microRNAs in the cardiovascular system have provided new perspectives on disease mechanisms and have revealed intriguing therapeutic targets, as well as diagnostics, for a variety of cardiovascular disorders.
Diseases of the cardiovascular system are the most common congenital birth defects and causes of adult morbidity and mortality1-3. Although the cellular mechanisms and gene mutations responsible for numerous cardiovascular disorders have been extensively studied, it has become apparent only recently that microRNAs (miRNAs) have key roles in cardiovascular development and disease4-8. The prominent functions of miRNAs in cardiovascular biology probably reflect the sensitivity of the cardiovascular system to relatively subtle perturbations in gene expression, which can result in severe and often fatal abnormalities.
A primary role of miRNAs seems to be the ‘fine-tuning’ of gene expression to control development and tissue homeostasis8. However, under conditions of stress, the functions of miRNAs become especially pronounced, underscoring their roles in disease. Highly specific patterns of miRNA expression correlate with different cardiovascular disorders (such as cardiac hypertrophy, heart failure9-12, post-myocardial infarction remodelling13,14 and vascular remodelling15,16), and gain- and loss-of-function miRNA studies in mice have revealed pathogenic and protective functions of miRNAs in vivo6,17. Correlation of the cellular targets of miRNA action with cardiovascular phenotypes illuminates new biological pathways and disease mechanisms. Especially intriguing is the ability to manipulate individual miRNAs in vivo using oligonucleotide-based inhibitors or miRNA mimics, thereby opening up possibilities for the therapeutic manipulation of miRNAs18.
In this Review, we describe the biology and mechanisms of action of miRNAs in the cardiovascular system, and consider the opportunities and challenges for the therapeutic modulation of miRNAs in cardiovascular disease. See refs 8, 19 and 20 for more detailed reviews of the biosynthesis and mechanisms of action of miRNAs.
Functional concepts of miRNA action
MicroRNAs are ~22-nucleotide single-stranded RNAs that inhibit the expression of specific mRNA targets through Watson–Crick base pairing between the miRNA ‘seed region’ and sequences commonly located in the 3′ untranslated regions (UTRs). The human genome is estimated to encode up to 1,000 miRNAs21, which are either transcribed as standalone transcripts, frequently encoding several miRNAs, or generated by the processing of introns of protein-coding genes21. The integration of miRNAs into introns of protein-coding genes serves to coordinate the expression of the miRNA with the mRNA encoded by that gene, without the necessity for a separate set of cis-regulatory elements to drive expression of the miRNA (Fig. 1a). It is not uncommon for intronic miRNAs to modulate the same biological processes as the protein encoded by the host gene22-26. The dual functions of such genes, encoding protein and miRNA, provide sophisticated feedback and feedforward regulatory networks, specific examples of which are highlighted throughout this Review.
Figure 1. Concepts of miRNA function.

The potential modes of miRNA-based regulation of gene expression are shown. a, Intronic miRNAs are encoded within an intron of a host gene. mRNA splicing generates a protein-coding transcript and an miRNA stem–loop. Intronic miRNAs often regulate similar processes to that of the protein encoded by the host gene. AAA, polyadenylated tail of the transcript; pre-miRNA, precursor miRNA. b, A common mechanism of miRNA function involves the modest repression of several mRNAs in a common biological process by a single miRNA. This mechanism reduces the dependence on a single miRNA-mRNA interaction and increases the robustness of the gene-regulatory network. TF, transcription factor. c, Many miRNAs may cooperatively or redundantly regulate a single biological process, by individually targeting many components of that process or by synergistically repressing a crucial component of a pathway. d, miRNAs may act as a ‘buffer’ against minor perturbations in a biological pathway. This is accomplished by the targeting of factors that positively and negatively influence a particular process, thereby insulating that process from environmental fluctuations.
Genetic deletions of miRNAs in organisms ranging from worms to mice have shown that few developmental processes are absolutely dependent on single miRNAs8,27. A recent study using compound mutant worms suggested there was significant redundancy within miRNA families, between unrelated miRNAs, and even between miRNAs and transcription factors, perhaps evolving as a buffer against deleterious variations in gene-expression programs28,29. The actions of miRNAs often become pronounced under conditions of physiological or pathological signalling, suggesting conditional activities of miRNAs that necessitate genetic perturbation or sensitizing agents to uncover their functions.
miRNAs typically exert modest inhibitory effects on many mRNAs, which often encode proteins that govern the same biological process — for example, the fibrotic response is inhibited by miR-29 (ref. 14), cardiac conduction by miR-1 (refs 30-32), actin cytoskeletal dynamics by miR-145 (ref. 16), the phosphatidylinositol-3-OH kinase (PI(3)K)–AKT pathway by miR-486 (ref. 33) and stem-cell pluripotency by miR-145 (ref. 34). The cumulative reduction in expression of several components of a molecular pathway reduces the importance of a single miRNA-mRNA interaction to elicit a biological response, and adds robustness to gene-regulatory networks (Fig. 1b). The multiplicity of miRNA targets may also promote combinatorial regulation by miRNAs that individually target various mRNAs whose protein products contribute to one particular regulatory axis (Fig. 1c). In this model, a biological response would be expected only after co-expression of several miRNAs that cooperatively target various components of a functional network or are all required to sufficiently repress a single target. By contrast, some miRNAs seem to reinforce an appropriately ‘balanced’ pathway by targeting both positive and negative regulatory components (for example, agonism and antagonism of Nodal signalling by miR-430)35 (Fig. 1d). This mode of action allows buffering against minor physiological variations. Clearly, miRNA biology is a complex and highly orchestrated mode of gene regulation, potentially impinging on nearly all biological processes in mammals and having particularly important roles in disease states.
Oligonucleotide modulation of miRNA function
The ability of miRNAs to modulate important biological pathways offers opportunities for the manipulation of miRNA function using oligonucleotide inhibitors (antimiRs) or miRNA mimics (Fig. 2). Antisense oligonucleotides directed against specific miRNA sequences are efficiently taken up by a variety of tissues and block miRNA function in the heart and vasculature18.
Figure 2. Oligonucleotide manipulation of miRNA function.
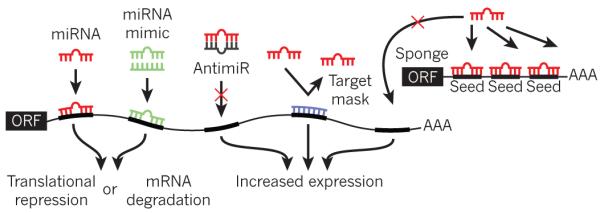
The various methods of artificially modulating miRNA expression or activity are shown. Endogenous miRNA (red) binds to complementary sequences in the 3′ UTR of a target gene, resulting in translational repression or mRNA degradation. An miRNA mimic (green) consists of an oligonucleotide duplex of the miRNA and a passenger strand. The miRNA mimic comprises the same nucleotide sequence as an endogenous miRNA, and is designed to target the same mRNAs as that miRNA. An antimiR (grey) is an oligonucleotide that is complementary to an endogenous miRNA, thereby designed to bind and inhibit its function. A target mask (blue) is an oligonucleotide designed to bind to a portion of an endogenous miRNA target without initiating mRNA degradation or translational inhibition. This strategy rescues one particular mRNA from miRNA-mediated repression. miRNA sponges consist of an open reading frame (ORF) linked to a 3′ UTR that contains several binding sites for a particular miRNA, acting as competitive inhibitors for miRNA binding.
Other oligonucleotide-based techniques involve ‘target protectors’ or ‘masks’, which block individual miRNAs from binding to their mRNA targets, thereby rescuing the mRNA from inhibition. Target protectors have been validated in zebrafish35 and in cultured cardiac myocytes36. miRNA ‘sponges’ or ‘decoys’ containing several miRNA-binding sites also act as competitive inhibitors for miRNA binding37,38. Although the results obtained from pharmacological knockdown or overexpression of miRNAs sometimes differ from those obtained using genetic mouse models, the development of these various technologies has greatly accelerated the rate at which basic biological questions can be answered in an experimental setting.
miRNAs in cardiovascular development
The requirement of miRNAs for cardiovascular development and function was initially demonstrated by tissue-specific deletion in mice of the Dicer gene, which encodes an enzyme that is essential for miRNA processing. Lethal phenotypes were observed after Dicer deletion in myocardial and vascular lineages39,40. Although these findings highlight the crucial roles of miRNAs in the cardiovascular system, no specific miRNA deletion has yet been found to cause fully penetrant embryonic lethality in mice, indicating significant redundancy of miRNA function28 and suggesting that the lethal consequences of Dicer deletion reflect the collective functions of many miRNAs rather than any single miRNA. The rapidly expanding number of miRNAs implicated in various aspects of cardiovascular biology precludes an in-depth review of all of them, so general principles of miRNA regulation and function are considered throughout this Review.
Roles of miRNAs in heart development
Heart formation requires precise and complex interactions among diverse cell types from several lineages — cardiomyocytes, endocardial, epicardial and vascular cells, fibroblasts and cells of the conduction system. Specific miRNAs are enriched in different cardiac cell types and, in some cases, have been found to participate in the specification of cell identity. Genetic ablation and antisense oligonucleotide-mediated knockdown studies have shown miRNA contributions to developmental processes as diverse as embryonic stem (ES)-cell differentiation, cardiomyocyte proliferation, contractility, ion-channel regulation and cardiac conduction (Fig. 3).
Figure 3. Functional role of miRNAs in the normal and diseased heart.

A normal and a hypertrophic heart are shown in schematic form, depicting miRNAs that contribute to normal function or pathological remodelling. The expression of selected miRNAs within the heart is shown, along with their corresponding functions. All arrows denote the normal action of each component or process. miR-1 and miR-133 are involved in the development of a normal heart (left) by regulating proliferation, differentiation and cardiac conduction. For example, proliferation is promoted by cell-cycle regulators, but miR-1 and miR-133 block these regulators, thus blocking proliferation. miR-208a also contributes to the regulation of the conduction system. After cardiac injury (right), various miRNAs contribute to pathological remodelling and the progression to heart failure. miR-29 and miR-21 block and promote cardiac fibrosis, respectively. miR-29 blocks fibrosis by inhibiting the expression of ECM components, whereas miR-21 promotes fibrosis by stimulating mitogen-activated protein kinase (MAPK) signalling. miR-208 controls myosin isoform switching, cardiac hypertrophy and fibrosis. miR-23a promotes cardiac hypertrophy by inhibiting ubiquitin proteolysis, which itself inhibits hypertrophy. Hypoxia results in the repression of miR-320 and miR-199, which promote and block apoptosis, respectively. ECM, extracellular matrix; LV, left ventricle; MHC, myosin heavy chain; RV, right ventricle.
Expression profiling has shown that the 18 most abundant miRNAs in the heart account for more than 90% of all cardiac miRNAs41. Because a threshold level of miRNA expression seems to be required for the efficient repression of target gene expression (typically >100 copies per cell)37,42, the regulation of heart development may depend on either a relatively discrete set of miRNAs or the combinatorial function of a larger array of miRNAs expressed at a low level. So far, a functional role in heart development has been ascribed to only the most enriched miRNAs, reflecting a dosage requirement, functional redundancy or both.
miR-1 is the most abundant miRNA in cardiac myocytes, and it was the first miRNA implicated in heart development30. miR-1 and the related miRNA miR-133 arise from a common precursor RNA, the expression of which in the embryonic heart is mediated by two separate enhancers that are regulated by the transcription factors SRF and MEF2 (refs 43, 44), integrating these miRNAs into well-characterized transcriptional networks. miR-1 and miR-133 seem to function cooperatively to promote mesoderm differentiation of ES cells and suppress endodermal and ectodermal cell fates45. By contrast, they have opposing roles later in the cardiac lineage when miR-1 promotes and miR-133 inhibits cardiomyocyte differentiation45. Neither miR-1 nor miR-133 is absolutely required for the specification of cardiac cell fates in vivo, as 50% of mice lacking either miRNA are viable30,46. This disparity between the functions of miRNAs as determined by in vitro assays versus in vivo loss-of-function studies is a common theme, and suggests that compensatory mechanisms that account for the unexpectedly mild phenotypes may be activated in genetic knockout mice. Zebrafish seem to be particularly sensitive to miRNA regulation, such that inhibition of specific miRNAs evokes more dramatic phenotypes than those seen in mutant mice. For example, antisense-mediated knockdown of miRNAs in zebrafish has revealed roles for miRNAs in the formation of the cardiac chambers and the atrioventricular canal47,48.
Roles of miRNAs in vascular and blood development
The formation and function of the vascular system requires the establishment and remodelling of a contiguous series of lumenized tubes made of endothelial cells. Concurrent recruitment of vascular smooth muscle cells (SMCs) to the endothelial plexus during vessel maturation imparts the necessary tone and contractility for proper blood flow. Numerous miRNAs have been shown to govern these processes during vascular development and disease (Fig. 4).
Figure 4. Functional role of miRNAs in the vascular system.

Blood vessel schematic showing the endothelial and smooth muscle layers, red blood cells and the proliferating SMCs of a neointimal lesion. The expression of select miRNAs is shown, along with their observed functional role. Hypoxia results in the activation of miR-210 and miR-92a, which promote and inhibit angiogenesis, respectively. miR-126, an endothelial-cell-enriched miRNA encoded by an intron of the Egfl7 gene, modulates atherosclerosis and angiogenesis by regulating MAPK and PI(3)K signalling. Angiogenesis is also regulated by miR-218, which is encoded by an intron of the Slit genes. miR-143 and miR-145 are expressed in SMCs and control blood pressure and vascular tone, and contribute to vascular remodelling. miR-21 is induced in SMCs after vascular injury, and promotes proliferation and neointima formation. miR-451 regulates the proliferation and differentiation of erythroid cells.
The endothelial-cell-specific miRNA miR-126 is encoded by an intron of the epidermal growth factor-like domain 7 (Egfl7) gene, which encodes an endothelial-cell-enriched growth factor involved in the control of cell migration49. miR-126 is induced by blood flow and controls angiogenic sprouting of aortic arch vessels by the stimulation of vascular endothelial growth factor signalling50. Mice lacking miR-126 are partially viable but have fragile and leaky blood vessels and defects in angiogenesis51,52. Antisense-oligonucleotide-mediated knockdown of miR-126 in zebrafish causes complete embryonic lethality owing to the loss of vascular integrity and haemorrhaging53. Vascular patterning in mouse retinas is also modulated by miR-218, which is encoded by an intron of the Slit1 and Slit2 genes and inhibits several components of the SLIT–ROBO signalling pathway54. This study is an important demonstration of the coordinated regulation of a biological process by an miRNA and its host gene.
miR-143 and miR-145, encoded by a bicistronic pre-miRNA, are expressed specifically in SMCs under the control of SRF and members of the myocardin family of co-activators. These miRNAs target numerous regulators of actin signalling, including Rho GTPases, sling-shot homologue 2, adducin, cofilin and actin itself16. miR-145 has been reported to be necessary and sufficient for SMC differentiation in vitro55. However, mice lacking both miR-143 and miR-145 are viable, suggesting that further mechanisms modulate their functions in vivo16,56,57. miR-145-mutant mice have reduced vascular tone, which contributes to a reduction in blood pressure16,57. Vascular SMCs from these mutant mice show diminished sensitivity to mechanical injury and an abnormality in phenotypic switching in response to injury that seems to reflect perturbations in actin signalling and SRF activation. Collectively, these studies demonstrate that miRNAs function as sensors of mechanical and environmental changes, thus linking dynamic physiological processes with the regulation of gene expression.
The differentiation of blood cells is also dependent on miRNA activity58. An miRNA expression signature has been described for haematopoietic stem-cell progenitors, which show dynamic regulation during differentiation59. Furthermore, the Ago2-(also known as Eif2c2-) null mouse has erythroid lineage defects60, and modulation of miRNA expression in erythroid progenitors suggests a role for miRNAs in their differentiation61,62. Indeed, loss-of-function studies in mice have also implicated miRNAs, including miR-223 and miR-451, in erythroid proliferation and differentiation63-66. For example, gene targeting and pharmacological knockdown of miR-451, which is enriched in erythroid cells, results in reduced baseline haematocrit levels and impaired erythroid expansion in response to oxidative stress63,65,66. Further analysis of miRNAs in circulating cells may reveal roles in functions such as oxygen delivery, angiogenesis and the inflammatory process.
miRNAs in cardiovascular disease
Heart failure and several cardiovascular diseases are associated with the re-expression of the fetal cardiac gene program, which may have causative or adaptive roles1 and includes a signature pattern of miRNAs10,12. Indeed, numerous cardiac-enriched miRNAs show dynamic regulation in human heart disease, suggesting their involvement in the regulation of cardiovascular disease9,11,12.
Roles of miRNAs in heart disease
The importance of individual miRNAs in the setting of heart disease has been shown by genetic deletion in mice subjected to various cardiovascular insults (Fig. 3). miRNAs are implicated in pathologies as diverse as arrhythmias (miR-1 (ref. 31), miR-133 (ref. 32) and miR-208a (ref. 67)), fibrosis (miR-21 (ref. 68) and miR-29 (ref. 14)), pressure-overload-induced remodelling (miR-208 (refs 67, 69) and miR-133 (ref. 70)), and metabolic disorders (miR-33 (ref. 24)).
One of the best-characterized examples of stress-dependent gene regulation by an miRNA involves a family of miRNAs encoded by myosin heavy chain (MHC) genes, referred to as MyomiRs67,69,71. This is also an example of intronic miRNAs participating in a process related to host gene function. Three members of this miRNA family, miR-208a, miR-208b and miR-499, are encoded by the α-MHC (also known as Myh6), β-MHC (Myh7) and Myh7b genes, respectively. These MyomiRs regulate a collection of transcriptional repressors and signalling molecules that govern MHC expression, as well as thyroid hormone activity and the stress-responsiveness of cardiac muscle cells. Deletion of Mir208a in mice abrogates the re-activation of the fetal β-MHC gene in response to haemodynamic cardiac stress, and protects the heart from pathological remodelling67,69. The MyomiR family thus constitutes an intricate regulatory circuit that controls myosin gene expression and cardiac stress responsiveness during adaptation to pathological signalling.
In addition to the miR-208 family, several other miRNAs have been implicated as either causative or protective in heart disease. NFATc3 is a transcriptional mediator of cardiac stress signalling that promotes pathological hypertrophy72. NFATc3 was recently shown to induce miR-23a expression in cardiomyocytes, and antagomir-based knockdown of miR-23a in mice abrogates isoproterenol-induced cardiac hypertrophy73. Conversely, acute knockdown of miR-133 was shown to induce pathological cardiac hypertrophy in mice70, suggesting a potential cardioprotective role for endogenous miR-133. However, these findings contrast with the phenotype of Mir133-null mice, which undergo a normal hypertrophic response46, highlighting the difference between pharmacological modulation of miRNA expression and genetic deletion studies.
The miR-29 family, which is downregulated after myocardial infarction, inhibits the expression of several collagens and extracellular matrix proteins, thereby contributing to scar formation and fibrosis14. Similarly, the miR-199 family is rapidly downregulated in cardiac myocytes under hypoxic conditions, relieving the repression of sirtuin 1 and hypoxia-inducible factor 1-α in a model of hypoxia preconditioning74.
The miRNA that repeatedly shows dynamic regulation after cellular stress is miR-21, which was shown to promote cardiac hypertrophy and fibrosis in response to pressure overload. Knockdown of miR-21 with a cholesterol-modified antagomir attenuated cardiac remodelling after thoracic aortic constriction68. This response was attributed to the derepression of the protein sprouty, which negatively regulates the profibrotic extracellular signal-regulated kinase-mitogen-activated protein kinase (ERK–MAPK) cascade in cardiac fibroblasts68. Paradoxically, however, neither genetic deletion nor tiny locked-nucleic-acid (LNA)-mediated knockdown of miR-21 in mice alters fibrosis or hypertrophy in response to thoracic aortic constriction or other cardiac stresses75. The contrasting conclusions of these studies emphasize the gaps in our understanding of the mechanisms of miRNA action and oligonucleotide-based targeting strategies for their inhibition.
Roles of miRNAs in vascular disease
The vessel wall is composed of endothelial cells and SMCs that must maintain a sealed barrier yet allow the exchange of oxygen and nutrients with adjacent tissues. Vessels can respond to injury or changes in the environment by undergoing phenotypic changes that promote endothelial cell migration or fragility, as well as SMC de-differentiation, proliferation and migration. Numerous miRNAs show marked alterations in expression during vascular injury and disease, and expression signatures have now been correlated with pathologies such as ischaemia, tumour angiogenesis, atherosclerosis and a proliferative thickening and obstruction of the vessel known as restenosis15. Some miRNAs have been shown to have causal roles in these disorders (Fig. 4).
Angiogenesis is a process of endothelial cell proliferation and vascular tube sprouting that is promoted in adulthood by various stimuli, including tumour growth, retinal damage and ischaemia. miR-21 can influence the function and migration of angiogenic progenitor cells during coronary artery disease76. Likewise, ischaemia-induced angiogenesis in adult tissues can be promoted or inhibited by antisense oligonucleotides directed against miR-92a (ref. 77) or miR-126 (ref. 78), respectively. miR-126 may also influence susceptibility to atherosclerosis, through the modification of endothelial cell function79,80.
Vessel injury, instigated by diverse factors such as atherosclerosis, hypertension and damage due to a mechanical stenting, results in SMC phenotypic changes indicative of a de-differentiated state. Such SMCs become proliferative and migratory, entering the vessel lumen and causing restenosis. Recent studies have implicated miRNAs as mediators of SMC phenotypic modulation and vessel remodelling. The expression of miR-21 and the miR-143/145 cluster are up- and downregulated, respectively, after mechanical injury of large vessels15, and restoration of miR-21 and miR-145 to normal levels prevents restenosis15,56,81.
miRNA mutations as the basis of disease
The pervasive influence of miRNAs on cardiovascular function and disease raises questions as to whether polymorphisms in miRNAs or their target sequences in mRNA transcripts affect human disease. Although mutations within the seed regions of evolutionarily conserved miRNAs are not common, single nucleotide polymorphisms (SNPs) within miRNA-binding sites in the 3′ UTRs of target mRNAs have been observed at a higher frequency82,83. A notable example is in the Texel breed of sheep, which develops extreme skeletal muscle hypertrophy owing to an SNP in the 3′ UTR of the mRNA encoding myostatin, a negative regulator of muscle growth84. This mutation creates a binding site for miR-1, resulting in repression of myostatin expression and unrestricted muscle growth. SNPs within potential miRNA-binding sites have also been identified in mRNAs associated with hypertension and cardiovascular disease85. SNPs in miRNAs or their targets that cause significant phenotypes seem unlikely to be a widespread occurrence, however, because of the substantial degeneracy allowed in miRNA-mRNA interactions and the redundant regulation of an individual mRNA by several unrelated miRNAs. It remains to be determined whether a causative link can be made between miRNA SNPs and human disease.
Clinical perspectives
Identifying the signature patterns of miRNAs associated with different cardiovascular disorders has opened up opportunities for miRNA diagnostics. miRNA profiling can discriminate between specific forms of heart disease10,12, such as dilated cardiomyopathy, ischaemic cardiomyopathy and heart failure, and disease-associated miRNA expression patterns in failing human hearts can be normalized by the stabilization of cardiac output11,86. Recently, several miRNAs have been detected in plasma and reported to be diagnostic for heart failure and myocardial infarction87-89. Whether circulating miRNAs are functionally relevant or are simply released from injured tissues remains to be determined. However, the cellular secretion of particular miRNAs by exosomes suggests specificity in the process of miRNA secretion.
In contrast to many cellular mediators of disease, which are difficult (or impossible) to modulate therapeutically, it is unquestionable that drugs can target miRNAs. Thus, the involvement of miRNAs in almost every aspect of cardiovascular disease raises exciting possibilities for the therapeutic manipulation of miRNA-regulated processes. Therapies based on antimiRs or miRNA mimics are now being developed to repress pathological miRNAs or overexpress protective miRNAs, respectively. Indeed, antimiR-based studies demonstrating efficacy in non-human primates have already been reported90,91 and have been advanced to human clinical trials.
The ability of individual miRNAs to modulate complex disease pathways through the targeting of several components of regulatory networks enables miRNAs to modulate tissue stress responses in a manner that is distinct from that of classical drugs. The multiplicity of miRNA targets also enables miRNAs to bypass mechanisms that render cells or tissues insensitive to certain drugs. For example, cells can develop insensitivity to single drugs through rare mutations in drug targets or desensitization of cell-surface receptors. Such mechanisms are unlikely to diminish sensitivity to miRNA inhibitors, which target several steps in a disease pathway.
Conversely, the targeting of large collections of mRNAs raises possibilities for off-target effects or even opposing effects of miRNAs in different tissues. Because the mechanistic basis of miRNA-based therapeutics is not clear, the possibility exists that modulating such a diverse set of target mRNAs will affect beneficial processes as well as the pathological condition. The heart takes up globally administered antimiR oligonucleotides less efficiently than the kidneys and liver, and the pharmacokinetics of miRNA-based therapies remain a hurdle. This issue may necessitate the development of new cardiovascular delivery systems for miRNA-based therapeutics, to limit uptake in healthy tissue. These methods would not be required for strategies involving knockdown of cardiac-specific miRNAs. Conjugation of antimiRs or miRNA mimics to homing molecules such as peptides, antibodies or other bioactive molecules might enrich uptake in cardiac tissue. This technology has not yet been successfully translated to the clinic, however, and other methods may improve tissue-specific uptake, such as direct administration by cardiac catheterization or a drug-coated stent.
Looking to the future
Despite recent advances in identifying miRNA contributions to cardiovascular development and disease, as well as in developing miRNA diagnostics and miRNA inhibitors, many gaps remain in our knowledge of miRNA-based regulation of gene expression in the normal and diseased heart and cardiovascular system. For example, the many potential target mRNAs for each miRNA pose significant challenges to the identification of those mRNAs that are relevant to a particular miRNA-regulated process. Compounding this difficulty is the apparent variability in miRNA function based on physiological context or cell type, making it necessary to define the potential disparate functions of individual miRNAs in different settings. Another important consideration is that combinatorial interactions between multiple miRNAs with common or coordinated target mRNAs are likely to have a major role in gene regulation and the control of physiological pathways. Thus, it will be crucial to identify sets of miRNAs acting cooperatively within the cardiovascular system. This information will be particularly relevant to the development of miRNA-based therapeutics, as cocktails of miRNA inhibitors may prove more efficacious than targeting a single miRNA.
With the current pace of advancements in deciphering the basic principles of miRNA action in cardiovascular development and disease, we foresee new therapeutic applications for the prevention and treatment of human pathologies based on miRNA biology in the relatively near future.
Acknowledgements
We apologize to all colleagues whose work could not be cited owing to space restrictions. We thank J. Cabrera for artwork and J. Brown for editorial assistance. E.N.O. was supported by grants from the National Institutes of Health, the Donald W. Reynolds Center for Clinical Cardiovascular Research, the Robert A. Welch Foundation, the Fondation Leducq’s Transatlantic Network for Excellence in Cardiovascular Research Program, the American Heart Association and the Jon Holden DeHaan Foundation. E.M.S. was supported by a scientist development grant from the American Heart Association.
Footnotes
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/nature.
References
- 1.Hill JA, Olson EN. Cardiac plasticity. N. Engl. J. Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 3.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–948. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- 4.Cordes KR, Srivastava D. MicroRNA regulation of cardiovascular development. Circ. Res. 2009;104:724–732. doi: 10.1161/CIRCRESAHA.108.192872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nature Rev. Cardiol. 2009;6:419–429. doi: 10.1038/nrcardio.2009.56. [DOI] [PubMed] [Google Scholar]
- 6.Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–1032. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J. Clin. Invest. 2007;117:2369–2376. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev. Cell. 2010;18:510–525. doi: 10.1016/j.devcel.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikeda S, et al. Altered microRNA expression in human heart disease. Physiol. Genomics. 2007;31:367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 10.van Rooij E, et al. A signature pattern of stress–responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl Acad. Sci. USA. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. This important paper describes the dynamic regulation of miRNA expression during cardiac stress.
- 11.Matkovich SJ, et al. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation. 2009;119:1263–1271. doi: 10.1161/CIRCULATIONAHA.108.813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thum T, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 13.Roy S, et al. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc. Res. 2009;82:21–29. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Rooij E, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl Acad. Sci. USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji R, et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ. Res. 2007;100:1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 16.Xin M, et al. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–2178. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang ZP, Neppl RL, Wang DZ. MicroRNAs in cardiac remodeling and disease. J. Cardiovasc. Transl. Res. 2010;3:212–218. doi: 10.1007/s12265-010-9165-y. [DOI] [PubMed] [Google Scholar]
- 18.van Rooij E, Marshall WS, Olson EN. Toward microRNA-based therapeutics for heart disease: the sense in antisense. Circ. Res. 2008;103:919–928. doi: 10.1161/CIRCRESAHA.108.183426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 21.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 22.Lutter D, Marr C, Krumsiek J, Lang EW, Theis FJ. Intronic microRNAs support their host genes by mediating synergistic and antagonistic regulatory effects. BMC Genomics. 2010;11:224. doi: 10.1186/1471-2164-11-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao G, et al. Intronic miR-301 feedback regulates its host gene, ska2, in A549 cells by targeting MEOX2 to affect ERK/CREB pathways. Biochem. Biophys. Res. Commun. 2010;396:978–982. doi: 10.1016/j.bbrc.2010.05.037. [DOI] [PubMed] [Google Scholar]
- 24.Najafi-Shoushtari SH, et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poliseno L, et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010;3:ra29. doi: 10.1126/scisignal.2000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barik S. An intronic microRNA silences genes that are functionally antagonistic to its host gene. Nucleic Acids Res. 2008;36:5232–5241. doi: 10.1093/nar/gkn513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarez-Saavedra E, Horvitz HR. Many families of C. elegans microRNAs are not essential for development or viability. Curr. Biol. 2010;20:367–373. doi: 10.1016/j.cub.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ambros V. MicroRNAs: genetically sensitized worms reveal new secrets. Curr. Biol. 2010;20:R598–R600. doi: 10.1016/j.cub.2010.05.054. [DOI] [PubMed] [Google Scholar]
- 29.Brenner JL, Jasiewicz KL, Fahley AF, Kemp BJ, Abbott AL. Loss of individual microRNAs causes mutant phenotypes in sensitized genetic backgrounds in C. elegans. Curr. Biol. 2010;20:1321–1325. doi: 10.1016/j.cub.2010.05.062. This paper suggests redundant and stress-responsive roles of miRNAs, through using miRNA mutants in Dicer-deficient C. elegans.
- 30.Zhao Y, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. This paper demonstrates an important role for an miRNA in heart development by genetic deletion in mice.
- 31.Yang B, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nature Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 32.Luo X, et al. Down-regulation of miR-1/miR-133 contributes to re-expression of pacemaker channel genes HCN2 and HCN4 in hypertrophic heart. J. Biol. Chem. 2008;283:20045–20052. doi: 10.1074/jbc.M801035200. [DOI] [PubMed] [Google Scholar]
- 33.Small EM, et al. Regulation of PI3-kinase/Akt signalling by muscle-enriched microRNA-486. Proc. Natl Acad. Sci. USA. 2010;107:4218–4223. doi: 10.1073/pnas.1000300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–658. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 35.Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- 36.Xiao J, et al. Novel approaches for gene-specific interference via manipulating actions of microRNAs: examination on the pacemaker channel genes HCN2 and HCN4. J. Cell. Physiol. 2007;212:285–292. doi: 10.1002/jcp.21062. [DOI] [PubMed] [Google Scholar]
- 37.Brown BD, Naldini L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nature Rev. Genet. 2009;10:578–585. doi: 10.1038/nrg2628. [DOI] [PubMed] [Google Scholar]
- 38.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nature Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen JF, et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl Acad. Sci. USA. 2008;105:2111–2116. doi: 10.1073/pnas.0710228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Albinsson S, et al. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler. Thromb. Vasc. Biol. 2010;30:1118–1126. doi: 10.1161/ATVBAHA.109.200873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao PK, et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res. 2009;105:585–594. doi: 10.1161/CIRCRESAHA.109.200451. Deep sequencing showed that the 18 most abundant cardiac miRNAs account for more than 90% of all miRNAs in the heart.
- 42.Brown BD, et al. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nature Biotechnol. 2007;25:1457–1467. doi: 10.1038/nbt1372. [DOI] [PubMed] [Google Scholar]
- 43.Liu N, et al. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl Acad. Sci. USA. 2007;104:20844–20849. doi: 10.1073/pnas.0710558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 45.Ivey KN, et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell. 2008;2:219–229. doi: 10.1016/j.stem.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu N, et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22:3242–3254. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deacon DC, et al. The miR-143-adducin3 pathway is essential for cardiac chamber morphogenesis. Development. 2010;137:1887–1896. doi: 10.1242/dev.050526. [DOI] [PubMed] [Google Scholar]
- 48.Morton SU, et al. microRNA-138 modulates cardiac patterning during embryonic development. Proc. Natl Acad. Sci. USA. 2008;105:17830–17835. doi: 10.1073/pnas.0804673105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt M, et al. EGFL7 regulates the collective migration of endothelial cells by restricting their spatial distribution. Development. 2007;134:2913–2923. doi: 10.1242/dev.002576. [DOI] [PubMed] [Google Scholar]
- 50.Nicoli S, et al. MicroRNA-mediated integration of haemodynamics and Vegf signalling during angiogenesis. Nature. 2010;464:1196–1200. doi: 10.1038/nature08889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhnert F, et al. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989–3993. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- 52.Wang S, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fish JE, et al. miR-126 regulates angiogenic signalling and vascular integrity. Dev. Cell. 2008;15:272–284. doi: 10.1016/j.devcel.2008.07.008. References 52 and 53 show a crucial role for miR-126 in angiogenesis.
- 54.Small EM, Sutherland LB, Rajagopalan R, Wang S, Olson EN. MicroRNA-218 regulates vascular patterning by modulation of Slit-Robo signaling. Circ. Res. 2010;107:1336–1344. doi: 10.1161/CIRCRESAHA.110.227926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cordes KR, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elia L, et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16:1590–1598. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boettger T, et al. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Invest. 2009;119:2634–2647. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao G, Yu D, Weiss MJ. MicroRNAs in erythropoiesis. Curr. Opin. Hematol. 2010;17:155–162. doi: 10.1097/MOH.0b013e328337ba6c. [DOI] [PubMed] [Google Scholar]
- 59.Georgantas RW, III, et al. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc. Natl Acad. Sci. USA. 2007;104:2750–2755. doi: 10.1073/pnas.0610983104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Carroll D, et al. A Slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev. 2007;21:1999–2004. doi: 10.1101/gad.1565607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu J, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev. Cell. 2008;14:843–853. doi: 10.1016/j.devcel.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Q, et al. MicroRNA miR-24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood. 2008;111:588–595. doi: 10.1182/blood-2007-05-092718. [DOI] [PubMed] [Google Scholar]
- 63.Rasmussen KD, et al. The miR-144/451 locus is required for erythroid homeostasis. J. Exp. Med. 2010;207:1351–1358. doi: 10.1084/jem.20100458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnnidis JB, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 65.Patrick DM, et al. Defective erythroid differentiation in miR-451 mutant mice mediated by 14-3-3ζ. Genes Dev. 2010;24:1614–1619. doi: 10.1101/gad.1942810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu D, et al. miR-451 protects against erythroid oxidant stress by repressing 14-3-3ζ. Genes Dev. 2010;24:1620–1633. doi: 10.1101/gad.1942110. References 65 and 66 show that miR-451 is required for proper erythroid differentiation, and suggest a potential therapeutic application for targeting miR-451 for degradation.
- 67.Callis TE, et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Invest. 2009;119:2772–2786. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thum T, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. This paper demonstrates an important role for miR-21 in cardiac remodelling using antagomir-mediated knockdown in mice.
- 69.van Rooij E, et al. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. The first paper to show a role for an miRNA, miR-208a, in the control of cardiac remodelling, using a genetic knockout.
- 70.Care A, et al. MicroRNA-133 controls cardiac hypertrophy. Nature Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 71.van Rooij E, et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell. 2009;17:662–673. doi: 10.1016/j.devcel.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Molkentin JD, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin Z, et al. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl Acad. Sci. USA. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rane S, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1α and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009;104:879–886. doi: 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patrick DM, et al. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J. Clin. Invest. 2010;120:3912–3916. doi: 10.1172/JCI43604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fleissner F, et al. Asymmetric dimethylarginine impairs angiogenic progenitor cell function in patients with coronary artery disease through a microRNA-21-dependent mechanism. Circ. Res. 2010;107:138–143. doi: 10.1161/CIRCRESAHA.110.216770. [DOI] [PubMed] [Google Scholar]
- 77.Bonauer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 78.van Solingen C, et al. Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. J. Cell. Mol. Med. 2009;13:1577–1585. doi: 10.1111/j.1582-4934.2008.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zernecke A, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal. 2009;2:ra81. doi: 10.1126/scisignal.2000610. [DOI] [PubMed] [Google Scholar]
- 80.Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl Acad. Sci. USA. 2008;105:1516–1521. doi: 10.1073/pnas.0707493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheng Y, et al. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ. Res. 2009;105:158–166. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saunders MA, Liang H, Li WH. Human polymorphism at microRNAs and microRNA target sites. Proc. Natl Acad. Sci. USA. 2007;104:3300–3305. doi: 10.1073/pnas.0611347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nature Genet. 2006;38:1452–1456. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 84.Clop A, et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nature Genet. 2006;38:813–818. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- 85.Sethupathy P, et al. Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3′ untranslated region: a mechanism for functional single-nucleotide polymorphisms related to phenotypes. Am. J. Hum. Genet. 2007;81:405–413. doi: 10.1086/519979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schipper ME, van Kuik J, de Jonge N, Dullens HF, de Weger RA. Changes in regulatory microRNA expression in myocardium of heart failure patients on left ventricular assist device support. J. Heart Lung Transplant. 2008;27:1282–1285. doi: 10.1016/j.healun.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 87.Voellenkle C, et al. MicroRNA signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiol. Genomics. 2010;42:420–426. doi: 10.1152/physiolgenomics.00211.2009. [DOI] [PubMed] [Google Scholar]
- 88.Fichtlscherer S, et al. Circulating microRNAs in patients with coronary artery disease. Circ. Res. 2010;107:677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- 89.Ji X, et al. Plasma miR-208 as a biomarker of myocardial injury. Clin. Chem. 2009;55:1944–1949. doi: 10.1373/clinchem.2009.125310. [DOI] [PubMed] [Google Scholar]
- 90.Elmen J, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 91.Lanford RE, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. The first report of therapeutically targeting an miRNA for the treatment of a disease in non-human primates.