

Abstract
Autophagy has been reported to be increased in irradiated cancer cells resistant to various apoptotic stimuli. We therefore hypothesized that induction of autophagy via mTOR inhibition could enhance radiosensitization in apoptosis-inhibited H460 lung cancer cells in vitro and in a lung cancer xenograft model. To test this hypothesis, combinations of Z-DEVD (caspase-3 inhibitor), RAD001 (mTOR inhibitor) and irradiation were tested in cell and mouse models. The combination of Z-DEVD and RAD001 more potently radiosensitized H460 cells than individual treatment alone. The enhancement in radiation response was not only evident in clonogenic survival assays, but also was demonstrated through markedly reduced tumor growth, cellular proliferation (Ki67 staining), apoptosis (TUNEL staining) and angiogenesis (vWF staining) in vivo. Additionally, upregulation of autophagy as measured by increased GFP-LC3-tagged autophagosome formation accompanied the noted radiosensitization in vitro and in vivo. The greatest induction of autophagy and associated radiation toxicity was exhibited in the tri-modality treatment group. Autophagy marker, LC3-II, was reduced by 3-methyladenine (3-MA), a known inhibitor of autophagy, but further increased by the addition of lysosomal protease inhibitors (pepstatin A and E64d), demonstrating that there is autophagic induction through type III PI3 kinase during the combined therapy. Knocking down of ATG5 and beclin-1, two essential autophagic molecules, resulted in radiation resistance of lung cancer cells. Our report suggests that combined inhibition of apoptosis and mTOR during radiotherapy is a potential therapeutic strategy to enhance radiation therapy in patients with non-small cell lung cancer.
Keywords: radiotherapy, autophagy, lung cancer, caspase, mTOR
Introduction
Lung cancer is the leading cause of cancer-related mortality in North America and throughout the world. Non-small cell lung cancer (NSCLC) accounts for ~75% of lung cancer. Standard therapies for advanced NSCLC include radiotherapy and chemotherapy, which confer palliative and moderate survival benefits with significant toxicities.1–3 A major limitation to the curative potential of current therapy is that many types of cancer, including lung cancer, are resistant to apoptotic stimuli of anti-neoplastic agents and become progressively incurable.4,5 Understanding the molecular mechanism behind the events responsible for the radiation-induced cell death is therefore crucial to developing improved strategies for the treatment of lung cancer.
Traditionally, apoptosis has been considered to be the predominant type of programmed cell death, and the major form of cell death induced by ionizing radiation. Advances in the understanding of autophagy in normal as well as pathological conditions established autophagic cell death as an alternative form of cell death, leading to the reclassification of programmed cell death into two types: Type I as apoptotic death and Type II as autophagic death.6,7 Autophagy is an evolutionarily conserved process of sequestering organelles and long-lived proteins in a double-membrane vesicle, the autophagosomes, for subsequent lysosomal degradation.8,9 This process can be triggered by physiologic stress, such as nutrient deprivation,10 or by inhibiting the mammalian target or rapamycin (mTOR) pathway.11 When stress conditions are excessive, autophagy can lead to cell death by cell-digestion. Prolonged autophagy in cancer cells can also lead to autophagic cell death, suggesting the possibility of utilizing autophagy activation for cancer therapy.12–15
Previous studies have shown that mouse embryonic fibroblasts (MEFs) deficient in Bax/Bak, gatekeepers of caspase-mediated apoptotic death, undergo increased cell death by treatment with etoposide,16 an inducer of apoptosis, and ionizing radiation.17,18 Furthermore, radiosensitization in the absence of Bax and Bak results in increased autophagy, and this radiosensitization response is blocked by inhibitors of autophagy such as 3-methyladenine (3-MA).18 In addition, inhibition of mTOR signaling via RAD001 in Bax/Bak−/− human breast and lung cancer cells with consequent upregulation of autophagy has been shown to significantly enhance radiosensitization.19 These reports suggest that activation of autophagy through inhibition of apoptosis can be utilized as an effective radiation therapy for cancer.
We therefore further investigated the concept of apoptosis inhibition as a way to sensitize lung cancer to radiation in vivo. To address this question, we utilized Z-DEVD, a specific inhibitor of terminal apoptotic effector, caspase-3 and RAD001, an mTOR inhibitor, in a H460 lung cancer xenograft model. Notably, we found that the combined administration of Z-DEVD and RAD001 not only enhanced radiosensitivity in clonogenic assays as compared to either agent alone, but it also led to a significant tumor growth delay in the in vivo model. Consistent with the in vitro data, combination treatment was associated with reduction in tumor volume, decreased cellular proliferation and apoptosis, as well as increased autophagosome formation in xenograft tumor section as seen by electron microscopy.
Results
Radiosensitization of H460 lung cancer cells by caspase-3 and mTOR inhibition
To determine if the combination of caspase-3 inhibition and autophagy induction leads to radiosensitization in H460 lung cancer cells, we used clonogenic assays. We examined the effects of Z-DEVD (an inhibitor of caspase-3), RAD001 (an inhibitor of mTOR) and the combination of Z-DEVD and RAD001 on cell survival. Cells were irradiated with doses ranging from 0 to 6 Gy. The DER in H460 lung cancer cells treated with Z-DEVD (50 μM for 24 hrs) compared to DMSO control was 1.1 (p < 0.01, Student’s t test, n = 3) (Fig. 1). The DER in the RAD001 (10 nM, for 1 hr) treatment group as compared to DMSO control was 1.28 (p < 0.009, Student’s t test, n = 3) (Fig. 1). Administration of both Z-DEVD and RAD001 resulted in a DER of 1.53 (p < 0.009, Student’s t test, n = 3) compared to DMSO control (Fig. 1). These findings suggest that caspase-3 inhibition by Z-DEVD leads to increased radiation sensitivity, and that combination of caspase-3 and mTOR inhibition can increase the radiation sensitivity in H460 lung cancer cells.
Figure 1.
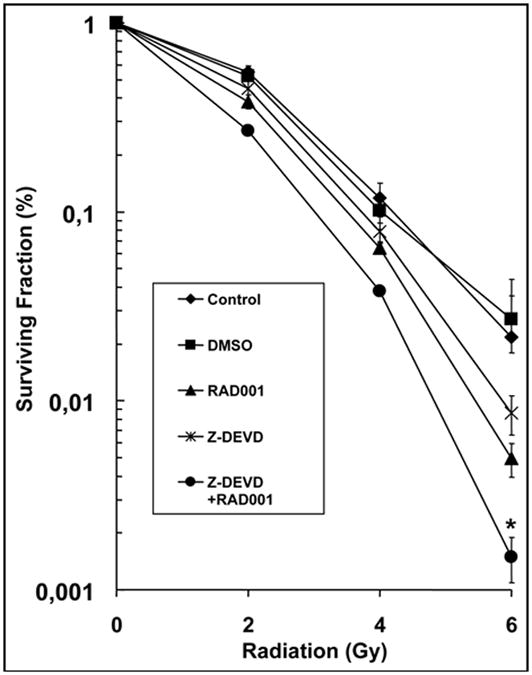
Combined inhibition of caspase-3 and mTOR reduces H460 lung cancer cells clonogenic survival. H460 NSCLC cells were treated with DMSO control, Z-DEVD (50 μM, for 24 hrs), RAD001 (10 nM, for 1 hr) or Z-DEVD and RAD001 with radiation. After 8 days, colonies were stained and the scored colonies were graphed. Survival curves for H460 cells ± Z-DEVD, mTOR or Z-DEVD and RAD001 treatment. The DER was calculated for each of the conditions with comparison to the DMSO control group. The Z-DEVD, RAD001 and combined Z-DEVD and RAD001 treatment groups had DERs of 1.1 (p < 0.01), 1.28 (p < 0.009) and 1.53 (p < 0.009), respectively. All statistical analyses were performed using Student’s t-test with comparison to the DMSO control group. Points, mean of three independent experiments; bars, S.D.
Delayed tumor growth and decreased tumor volume in H460 xenograft tumors treated with Z-DEVD, RAD001 and/or radiation in vivo
To validate our in vitro results, we performed xenograft experiments to demonstrate that combined treatment with Z-DEVD/ RAD001 can be used as radiosensitizing agents in animal models. H460 lung cancer cells were implanted subcutaneously in athymic mice and grown for approximately 7 days to an average volume of 0.18 cm3 prior to therapy. The treatment groups consisted of a vehicle control, Z-DEVD (2 mg/kg, i.p.), RAD001 (2.5 mg/kg, p.o.), and Z-DEVD/RAD001 treated with radiation as described in the Material and Methods section. Growth delay was calculated as the number of days required to reach a tumor volume of 2 cm3 for treatment groups relative to control. As shown in Figure 2A, as compared to DMSO control, radiation therapy delayed tumor growth by approximately 11 days. In comparison to the group with RT alone, a greater prolongation in tumor growth was observed with RAD001 + RT treatment (5 days, p = 0.004) or with Z-DEVD + RT treatment (3 days, p = 0.008) (Fig. 2A). The combination therapy exerted an additive effect and induced a statistically significant tumor growth delay when compared with radiation alone (32 vs. 23 days, p = 0.002) (Fig. 2A). In addition, combination treatments were very well tolerated, with minimal weight loss at 16 days in non-irradiated and irradiated groups relative to control (Fig. 2B). The combination therapy yielded the least increase in body weight. The reversal of tumor growth delay, as measured by increasing body weight, occurred approximately at day 13 for all treatment groups (Fig. 2B).
Figure 2.
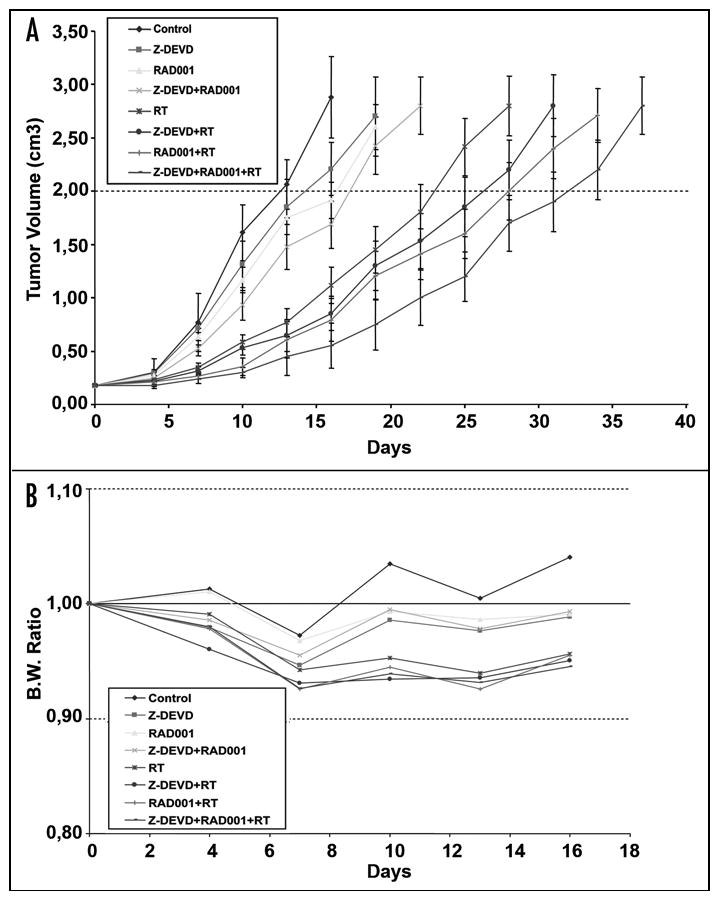
Improved tumor growth delay with combined Z-DEVD/RAD001 and radiotherapy at well-tolerated doses in H460 xenograft model. H460 lung cancer cells were implanted subcutaneously in athymic mice. After 6–8 days, mice were treated daily for 7 days, with vehicle control, Z-DEVD (2 mg/kg, i.p), RAD001 (2.5 mg/kg, p.o.), and/or combined Z-DEVD and RAD001, and then were treated 1 hr after drug treatment with 2 Gy of radiation, daily over 5 consecutive days. Tumors were excised when they reached approximately 2.5–3.0 cm3, and tumor growth delay was defined by the number of days required to reach a tumor volume of 2 cm3 was measured. (A) Tumors were measured regularly and growth delay was calculated for treatment groups relative to control tumors. The combined tri-modality therapy (*) induced a marked tumor growth delay when compared with radiation alone (32 vs. 23 days, p = 0.002, Student’s t-test). Points, mean value of tumor volumes in 5 mice; bars, S.D. (B) Body weights were measured every 5 days and body weight ratio was calculated relative to baseline measurement.
Decreased cell proliferation, apoptosis and vascular density in irradiated H460 xenograft tumors as a result of caspase-3 and mTOR inhibition
To delineate the mechanism underlying the observed effects in xenograft model, we examined the marker for cell proliferation (Ki67, Fig. 3A), for apoptosis (TUNEL, Fig. 3C), and for angiogenesis (vWF, Fig. 4A) by performing immunohistochemistry on xenografted tumor sections in each treatment groups. As shown in Figure 3B, DMSO-treated control exhibited 140 ± 2.5 Ki67 stained cells, while RT-treated group resulted in dramatic reduction of the proliferation index (38 ± 1.5). When radiation was combined with Z-DEVD, a slight decrease in Ki67 index (32 ± 2) was observed. Combining RAD001 with irradiation caused a further decrease to 23 ± 2 stained cells. The greatest reduction in cell proliferation (13 ± 2) resulted from the combined treatment with RAD001/Z-DEVD and radiation (13 ± 2 vs. 37.6 ± 1.5 Ki-67 positive cells per field, p = 0.0001, Student’s t-test) (Fig. 3B). To test whether the increased response to radiation seen in vitro and in vivo is associated with apoptosis inhibition, the level of apoptosis in xenografts were measured by TUNEL staining (Fig. 3C). Radiation therapy alone increased positive TUNEL staining by ~8-fold compared to control group (Fig. 3D). All non-irradiated groups showed insignificant increase in apoptosis level, although RAD001 had the most effect relatively. As expected of its action in inhibiting caspase-3, Z-DEVD and radiation decreased positive TUNEL staining by 3-fold when compared to radiation alone. RAD001 and radiation decreased the number of positive TUNEL cells to a lesser degree, by 2-fold in comparison to radiation alone (Fig. 3D). In the combined treatment of Z-DEVD/ RAD001 with radiation, we observed that positive TUNEL cell returned to the level similar to the untreated control group (2 ± 1 vs. 13 ± 1.5 TUNEL-positive cells per field, p = 0.0008, Student’s t-test, n = 3). This suggests that combination therapy is a potent inhibitor of apoptosis and that radiosensitization can indeed occur amidst the significantly reduced level of apoptosis.
Figure 3.
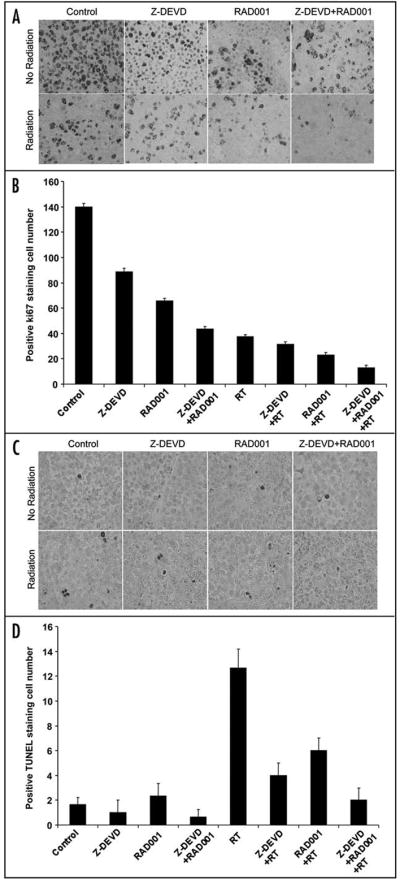
Z-DEVD/RAD001 with radiation reduces Ki67 proliferative marker and increases apoptotic index in H460 xenograft model. Histologic sections were obtained from the tumors of the mice in each treatment group from the tumor volume study. Number of positive cells were scored and graphed by averaging three repeated assessments. (A) Representative images of histological section following standard Ki67. (B) Average Ki67 proliferative index of each treatment group was determined by counting positive cells per microscopic field. The greatest reduction in cell proliferation (~10) resulted from the combined treatment with RAD001/Z-DEVD and radiation (13 ± 2 vs. 37.6 ± 1.5 Ki-67 positive cells per field, p = 0.0001, Student’s t-test). Columns, mean of Ki67-positive cells of three random microscopic fields; bars, S.D. (C) TUNEL staining was also performed on tumor sections, as shown by the representative histological images. (D) Apoptotic index was similarly calculated by counting positive TUNEL stained cells per microscopic field. In the combined treatment of Z-DEVD/RAD001 with radiation, we observed that positive TUNEL cell returned to the level similar to the untreated control group (2 ± 1 vs. 13 ± 1.5 TUNEL-positive cells per field, p = 0.0008, Student’s t-test, n = 3). Columns, mean of TUNEL positive cells in 3 random microscopic fields; bars, S.D.
Figure 4.

Z-DEVD/RAD001 sensitizes vascular endothelial in vitro model to ionizing radiation and reduces in vivo vascular density in irradiated H460 tumors. (A) Representative histological section following von Willebrand factor (vWF) staining. Histologic sections were obtained from the tumors of the mice in each treatment group from the in vivo tumor volume study, and stained for blood vessels using an antibody for vWF. (B) Blood vessels were quantified by randomly selecting 400X fields and counting the number of blood vessels per field. This was done in triplicate and the average of the three counts was calculated. Columns, mean number of vessels counted per microscopic field; bars, S.D. (C) Human umbilical vein endothelial cells (HUVECs) were treated with 5 μmol/L Z-DEVD/RAD001 and then immediately irradiated with either 0 or 5 Gy. Six hours later, cells were trypsinized and re-plated on 24-well plates coated with Matrigel. After 24 hrs, cells were fixed and stained with H&E. The slides were examined by microscopy (×100), and representative fields are shown. (D) Stained tubules were then counted in three separate, randomly selected fields. Columns, mean number of tubules counted per microscopic field; bars, S.D.
To determine the effects of radiation with Z-DEVD and/or RAD001 in vivo, we next examined the vascular density as detected by vWF staining. This was prompted by the established function of RAD001 in angiogenesis inhibition.20 Moreover, markers of angiogenesis have been shown to have prognostic significance in solid tumors.21 vWF staining in the combination treatment group yielded a more than 10-fold reduction in the number of staining vessels in comparison to controls, and over a 4-fold reduction relative to radiation therapy alone (Fig. 4A and B) (0.83 ± 0.3 vs. 3.7 ± 0.6 vessels per field, p < 0.004, Student’s t-test). Likewise, apoptosis and mTOR inhibitors may exert antiangiogenic effects that can possibly be correlated with their in vivo antitumor effects.
Marked reduction of endothelial tubule formation observed in vitro under tri-modality treatment
To explore the effects of Z-DEVD/RAD001 and radiation observed on vasculature, we tested the in vitro effects of caspase-3 and mTOR inhibition on endothelial tubule formation, which is a critical step in angiogenesis. HUVECs were treated with DMSO, Z-DEVD (50 μM for 24 hrs), RAD001 (10 nM for 2 hrs) or Z-DEVD + RAD001, and then cells were treated with 5 Gy, after which the endothelial tubule formation was observed. Representative photographs are shown in Figure 4C, and the mean number of counted tubules in three separate (×100) fields is shown in Figure 4D. Compared to DMSO control (17 ± 1 tubules/field), treatment of HUVEC with ionizing radiation alone resulted in dramatic reduction of tubule formation (11 ± 1 tubules/field). The same antiangiogenic pattern amongst treatment groups was observed between non-irradiated and irradiated groups in that the most potent antiangiogenic effects were exerted by the combined group. Under irradiation, more pronounced reduction in endothelial tubule formation was observed, especially in the combined group showing less than 1.3 ± 0.6 tubules/field (Fig. 4C and D). Taken together, these results suggest that the tumor growth delay effect induced by caspase-3/mTOR inhibition is at least associated with, if not partly due to, their potent antiangiogenic properties.
Increased autophagy induction in irradiated H460 tumors in vitro and in vivo following the administration of Z-DEVD and RAD001
We have previously shown that MEFs deficient in Bax/ Bak demonstrated increased radiation sensitivity in correlation with upregulated autophagosome formation.17,22 In order to assess whether radiation sensitivity in Z-DEVD/RAD001 treated cells is also correlated with autophagosome formation, we performed trans-fection experiments with GFP-LC3 and monitored GFP expression in control, Z-DEVD, RAD001 and combined Z-DEVD/RAD001 treatments with irradiation. Microtubule associated protein-1 light-chain 3 (LC3) is an important constituent of autophagosomes, and GFP-LC3 has been demonstrated to be an specific marker of mammalian autophagy.23 After H460 cells exposure to Z-DEVD, RAD001 and combination of Z-DEVD/RAD001 followed by 5 Gy of radiation, we observed changes in cellular localization of punctate GFP expression from predominant diffuse cytoplasmic expression to a punctate pattern characteristic of autophagosome formation. Quantification of the percentage of punctate GFP expression in each treatments (Fig. 5A) showed that single treatment with Z-DEVD or RAD001 resulted in approximately 30% and 35% of punctate GFP expression, respectively, compared to ~5% of punctate GFP expression in DMSO control. However, when Z-DEVD and RAD001 were combined, we observed that over 50% of the cells had punctate GFP expression pattern (Fig. 5A), suggesting that combination treatment is a more potent inducer of autophagy than either drug alone. The presence of autophagosomes was further confirmed in electron microscopy of tumor sections obtained from our in vivo tumor volume study upon combination treatment (Fig. 5B).
Figure 5.

Z-DEVD/RAD001 with radiation increased autophagy induction in H460 tumor model. (A) GFP-LC3-transfected H460 cells were treated with either DMSO control, Z-DEVD (50 μM, for 24 hrs), RAD001 (10 nM, for 1 hrs) or Z-DEVD + RAD001 with 0 or 5 Gy radiation, and subsequently examined by fluorescence microscopy after 48 hrs. The percentage of cells with punctate GFP-LC3 fluorescence was calculated relative to all GFP-positive cells. Columns, mean percentage of GFP-LC3 expressing punctate cells in 3 microscopic fields; Bars, S.D. (B) Representative electron micrograph image showing autophagic vacuoles with content (black arrows) following Z-DEVD/RAD001 treatment with radiation in an in vivo H460 xenograft model. (C) H460 cells were treated with Z-DEVD + RAD001 + 5 Gy. They were then treated with either 3MA (200 uM) or pepstatin A (10 μg/ml) and E64d (10 μg/ml) for 2 hrs. Cells were harvested in one or two days for western analyses for LC3-II.
Though LC3 expression is the most commonly used marker for autophagy induction, accumulation of this protein can occur without activation of autophagy.24,25 To determine whether the observed H460 cells with GFP punctates, following the triple treatment, resulted from autophagy induction or sluggish autophagy flux, we used 3MA to inhibit autophagy induction and used pepstatin A and E64d, lysosomal protease inhibitors, to inhibit the autophagy flux. Cells were treated in the following sequences: Z-DEVD/RAD001/RT, Z-DEVD/RAD001/ RT + 3MA or Z-DEVD/RAD001/RT + pepstatin A/ E64d. We detected increased protein levels of autophagic marker, LC3-II, after the triple therapy, which was attenuated by 3MA, which inhibits type III PI3K, initiator of autophagy (Fig. 5C). LC3-II levels were however elevated by pepstatin A/E64d, which blocked the flux of autophagy (Fig. 5C). These results suggest that the measured autophagic cells following Z-DEVD + RAD001 + RT resulted mainly from autophagy induction, rather than from reduced autophagy flux.
Radioresistance of irradiated H460 cells with knockdown of key autophagy proteins
We demonstrated in the previous section that irradiation of H460 tumors resulted in increased autophagosome production, and this was associated with decreased tumor growth kinetics. To determine if radiation-induced cell death is mediated by autophagy, we knocked down the expression of two essential autophagy proteins, ATG5 and Beclin-1. We transfected H460 cells with 12.5 nM of siRNA to ATG5 and Beclin-1, which resulted in a significant decrease in the expression levels of protein (Fig. 6A). These cells were irradiated with doses ranging from 0 to 6 Gy. The DER in H460 cells treated with siRNA ATG5/Beclin-1 was 0.85 (p < 0.021, Student’s t-test, n = 3) as compared to the siRNA control, and siRNA control was not significantly different from untransfected H460 cells (Fig. 6B). These findings suggest radiation-induced cell death is at least partially mediated by autophagy.
Figure 6.
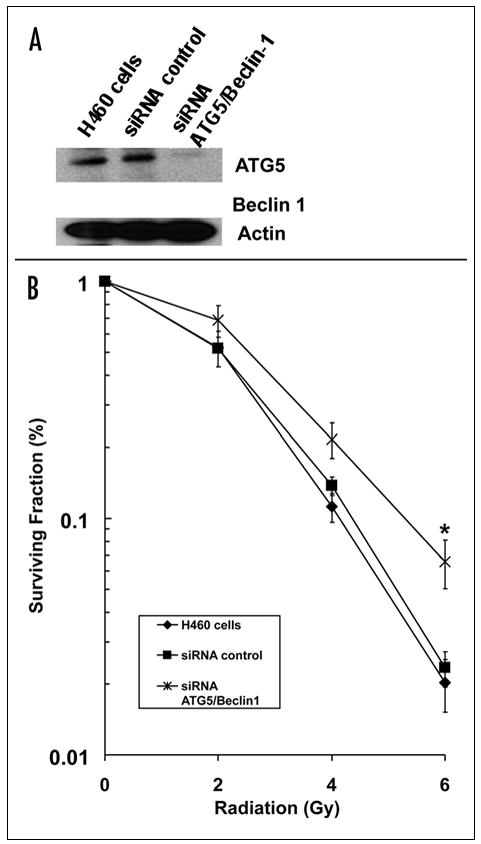
Inhibition of autophagy by targeting siRNA to ATG5 and Beclin-1 confers radioresistance in H460 lung cancer cells. (A) ATG5/Beclin-1 expression was determined by Western blotting using lysates from H460 lung cancer cells treated with siRNAs against ATG5/Beclin-1. Actin was probed to demonstrate equal loading. (B) H460 lung cancer cells were treated with DMSO control, siRNA control and siRNA ATG5/Beclin-1 (12.5 nM) under irradiation with doses ranging from 0 to 6 Gy. After 8 days, colonies were stained and the scored colonies were graphed. The DER in H460 cells treated with siRNA ATG5/Beclin-1 was 0.85 (p = 0.021, Student’s t-test). Points, mean of 3 independent experiments; bars, S.D.
Discussion
The present study addresses the potential application of inhibiting both apoptosis and mTOR signaling to improve the efficacy of radiation therapy. A combined administration of the caspase-3 inhibitor Z-DEVD and the mTOR inhibitor RAD001 in xenografted H460 lung tumors resulted in superior radiation cytotoxicity than by either agent alone (Fig. 2A). Enhanced radiosensitiza-tion was accompanied by delayed tumor growth, reduced cellular proliferation, and near abrogation of angiogenesis, all of which did not adversely affect the treatment tolerability in vivo (Figs. 2B, 3 and 4). Consistent with our previous study targeting upstream apoptotic regulators,19 increased radiation-induced tumor cytotoxicity was associated with autophagy. Taken together, the combination therapy that activates autophagy may represent a novel approach to improve local tumor control by radiotherapy.
Apoptosis, regarded as the main death execution machinery, may play only a minor role in radiation-induced cell death. Some have argued that apop-tosis accounts for less than 20% of radiation-induced cell death,26 and that alternative modes of radiation sensitivity are bound to exist in apoptosis-resistant malignant tumors.27 Manipulation of alternative types of cell death appears crucial in targeting malignant tumors frequently resistant to apoptotic stimuli. Many studies have suggested autophagy as an alternative death pathway adopted by irradiated cancer cells.19,28 We have previously shown that inhibition of apoptosis with the pan-caspase inhibitor Z-VAD or siRNA knockout of Bax/ Bak upregulates autophagy in radio-sensitized prostate cancer cell lines.19 Consistent with these results, Demasters et al.,29 reported the potentiation of radiation sensitivity in breast tumor cells by EB 1089, a vitamin D3 analogue, via enhanced promotion of autophagic cell death.29 Given its association with the radiosensitization response, autophagic activation may play a role in radiation-induced cell death.30
To confirm the observed link between autophagy and radiosensitization, we applied RAD001 in addition to Z-DEVD on H460 lung cancer cells to see if an enhancement of autophagic activity would augment radiation toxicity. mTOR inactivation has been shown to initiate autophagy31,32 while its activation under nutrient-rich conditions represses autophagy.33,34 The capacity to achieve radiosensitization through mTOR inhibition has been shown before,31,35,36 and we analogously demonstrate that the combined administration of RAD001 and Z-DEVD radiosensitizes H460 cancer cells to a greater extent (DER = 1.53, p < 0.009) than with Z-DEVD (DER = 1.1, p < 0.01) or RAD001 alone (DER = 1.28, p < 0.009) (Fig. 1). Combined therapy not only delayed tumor growth (32 vs. 23 days, p = 0.002) (Fig. 2A) and markedly decreased cellular proliferation (Fig. 3A and B), but was also well-tolerated in mice with minimal body weight change (Fig. 2B). These encouraging results suggest the potential application of mTOR inhibition in apoptosis-inhibited tumors to improve radiation toxicity in clinical setting.
Since radiotherapy is known to cause antiangiogenic in addition to its direct cytotoxic effects, we examined whether alterations in angio-genesis has any bearing to the enhanced radiation response. mTOR inhibitors such as rapamycin and rapamycin-derivative RAD001 (Everolimus) are known for their antiangiogenic effects20,37,38 that can sensitize cancer cells to radiation effects.35,39 For instance, rapamycin at concentrations as low as 0.01 μg/ml have been demonstrated to suppress angiogenesis in mice by decreasing serum VEGF as well as the receptor responsiveness to VEGF.20 The documented antiangiogenic effects of rapamycin was not due to the inhibition of tumor proliferation, since no apparent anti-proliferative effect was seen at the therapeutically relevant RAPA level (<1.0 μg/ml). Our results also support a reduction of angiogenesis following RAD001 and radiation treatment. Of note, Z-DEVD resulted in an attenuation in vascular density similar to RAD001 The most prominent antiangiogenic effects, however, were observed with the combination of Z-DEVD, RAD001 and RT, resulting in almost no vessel formation in vivo (Fig. 4A and B). This phenomenon was also observed in vitro with HUVEC cells (Fig. 4C and D). Interestingly, the number of tubules after treatment with Z-DEVD and radiation was similar to the RAD001 and radiation group, suggesting that the inhibition of caspase-3 could affect endothelial function via autophagy (Fig. 4). The marked reduction in angiogenesis observed in the tri-modality group may therefore be attributable to the events following apoptosis inhibition in addition to mTOR inactivation.
We then hypothesized autophagy to be the additional radiosensitizing mechanism in the poorly vascularized lung tumors. The temporal association between autophagic upregulation and increased tumor kill suggest the possibility that autophagy may play a role in radiation-induced cell death.40–42 In our study, the greatest induction of GFP-LC3 labeled autophagosome formation occurred under the tri-combined therapy in vitro (Fig. 5A) and in vivo (Fig. 5B). Although autophagy is difficult to detect in vivo, a punctate distribution of GFP-tagged light-chain 3 (LC3) on the membranes of autophagosomes has been used as an indicator of autophagy.43 To the author’s knowledge, this is the first study to show that inhibiting both apoptotic effector and mTOR signaling can effectively amplify the autophagic response detectable in vivo.
Examining GFP-LC3 accumulation in response to irradiation is a qualitative assay. To more directly evaluate the role of autophagy in the radiosensitization of H460 cells, we targeted siRNA to autophagic genes, ATG5 and Beclin-1. The siRNA transfected cells demonstrated a significant knockdown of ATG5 and Beclin-1 expression (Fig. 6A), and this associated with a statistically significant increased in clonogenic survival (Fig. 6B). This is strong evidence that radiation-induced cell death is mediated at least in part through activation of autophagy.
With regard to the radiation-associated autophagic response, some have hypothesized that excessive autophagy or uncontrolled self-degradation may directly result in cell death.44 In fact, inhibition of autophagy by PI3 kinase inhibitors 3-methyladenine (3-MA) or wortmanin in apoptosis-deficient Bax/Bak−/− MEFs has been shown to improve cell viability.16 Alternatively, it has been shown that autophagic activation can indirectly cause cell death by activating other IR-induced death pathways. For instance, Z-VAD administration has been documented to induce necrotic death45 and even sensitize some cell lines to tumor-necrosis factor-α (TNFα)-induced necrotic death.46 Nonetheless, whether autophagy assumes an active or conducive role in radiation-triggered cell death remains controversial.
Given the evolutionarily conserved cytoprotective role of autophagy,47 however, increased autophagosome formation may signify a survival attempt by cancer cells subjected to genotoxic stress. In this view, a transient activation of autophagy may bring about a gradual adaptation to stress,48 as noted in our gradual reversal of tumor growth delay in vivo (Fig. 2A). Autophagy may indeed play a protective role in irradiated cancer cells by preventing the nutrient-limiting and damaging effects of ionizing radiation.28,49 In normal cells, autophagy has recently been found to confer cellular protection against genomic instability during metabolic stress.50 When this protective mechanism is inhibited via knockout of Beclin-1 or constitutive activation of Akt protein kinase in apoptosis-deficient tumors, necrotic death has been documented to ensue.51 Autophagy may therefore be a highly preserved survival mechanism adopted by cancer cells under radiation stress.
GFP-LC3 accumulation is a surrogate for autophagy induction, but induction of LC3 does not necessarily correlate to activation of the autophagocytosis. The increased GFP-LC3 can represent either increased autophagosome production or a diminished clearance of intra-autophagosomal products.52–54 A higher fidelity method for determining autophagic flux is observing the change in LC3 expression in the presence and absence of lysosomal protease inhibitors.24,25 We used pepstatin A, an inhibitor of cathepsins D,55–57 and E64d, a cell permeable inhibitor of cathepsins B, H and L55,58,59 to evaluate autophagic flux in our model. 3MA eliminated the induction of LC3-II in the triple agent-treated H460 cells (Fig. 5C), whereas inhibition of lysosomal turnover with pepstatin A and E64d further elevated the autophagy marker, LC3-II (Fig. 5C). These findings suggest a direct role for autophagic induction and autophagic activation in the mediation of the response of these cells to combined treatments.
Our results may also suggest that increased autophagosome formation detected in dying or dead cells signifies a failed survival attempt (reviewed in ref. 60). Interestingly, autophagic failure and consequent accumulation of misfolded proteins have been documented to induce apoptosis61 and to play a critical role in inducing apoptotic clearance of cells during mammalian development.62 From this evolutionary perspective, it would be worth exploring whether autophagy as a highly preserved survival mechanism mediates all radiation-induced cell death pathways, including necrosis.
Although association between autophagy and radiosensitization was demonstrated, the precise role of autophagy in relation to cell death is hard to define. To understand the downstream effects of autophagy, it would be necessary to examine other death pathways in action when both apoptosis and autophagy are blocked. Nevertheless, this study examined the enhancement of radiotherapy efficacy via autophagy induction, and suggests a clinical potential to better the current treatment for patients presenting NSCLC. We showed the efficacy of combining radiation with Z-DEVD and RAD001 to significantly sensitize H460 lung cancer in vivo. Further steps in the improvement of anticancer therapy using the concept of apoptosis inhibition include the development of novel agents that can selectively target major effectors of apoptosis with good pharma-cokinetic characteristics for the use in a clinical setting.
Materials and Method
Cell culture and chemicals
H460 lung cancer cells were cultured in RPMI 1640 (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin and incubated at 37°C and humidified 5% CO2. Human umbilical vein endothelial cells (HUVECs) were obtained from Clonetics (now Lonza Inc., Allendale, NJ) and were maintained in EBM-2 medium supplemented with EGM-2 MV single aliquots (BioWhittaker, Walkersville, MD). Z-DEVD-FMK (Z-DEVD) was purchased from R&D System, Inc., (Minneapolis, MN), and RAD001 from Novartis Pharmaceutical (East Hanover, NJ).
Clonogenic assay
H460 lung cancer cells were treated with DMSO, Z-DEVD (50 μM, for 24 hrs), RAD001 (10 nM for 1 hr), or a combination of Z-DEVD and RAD001. Cells were irradiated with 0 to 6 Gy as indicated at a dose rate of 1.8 Gy/min utilizing a 137Cs irradiator (J.L. Shepherd and Associates, Glendale, CA). Following irradiation, cells were incubated at 37°C for 8–10 days. Cells were fixed for 15 minutes with 3:1 methanol: acetic acid and stained for 15 minutes with 0.5% crystal violet (Sigma, St. Louis, MO) in methanol. After staining, colonies were counted using a cutoff of 50 viable cells. Surviving fraction (SF) was calculated as (mean colony counts)/(cells inoculated) × (plating efficiency (PE)), where PE was defined as (mean colony counts)/(cells inoculated for non-irradiated controls). The radiation dose enhancement ratio (DER) by drug (Z-DEVD, RAD001 or both) was calculated as (SF at an indicated dose of radiation alone)/(SF at an indicated dose of radiation + drug). Radiosensitization is defined as the term used when drug increases the sensitivity of cells to radiation (as assessed by clonogenic inhibition), and is represented in the form of DER. A DER equal to 1 suggests an additive radiation effect and a DER > 1 indicates a supra-additive effect. Experiments were conducted in triplicate and mean, SD, and p values (for Z-DEVD, RAD001 or combined Z-DEVD and RAD001 treated cells versus DMSO control, using a Student’s test) were calculated.
siRNA transfection
siRNA Beclin-1 (mouse) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). siRNA ATG5 (mouse) was synthesized by Dharmacon Research (Lafayette, CO). H460 cells were transfected with 12.5 nM of each of the siRNAs utilizing Lipofectamine 2000 (Invitrogen, Life Technologies, Carlsbad, CA). After 24 hrs, the transfected cells were ready for experimental use.
Autophagy assay
H460 lung cancer cells (2 × 105 cells) were seeded in a 6-well tissue culture plate overnight and subsequently transfected with 2 μg of GFP-LC3 expression plasmid (a gift from Dr. Noboru Mizushima, Tokyo, Japan) using the Lipofectamine 2000 reagent. After 12 hrs, cells were treated with DMSO, Z-DEVD (50 μM for 24 hrs), RAD001 (10 nM for 1 hr), or a combination of Z-DEVD and RAD001. Cells were then treated with 5 Gy, after which they were incubated for 48 hrs at 37°C. The fluorescence of GFP-LC3 was observed under a confocal fluorescence microscope. Three fields were captured, and 100 cells were scored in each field. The percent of punctate GFP cells per total GFP transfected cells were calculated and experiments were conducted at least three times.
Quantification of autophagic flux
In order to distinguish between autophagic induction and autophagic flux, we detected the levels of LC3-II following the treatment of either 3MA, a type III PI3K inhibitor, or lysosomal protease inhibitors. Our lysosomal protease inhibitors were E64d (10 μg/ml, Sigma, St. Louis, MO) and peptastin A (10 μg/ml, Sigma, St. Louis, MO). H460 lung cancer cells (2 × 105 cells) were again seeded in tissue culture plates overnight and treated with Z-DEVD (50 μM for 24 hrs) and RAD001 (10 nM for 1 hr). These cells were irradiated with 5 Gy and then treated either with 200 uM 3MA or with pepstatin A (10 μg/ml) and E64d (10 μg/ml), for a period of 2 hrs. Cells were then collected in 24 hrs and 48 hrs for western analyses.
Tumor volume assessment
H460 lung cancer cells were used as a xenograft model in female athymic nude mice (nu/nu, 5 to 6 weeks old [Harlan Sprague Dawley Inc., Indianapolis, IN]). A suspension of 1 × 106 cells in 100 μL volume was injected subcutaneously into the right flank of mice using a 1-cc syringe with 27½-gauge needle. Tumors were grown for 6 to 8 days until average tumor volume reached 0.18 cm3. Treatment groups consisted of vehicle control (in DMSO), Z-DEVD, RAD001, combined Z-DEVD and RAD001, vehicle plus radiation, Z-DEVD plus radiation, RAD001 plus radiation, combined Z-DEVD and RAD001 plus radiation Each treatment group contained 5 mice. Vehicle control, Z-DEVD (intra-peritoneal (i.p.)) at doses of 2 mg/kg, and RAD001 (per orum (p.o.)) at doses of 2.5 mg/kg were administered for 7 consecutive days. In the case of combination treatment, vehicle or drug was given for 2 days prior to the first dose of irradiation. Mice in radiation groups were irradiated 1 hr after Z-DEVD and RAD001 treatment with 2 Gy daily over 5 consecutive days. Tumors on the flanks of the mice were irradiated using an X-ray irradiator (Therapax, AGFA NDT, Inc., Lewis Town, PA). The non-tumor parts of the mice were shielded by lead blocks. Tumors were measured 2 or 3 times weekly in 3 perpendicular dimensions using a Vernier caliper. Tumor Volumes were calculated using the formula for volume of ellipsoid: L × W × H/2, where L = length, W = width and H = height. Growth delay was calculated as the number of days required to reach a tumor volume of 2 cm3 for treatment groups relative to control tumors.
Histological sections, vWF, Ki-67 and TUNEL staining
Mice were implanted with H460 lung cancer cells as described above in the tumor volume studies. After 6–8 days, mice in the drug treatment group were treated with 2 mg/kg Z-DEVD (i.p.) and 2.5 mg/kg RAD001 (p.o.) daily for 7 days. Mice in the radiation treatment group were treated with 2 Gy daily fractions for 5 days, administered by use of an X-ray irradiator as described above in the tumor volume studies. After 7 days of daily treatments, excised tumors were paraffin fixed, and prepared in ultra-thin (90 nm) sections. Slides from each treatment group were then stained for vWF using anti-vWF polyclonal antibody (Chemico, Pontiac, MI). Blood vessels were quantified by randomly selecting 400X fields and counting the number of blood vessels per field. Ki67 and TUNEL staining were performed in our pathology core laboratory. The samples were examined with JEOL-1010 electron microscope (JEOL) operated at 80 kv. The number of positive cells were scored and graphed by averaging the three fields scored.
Endothelial cell morphogenesis assay: tubule formation
Human umbilical vein endothelial cells (HUVECs) grown to ~70% confluency were treated with DMSO, Z-DEVD (50 μM for 24 hrs), RAD001 (10 nM for 2 hrs) or combined Z-DEVD with RAD001, and then cells were treated with 5 Gy. Cells were then trypsinized and counted. They were seeded at 48,000 cells per well on 24-well plates coated with 300 μl of Matrigel (BD Biosciences, San Jose, CA). These cells undergo differentiation into capillary-like tube structures and were periodically observed by microscope. One day later, cells were stained with haematoxylin and eosin (H&E) and photographs were taken via microscope. The average number of tubes was calculated from examination of three separate microscopic fields (100×) and representative photographs were taken.
Western blot analysis
H460 cells were treated with DMSO (vehicle), siRNA control, and 12.5 nM siRNA ATG5/Beclin-1 for 24 hrs. The cells were collected, washed twice with ice-cold phosphate-buffered saline before the addition of lysis buffer (24 mM Tris-HCL, pH 7.4, 150 mM NaCl, 24 mM EDTA, 1% Nonidet P-40, 50 mM NaF, 1 mM Na3Vo4, 1 mM NaMO4 and mixture inhibitor (5 μl/ml, Sigma, St. Louis, MO). Protein concentrations were quantified using the Bio-Rad method. Equal amounts of protein were loaded into each well, resolved on a 12.5% SDS-PAGE gel and transferred to polyvi-nylidene difluoride membranes (Bio-Rad, Hercules, CA). Membranes were blocked with 5% nonfat dry milk in phosphate-buffered saline-Tween 20 for 1 hr at room temperature. The blots were then incubated overnight at 4°C with antibodies to ATG-5 (gift from Dr. Norubo Mizushima), Beclin-1 (Santa Cruz Biotechnology, Santa Cruz, CA), LC3 (MRL, Woburn, MA) and actin (Santa Cruz Biotechnology, Santa Cruz, CA). Goat anti-rabbit IgG secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was incubated for 45 minutes at room temperature. Western blots were analyzed using the chemiluminescence detection system (Perkin-Elmer Life Sciences, Waltham, MA) according to the manufacturer’s protocol.
Acknowledgments
This work is supported in part by DOD PC031161 and NCI 1R01 CA125842-01A1.
References
- 1.Ferreira CG, Huisman C, Giaccone G. Novel approaches to the treatment of non-small cell lung cancer. Crit Rev Oncol Hematol. 2002;41:57–77. doi: 10.1016/s1040-8428(01)00197-4. [DOI] [PubMed] [Google Scholar]
- 2.Feng X, Bai YJ, Lu B, Wang CG, Qi YX, Liu YX, Geng GL, Li L. Low temperature induced synthesis of TiN nanocrystals. Inorg Chem. 2004;43:3558–60. doi: 10.1021/ic049841n. [DOI] [PubMed] [Google Scholar]
- 3.Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–8. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 4.Nishio K, Nakamura T, Koh Y, Suzuki T, Fukumoto H, Saijo N. Drug resistance in lung cancer. Curr Opin Oncol. 1999;11:109–15. doi: 10.1097/00001622-199903000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Andriani F, Perego P, Carenini N, Sozzi G, Roz L. Increased sensitivity to cisplatin in non-small cell lung cancer cell lines after FHIT gene transfer. Neoplasia. 2006;8:9–17. doi: 10.1593/neo.05517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baehrecke EH. How death shapes life during development. Nat Rev Mol Cell Biol. 2002;3:779–87. doi: 10.1038/nrm931. [DOI] [PubMed] [Google Scholar]
- 7.Reggiori F, Klionsky DJ. Autophagosomes: biogenesis from scratch? Curr Opin Cell Biol. 2005;17:415–22. doi: 10.1016/j.ceb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–81. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 10.Mortimore G, Poso AR. Amino acid control of intracellular protein degradation. Methods Enzymol. 1988;166:461–76. doi: 10.1016/s0076-6879(88)66060-5. [DOI] [PubMed] [Google Scholar]
- 11.Kelekar A. Autophagy. Ann N Y Acad Sci. 2005;1066:259–71. doi: 10.1196/annals.1363.015. [DOI] [PubMed] [Google Scholar]
- 12.Levine B. Unraveling the role of autophagy in cancer. Autophagy. 2006;2:65–6. doi: 10.4161/auto.2.2.2457. [DOI] [PubMed] [Google Scholar]
- 13.Nelson DA, White E. Exploiting different ways to die. Genes Dev. 2004;18:1223–6. doi: 10.1101/gad.1212404. [DOI] [PubMed] [Google Scholar]
- 14.Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol. 2007;78:217–45. doi: 10.1016/S0070-2153(06)78006-1. [DOI] [PubMed] [Google Scholar]
- 15.Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy. 2006;2:85–90. doi: 10.4161/auto.2.2.2463. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 17.Buytaert E, Callewaert G, Vandenheede JR, Agostinis P. Deficiency in apoptotic effectors bax and bak reveals an autophagic cell death pathway initiated by photodamage to the endoplasmic reticulum. Autophagy. 2006;2:238–40. doi: 10.4161/auto.2730. [DOI] [PubMed] [Google Scholar]
- 18.Motzer RJ, Rini BI, Bukowski RM, Curti BD, George DJ, Hudes GR, Redman BG, Margolin KA, Merchan JR, Wilding G, Ginsberg MS, Bacik J, Kim ST, Baum CM, Michaelson MD. Sunitinib in patients with metastatic renal cell carcinoma. Jama. 2006;295:2516–24. doi: 10.1001/jama.295.21.2516. [DOI] [PubMed] [Google Scholar]
- 19.Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, Gi YJ, Lu B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosen-sitizes PTEN null prostate cancer cells. Cancer Res. 2006;66:10040–7. doi: 10.1158/0008-5472.CAN-06-0802. [DOI] [PubMed] [Google Scholar]
- 20.Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW, Geissler EK. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8:128–35. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]
- 21.Poon RT, Fan ST, Wong J. Clinical implications of circulating angiogenic factors in cancer patients. J Clin Oncol. 2001;19:1207–25. doi: 10.1200/JCO.2001.19.4.1207. [DOI] [PubMed] [Google Scholar]
- 22.Kim KW, Mutter RW, Cao C, Albert JM, Freeman M, Hallahan DE, Lu B. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J Biol Chem. 2006;281:36883–90. doi: 10.1074/jbc.M607094200. [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 25.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 26.Verheij M, Bartelink H. Radiation-induced apoptosis. Cell Tissue Res. 2000;301:133–42. doi: 10.1007/s004410000188. [DOI] [PubMed] [Google Scholar]
- 27.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–7. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 28.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–44. [PubMed] [Google Scholar]
- 29.Demasters G, Di X, Newsham I, Shiu R, Gewirtz DA. Potentiation of radiation sensitivity in breast tumor cells by the vitamin D3 analogue, EB 1089, through promotion of autoph-agy and interference with proliferative recovery. Mol Cancer Ther. 2006;5:2786–97. doi: 10.1158/1535-7163.MCT-06-0316. [DOI] [PubMed] [Google Scholar]
- 30.Gewirtz DA. Autophagy as a mechanism of radiation sensitization in breast tumor cells. Autophagy. 2007;3:249–50. doi: 10.4161/auto.3723. [DOI] [PubMed] [Google Scholar]
- 31.Paglin S, Lee NY, Nakar C, Fitzgerald M, Plotkin J, Deuel B, Hackett N, McMahill M, Sphicas E, Lampen N, Yahalom J. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005;65:11061–70. doi: 10.1158/0008-5472.CAN-05-1083. [DOI] [PubMed] [Google Scholar]
- 32.Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphati-dylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–46. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 33.Lee SB, Kim S, Lee J, Park J, Lee G, Kim Y, Kim JM, Chung J. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007;8:360–5. doi: 10.1038/sj.embor.7400917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–13. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shinohara ET, Cao C, Niermann K, Mu Y, Zeng F, Hallahan DE, Lu B. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene. 2005;24:5414–22. doi: 10.1038/sj.onc.1208715. [DOI] [PubMed] [Google Scholar]
- 36.Albert JM, Kim KW, Cao C, Lu B. Targeting the Akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol Cancer Ther. 2006;5:1183–9. doi: 10.1158/1535-7163.MCT-05-0400. [DOI] [PubMed] [Google Scholar]
- 37.Leon SP, Folkerth RD, Black PM. Microvessel density is a prognostic indicator for patients with astroglial brain tumors. Cancer. 1996;77:362–72. doi: 10.1002/(SICI)1097-0142(19960115)77:2<362::AID-CNCR20>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 38.Wesseling P, Ruiter DJ, Burger PC. Angiogenesis in brain tumors; pathobiological and clinical aspects. J Neurooncol. 1997;32:253–65. doi: 10.1023/a:1005746320099. [DOI] [PubMed] [Google Scholar]
- 39.Lu B, Shinohara ET, Edwards E, Geng L, Tan J, Hallahan DE. The use of tyrosine kinase inhibitors in modifying the response of tumor microvasculature to radiotherapy. Technol Cancer Res Treat. 2005;4:691–8. doi: 10.1177/153303460500400614. [DOI] [PubMed] [Google Scholar]
- 40.Arafat WO, Buchsbaum DJ, Gomez Navarro J, Tawil SA, Olsen C, Xiang J, El-Akad H, Salama AM, Badib AO, Stackhouse MA, Curiel DT. An adenovirus encoding proapoptotic Bax synergistically radiosensitizes malignant glioma. Int J Radiat Oncol Biol Phys. 2003;55:1037–50. doi: 10.1016/s0360-3016(02)04488-7. [DOI] [PubMed] [Google Scholar]
- 41.Pataer A, Fang B, Yu R, Kagawa S, Hunt KK, McDonnell TJ, Roth JA, Swisher SG. Adenoviral Bak overexpression mediates caspase-dependent tumor killing. Cancer Res. 2000;60:788–92. [PubMed] [Google Scholar]
- 42.Shinoura N, Saito K, Yoshida Y, Hashimoto M, Asai A, Kirino T, Hamada H. Adenovirus-mediated transfer of bax with caspase-8 controlled by myelin basic protein promoter exerts an enhanced cytotoxic effect in gliomas. Cancer Gene Ther. 2000;7:739–48. doi: 10.1038/sj.cgt.7700158. [DOI] [PubMed] [Google Scholar]
- 43.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophago-some membranes after processing. Embo J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer. 2004;4:592–603. doi: 10.1038/nrc1412. [DOI] [PubMed] [Google Scholar]
- 45.Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol. 1999;9:967–70. doi: 10.1016/s0960-9822(99)80425-4. [DOI] [PubMed] [Google Scholar]
- 46.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–85. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 48.Rose TL, Bonneau L, Der C, Marty-Mazars D, Marty F. Starvation-induced expression of autophagy-related genes in Arabidopsis. Biol Cell. 2006;98:53–67. doi: 10.1042/BC20040516. [DOI] [PubMed] [Google Scholar]
- 49.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–7. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 50.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–81. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Degenhardt K, Mathew Robin, Beaudoin Brian, Bray Kevin, Anderson Diana, Chen Guanghua, Mukherjee Chandreyee, Shi Yufang, Gelinas Celine, Fan Yongjun, Deirdre A, Nelson, Jin Shengkan, White Eileen. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brady NR, Hamacher Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. Febs J. 2007;274:3184–97. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 53.Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgo J, Diaz J, Lavandero S, Harper F, Pierron G, di Stefano D, Rizzuto R, Szabadkai G, Kroemer G. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–39. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]
- 54.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 55.Kirschke H, Barrett A. Chemistry of lysosomal proteases. London: Academic Press; 1987. pp. 193–238. [Google Scholar]
- 56.Ueno T, Kominami E. Mechanism and regulation of lysosomal sequestration and proteoly-sis. Biomed Biochim Acta. 1991;50:365–71. [PubMed] [Google Scholar]
- 57.Umezawa H, Aoyagi T, Morishima H, Matsuzaki M, Hamada M. Pepstatin, a new pepsin inhibitor produced by Actinomycetes. J Antibiot (Tokyo) 1970;23:259–62. doi: 10.7164/antibiotics.23.259. [DOI] [PubMed] [Google Scholar]
- 58.Tamai M, Matsumoto K, Omura S, Koyama I, Ozawa Y, Hanada K. In vitro and in vivo inhibition of cysteine proteinases by EST, a new analog of E-64. J Pharmacobiodyn. 1986;9:672–7. doi: 10.1248/bpb1978.9.672. [DOI] [PubMed] [Google Scholar]
- 59.Tamai M, Yokoo C, Murata M, Oguma K, Sota K, Sato E, Kanaoka Y. Efficient synthetic method for ethyl (+)-(2S,3S)-3-[(S)-3-methyl-1-(3-methylbutylcarbamoyl)butylcarbamoy l]-2-oxiranecarb oxylate (EST), a new inhibitor of cysteine proteinases. Chem Pharm Bull (Tokyo) 1987;35:1098–104. [PubMed] [Google Scholar]
- 60.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 61.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 62.Qu X, Zou Z, Sun Q, Luby Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–46. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]