

Abstract
Toxoplasma gondii is an excellent model organism for studies on the biology of the Apicomplexa due to its ease of in vitro cultivation and genetic manipulation. Large-scale reverse genetic studies in T. gondii have, however, been difficult due to the low frequency of homologous recombination. Efforts to ensure homologous recombination have necessitated engineering long flanking regions in the targeting construct. This requirement makes it difficult to engineer chromosomally targeted epitope tags or gene knock out constructs only by restriction enzyme mediated cloning steps. To address this issue we employed multisite Gateway® recombination techniques to generate chromosomal gene manipulation targeting constructs. Incorporation of 1.5 to 2.0 kb flanking homologous sequences in PCR generated targeting constructs resulted in 90% homologous recombination events in wild type T. gondii (RH strain) as determined by epitope tagging and target gene deletion experiments. Furthermore, we report that split marker constructs were equally efficient for targeted gene disruptions using the T. gondii UPRT gene locus as a test case. The methods described in this paper represent an improved strategy for efficient epitope tagging and gene disruptions in T. gondii.
Keywords: Toxoplasma gondii, epitope tagging, PCR product mediated transfection, Gateway vectors, gene deletion, UPRT knock out
1. Introduction
T. gondii is an obligate intracellular protozoon parasite transmitted by ingestion of either food, containing bradyzoites or oocysts, or water, containing oocysts. It has a broad host range and geographical distribution with estimates that infection is present in at least 60 million people in the United States. T. gondii causes symptomatic infections in both immune competent and immune compromised individuals. The most commonly recognized infections are encephalitis in patients with HIV infection and chorioretinitis in the setting of congenital infection. In addition to its clinical significance, T. gondii is an excellent model organism for studies on the biology of the Apicomplexa due to its ability to be grown in tissue culture, the well developed genetic manipulation techniques for this organism (Kim and Weiss, 2004) and the available of extensive genome sequence data (Gajria et al., 2008).
Functional analysis of genes identified in genome sequencing projects is one of the prime research areas of the “post-genomic era”. Epitope tagging has proven to be an important tool for the analysis of protein function, protein interaction and sub cellular distribution. These enable researchers to perform quickly experiments that previously were only feasible with the production of monoclonal or polyclonal antibodies. In addition, antibody production can be expensive, time consuming, yielding reagents of variable quality requiring extensive purification and characterization. In T. gondii plasmid based expression of epitope tagged proteins has been employed in several studies (Binder and Kim, 2004, Binder et al., 2008, Stedman et al., 2003, Striepen et al., 1998), but expression of tagged genes from a plasmid can cause artifacts due to the lack of regulated expression of the gene interest (as would be seen at the endogenous gene locus).
Another powerful genetic strategy is gene deletion which can be performed on a genome wide scale as reported in the yeast Saccharomyces cerevisiae (Mnaimneh et al., 2004, Winzeler et al., 1999). T. gondii, being haploid is amenable for gene knockout studies and the essentiality of several T. gondii genes has been tested using this genetic approach (Binder et al., 2008, Donald and Roos, 1995, Donald and Roos, 1998, Zhang et al., 1999). Generation of gene specific knock out constructs often involves several laborious time consuming cloning steps and is not scalable. In bacteria and the yeast S. cerevisiae epitope tagging and gene deletion are of special interest because both of these processes are achievable at chromosomal loci by a simple PCR based strategy (Knop et al., 1999, Wach et al., 1997). Therefore, these procedures have become routine even on a genome wide scale (Ghaemmaghami et al., 2003, Huh et al., 2003). Gene replacement or modification requires homologous recombination at the target gene locus. In yeast due to the high frequency of homologous recombination only about 50bp of homologous DNA sequences are sufficient for targeted gene manipulation (Wach et al., 1994).
The relative frequency of the homologous versus non homologous random recombination depends on the size of the flanking homology regions used in the engineered construct used for transfection. In organisms with a poor frequency of homologous recombination, including T. gondii, a large region of identical sequences is required in the targeting construct to induce homologous recombination. Recently, T.gondii strains lacking the Ku80 protein which is required for the non homologous end joining of double strand DNA breaks, have been engineered (Fox et al., 2009, Huynh and Carruthers, 2009). Deletion of the Ku80 gene in various organisms has significantly enhanced homologous recombination, thereby requiring smaller regions for successful targeting (Ninomiya et al., 2004, Choquer et al., 2008, Goins et al., 2006). Absence of Ku80 protein in T. gondii led to substantial increase in the efficiency of homologous recombination, by decreasing the chance of random integration. For gene deletion studies, DNA flanks of 500 to 1000 bp were sufficient for homologous recombination (Huynh and Carruthers, 2009).
Ku80 knock out (Δku80) strains were found to exhibit similar growth rate and virulence as compared to the wild type T. gondii strains (Fox et al., 2009, Huynh and Carruthers, 2009). However, lack of Ku80 rendered these parasite strains more susceptible to double strand DNA breaks (Fox et. al. 2009). Ku80 proteins have been implicated in a range of cellular activities such as telomere maintenance, tumor suppression, gene transcription regulation, heat shock induced response, and apoptosis (Fisher and Zakian, 2005). Because of these concerns, in some cases it may be useful to target wild type T. gondii strains, which still have Ku80. In other cases, researchers may need to perform transfection of strains for which Δku80 is not available. In such wild type strains longer regions of homologous isogenic DNA are required for inducing homologous recombination.
Classical cloning techniques used for the engineering of tagging or gene knock out constructs require extensive sequence information of the target gene with compatible restriction sites. These requirements complicate the construction of the parent plasmid backbone when one wants to tag or delete more than one gene at a time.
To alleviate these “bottle-necks” we have devised an approach with the aid of Gateway® technology to efficiently, either to delete or epitope tag a target gene at the chromosomal locus in a wild type T. gondii strain. This technology is a flexible and universal cloning approach based on lambda phage site specific recombination (Hartley et al., 2000). We used restriction enzyme mediated cloning with a multisite Gateway® system to tag T. gondii predicted gene 25.m01787 (coding for a homolog of yeast RNA polymerase II transcription factor Brf1) at the endogenous locus. The multisite Gateway® system with a PCR amplified construct can also be used to engineer deletion constructs with long regions of flanking sequences more efficiently. Finally, using UPRT as a test case, we demonstrate that gene deletion is simple and efficient by employing a split marker strategy.
2. Materials and methods
2.1. Parasite culture and purification
Toxoplasma gondii RH strain was maintained by serial passage at 37 °C 5% CO2 in human foreskin fibroblasts (HFF) in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine and 5 mM penicillin/streptomycin (Invitrogen-GIBCO, Life Technology, Carlsbad, California). Infected cultures were lysed by passage through a 25-gauge needle, the released parasites purified by filtration through a 3μm Nucleopore membrane, and then pelleted by centrifugation as published (Roos et al., 1994).
2.2. Generation of a plasmid template for the amplification of ORF25.m01787 3HA tagging cassette
The pTetO7SAG4SUB2HA plasmid, in which TgSUB2 gene is fused at the C-terminus with a single influenza virus hemagglutinin (HA) epitope sequence at a Nhe1 restriction enzyme site was used as the parent plasmid for manipulation (Binder and Kim, 2004). The Gateway® cassette reading frame C.1 (RFC.1, Invitrogen, CA) was used as the template to amplify RFC.1 with oligonucleotides (RFC.1Nsi1For and RFC.1NheRev) containing Nsi1 and Nhe1 restriction enzyme sites. The 1.7 Kb PCR product was ligated to the vector pTetO7SAG4SUB2HA plasmid at Nsi1 and Nhe1 replacing SUB2HA fragment. The new plasmid pTetO7SAG4RFC.1 was grown in ccdB survival bacterial cells (Invitrogen, CA) and selected in the presence of carbenicillin and chloromphenicol.
Two oligonucleotides encoding three tandem repeats of the HA epitope were designed with Nhe1 and Pac1 restriction sites and a stop codon in frame with the epitope encoding sequence before the Pac1 restriction site. Equimolar concentrations of the sense and antisense primers were mixed and then denatured at 94°C and annealed at 25°C. The double stranded oligonucleotide epitope module, allowing a C-terminal translational fusion of 3HA (YPYDVPDYA), was then inserted using a ligase reaction into the digested pTetO7SAG4RFC.1 vector to generate pTetO7SAG4RFC.13HA (Fig. 1A).
Figure 1. Design of a Gateway epitope tagging vector for T. gondii and the strategy for chromosomal C-terminal epitope tagging of ORF25.m01787.

A: Schematic map of the Gateway plasmid pTetO7SAG4RFC.13HA: Various restriction sites present in the fragments RFC.1 and TetO7SAG4 and the vector backbone can be used for target gene cloning. Multiple restriction sites are available in the vector after SacII for cloning the 3’UTR of the target gene.
B: Cloning of part of ORF25.m01787 genomic region into pTetO7SAG4RFC.13HA vector to generate a C-terminal epitope tag fusion: Location of the primer in the genomic region of ORF25.m01787 used for epitope insertion and cloning to pTetO7SAG4RFC.13HA vector. Solid dark bar represents chromosome.
C: Generation of gateway entry vectors: Location of the attB hybrid PCR primers designed to amplify PCR fragments for BP clonase mediated generation of three entry vectors. Each PCR fragment is flanked with unique attB sequences that are designed for the three fragment multisite Gateway® reaction.
D: Structure of the final PCR product containing the epitope tagging DNA module with regions identical to chromosome for inducing homologous recombination: T. gondii CAT cassette is flanked on the 5′ side by a 2.4 kb of 3HA tag inserted genomic region of ORF25.m01787 cloned in frame with a 3HAA tag and on the 3′ side by 1.5kb of genomic 3′ flanking region of the ORF25.m01787 target locus. Transfection of the PCR product results in the homologous recombination at the target gene locus and insertion of a C-terminal 3HA epitope sequence in the genome.
E: A schematic view of the altered target gene chromosomal locus with its C-terminal HA tag: The various primers designed and used for verification of the double cross over event in the target gene locus by diagnostic PCR are shown in the figure.
The plasmid template for genomic tagging of ORF 25.m01787 (renamed TgME49_007900 in Release 6 www.Toxodb.org ) with the 3HA epitope was then engineered using three steps. In the first step a 1.5-kb genomic region of ORF25.m01787 gene upstream of the predicted stop signal was PCR amplified using forward and reverse primers (Kpn1BRF1For and BRF1NheRev) with built in Kpn1 and Nhe1 sites as shown in Fig. 1B. After double digestion with the enzymes, the genomic DNA fragment was ligated into the pTetO7SAG4RFC.13HA vector and then transformed into TOP10 chemically competent E. coli cells for selection in the presence of carbenicillin to yield a plasmid pORF3HA1. An 0.8-kb 3′ untranslated/flanking region (UTR) of ORF 25.m01787 immediately after the predicted stop codon was amplified with primers (Pac1BRF1For and BRF1SacIIRev) containing Pac1 and SacII restriction enzyme sites (Fig. 1B) using RH genomic DNA as the template. The purified PCR product was then ligated at the Pac1 and SacII sites of pORF3HA1 to generate pORF3HA2.
In the second step, three entry vectors were generated using the Multisite Gateway Technology kit (Invitrogen, CA) by following the protocols supplied with the reagent. In brief, three PCR fragments: A, B and C each flanked by specific attB sites were generated (Fig. 1C). For the PCR fragment A, sense primer was a chimera of attB4 sequences (attB4BRF1For) followed by the genomic region of the ORF 25m.01787 just after the Kpn1 restriction site in the plasmid pORF3HA2. The antisense primer attB1BRF1Rev was designed to have attB1 sequences and a region of 3′UTR of the ORF25.m01787 just before the SacII restriction site in the plasmid pORF3HA2. Using the above primer pair and pORF3HA2 as a template a 2.4kb PCR product was then generated. The fragment B was a PCR product flanked by attB1 and attB2 sites and was amplified from a plasmid containing chloromphenicol acetyl transferase (CAT) gene cloned under the control of T. gondii tubulin promoter as a template using attB1CATFor and attB2CATRev primer pair (Soldati and Boothroyd, 1993). The DNA fragment C was a 1.5kb genomic region downstream of 0.8 kb 3′ UTR of the target ORF 25.m01787 (Fig. 1C) and was generated using attB2BRF1For and attB3BRF1Rev primers and thus is flanked by attB2 and attB3 sequences. Around 100ng each of the purified PCR products were incubated with 150 ng of appropriate pDONR vectors respectively in the presence of 2ul of BP clonase II enzyme mix. The BP clonase mediated recombination cloning yielded three kanamycin selectable Gateway entry vectors namely pENTR1, pENTR2 and pENTR3 containing fragments A, B and C respectively.
In step 3 of the generation of ORF 25.m01787 tagging construct, 20–25 fmoles of the three entry vectors generated in the second step viz; pENTR1, pENTR2 and pENTR3 were incubated with 60ng of the pDEST R4-R3 in the presence of LR clonase Plus enzyme mix (Invitrogen, CA) in a 10ul final reaction mixture overnight at 25°C. The reaction was then terminated by the addition of proteinase K. The LR clonase plus enzyme mixture was then transformed to TOP10 chemically competent cells and the transformants were selected on plates containing carbenicillin. Positive clones were identified by colony PCR and DNA sequencing to obtain the final gene tagging construct p25.m01787MSG. This plasmid contains the C-terminal ORF25.m01787 tagging module with T. gondii CAT cassette flanked by 2.3 kb of the chromosomal region of the target gene with in frame 3HA tag and 1.5kb of the genomic region of the target gene downstream of the 3’UTR of the gene (Fig. 1D). The primer pair BRF1HAIntFor and BRF1HAIntRev was used to amplify the 5.3kb PCR product using p25.m01787MSG as the template. The homogeneous PCR product was purified by phenol chloroform extraction and concentrated by ethanol precipitation and the concentrated PCR fragment was used directly for T. gondii transfection.
2.3. Engineering of plasmid template for the amplification of UPRT gene disruption cassette
The Gateway system, as described above, was utilized for construction of the knockout vectors. To construct a UPRT knockout plasmid template, 2kb of the 5′ and 3′ flanking genomic region flanking the predicted coding region of UPRT was used. The 5′ region PCR product was amplified with primers containing attB4 and B1 sequences (attB4UPRT2kbFor and attB1UPRT2kbRev) was cloned into P4-P1R vector to yield pENTR-A. The 3′ region was amplified with chimeric primers containing attB2 and attB3 sequence (attB2UPRT2kbFor and attB3UPRT2kbRev) and the PCR product was cloned into P2RP3 vector to give plasmid pENTR-B. Use of LR reactions containing vectors pENTR-A, pENTR-B, pENTR2 (see above in section 2.2) and the destination vector pDSET-R4R3 followed by an LR clonase plus reaction resulted in a plasmid containing T. gondii CAT expression fragment flanked by 2kb of 5′ and 3′ genomic region of UPRT respectively. This plasmid was named pUPRTDISMSG.
2.4. Routine Transfection
Electroporation was used for transfection of T. gondii employing either circular plasmid DNA or linear DNA fragments generated by PCR. Depending on the experiment, either a 4 mm or 1 mm cuvette was utilized for transfection. For a 4mm cuvette 107 parasites were used in a total of 0.8ml of transfection medium containing 15 to 20 ηmoles of DNA. For a 1mm cuvette, 106 parasites were electroporated with 3 to 5 ηmoles of transforming DNA in total of 0.1ml of transfection medium. For every transfection either plasmid DNA or PCR-generated DNA was ethanol precipitated and the dried DNA pellet was resuspended in cytomix buffer. Electroporation was carried out by a single pulse using a BTX Electro Cell Manipulator 630, with a resistance of 50 ohms and with a charging voltage of 2.0 kV for the 4mm cuvette and 0.5kV for the 1mm cuvette. After transfection, parasites were subjected to drug selection in the presence of 20μM of chloramphenicol after one round of host cell lysis (Kim et al., 1993). At this concentration of the drug, the parasites transfected with no DNA control were killed within two rounds of selection. After three rounds of selection in the presence of drug, parasites were cloned by limiting dilution in a 96 well plate and the drug resistant parasite clones were expanded in T25 flasks for subsequent experiments.
2.5. AMAXA transfection
For each transfection, 106 parasites suspended in cytomix were mixed with 1 to 3 ηmoles of linear PCR-generated DNA in a final reaction volume of 100μl. The parasite DNA mixture was transferred to the 2 mm gap AMAXA cuvette, then nucleofected using a pre-programmed T16 setting on an AMAXA Nuceleofector® system. Infection of the host cells and the subsequent drug selection was carried out as described above.
2.6. Southern blot analysis
T. gondii genomic DNA was isolated using a DNAeasy mini kit (Qiagen, San Leandro, California) following the manufacturer’s instructions. A total of 10ug of genomic DNA from each clone was digested with appropriate enzymes, loaded onto 1% agarose gels, transferred to Hybond N+ membrane (Amersham Biosciences Inc., Piscataway, NJ) and cross-linked at 1,200 J using Spectrolinker XL-1000 UV cross-linker. Blots were then hybridized sequentially with 500 bp of gene specific probe and 580 bp of CAT selection marker specific probe respectively using Rapid-Hyb buffer (Roche Applied Science, IN). Probes (gene specific and CAT) were generated by using a PCR DIG probe synthesis kit (Roche Applied Science, IN) following the manufacturer’s instructions. For the BRF1gene specific probe a 500bp genomic region corresponding to the 3′UTR region of the gene was amplified using genomic DNA as the template with the BRF1ProbeFor and BRF1ProbeRev primer pair. For the drug selection cassette specific probe a 588bp region of the CAT gene was amplified using the CATProbeFor and CATProbeRev primers.
2.7. Preparation of cDNA and RT-PCR
Total RNA was prepared from RH tachyzoites using Trizol (Invitrogen) following the protocol supplied with the reagent. Total RNA was subjected to DNaseI (Roche Applied Science, IN) treatment and purified with RNeasy Protect Mini Kit (Qiagen, San Leandro, California, US). A 0.5ug aliquot of total RNA was used as a template in a 10ul reaction for preparing cDNA using the RNA LA PCR kit Ver 1.1 (TaKaRa Bio Inc, Madison, WI). cDNA (1μl) was then used as the template for RT-PCR using primers specific to UPRT gene (UPRTRTFor and UPRTRTRev) and Actin gene (ActinRTFor and ActinRTRev) by following the protocol supplied with RNA LA PCR kit.
2.8. Immunoblot and immunofluorescence analysis
Total parasite lysates were prepared from tachyzoites after 48 h post infection in sodium dodecyl polyacrylamide gel electrophoresis sample buffer containing ®-mercaptoethanol. Proteins were then separated by SDS-PAGE (10%), transferred to nitrocellulose membrane and the blots were probed with anti-HA mouse monoclonal antibodies conjugated to horseradish peroxidase (3F10, Roche Applied Science, IN) and detected using an enhanced chemiluminiscent reagent (Thermo Fisher Scientific, Waltman, MA) using standard procedures.
For immunofluorescence analysis (IFA) a confluent HFF monolayer was grown on a cover slip in a 24 well plate and was then infected with parasites. Twenty four hours post infection the cells were washed with PBS, fixed for 20 min in 3% paraformaldehyde, permeabilized for 10 min in 0.2% Triton-X-100 in PBS and then blocked in PBS1×/BSA3%/Triton-X-100 0.2% solution. The coverslip was then incubated with rat anti-HA 1:200 dilution (Roche Applied Science, Indianapolis, IN), washed three times with PBS1×/BSA3%/Triton-X-100 0.2%, incubated with Alexa Fluor-conjugated secondary antibodies 1:2000 dilution (Invitrogen-Molecular Probes), washed twice with PBS1×/BSA3%/Triton-X-100 0.2%, and mounted on slides with ProLong gold antifade reagent (Invitrogen-Molecular Probes). Slides were then examined and photographed using an Olympus Digital Microscope.
2.9. Bacterial Transformation
Bacterial transformations were carried out using Mach1 frozen competent cells (Invitrogen). Mach1 cells were used to select against plasmids containing the ccdB gene while ccdB survival cells were used to allow the replication of plasmids containing the ccdB gene.
2.10. Primer Sequences
All of the primer sequences used are provided in Table 1.
Table 1. Primer Sequences.
Restriction enzyme sequences are underlined, attB sequences are indicated by bold stype
| RFC.1Nsi1For | GGCCATGCATCAAACAAGTTTGTACAAAAAAGCTGAACG |
|---|---|
| RFC.1NheRev | CAGGCTAGCCAATCGAACCACTTTGTACAAGAAAGC |
| Kpn1BRF1For | ATCGGTACCTGCAGGAACAAATTCCCGTATACGCGAG |
| BRF1NheRev | GTAGCTAGCGTTCCCGTTTTGCTCAGTGACTCTTCGTACAGG |
| Pac1BRF1For | CGTTAATTAAAAGAGTAGCAGACTCTGTGACGAGCGAAGACC |
| BRF1SacIIRev | CGACCGCGGAGTCAAATTGCTGAGGCCTCGCAGGACAGTGCG |
| attB4BRF1For | GGGGACAACTTTGTATAGAAAAGTTGTCTGCAGGAACAAATTCCCGTATACGCGAG |
| attB1BRF1Rev | GGGGACTGCTTTTTTGTACAAACTTGCAGTCAAATTGCTGAGGCCTCGCAGGACAGTGCG |
| attB2BRF1For | GGGGACAGCTTTCTTGTACAAAGTGGCGTCTGCTCAACACAGAGTGTACAACCGG |
| attB3BRF1Rev | GGGGACAACTTTGTATAATAAAGTTGCCAAGAGCCTCTCGCTGGAGTTCCAGG |
| attB1CATFor | GGGGACAAGTTTGTACAAAAAAGCAGGCTCCGATAAGCTTGATGGCGATGCATGTCC |
| attB2CATRev | GGGGACCACTTTGTACAAGAAAGCTGGGTCGTGTTAACCGGTTCGACTAAAACAAC |
| BRF1HAIntFor | GATGCTTTCTGGTTATCCGAGCTTATGTCGACTTCAGCG |
| BRF1HAIntRev | GTTGACCATTGATCTACCGTACTTTGTCCG |
| DP1 | CACTTGGTCCTTTGCAGAATTTTGTACGCCC |
| DP2 | CATCATATGGATAGCTAGCGTTCCCGTTTTGC |
| DP3 | GGACATGCATCGCCATCAAGCTTATC |
| DP4 | CTTGTTGTGCTTCTGTCAAGCACCTCGGC |
| BRF1DP5 | CTGGAT ATA CCACCGTTGATATATCCC |
| BRF1DP6 | TCTTCGCCCCCGTTTTCACCATGGG |
| attB4UPRT2kbFor | GGGGACAACTTTGTATAGAAAAGTTGTCCGTGTACCTGCCGGGCCTTGCATCC |
| attB1UPRT2kbRev | GGGGACTGCTTTTTTGTACAAACTTGCCGTAGAAGCCGGGGCCGCTACAAGG |
| attB2UPRT2kbFor | GGGGACAGCTTTCTTGTACAAAGTGGCGGACAGACCGCTGACGGAATCGCGG |
| attB3UPRT2kbRev | GGGGACAACTTTGTATAATAAAGTTGCCCTGCCGGTGCGTTTGCGCTCTTC |
| UPRT2kbFor(DisFLF) | CTCGCCTCAGACAATTTGTCAACTGC |
| UPRT2kbRev(DisFLR) | CTGGTGCGTACTTCTGTATGTAGCC |
| UPRT1kbFor(DisFLF) | GCGCCACCCGCTGTGCCTAGTATCG |
| UPRT1kbRev(DisFLR) | CGGGAATCAGACCCTCGTCTCCGGTGG |
| UPRT0.5kbFor(DisFLF) | GTGACTGATTTTCTGCACGTTGGC |
| UPRT0.5kbRev(DisFLR) | GTAACGTGGACCATTCTTCACATTGC |
| DisSplit For | CCACCGTTGATATATCCCAATGGCATCG |
| DisSplitRev | GCATTCTGCCGACATGGAAGCCATCACAAA |
| DP7 | TGCACGGATCCACAGGAGACTTTATCTCGC |
| DP8 | AATTCCCTACGGCAGGACACCGTTGTTCTTGC |
| DP9 | GCTCCTTGTCGATCCCCGATATTCGAC |
| DP10 | ATTTCGGTGACCGGTACTTTGGAACCATG |
| DP11 | CCACCGTTGATATATCCCAATGGCATCG |
| DP12 | GCATTCTGCCGACATGGAAGCCATCACAAA |
| Brf1ProbeFor | ACGCAGCTCCCTCGAAAAGGAAGGG |
| Brf1ProbeRev | AACGGTCGAGGGAACTCGAAACGCC |
| CATProbeFor | CCACCGTTGATATATCCCAATGGCATCG |
| CATProbeRev | GCATTCTGCCGACATGGAAGCCATCACAAA |
| UPRTRTFor | GCGCAGGTCCCAGCGAGCGGAAAG |
| UPRTRTRev | CATGGTTCCAAAGTACCGGTCACCG |
| Actin RTFor | GCGGATGAAGAAGTGCAAGCCTTGG |
| ActinRTRev | GAAGCACTTGCGGTGGACGATGCTCGGG |
3. Results
3.1. Construction of a plasmid template for inserting a C-terminal epitope tag for a target gene
To validate the strategy of epitope tagging of T. gondii genes at their chromosomal loci, a putative T. gondii gene 25.m01787 (renamed TGME_007900 in release 6 of the genome), identified by the T. gondii sequencing project (www.Toxodb.org), was selected and tagged with a 3HA epitope. To expedite the insertion of an epitope at the C-terminus of a target gene and reduce the number of cloning and ligation steps associated with restriction enzyme cloning, we developed a Gateway® cloning strategy. To this end, we converted a T. gondii expression vector into a Gateway compatible destination vector by cloning the reading frame C.1 (RFC.1, Invitrogen) fragment into the pTetO7SAG4SUB2HA plasmid, replacing the SUB2 gene to yield pTetO7SAG4RFC.13HA (Fig. 1A). We replaced the single HA epitope sequence in this plasmid with three tandom repeats of HA sequence. After an initial two restriction enzyme mediated cloning procedures our method utilizes the Multisite Gateway® technology. As shown in the Figure 1, in the final plasmid used as a PCR template to amplify the ORF25.m01787 genomic epitope tagging module, HA3 epitope sequences have been inserted to the C-terminus of the predicted protein with a minimal sequence alteration in its neighborhood in the chromosome.
3.2. Transfection, cloning and verification of integration
To test the feasibility of using the PCR generated DNA fragments for chromosomal gene manipulation, we PCR-amplified a 5.3kb fragment from the p25.m01787MSG plasmid. After transfection, drug selection and cloning by limiting dilution we selected 36 clones for further analysis by diagnostic PCR and immunoblotting. Both homologous gene targeting and non-homologous random integration are distinguishable by PCR analysis using primers at each of the 5′ and 3′ junctions of the altered gene. A schematic map of the genomic region of the gene with integrated C-terminal epitope tag and the location of the diagnostic primers is displayed in Figure 1E. The diagnostic primer DP1 anneals at the target gene sequence in the chromosome, which is outside the region of alteration; therefore this primer doesn’t hybridize to any region either in the plasmid p25.m01787MSG used as the template for the final PCR or in the final PCR fragment used for the transfection. Primer DP2 encompasses regions of the target gene and the epitope encoding region. Primer DP3 is within the selection marker module. Therefore neither DP2 nor DP3 are expected to give any PCR product when used with DP1 primer from the wild type genomic DNA as the template. When primer DP1 was used with DP2 and DP3 a specific diagnostic PCR product of 1.6kb (Fig. 2A, lanes 1 and2) and 2.5 kb (Fig. 2B lanes 1 and 2), respectively, was obtained from the genomic DNA of the positive clones B6 and C4. The absence of this specific PCR product in clone B7 (Figs 2 A and B lane 3) indicates that transformation module has integrated randomly in the genome resulting in the emergence of chlorampehnicol positive parasites. The absence of PCR product from the wild type RH genomic DNA (Figs 2 A and B, lane 4) and plasmid p25.m01787MSG (Figs 2A and B, lane 5) confirms the specificity of the reaction.
Figure 2. Verification of the C-terminal epitope tagging by diagnostic PCR.

Confirmatory PCR was performed using genomic DNA as the template from wild type and transfected parasite clones. PCR products obtained at the 5′ junction from (A) primers DP1::DP2; (B) DP1::DP3; and at the 3′ junction of the altered chromosome with primers; (C) DP4::DP5; and (D) DP4::DP6. Lane 1: positive clone B6, Lane 2: positive clone C4, Lane 3: negative clone B7, Lane 4: wild type RH genomic DNA, Lane 5: p25.m01787MSG plasmid, Lane 6: No DNA control, and Lane 7: DNA ladder.
Similarly, at the 3′ junction, three independent primers were designed as shown in Figure 1E. Primer DP4 is in the chromosome outside of the region of homology in the targeting construct. Primers DP5 and DP6 are within the chloramphenicol selection cassette. Primers DP4 and DP5 yielded a specific PCR product of 2.5 kb (Fig 2C; lanes 1 and 2) only from clones B6 and C4 confirming that the epitope tagging construct has recombined at the target gene genomic locus by double crossover recombination. Similar results are seen with primer pair DP4 and DP6, which yielded a diagnostic PCR product of 2kb (Fig 2C; lanes 8 and 9) from positive clones. The absence of PCR product when genomic DNA either from RH wild type parasite (Fig 2C; lanes 4 and 11) or from plasmid p25.m01787MSG (Fig 2C; lanes 5 and 12) was used validates the specificity of the reaction. Out of 36 chlorampehnicol positive clones, 34 yielded specific diagnostic products at the 5′ and 3′junction of the altered gene for a calculated efficiency of more than 90% of homologous recombination using the PCR products for transfection.
3.3. Verification of epitope tag integration using immunoblot analysis and IFA
Successful integration of the epitope tagging construct at the target gene locus should result in the expression of an epitope tagged protein product. Parasite lysates examined by immunoblot demonstrated the expression of the HA-tagged protein in parasites in which diagnostic PCR had confirmed correct integration of the cassette in the target gene locus (Fig 3A, lanes 1 and 2). There was no signal from lanes containing lysates of either wild type parasite or parasite clone B7 (integration at an unrelated site) (Fig 3A, lanes 3 and 4). When the immunoblot was stripped and probed with antibody to tubulin, a tubulin protein specific signal was obtained in all the lanes confirming the specificity of HA epitope specific antibody.
Figure 3. Immunoblot demonstrating expression of epitope tagged ORF25.m01787.
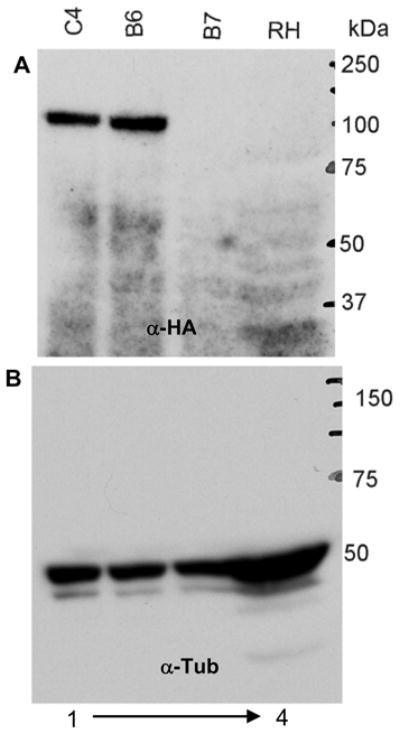
Parasite lysates from wild type RH and chloramphenicol resistant parasite clones were analyzed by immunoblot using (A) peroxidase conjugated HA peptide specific monoclonal antibody; and (B) tubulin specific antibody (control antibody). The C-terminal 3HA tagged ORF25.m01787 was detected in the lysates of clones B6 and C4 (positive clones), but not in the lysates of either RH wild type or clone B7 (negative clone). The tubulin blot staining demonstrates equal loading in all lanes.
IFA of clone B6 (epitope tagged clone) demonstrated nuclear localization of the protein (Fig. 4), consistent with the identification of ORF 25.m01787 as an RNA polymerase III transcription factor Brf1. This is similar to the localization seen in Brf1 in S. cerevisiae. A HA antibody specific signal was absent in clone B7 (non-specific integration site clone). In figure 4 the B7 HA signal panel was overexposed to illustrate parasites, however, the B7 merged parasites panel demonstrates similar levels of exposure for both the HA and DAPI signal to that seen in the B6 merged panel.
Figure 4. Immunofluorescence (IFA) of clones B6 and B7 using anti HA antibody.
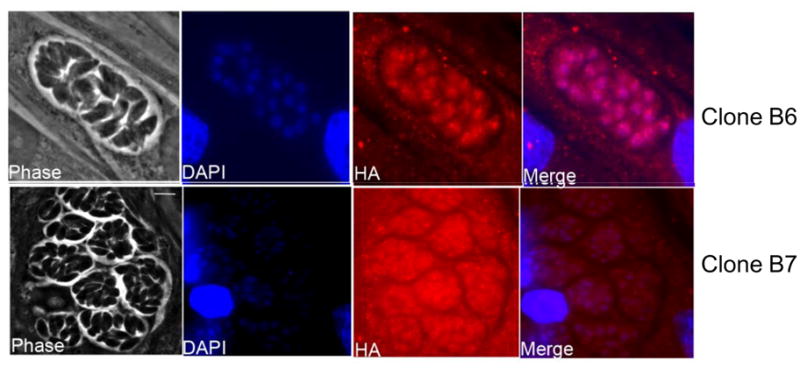
Phase, DAPI (DNA, blue), HA (red) and merged (blue and red) images of B6 (HA-positive) and B7 (HA-negative) transfectants. ORF25.m01787 is predicted to be a homolog of RNA polymerase III transcription factor and expected to localize to the nucleus. As demonstrated B6 exhibits specific nuclear location. The absence of antibody specific signal in clone B7 proves the specificity of the antibody (HA signal is overexposed in the HA panel of B7 illustrating that nuclear localization is not present; merged panel shows similar exposures of DAPI and HA for B6 and B7).
3.4. Confirmation of the target gene specific integration by Southern blot hybridization
Southern analysis of BgIII digested T.gondii RH strain genomic DNA probed with a 25.m01787 gene specific probe resulted in demonstration of a single band of 2.9kb (Fig. 5C, lane 12). There was no reaction when this blot was stripped and reprobed with a CAT specific probe (Fig 5C, lane 6). Southern analysis of Bglll digested T. gondii B6 DNA demonstrated a 4.2kb fragment when probed with the 25.m01787 gene specific probe (Fig 5C, lane 10) or with the CAT specific probe (Fig 5C, lane 4), respectively, confirming the integration of the PCR fragment at the target gene locus by homologous recombination by a double cross over event. In clone B7 which is resistant to chloramphenicol there is both a 2.9 and a 4.2 kb band (Fig 5C; lane 11), indicating that the target gene locus is indeed intact and that at least 2 copies of the construct integrated. This is consistent with the results obtained with both the diagnostic PCR and immunoblots. Similar Southern blot results were also seen when digesting genomic DNA with EcoRV and Sac1 restriction enzymes.
Figure 5. Homologous integration of the epitope tagged PCR product at the target gene locus.

Position of the target gene specific (open double sided arrow) and CAT gene (striped double sided arrow) specific probes in the genomic context have been depicted in the wild type (Fig A) and positive transformant (Fig B). Southern blot analysis of the BglII and EcoRV plus Sac1 (RV+S) digested genomic DNA from wild type RH, positive parasite clone B6 and negative clone B7. Blots were sequentially probed with DIG labeled gene specific (open double headed arrow) and CAT (chloramphenicol) selection cassette specific (striped double headed arrow) PCR probes (Fig C).
3.5. PCR product mediated gene deletion of T. gondii UPRT gene
To validate the strategy for chromosomal gene deletion using Gateway® assisted PCR generated DNA fragments, we chose to delete a T. gondii non-essential single copy gene encoding uracil phosphoribosyltransferase (UPRT) (Donald and Roos, 1995). UPRT is an enzyme of the pyrimidine salvage pathway. As UPRT is not found in mammalian host cells, it can be used as a negative selectable marker for genetic manipulation in T. gondii. 5-fluoro-2-deoxyuridine is converted by T. gondii to 5-fluorouracil, which is then converted to 5-fluorouridine by UPRT, and then to 5-fluorodeoxyuridine monophosphate, which is lethal to the parasite because it inhibits the synthesis of thymidine monophosphate. Selection for resistance to 5-fluoro-2-deoxyuridine permits the identification of parasites in which homologous recombination at the target gene locus resulted in the replacement of the active UPRT with CAT selection cassette fragment.
Since we observed successful homologous recombination for epitope tagging by using approximately 2kb of flanking sequences, we used 2kb of UPRT gene flanking sequence to generate a plasmid template for amplifying a UPRT deletion cassette fragment. Use of a multi-site Gateway reaction strategy reduced the steps involved in generating the final plasmid construct for deletion to only two steps of clonase-mediated reactions as described in the materials and methods section.
To PCR-amplify a gene specific deletion cassette using pUPRTDISMSG we employed two procedures. In one method, we amplified a full-length UPRT gene disruption cassette using the primers DisFLF and DisFLR (Fig 6A). In the second method, the full-length deletion cassette was split into two fragments UPRTDISCAS-A and UPRTDISCAS-B (Fig. 6B). UPRTDISCAS-A was amplified using primers DisFLF and DissplitR and contained 2 kb of 5′ genomic region of the UPRT coding region and part of the CAT selection cassette. The UPRTDISCAS-B fragment was amplified using primers DisFLR and DissplitF and contained the 3′ genomic region of the UPRT coding region and part of the CAT selection cassette (see Fig. 6B). Both the PCR products have part of CAT selection region, but neither has a functional full length CAT and each product has a different UPRT genomic region. This split PCR strategy should favor homologous recombination and limit random integration as a functional CAT selection cassette is dependent on homologous recombination in this setting.
Figure 6. Schematic representation of the PCR product mediated UPRT gene deletion in T. gondii.

A: Full-length disruption cassette amplified using DisFLF and DisFLR primers contains the T. gondii CAT selection cassette flanked at each side by ~ 2kb of genomic DNA from 5′ and 3′ region of UPRT gene. B: Strategy for split cassette mediated gene disruption in T. gondii. Combination of primers DisFLF and DisSplitR yielded UPRTDISCAS-A fragment which contains 5′ genomic region of the UPRT gene. Amplification using DisFLR and DisSpiltF using pUPRTDISMSG as the template resulted in the production of the UPRTDISCAS-B fragment. C: Three independent homologous recombination events are required for the reciprocal replacement of UPRT coding region in the genome with split cassette fragments: UPRIDSCAS-A and UPRTDISC-B. UPRT knock out parasites were enriched by growing the transfected parasites initially in the presence of chloramphenicol and then in the presence of 5mM 5-fluoro-2-deoxyuridine.
Equimolar concentrations of the UPRTDISCAS-A and B were mixed and used for the transformation of T. gondii parasites. As shown in the figure 6C, three independent homologous recombination events are required for the successful integration of the split cassette at the UPRT gene locus thereby to yield UPRT disrupted CAT positive transformants. After three rounds of chloramphenicol selection, we placed parasites under 5mM 5-fluoro-2-deoxyuridine selection to identify those in which homologous recombination at UPRT gene locus had resulted in disruption of this gene locus.
3.6. Confirmation of the UPRT gene deletion
To confirm the deletion of the UPRT gene we isolated genomic DNA from parasites that had been mock transfected (i.e. no DNA) and from transfected parasites resistant to 5-fluoro-2-deoxyuridine. We examined these clones by PCR analysis using specific primers designed to both 5 and 3′ junctions of the modified UPRT locus. The positions of the various diagnostic primers are demonstrated in Figure 6C. Primers DP7 and DP8 annealed to the chromosome outside of the recombination sites at the 5′ and 3′ region of the UPRT gene respectively, primers DP9 and DP10 annealed in the UPRT coding region and primers DP11 and DP12 annealed within the CAT selection cassette. When we performed PCR using a mixture of DP7, DP9 and DP12 (5″ region) as shown in figure 7A a band of 2.2 kb could be demonstrated in wild type T. gondii indicating the presence of an intact UPRT gene. In the knockout parasites, transfected with either the full-length cassette (Fig 7A; FL) or with the split cassette fragments (Fig 7A, split), a PCR product of 3.4kb was obtained as expected. This confirmed that replacement of the UPRT coding region in the genome by the CAT selection fragment was successful using both strategies. Similarly, PCR using primers DP8, DP10 and DP11 (3′ region) demonstrated (Figure 7A) a 2.2kb PCR product with DP8 and DP10 from wild type T.gondii and a 3kb band with DP8 and DP11 in the knockouts (figure 7A lane FL and Split 3′ junction). When the final Multisite gateway plasmid pUPRTDISMSG, which was the source for amplification of UPRT disruption module, was used as a template, no PCR products were obtained confirming the specificity of the diagnostic primers for the genomic locus.
Figure 7. Confirmation of the UPRT gene deletion using split cassette and full length gene deletion strategies.

A: Genomic DNA from wild type RH and drug resistant parasites obtained after transfection using full-length UPRT disruption cassette (FL) and Split deletion cassette (Split) were subjected to PCR analysis using primers DP7, DP9 and DP12 (5′ junction) and primers DP8, DP11 and DP12 (3′ junction). Plasmid pUPRTDISMSG was used as the negative control. This demonstrates the effectiveness of both split and FL constructs in obtaining the UPRT deletion.
B: RT-PCR experiments using cDNA isolated from wild type RH and UPRT deletion clones obtained by either the full-length (FL) or by split cassette (Split) deletion strategy. Primers designed for the T.gondii actin gene resulted in the amplification of a 1.1kb gene transcript from all of the parasites. UPRT gene specific primers amplified a 760bp transcript only from the wild type (RH strain) parasite cDNA and not from either UPRT knock out parasite strains.
3.7 RT-PCR experiments demonstrate the absence of UPRT transcripts in the knock out parasites
Further confirmation of UPRT gene deletion was obtained by RT-PCR, a 760 bp transcript corresponding to the coding region of the UPRT gene was seen with cDNA from wild type parasites, but UPRT transcripts were completely absent in the knock out parasites (Fig. 7B lane FL and Split under UPRT). A T. gondii actin gene (1.1kb) was used as a positive control in these experiments (Fig 7B, Actin). All the RNA samples were subjected to DNAse I treatment and the removal of genomic DNA contamination was verified by appropriate no RT controls (data not shown).
3.8. Minimum region of homology required for inducing homologous recombination at the target gene locus
To determine the minimum region of homology required for inducing efficient homologous recombination at the target gene locus, we prepared UPRT gene disruption modules in which CAT selection cassette was flanked by 0.5kb, 1kb and 2kb of genome sequence from the 5′ and 3′ region of the UPRT coding sequence respectively. These constructs were produced using the split cassette strategy as discussed above. Parasites were transfected in 1mm gap cuvette in 100 ul of cytomix buffer with 5 ηmoles (corresponds to 5 to 10 μg of DNA) of the disruption module. After transfection and drug selection, we could identify 5-fluoro-2-deoxyuridine resistant parasites from parasites transfected with either 1kb or 2kb of flanking regions; however, parasites transfected with 0.5kb of flanking regions did not produce 5-fluoro-2-deoxyuridine resistant parasites. This suggests that 0.5 kb of genomic homology is not sufficient for homologous gene replacement at a target gene locus in wild type parasites. In these experiments, all of the identified UPRT disruptants (5-fluoro-2-deoxyuridine resistant clones) were confirmed by diagnostic PCR as described in section 3.6 (data not shown).
3.9. Comparison of traditional electroporation to AMAXA nucleofector based transfection for PCR product mediated genome manipulation
Our electroporation experiments described above were performed with a BTX electroporator; however, we routinely use an AMAXA based nucleofector transfection system for Plasmodium yoelii, a member of Apicomplexa (Jongco et al., 2006). We had previously determined that the use of cytomix buffer in combination with T16 program was optimal for T. gondii transfection using a luciferase based reporter assay (K Kim; unpublished results). We, therefore, tested and compared the feasibility of using this system for genetic manipulation of T. gondii employing our current vectors (summarized in Table 2). For C-terminal epitope tagging, we used 1 to 2.5 ηmoles (corresponding to 4 to 10 μg of the transforming DNA cassette) and the parasites were transfected in cytomix buffer using the AMAXA T16 program. We found chloramphenicol resistant parasites only when parasites were transfected with more than 2 nmoles of PCR product. The efficiency of the homologous recombination at the target gene locus as determined after the diagnostic PCR of the genomic DNA of the drug resistant clones was ~90% (similar to that seen with traditional BTX electroporation using much larger quantities of DNA). Similarly, for the UPRT deletion we used three different deletion fragments with varying length of flanking homology sequences and 1.5 or 3.0 nmoles of UPRT gene disruption cassette using the split cassette strategy as described above. After transfection and drug selection, we found 5-fluoro-2-deoxyuridine resistant parasites only if the parasites were transfected with 3.0 ηmoles of disruption cassette. Similar to BTX electroporation for disruption the AMAXA nucleofector system also required a minimum of 1kb of flanking genomic sequences in wild type parasites for homologous recombination at the UPRT gene locus. All of the positive transformants had confirmatory diagnostic PCRs at both the 5′ and 3′ junctions (data not shown). In the transfection experiments described parasites transfected with no DNA were used as the positive control. In addition, parasites transfected with various DNA fragments were grown in the absence of any drug selection to assess the influence of transfection conditions on the viability of the parasites.
Table 2.
Summary of the AMAXA transfection condition used and the results
| Epitope tagging | Amount of DNA used for transfection (ηmoles) | Emergence of chloramphenicol resistance parasites | Diagnostic PCR confirmation |
|---|---|---|---|
| 1 | No | - | |
| 2 | Yes | Yes | |
| 2.5 | Yes | Yes | |
| UPRT deletion | Amount of DNA used for transfection (ηmoles) | Emergence of 5-fluoro-2-deoxyuridine/chloramphenicol resistant parasites | Confirmation by diagnostic PCR |
| Length of flanking homology sequence | |||
| 0.5kb | 3 | no | - |
| 3.0 | no | - | |
| 1.0kb | 1.5 | no | - |
| 3.0 | Yes | Yes | |
| 2.0kb | 1.5 | No | - |
| 3 | Yes | Yes |
4. Discussion
The development of the Gateway compatible T. gondii expression vector pTetO7SAG4RFC.13HA facilitates the cloning of target genes in frame with a C-terminal epitope tag. This vector, pTetO7SAG4RFC.13HA, has additional unique restriction sites that will permit the cloning other of epitopes such as c-myc, FLAG, or GFP and its variants (Fig 1A). We originally developed pTetO7Sag4RFC.13HA for use in transient transfection to clone target genes for ectopic expression as an epitope fused protein product; however, we found this redesigned vector was much more useful as a shuttle vector for epitope tagging. One of the advantages of using a plasmid containing the RFC.1 fragment for initial cloning is that selective killing of cells containing either parent vector or inefficiently digested vectors during cloning occurs due to the sensitivity of commonly used E. coli stains to the ccdB gene residing in the RFC.1 fragment. The product of ccdB is a potent inhibitor of E. coli DNA gyrase, thereby, killing the growth of most E. coli strains. Plating of transformants on LB agar containing carbenicillin allows for the specific selection of bacterial clones containing a vector in which a genomic fragment of the target gene replaces RFC.1. Multiple restriction sites are available in pTetO7SAG4RFC.13HA that permit the cloning of genomic fragments of target genes in conjunction with Nhe1. To facilitate the cloning of a 3′ flanking region there are several restriction sites available in conjunction with Pac1 in pTetO7SAG4RFC.13HA. Since this vector is only used as a shuttle vector to generate the epitope tagged genomic fragment of a target gene any restriction sites present in the tetO7SAG4 promoter and RFC.1 fragment can be potentially used for cloning.
In designing the tagging vector we cloned ~0.8kb of genomic DNA of ORF25.m01787 after the predicted stop codon into pTetO7SAG4RFC.13HA following the epitope tag. We believe that this region should contain any sequence elements involved in transcriptional regulation of the gene and once integrated will produced a tagged version of the gene that is similar to its native gene in genomic context (compare figure 1D and 1E). The presence of an epitope tagged protein product in the nucleus, consistent with the nuclear localization S. cerevisiae, BRF1, indicates that insertion of an epitope tag sequence in the genome has not interfered with alternate splicing of the gene or nuclear localization of the protein.
When we compared the sequence of cDNA of ORF25.m01787 obtained after RT-PCR and the predicted gene sequenced (www.toxodb.org) we found errors in the splice site predictions of the annotated gene. The genome epitope tagging method eliminates the need for identifying and confirming the location of the first authentic ATG in the gene or the intron/exon junctions of the gene. The only requirement is the identification of the predicted stop codon in the genome sequence to provide an accurate C-terminal epitope tag. This method could also be used to provide an internal tag of the genes encoding higher molecular weight proteins. Genome tagging eliminates many of the difficulties of cloning and characterization full-length genes prior to their cellular and biochemical characterization. Some precautions, however, need to be taken when a new gene is being investigated by this method. First, specific criteria must be applied to assess the functionality of the protein. Second, one has to be aware that an epitope-tagged protein is always different from its wild type counterpart. Therefore, independent verification of results obtained with the tagged derivatives is always desirable.
Our results demonstrate that PCR-generated DNA fragments can be used for gene disruption in T. gondii and provides a new simple and attractive method for gene manipulation in this pathogen. The generation of the gene disruption cassette by PCR eliminates many complex restriction enzyme mediated cloning reactions and is only dependent on two clonase mediated reactions. All the reagents for this method are commercially available. We have demonstrated using the UPRT locus that 1kb of flanking genomic region is sufficient for inducing homologous recombination. It should be appreciated, however, that the minimal length required by homologous recombination may depend on the specific recombinogenic properties of that region of the chromosome. We have found cloning longer genomic region of homology in the first BP clonase reaction may be desirable since disruption modules with required flanking sequences can always be amplified by PCR reaction with specific set of primers. The only limitation of the use of a longer region of homologous sequence in the final disruption module is the additional time to generate these fragments by PCR. However, the split cassette strategy is a useful method for overcoming these limitations. We found that the split cassette strategy was very efficient in generating a UPRT knockout. This is consistent with the reports of use of a split cassette strategy to efficiently to disrupt genes in Cryptococcus neoformans, a fungal human pathogen with a low efficiency of homologous integration (Fu et al., 2006).
Acknowledgments
This work was supported by NIH Grant AI 39454 (LMW), AI 087625 (KK), AI 046985 (KK) and NIH/NIAID Contract HHSN266200400054C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Binder EM, Kim K. Location, location, location: trafficking and function of secreted proteases of Toxoplasma and Plasmodium. Traffic. 2004;5:914–924. doi: 10.1111/j.1600-0854.2004.00244.x. [DOI] [PubMed] [Google Scholar]
- Binder EM, Lagal V, Kim K. The prodomain of Toxoplasma gondiiGPI-anchored subtilase TgSUB1 mediates its targeting to micronemes. Traffic. 2008;9:1485–1496. doi: 10.1111/j.1600-0854.2008.00774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquer M, Robin G, Le Pecheur P, Giraud C, Levis C, Viaud M. Ku70 or Ku80 deficiencies in the fungus Botrytis cinereafacilitate targeting of genes that are hard to knock out in a wild-type context. FEMS Microbiol Lett. 2008;289:225–232. doi: 10.1111/j.1574-6968.2008.01388.x. [DOI] [PubMed] [Google Scholar]
- Donald RG, Roos DS. Insertional mutagenesis and marker rescue in a protozoan parasite: cloning of the uracil phosphoribosyltransferase locus from Toxoplasma gondii. Proc Natl Acad Sci U S A. 1995;92:5749–5753. doi: 10.1073/pnas.92.12.5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald RG, Roos DS. Gene knock-outs and allelic replacements in Toxoplasma gondii: HXGPRT as a selectable marker for hit-and-run mutagenesis. Mol Biochem Parasitol. 1998;91:295–305. doi: 10.1016/s0166-6851(97)00210-7. [DOI] [PubMed] [Google Scholar]
- Fisher TS, Zakian VA. Ku: a multifunctional protein involved in telomere maintenance. DNA Repair (Amst) 2005;4:1215–1226. doi: 10.1016/j.dnarep.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. Efficient gene replacements in Toxoplasma gondiistrains deficient for nonhomologous end joining. Eukaryot Cell. 2009;8:520–529. doi: 10.1128/EC.00357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Hettler E, Wickes BL. Split marker transformation increases homologous integration frequency in Cryptococcus neoformans. Fungal Genet Biol. 2006;43:200–212. doi: 10.1016/j.fgb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Gajria B, Bahl A, Brestelli J, Dommer J, Fischer S, Gao X, Heiges M, Iodice J, Kissinger JC, Mackey AJ, Pinney DF, Roos DS, Stoeckert CJ, Jr, Wang H, Brunk BP. ToxoDB: an integrated Toxoplasma gondiidatabase resource. Nucleic Acids Res. 2008;36:D553–556. doi: 10.1093/nar/gkm981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Goins CL, Gerik KJ, Lodge JK. Improvements to gene deletion in the fungal pathogen Cryptococcus neoformans: absence of Ku proteins increases homologous recombination, and co-transformation of independent DNA molecules allows rapid complementation of deletion phenotypes. Fungal Genet Biol. 2006;43:531–544. doi: 10.1016/j.fgb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Hartley JL, Temple GF, Brasch MA. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Huynh MH, Carruthers VB. Tagging of endogenous genes in a Toxoplasma gondiistrain lacking Ku80. Eukaryot Cell. 2009;8:530–539. doi: 10.1128/EC.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongco AM, Ting LM, Thathy V, Mota MM, Kim K. Improved transfection and new selectable markers for the rodent malaria parasite Plasmodium yoelii. Mol Biochem Parasitol. 2006;146:242–250. doi: 10.1016/j.molbiopara.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Kim K, Soldati D, Boothroyd JC. Gene replacement in Toxoplasma gondiiwith chloramphenicol acetyltransferase as selectable marker. Science. 1993;262:911–914. doi: 10.1126/science.8235614. [DOI] [PubMed] [Google Scholar]
- Kim K, Weiss LM. Toxoplasma gondii:the model apicomplexan. Int J Parasitol. 2004;34:423–432. doi: 10.1016/j.ijpara.2003.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15:963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Mnaimneh S, Davierwala AP, Haynes J, Moffat J, Peng WT, Zhang W, Yang X, Pootoolal J, Chua G, Lopez A, Trochesset M, Morse D, Krogan NJ, Hiley SL, Li Z, Morris Q, Grigull J, Mitsakakis N, Roberts CJ, Greenblatt JF, Boone C, Kaiser CA, Andrews BJ, Hughes TR. Exploration of essential gene functions via titratable promoter alleles. Cell. 2004;118:31–44. doi: 10.1016/j.cell.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Ninomiya Y, Suzuki K, Ishii C, Inoue H. Highly efficient gene replacements in Neurosporastrains deficient for nonhomologous end-joining. Proc Natl Acad Sci U S A. 2004;101:12248–12253. doi: 10.1073/pnas.0402780101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos DS, Donald RG, Morrissette NS, Moulton AL. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 1994;45:27–63. doi: 10.1016/s0091-679x(08)61845-2. [DOI] [PubMed] [Google Scholar]
- Soldati D, Boothroyd JC. Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science. 1993;260:349–352. doi: 10.1126/science.8469986. [DOI] [PubMed] [Google Scholar]
- Stedman TT, Sussmann AR, Joiner KA. Toxoplasma gondiiRab6 mediates a retrograde pathway for sorting of constitutively secreted proteins to the Golgi complex. J Biol Chem. 2003;278:5433–5443. doi: 10.1074/jbc.M209390200. [DOI] [PubMed] [Google Scholar]
- Striepen B, He CY, Matrajt M, Soldati D, Roos DS. Expression, selection, and organellar targeting of the green fluorescent protein in Toxoplasma gondii. Mol Biochem Parasitol. 1998;92:325–338. doi: 10.1016/s0166-6851(98)00011-5. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Alberti-Segui C, Rebischung C, Philippsen P. Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast. 1997;13:1065–1075. doi: 10.1002/(SICI)1097-0061(19970915)13:11<1065::AID-YEA159>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the Scerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Kim K, Ma YF, Wittner M, Tanowitz HB, Weiss LM. Disruption of the Toxoplasma gondiibradyzoite-specific gene BAG1 decreases in vivo cyst formation. Mol Microbiol. 1999;31:691–701. doi: 10.1046/j.1365-2958.1999.01210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]