

Abstract
A new paradigm for drug activity is presented, which includes both recognition and subsequent irreversible inactivation of therapeutic targets. Application to both RNA and enzyme biomolecules has been demonstrated. In contrast to RNA targets that are subject to strand scission chemistry mediated by ribose H-atom abstraction, proteins appear to be inactivated through oxidative damage to amino acid side chains around the enzyme active site.
Keywords: metallodrug, metallotherapeutics, RNA, protease, enzyme, catalytic, cleavage, modification
Introduction
The use of metal ions and complexes as tools for investigation of protein properties and interactions,[1, 2] as biomarkers for cellular chemistry, and as candidates for therapeutic intervention,[3–21] have all emerged as major new focal points of investigation in inorganic chemistry. Early applications included the design of artificial nucleases and proteases, making use of site-selective binding to amino acid residues and metal-assisted proteolysis.[22–32] However, redox active metals were also found to promote oxidative modification of amino acid sidechains, including hydroxylation[33, 34] and carbonylation of amino acid side chains,[35, 36] and protein cross-linking.[33, 37, 38] Oxidative damage of nucleic acid bases and sugars, with the potential for subsequent strand cleavage, has also been observed.[12, 39–43] While many pathways to improving such modification chemistry have been explored, studies suggest that improvement of metal complex binding to the reactant molecule is an important, if not the major factor in promoting efficient chemistry.[6, 7] In this article we present a novel application of transition metal complexes as catalytic metallodrugs. The concept of a metallodrug builds on the experimental foundation described in the preceding remarks.
Concept
Blocking the functional activity of a therapeutic target by classical competitive inhibition is well documented, and almost all drugs function through the binding of a small molecule inhibitor to a biomolecular target that is typically a protein. The binding is reversible and the target remains functional after release of the drug. By contrast, a distinct and novel strategy involving irreversible catalytic inactivation of target RNA’s or proteins by transition metal complexes has now been demonstrated.[10, 13] Disruption of protein and nucleic acid structure and function that results from metal-promoted damage can be used to advantage in the design of new forms of therapeutic agent. Such metallodrugs include both a metal binding domain (to catalyze redox and Lewis acid chemistry) and a target recognition domain (Figure 1). Juxtaposition of a recognition element with a catalytic degradative element produces a molecule with properties that are superior to the sum of the individual component parts, and provides a novel design platform for drug development. While such molecules may retain their inhibitory properties, they also show the potential for catalytic degradation of the selected biomolecule (Figure 2). In this review, we outline the concept of a catalytic metallodrug and its application toward therapeutic targets
Figure 1.
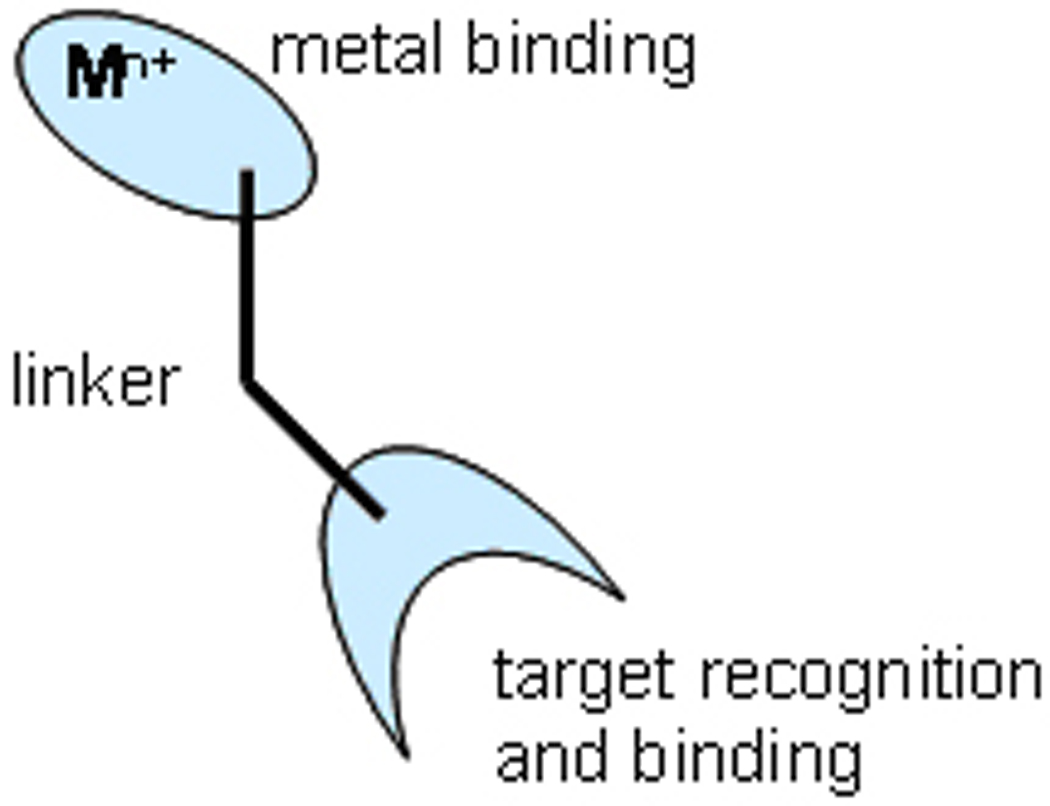
Metallodrug design, highlighting the metal binding and targeting domains
Figure 2.
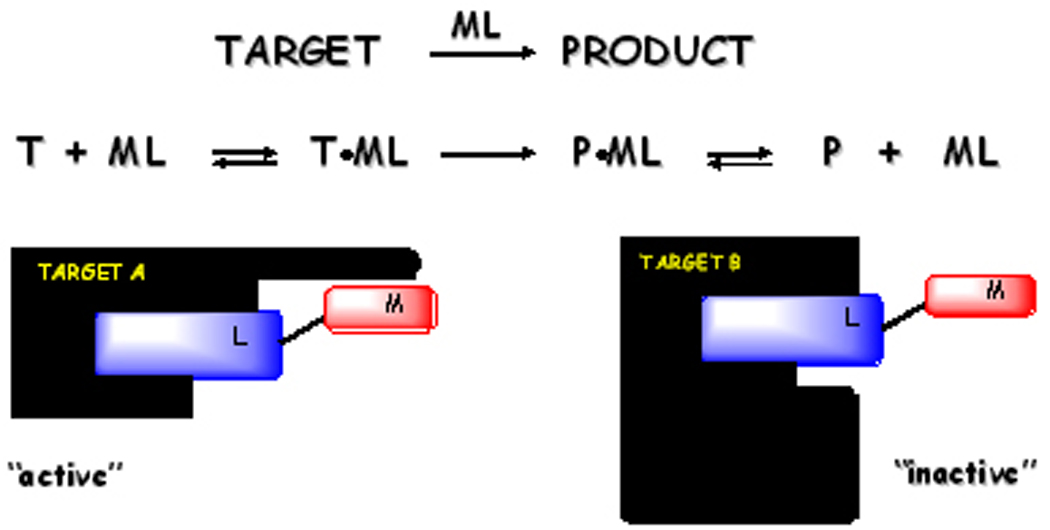
In a traditional approach to drug design a molecule with high binding affinity for a select protein target acts by reversible inhibition of protein function. An excess of molecule is required to ensure saturation of target. The approach described herein employs a substoichiometric concentration of drug that executes catalytic irreversible inactivation of a select protein target. Selectivity is based both on target recognition and productive orientation of metallodrug to execute irreversible cleavage or damage of the target. Multiple turnover also distinguishes this approach from suicide inhibition.
Potential advantages of a catalytic drug
Irreversible destruction of target also affords the potential for sub-stoichiometric administration of drug, with the promise of a significant lowering of dosage and a commensurate decrease or elimination of side effects or toxicity.[6, 7] This key point differentiates the activity of catalytic metallodrugs relative to the high affinity binding that is essential for the classical inhibitory mechanism of drugs currently on the market. High affinity binding of the targeting domain may not be desirable from the viewpoint of facile release of the metallodrug following inactivation of the target. Optimization of the binding affinity of the targeting domain is an issue that will need to be considered on a case-by-case basis. High affinity binding clearly has desirable traits but may be unnecessary with the metallodrug concept described here
Reduction in side effects or toxicity is a consequence of a double-filter mechanism for target recognition that is illustrated in Figure 2. If two proteins, A and B, are recognized by the targeting domain of the drug, but only protein A has a suitable orientation for chemical inactivation by the catalytic metal domain, then only protein A would be irreversibly inactivated by the metallodrug (Figure 2). The proximity and orientation of the metallodrug toward scissile bonds is of key importance,[44, 45] while the use of a subsaturating concentration of drug ensures that the majority of protein B is not influenced by the metallodrug.
Mechanistic considerations
Metal-mediated oxidative degradation is typically achieved in vitro by addition of physiologically relevant reducing agents such as ascorbic acid, glutathione or peroxide. Formation of reactive oxygen species presumably arises through Fenton-type chemistry that gives rise to a copper associated reactive oxygen species that mediates oxidative damage to target molecules (Figure 3). Formation of diffusible hydroxyl radicals has been observed by use of the common rhodamine B assay, and prior studies of RNA and DNA cleavage revealed a significant reduction in diffusible hydroxyl radical concentration when substoichiometric catalyst was used to mediate damage to a target protein or nucleic acid.[12] This is consistent with formation and immediate reaction of a metal-associated reactive oxygen species. Numerous discussions of pathways for oxidative damage to proteins and nucleic acids mediated by metal-catalyzed Fenton-type chemistry can be found in the literature.[36, 46]
Figure 3.
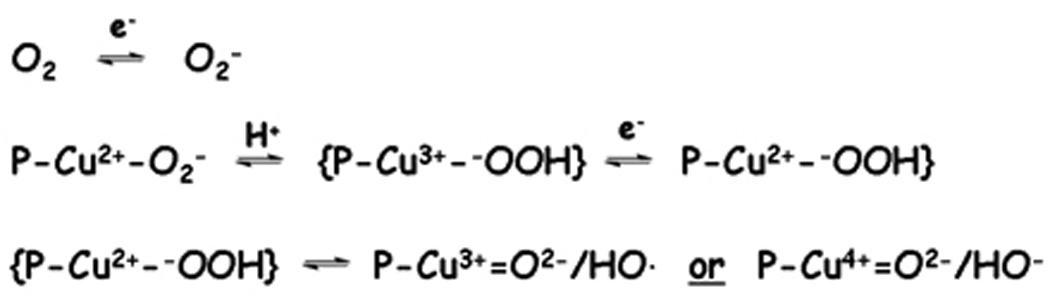
Likely pathways for formation of copper-associated reactive oxygen species that can promote oxidative cleavage of phosphoribose chains in nucleic acids, or sidechain oxidation of proteins. P is the peptide ligand.
Suh and coworkers have developed cobalt(III) cyclen complexes as mediators of hydrolytic cleavage of the peptide backbone of myoglobin[47] and peptide deformylase that most likely function through a mechanism of the type illustrated in Figure 4A.[3]
Figure 4.
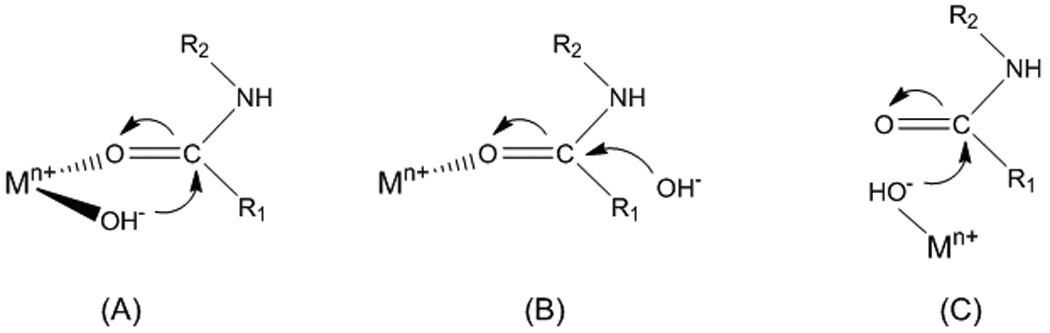
Possible mechanisms for metal-promoted hydrolysis of a peptide bond showing (A) template effect, (B) Lewis acid activation of the electrophilic center, and (C) Lewis acid activation of the nucleophile.
Illustrative examples
Metal-catalyzed oxidative modification of amino acid sidechains have been shown to result in the inactivation of disease-related proteins and enzymes such as angiotensin converting enzyme (ACE), thermolysin (TLN), carbonic anhydrase (CA-I), while the hydrolysis of peptide bonds has more recently been demonstrated for amyloid peptides.[3–14, 16–21] Inactivation is promoted either through oxidative damage of active site residues, or cleavage of the protein/peptide backbone. In our laboratory we have employed the amino terminal Cu and Ni motif (ATCUN)[48] as a metal binding domain that is coupled to an enzyme-recognition moiety. In other studies the cyclen motif has been used to incorporate cobaltic ion.[3, 14, 19, 20] The conceptual basis for a catalytic metallodrug, and its reduction to practice, is illustrated through several examples stemming from our recent efforts in this area, as well as studies from other laboratories on artificial proteases.
(1) Angiotensin converting enzyme
Angiotensin converting enzyme is one of three important protease enzymes that regulate blood pressure and other vascular chemistry, and all have been implicated in cardiovascular disease. Current treatments utilize inhibitors such as lisinopril (Figure 5).[49] Metal-peptide complexes that appear to mimic some of the recognition features of lisinopril, such as the Lys chain in binding pocket S1’ display both classical inhibitory behavior (Figure 5) as well as catalytic inactivation of ACE in the presence of dioxygen and an electron source (Figure 6).
Figure 5.
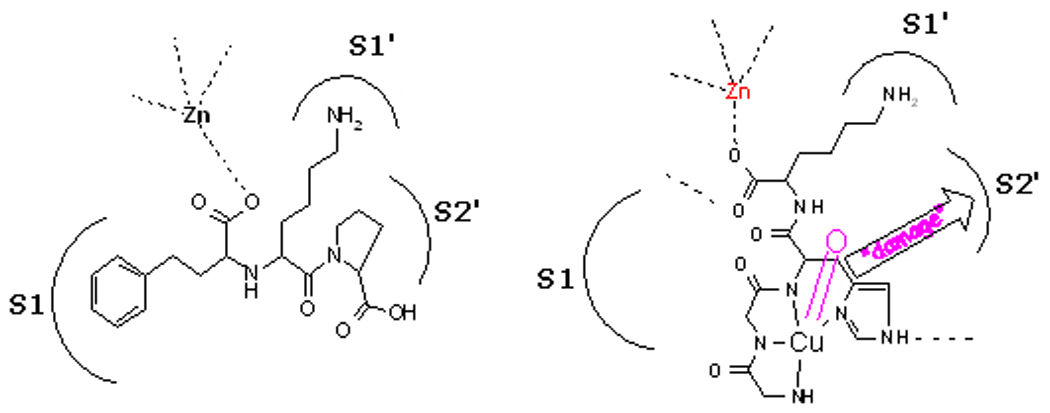
(left) ACE active site showing the association of lisinopril (inhibitor) with enzyme sub-sites. (right) A schematic illustration of the proposed mechanism of action. A bound metallopeptide activates dioxygen and the resulting reactive oxygen species mediates irreversible damage to the target enzyme.
Figure 6.
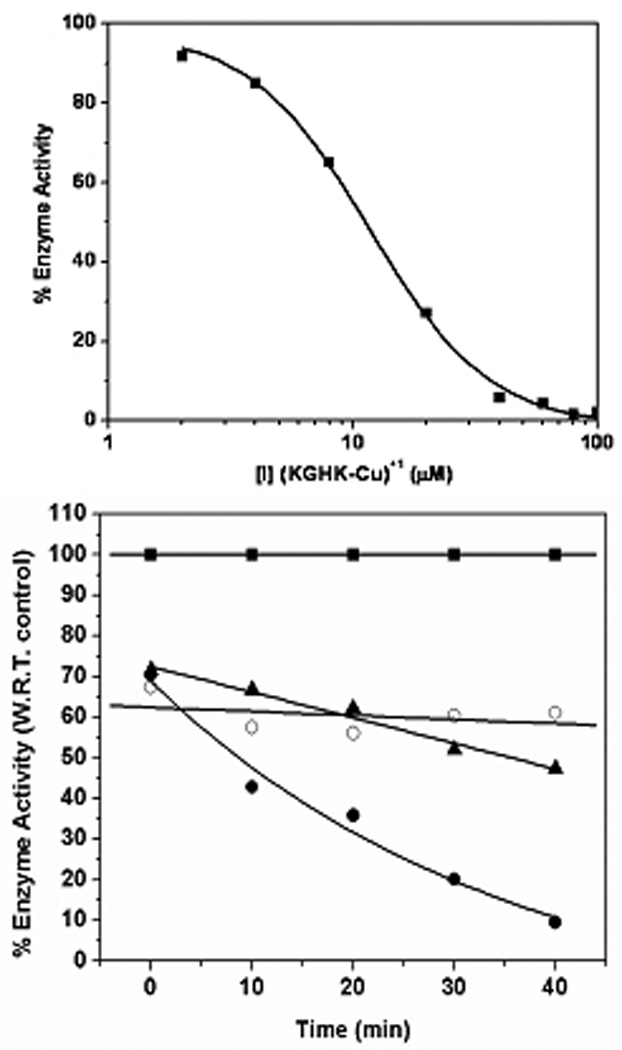
(top) Dose dependent inhibition of ACE by [Cu(KGHK)]+ under hydrolytic conditions.[10, 11] (bottom) Enzyme activity was determined at the given time intervals by taking aliquots of enzyme and determining the activity under initial velocity conditions with 10 µM substrate and 10 µM ZnCl2 in 50 mM HEPES (pH = 7.4) containing 300 mM NaCl, and 1 nM ACE. Conditions included (■) no inhibitor 1 mM ascorbate; (●) with [KGHK-Cu]+ (4.4 µM), 1 mM ascorbate; (○) with [KGHK-Cu]+ (4.4 µM), no ascorbate; and (▲) with Cu2+(aq) (4 µM), 1 mM ascorbate. Fluorogenic substrate Mca-Arg-Pro-Pro-Gly-Phe-Ser-Ala-Phe-Lys(Dnp)-OH is used in all cases to determine enzyme activity.
(2) Thermolysin
Thermolysin belongs to the same family of metalloproteases that regulate the cardiovascular system in human physiology, including neprilysin (NEP),[50] endothelin connverting enzyme I (ECE-I)[51] and angiotensin converting enzyme (ACE).[52] These enzymes possess a consensus sequence HEXXH that constitutes the zinc-containing catalytic domain.[50, 53] Crystallographic data for TLN[54] and various TLN-inhibitor complexes have been used in efforts to model the NEP active site,[55, 56] and TLN serves as a test vehicle to identify proposed inhibitor interactions within the active site of zinc metallo-proteases containing the HEXXH motif.[56, 57] Metallopeptide [Cu2+•Cys-Gly-His-Lys] was observed to stimulate thermolysin activity at low concentration (< 20 µM) and inhibit the enzyme at higher concentration (Figure 7), with binding affinities of 2.0 µM and 4.9 µM, respectively. The N-terminal cysteine is available as a zinc anchoring residue and appears to play a critical functional role, since the [Cu2+•Lys-Gly-His-Lys]+ homologue exhibits neither stimulation nor inhibition of TLN. Under oxidizing conditions (ascorbate/O2) the catalyst is shown to mediate the complete irreversible inactivation of TLN at concentrations where enzyme activity would otherwise be stimulated.
Figure 7.
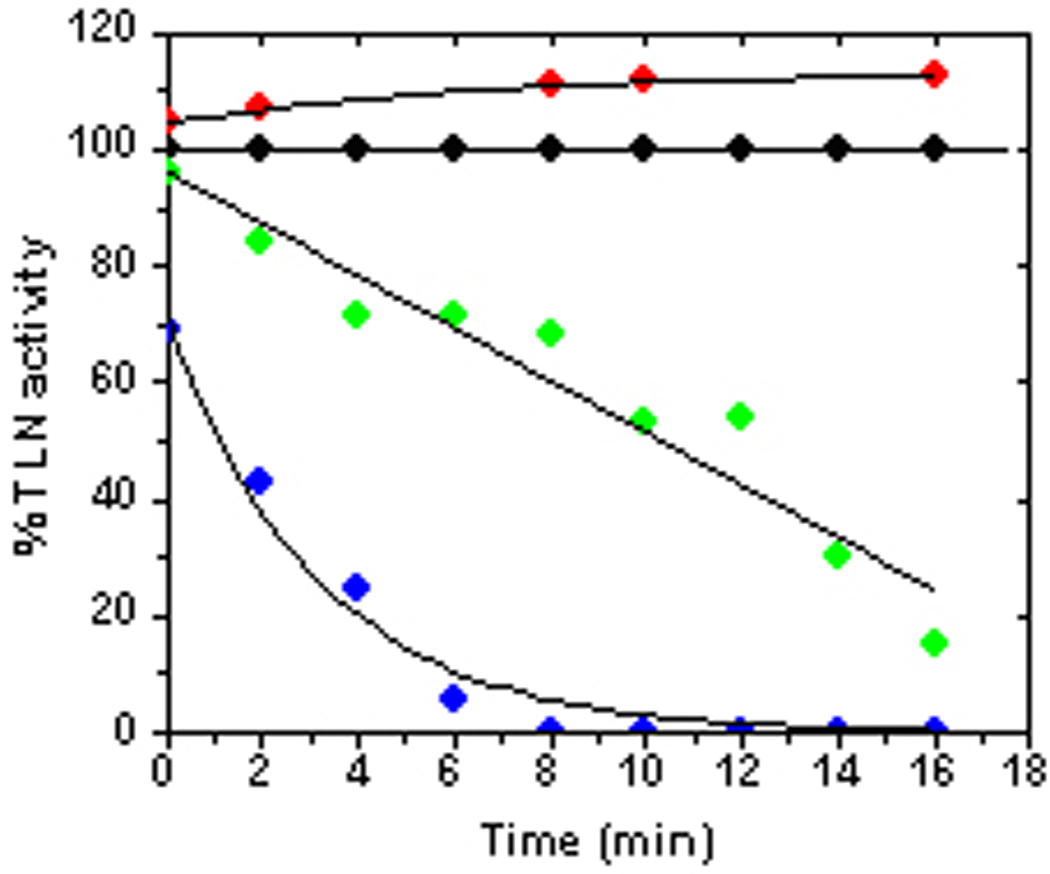
TLN activity[8] was determined from initial velocity measurements with 10 µM substrate in 50 mM HEPES buffer (pH 7.4) containing 10 mM CaCl2, 5 mM NaCl and 10 µM ZnCl2 (final reaction volume 100 µL, final enzyme concentration 5 nM). The fixed time assay involved incubation of substrate with TLN for 30 min and the relative fluorescence units data (RFU) obtained was converted to % TLN activity and plotted against the time aliquots were withdrawn. Conditions include: (●) no inhibitor, 0.5 mM ascorbate; (●) 1 µM [Cu2+•Cys-Gly-His-Lys], no ascorbate; (●) 1 µM [Cu2+•Cys-Gly-His-Lys], 0.5 mM ascorbate (●) 1 µM Cu2+(aq), 0.5 mM ascorbate.
(3) Carbonic Anhydrase
Carbonic anhydrases mediate a number of biosynthetic reactions including the reversible hydration of CO2 to form bicarbonate,[58] and have been implicated in a multitude of medical disorders, including glaucoma, kidney problems, epilepsy, gastric and duodenal ulcers, neurological disorders, and osteoporosis.[58–61] These enzymes are inhibited by sulfonamides,[58–62] which provide a targeting domain for metallodrug design (Figures 8 and 9). Coupling of sulfonamide inhibitors of carbonic anhydrase to the GGH ATCUN motif yielded a complex[9] that directs a Cu2+-bound GGH complex to mediate oxidative damage within the active site of the enzyme, targeting residues prone to such reactivity.[35, 36, 63] This work also established non-peptide moieties as targeting agents when coupled to a copper-ATCUN motif,[48] The most likely active species is a copper associated reactive oxygen species (Figure 3).[8, 10, 11, 64–66]
Figure 8.

Summary of residues that are susceptible to oxidative in human CA-I (PDB: 1CZM) following interaction with Cu-GGHSLN in the presence of ascorbate.[9] Distances from the oxidized residues to the catalysts are listed under each label.
Figure 9.
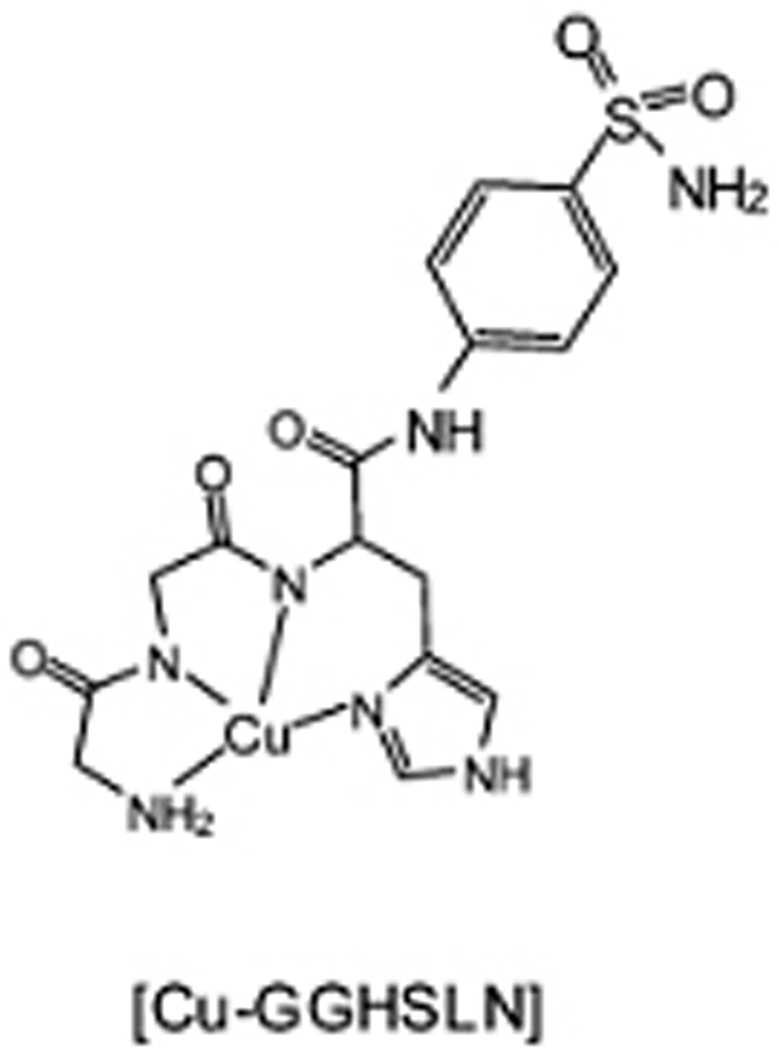
Catalytic complex for inactivation of carbonic anhydrase.
Incubation of CA with Cu-GGHSLN under oxidative conditions yielded clear evidence for oxidation of 1 or 2 residues per protein molecule. More extensive oxidation of predominantly surface residues was observed with Cu2+(aq), although the relatively slow inactivation chemistry suggested that surface residues were the most probably target sites for free copper ion. Characterization of oxidized residues was achieved by enzymatic digestion using both chymotrypsin and trypsin, and the modified residues were mapped by mass spectrometric analysis (Figure 8). Clear evidence for specific amino acid residue modification in the proximity of the enzyme active site was obtained with the most prominent oxidation observed for select histidine residues (H40, H64, H67, H94, H96, H103, H200, and H243). Prior reports indicate that histidine residues are effectively converted to 2-oxo-histidine upon exposure to Cu-ATCUN derived oxygen based radicals.[46, 67–70] Tryptophan was also found to be selectively oxidized (W97, W123).[71] Significantly, none of the zinc bound histidine residues were oxidized, and so the time dependent inhibition of CA-I by Cu-GGHSLN does not involve a random oxidation pathway, but seems to be specific to residues that lie close to the Cu-GGH domain of the metallopeptide-drug conjugate. Localization of modified residues around the active site supports the non-diffusive nature of the oxygen species, and association with the catalytic copper center, and three of the modified residues sites (His64, His67, and His200) have been implicated in rate limiting proton transfer within the active site.[72, 73]
(4) Peptide Deformylase and Amyloid Peptides
Building on earlier reports of cyclen-based catalysts for myoglobin cleavage,[21, 47] Suh and coworkers have developed related families of cobalt cyclen derivatives that mediate cleavage of peptide deformylase (PDF),[3] an antimicrobial target, with a reported kcat of 0.05 h−1. Ligand selection to promote binding utilized a library approach, with a cleavage yield in the range of 10–30%. These workers have also developed cobalt cyclen ligand complexes that associate with plaques containing amyloid peptides and promote hydrolytic cleavage (Figure 4A) of these peptides thereby solubilizing the amyloid fibrils (Figure 10).[14, 19, 20]
Figure 10.

(top) Example of cleavage agent for human islet amyloid polypeptide (h-IAPP), which has been implicated in β-cell apoptosis with resulting type II diabetes milletus (T2DM),[19] amyloid β (Aβ42 and Aβ40), an Alzheimer’s’ target,[20] and α-synuclein, a Parkinson’s disease target,[14] as well as (bottom) a peptide deformylase cleavage agent.[3]
(5) HIV rev response element (RRE) RNA
The concept of a catalytic metallodrug, capable of irreversible, multiturnover degradation of a therapeutic target has also been tested against HIV RRE RNA, both in vitro[13, 16] and in cellular assays.[5] Figure 11 illustrates a family of N-terminal metal binding ATCUN motifs[74] that are extended to include the RRE RNA recognition peptide through a variable length glycine linker sequence (Gx, x = 0, 1, 2, 4, 6).
Figure 11.

Peptide design showing metal binding, linker, and RRE RNA targeting sequences. A control Rev peptide corresponds to the targeting domain alone.
Metallopeptide-mediated oxidative cleavage of RRE RNA was effective even under sub-stoichiometric conditions of RRE RNA and Cu-peptide complex in the presence of ascorbate. Cleavage was not random, but rather three specific cleavage sites were observed within the likely the binding pocket for the metallopeptide complex (Figure 12). The accurate masses obtained for the cleavage products are different from the calculated mass values expected for products from hydrolysis, but is consistent with those expected following strand scission by a C-1’H or C-4’H oxidative cleavage path in the presence of ascorbate.[13]
Figure 12.
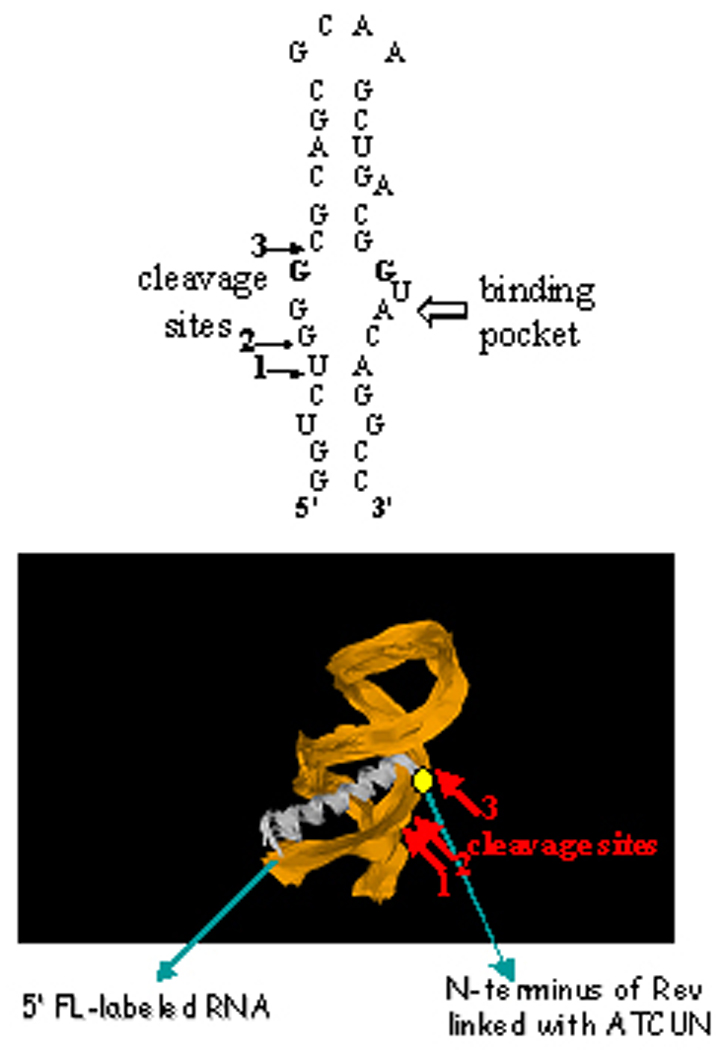
(top) A schematic illustration of the stem loop structure adopted by RRE RNA IIB,[75] showing the cleavage sites identified by mass spectrometry studies. (bottom) The positions of the cleavage sites are consistent with expectations from the solution structure.[75]
By use of a plasmid encoding a target RNA sequence fused to the C-terminus of the green fluorescent protein (GFP), the intracellular delivery and cleavage of the RRE recognition motif was assessed by monitoring the fluorescence from GFP.[5, 76] These metallopeptides were designed to explicitly address the need for cellular and nuclear uptake, possessing both an arginine rich motif (ARM) that promotes cellular uptake, and a nuclear import sequence, and results obtained by targeting the HIV RRE RNA target sequence in human Jurkat cells establishes the fact that the metallopeptides can effectively target cognate RNA’s in cellular assays.[76]
Concluding remarks
To address the need for novel therapeutics, an issue that reflects the increasing emergence of drug resistance as a critical problem in viral and bacterial disease, we have developed and continue to explore the concept of catalytic metallodrugs. Such a strategy encompasses both novel molecular frameworks, by inclusion of metal ions and the potential for structural variability through coordination chemistry, but also develops a novel mode of action through irreversible catalytic inactivation of a therapeutic target that can result in multiple turnovers from a single catalytic drug. This requires new design strategies, where high drug affinity is not necessarily an advantage, and target selectivity reflects both drug binding and the stereochemical requirements for productive chemistry on the target. Additional examples of metallodrugs that lack the targeting domain, but mediate formation of cellular reactive oxygen species, have recently shown promise as potential anticancer agents,[77] and other candidates and strategies of promise will surely emerge. Finally, it seems reasonable to predict that the irreversible nature of such chemistry should have a positive impact on the emergence of resistance strains. Such a prediction, and the long term prospects for “catalytic metallodrugs” await future investigation in what is likely to be a rapidly evolving field of study.
Acknowledgements
We acknowledge the National Institutes of Health for financial support of this work (GM 63740).
References
- 1.Thomas JJ, Bakhtiar R, Siuzdak G. Acc. Chem. Res. 2000:179–187. doi: 10.1021/ar9801200. [DOI] [PubMed] [Google Scholar]
- 2.Heyduk T, Baichoo N, Heyduk E. Met. Ions. Biol. Syst. 2001;38:255–287. [PubMed] [Google Scholar]
- 3.Chae PS, Kim MS, Jeung CS, Lee SDu, Park H, Lee S, Suh J. J. Am. Chem. Soc. 2005;127:2396–2397. doi: 10.1021/ja044043h. [DOI] [PubMed] [Google Scholar]
- 4.Chei WS, Suh J. Prog. Inorg. Chem. 2007;55:79–142. [Google Scholar]
- 5.Chen C, Cowan JA. Chem. Commun. 2002:196–197. doi: 10.1039/b108439a. [DOI] [PubMed] [Google Scholar]
- 6.Cowan JA. Curr. Op. Chem. Biol. 2001;5:634–642. doi: 10.1016/s1367-5931(01)00259-9. [DOI] [PubMed] [Google Scholar]
- 7.Cowan JA. Pure. Appl. Chem. 2008;80:1799–1810. [Google Scholar]
- 8.Gokhale NH, Bradford S, Cowan JA. J. Biol. Inorg. Chem. 2007;12:981–987. doi: 10.1007/s00775-007-0270-6. [DOI] [PubMed] [Google Scholar]
- 9.Gokhale NH, Bradford S, Cowan JA. J. Am. Chem. Soc. 2008;130:2388–2389. doi: 10.1021/ja0778038. [DOI] [PubMed] [Google Scholar]
- 10.Gokhale NH, Cowan JA. Chem. Commun. 2005:5916–5918. doi: 10.1039/b511081e. [DOI] [PubMed] [Google Scholar]
- 11.Gokhale NH, Cowan JA. J. Biol. Inorg. Chem. 2006;11:937–947. doi: 10.1007/s00775-006-0145-2. [DOI] [PubMed] [Google Scholar]
- 12.Jin Y, Cowan JA. J. Am. Chem. Soc. 2005;127:8408–8415. doi: 10.1021/ja0503985. [DOI] [PubMed] [Google Scholar]
- 13.Jin Y, Cowan JA. J. Am. Chem. Soc. 2006;128:410–411. doi: 10.1021/ja055272m. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, Yoo SH, Jeong K, Lee TY, Ahn JY, Suh J. Bull. Korean Chem. Soc. 2008;29:882–884. [Google Scholar]
- 15.Sigman DS, Bruice TW, Mazumder A, Sutton CL. Acc. Chem. Res. 1993;26:98–104. [Google Scholar]
- 16.Sreedhara A, Patwardhan A, Cowan JA. Chem. Commun. 1999:1147–1148. [Google Scholar]
- 17.Suh J. Acc. Chem. Res. 2003;36:562–570. doi: 10.1021/ar020037j. [DOI] [PubMed] [Google Scholar]
- 18.Suh J, Chei WS. Curr. Op. Chem. Biol. 2008;12:207–213. doi: 10.1016/j.cbpa.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 19.Suh J, Chei WS, Lee TY, Kim MG, Yoo SH, Jeong K, Ahn JY. J. Biol. Inorg. Chem. 2008;13:693–701. doi: 10.1007/s00775-008-0354-y. [DOI] [PubMed] [Google Scholar]
- 20.Suh J, Yoo S, Kim M, Jeong K, Ahn JY, Kim MS, Chae PS, Lee TY, Lee J, Jang YA, Ko EH. Angew. Chem. Int. Ed. 2007;46:7064–7067. doi: 10.1002/anie.200702399. [DOI] [PubMed] [Google Scholar]
- 21.Yoo SH, Lee BJ, Kim H, Suh J. J. Am. Chem. Soc. 2005;127:9593–9602. doi: 10.1021/ja052191h. [DOI] [PubMed] [Google Scholar]
- 22.Chen XH, Luo XM, Song YC, Zhou SZ, Zhu LG. Polyhedron. 1998;17:2271–2278. [Google Scholar]
- 23.de Oliveira MCB, Scarpellini M, Neves A, Terenzi H, Bortoluzzi AJ, Szpoganics B, Greatti A, Mangrich AS, de Souza EM, Fernandez PM, Soares MR. Inorg. Chem. 2005;44:921–929. doi: 10.1021/ic0485864. [DOI] [PubMed] [Google Scholar]
- 24.Djuran MI, Milinkovic SU. Polyhedron. 1999;18:3611–3616. [Google Scholar]
- 25.Kassai M, Grant KB. Inorg. Chem. Commun. 2008;11:521–525. [Google Scholar]
- 26.Korneeva EN, Ovchinnikov MV, Kostic NM. Inorg. Chim. Acta. 1996;243:9–13. [Google Scholar]
- 27.Luo XM, He WJ, Zhang Y, Guo ZJ, Zhu LG. Chin. J. Chem. 2000;18:855–862. [Google Scholar]
- 28.Milovic NM, Kostic NM. J. Am. Chem. Soc. 2003;125:781–788. doi: 10.1021/ja027408b. [DOI] [PubMed] [Google Scholar]
- 29.Parac TN, Kostic NM. J. Am. Chem. Soc. 1996;118:51–58. [Google Scholar]
- 30.Sutton PA, Buckingham DA. Acc. Chem. Res. 1987;20:357–364. [Google Scholar]
- 31.Takarada T, Yashiro M, Komiyama M. Chem. Eur. J. 2000;6:3906–3913. doi: 10.1002/1521-3765(20001103)6:21<3906::aid-chem3906>3.3.co;2-a. [DOI] [PubMed] [Google Scholar]
- 32.Zhu LG, Kostic NM. Inorg. Chim. Acta. 1994;217:21–28. [Google Scholar]
- 33.Garrison WM. Chem. Rev. 1987;87:381–398. [Google Scholar]
- 34.Schuessler H, Schilling K. Int. J. Radiat. Biol. 1984;45:267–281. doi: 10.1080/09553008414550381. [DOI] [PubMed] [Google Scholar]
- 35.Stadtman ER. Free Radic. Biol. Med. 1990;9:315–325. doi: 10.1016/0891-5849(90)90006-5. [DOI] [PubMed] [Google Scholar]
- 36.Stadtman ER, Berlett BS. J. Biol. Chem. 1991;266:17201–17211. [PubMed] [Google Scholar]
- 37.Ali FE, Barnham KJ, Barrow CJ, Separovic F. J. Inorg. Biochem. 2004;98:173–184. doi: 10.1016/j.jinorgbio.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Brown KC, Yang SH, Kodadek T. Biochemistry. 1995;34:4733–4739. doi: 10.1021/bi00014a030. [DOI] [PubMed] [Google Scholar]
- 39.Brittain IJ, Huang XF, Long EC. Biochemistry. 1998;37:12113–12120. doi: 10.1021/bi9806605. [DOI] [PubMed] [Google Scholar]
- 40.Liang Q, Ananias DC, Long EC. J. Am. Chem. Soc. 1998;120:248–257. [Google Scholar]
- 41.McLachlan GA, Muller JG, Rokita SE, Burrows CJ. Inorg. Chim. Acta. 1996;251:193–199. [Google Scholar]
- 42.Muller JG, Zheng P, Rokita SE, Burrows CJ. J. Am. Chem. Soc. 1996;118:2320–2325. [Google Scholar]
- 43.Nagane R, Koshigoe T, Chikira M. J. Inorg. Biochem. 2003;93:204–212. doi: 10.1016/s0162-0134(02)00619-0. [DOI] [PubMed] [Google Scholar]
- 44.Groves JT, Baron LA. J. Am. Chem. Soc. 1989;111:5442–5448. [Google Scholar]
- 45.Rana TM, Meares CF. Proc. Natl. Acad. Sci. USA. 1991;88:10578–10582. doi: 10.1073/pnas.88.23.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hong J, Schoneich C. Free Radic. Biol. Med. 2001;31:1432–1441. doi: 10.1016/s0891-5849(01)00722-5. [DOI] [PubMed] [Google Scholar]
- 47.Jeon JW, Son SJ, Yoo CE, Hong IS, Song JB, Suh J. Org. Letts. 2002;4:4155–4158. doi: 10.1021/ol0269300. [DOI] [PubMed] [Google Scholar]
- 48.Harford C, Sarkar B. Acc. Chem. Res. 1997;30:123–130. [Google Scholar]
- 49.Natesh R, Schwager SLU, Sturrock ED, Acharya KR. Nature. 2003;421:551–554. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- 50.Roques BP. Biochem. Soc. Trans. 1993;21:678–685. doi: 10.1042/bst0210678. [DOI] [PubMed] [Google Scholar]
- 51.Cheng X-M, Ahn K, Haleen SJ. Ann. Rep. in Med. Chem. 1997;32:61–70. [Google Scholar]
- 52.Wyvratt MJ, Patchett AA. Med. Res. Rev. 1985;5:483–531. doi: 10.1002/med.2610050405. [DOI] [PubMed] [Google Scholar]
- 53.Matthews BW. Acc. Chem. Res. 1988;21:333–340. [Google Scholar]
- 54.Holmes MA, Matthews BW. J. Mol. Biol. 1982;160:623–639. doi: 10.1016/0022-2836(82)90319-9. [DOI] [PubMed] [Google Scholar]
- 55.Gaucher JF, Selkti M, Tiraboschi G, Prange T, Roques BP, Tomas A, Fournie-Zaluski MC. Biochemistry. 1999;38:12569–12576. doi: 10.1021/bi991043z. [DOI] [PubMed] [Google Scholar]
- 56.Bohacek R, De Lombaert S, McMartin C, Priestle J, Gruetter M. J. Am. Chem. Soc. 1996;118:8231–8249. [Google Scholar]
- 57.Le Moual H, Roques BP, Crine P, Boileau G. FEBS Letters. 1993;324:196–200. doi: 10.1016/0014-5793(93)81392-d. [DOI] [PubMed] [Google Scholar]
- 58.Supuran CT, Scozzafava A, Casini A. Med. Res. Rev. 2003;23:146–189. doi: 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
- 59.Clare BW, Supuran CT. Expert Opin. Drug Metab. Toxicol. 2006;2:113–137. doi: 10.1517/17425255.2.1.113. [DOI] [PubMed] [Google Scholar]
- 60.Di Fiore A, De Simone G, Menchise V, Pedone C, Casini A, Scozzafava A, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:1937–1942. doi: 10.1016/j.bmcl.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 61.Khadikar PV, Sharma V, Karmarkar S, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:923–930. doi: 10.1016/j.bmcl.2004.12.056. [DOI] [PubMed] [Google Scholar]
- 62.Chakravarty S, Kannan KK. J. Mol. Biol. 1994;243:298–309. doi: 10.1006/jmbi.1994.1655. [DOI] [PubMed] [Google Scholar]
- 63.Stadtman ER. Am. J. Clin. Nutr. 1991;54:1125S–1128S. doi: 10.1093/ajcn/54.6.1125s. [DOI] [PubMed] [Google Scholar]
- 64.McDonald MR, Fredericks FC, Margerum DW. Inorg. Chem. 1997;36:3119–3124. doi: 10.1021/ic9608713. [DOI] [PubMed] [Google Scholar]
- 65.Margerum DW, Owens GD. Met. Ions. Biol. Syst. 1981;12:75–132. [Google Scholar]
- 66.Bossu FP, Margerum DW. J. Am. Chem. Soc. 1976;98:4003–4004. doi: 10.1021/ja00429a047. [DOI] [PubMed] [Google Scholar]
- 67.Hovorka SW, Biesiada H, Williams TD, Huhmer A, Schoneich C. Pharm. Res. 2002;19:530–537. doi: 10.1023/a:1015164200431. [DOI] [PubMed] [Google Scholar]
- 68.Hovorka SW, Williams TD, Schoeneich C. Anal. Biochem. 2002;300:206–211. doi: 10.1006/abio.2001.5447. [DOI] [PubMed] [Google Scholar]
- 69.Schoneich C. J. Pharm. Biomed. Anal. 2000;21:1093–1097. doi: 10.1016/s0731-7085(99)00182-x. [DOI] [PubMed] [Google Scholar]
- 70.Uchida K, Kawakishi S. Arch. Biochem. Biophys. 1990;283:20–26. doi: 10.1016/0003-9861(90)90606-y. [DOI] [PubMed] [Google Scholar]
- 71.Griffiths HR. Free Radic. Res. 2000;33:S47–S58. [PubMed] [Google Scholar]
- 72.Engstrand C, Jonsson B-H, Lindskod S. Eur. J. Biochem. 1995;229:696–702. doi: 10.1111/j.1432-1033.1995.tb20516.x. [DOI] [PubMed] [Google Scholar]
- 73.Mohanty AK, Kannan KK, Mahajan SK. J. Biosci. 1998;23:235–246. [Google Scholar]
- 74.Lau S-J, Kruck TPA, Sarkar B. J. Biol. Chem. 1974;249:5878–5884. [PubMed] [Google Scholar]
- 75.Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, Frankel AD, Williamson JR. Science. 1996;273:1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- 76.Jin Y, Cowan JA. J. Biol. Inorg. Chem. 2007;12:637–644. doi: 10.1007/s00775-007-0221-2. [DOI] [PubMed] [Google Scholar]
- 77.Dougan SJ, Habtemariam A, McHale SE, Parsons S, Sadler PJ. Proc. Natl. Acad. Sci. USA. 2008;105:11628–11633. doi: 10.1073/pnas.0800076105. [DOI] [PMC free article] [PubMed] [Google Scholar]