

Abstract
Reactive oxygen species (ROS) such as superoxide are emerging as important signaling molecules in physiological plasticity but also in peripheral and spinal cord pain pathology. Underlying mechanisms and pain-related ROS signaling in the brain remain to be determined. Neuroplasticity in the amygdala plays a key role in emotional-affective pain responses and depends on group I metabotropic glutamate receptors (mGluRs) and protein kinases. Using patch-clamp, live-cell imaging, and behavioral assays, we tested the hypothesis that mitochondrial ROS links group I mGluRs to protein kinase activation to increase neuronal excitability and pain behavior. Agonists for mGluR1/5 (DHPG) or mGluR5 (CHPG) increased neuronal excitability of neurons in the laterocapsular division of the central nucleus of the amygdala (CeLC). DHPG effects were inhibited by an mGluR5 antagonist (MTEP), IP3 receptor blocker (xestospongin C), or ROS scavengers (PBN, tempol), but not by an mGluR1 antagonist (LY367385) or NO synthase inhibitor (l-NAME). Tempol inhibited the effects of IP3 but not those of a PKC activator, indicating that ROS activation was IP3 mediated. Live-cell imaging in CeLC-containing brain slices directly showed DHPG-induced and synaptically evoked mitochondrial superoxide production. DHPG also increased pain-related vocalizations and spinal reflexes through a mechanism that required mGluR5, IP3, and ROS. Combined application of inhibitors of ERK (U0126) and PKA (KT5720) was necessary to block completely the excitatory effects of a ROS donor (tBOOH). A PKC inhibitor (GF109203X) had no effect. Antagonists and inhibitors alone did not affect neuronal excitability. The results suggest an important role for the novel mGluR5- IP3-ROS-ERK/PKA signaling pathway in amygdala pain mechanisms.
Introduction
Reactive oxygen species (ROS), such as superoxide and hydrogen peroxide, are emerging as important signaling molecules in physiological plasticity (Klann, 1998; Hu et al., 2006; Kishida and Klann, 2007) but also in pain pathophysiology (Chung, 2004). Biochemical and behavioral data implicate peripheral (Twining et al., 2004; Keeble et al., 2009) and spinal (Kim et al., 2004, 2009; Wang et al., 2004; Gao et al., 2007; Schwartz et al., 2008, 2009) ROS in inflammatory and neuropathic pain, but direct electrophysiological evidence is sparse. ROS scavengers inhibited the responses of spinal dorsal horn neurons in the capsaicin pain model (Lee et al., 2007) and C fiber-induced long-term potentiation in spinal cord slices (Lee et al., 2010).
Mechanisms of pain-related ROS signaling, ROS activation, and downstream targets remain to be determined. Pain-related functions of ROS in the brain are largely unknown. The present study focused on the amygdala, a key player in emotions and affective disorders (Maren, 2005; Phelps and Ledoux, 2005; Seymour and Dolan, 2008). Neuroplasticity in the central nucleus of the amygdala (CeA), particularly its laterocapsular division (CeLC), contributes critically to the emotional-affective component of pain and pain modulation (Gauriau and Bernard, 2002; Neugebauer et al., 2004, 2009; Minami, 2009). Pharmacologic inhibition of amygdala hyperactivity decreases nocifensive and affective responses in animal pain models (Neugebauer et al., 2004; Carrasquillo and Gereau, 2007; Pedersen et al., 2007; Ansah et al., 2010; Myers and Greenwood-Van Meerveld, 2010). Conversely, pharmacologic CeA activation produces pain behavior in the absence of tissue injury (Myers et al., 2007; Ansah et al., 2009; Han et al., 2010; Kolber et al., 2010).
Group I metabotropic glutamate receptors (mGluRs) play a critical role in pain-related hyperactivity of amygdala neurons (Neugebauer et al., 2003; Li and Neugebauer, 2004) and amygdala-mediated pain behaviors (Han and Neugebauer, 2005; Ansah et al., 2010; Kolber et al., 2010). A recent in vivo study suggests that facilitatory effects of group I mGluRs on nociceptive processing in CeLC neurons involve ROS (Ji and Neugebauer, 2010). The mechanistic link between mGluRs and ROS signaling, cellular site of ROS action, downstream targets, and behavioral consequences remain to be determined and are addressed here. Inositol-1,4,5-trisphosphate (IP3)-mediated calcium release increases mitochondrial calcium uptake and ROS production (Hajnoczky et al., 2006; Wu et al., 2007). Group I mGluRs typically couple to IP3 and protein kinase C (PKC) signaling (Varney and Gereau, 2002; Lesage, 2004; Neugebauer, 2007) and therefore are well positioned to activate ROS.
Effector mechanisms of ROS may include protein kinases (Kishida and Klann, 2007; Huddleston et al., 2008). Extracellular signal-regulated kinase (ERK) (Carrasquillo and Gereau, 2007; Fu et al., 2008; Kolber et al., 2010) and PKA rather than PKC (Bird et al., 2005; Fu et al., 2008) play an important role in pain-related amygdala functions. Group I mGluRs, including mGluR5, can activate ERK (Karim et al., 2001; Kolber et al., 2010), but the coupling mechanism remains to be determined. ERK inhibition does not completely block mGluR5 agonist effects in the amygdala (Kolber et al., 2010). Evidence from expression systems suggests that group I mGluRs can also activate PKA (Joly et al., 1995). Here, we report a novel mGluR5→IP3→mitochondrial ROS→ERK and PKA signaling cascade in the amygdala that can increase neuronal excitability and pain responses.
Materials and Methods
Male Sprague Dawley rats (150–350 g) were housed in a temperature-controlled room and maintained on a 12 h day/night cycle. Water and food were available ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Medical Branch (UTMB), Galveston, TX, and conform to the guidelines of the International Association for the Study of Pain, Seattle, WA, and the National Institutes of Health (NIH), Bethesda, MD.
Electrophysiology in brain slices
Slice preparation.
Coronal brain slices (300–500 μm) containing the CeLC of the right hemisphere were obtained from normal untreated rats (150–250 g) as described previously (Han et al., 2005b; Fu and Neugebauer, 2008; Ji et al., 2010). Rats were decapitated without the use of anesthesia to avoid chemical contamination of the tissue. A single brain slice was transferred to the recording chamber and submerged in artificial CSF (ACSF; 31 ± 1°C), which superfused the slice at ∼2 ml/min. ACSF contained the following (in mm): 117 NaCl, 4.7 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 11 glucose. The ACSF was oxygenated and equilibrated to pH 7.4 with a mixture of 95% O2/5% CO2. Only one or two brain slices per animal were used, one neuron was recorded in each slice, and a fresh slice was used for each new experimental protocol. Numbers in the manuscript refer to the number of neurons tested for each parameter.
Patch-clamp recording.
Whole-cell patch-clamp recordings were obtained from CeLC neurons using the “blind” patch technique as described previously (Neugebauer et al., 2003; Han et al., 2005b; Fu and Neugebauer, 2008). The boundaries of the different amygdalar nuclei are easily discerned under light microscopy [see Fu and Neugebauer (2008), their Fig. 1]. Recording pipettes (3–5 MΩ tip resistance) made from borosilicate glass (1.5 and 1.12 mm, outer and inner diameter, respectively; Drummond) were filled with intracellular solution containing the following (in mm): 122 K-gluconate, 5 NaCl, 0.3 CaCl2, 2 MgCl2, 1 EGTA, 10 HEPES, 5 Na2-ATP, and 0.4 Na3-GTP; pH was adjusted to 7.2–7.3 with KOH and osmolarity to 280 mOsm/kg with sucrose. Data acquisition and analysis of voltage and current signals were done using a dual four-pole Bessel filter (Warner Instruments), low-noise Digidata 1322 interface (Axon Instruments), Axoclamp-2B amplifier (Axon Instruments), Pentium PC, and pClamp9 software (Axon Instruments). Signals were low-pass filtered at 1 kHz and digitized at 5 kHz. Head-stage voltage was monitored continuously on an oscilloscope to ensure precise performance of the amplifier. High (>2 GΩ) seal and low (<20 MΩ) series resistances were checked throughout the experiment (using pClamp9 membrane test function) to ensure high-quality recordings. If series resistance (monitored with pClamp9 software, Axon Instruments) changed >10%, the neuron was discarded. Neurons were recorded at −60 mV. Using concentric bipolar stimulating electrodes (22 kW; SNE-100; Kopf Instruments), monosynaptic EPSCs (in voltage clamp), or action potentials (in current clamp) were evoked in CeLC neurons by focal electrical stimulation (Grass S88 stimulator) of inputs from the parabrachial area (PB). For stimulation of the PB-CeLC synapse, the electrode was positioned under microscopic control on the afferent fiber tract from the lateral PB, which runs dorsomedial to the central nucleus (CeA) and ventral to but outside of the caudate-putamen (Han et al., 2005b; Fu and Neugebauer, 2008). In the vicinity of this tract, no other afferents to the CeA have been described (Schwaber et al., 1988; Harrigan et al., 1994). For basal synaptic stimulation, electrical stimuli (200 μs square-wave pulses) were delivered at a frequency of 0.033 Hz. For high-frequency stimulation, trains of five stimuli delivered at 100 Hz (intertrain interval of 20 s) were applied. In current-clamp experiments designed to measure synaptically evoked action potentials, the stimulus intensity was set to just above the threshold for evoking spikes (typically 10 V). This stimulus intensity was also used in live-cell imaging studies (see below, Live-cell imaging of superoxide formation in brain slices). Neuronal excitability was measured by recording action potentials generated by intracellular current injections (500 ms) of increasing magnitude while the cell was held at a starting membrane potential of −60 mV.
Figure 1.
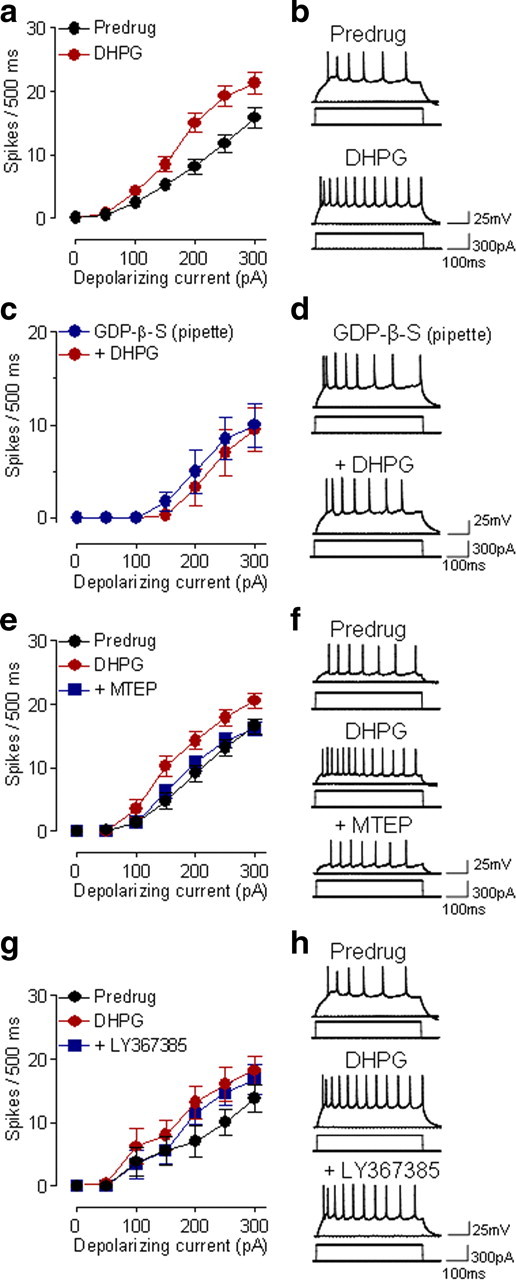
Group I mGluR5 activation increases excitability of CeLC neurons. a, DHPG (10 μm, 10–15 min) increased the input–output function of neuronal excitability of CeLC neurons (n = 31) significantly (p < 0.0001, main effect of drug, two-way ANOVA). Symbols show mean (± SE) number of spikes per 500 ms calculated from recordings like those in b. Action potentials were generated by depolarizing current injections of increasing magnitude (50 pA steps). b, Individual traces of action potentials generated by intracellular current injections (300 pA, 500 ms) before and during DHPG (10 μm). c, DHPG had no significant effect on neuronal excitability when a G protein inhibitor (GDP-β-S, 1 mm) was included in the patch pipette (n = 4 neurons; p > 0.05, two-way ANOVA). d, Individual example of action potentials evoked in a CeLC neuron with GDP-β-S included in the patch pipette (10 min after rupturing the cell membrane) and during the addition of DHPG. e, MTEP (1 μm, 15 min) decreased the facilitatory effect of DHPG (10 μm) significantly (n = 6 neurons; p < 0.0001, main effect of drug, two-way ANOVA). f, Action potentials evoked in a CeLC neuron in control (predrug), during DHPG, and during combined application of DHPG and MTEP. g, LY367385 (10 μm) had no significant effect on increased excitability induced by DHPG (10 μm) (n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA). h, Action potentials evoked in a CeLC neuron in control (predrug), during DHPG, and during combined application of DHPG and LY367385. a–g, DHPG, MTEP and LY367385 were superfused onto the brain slices. See Results for details of the statistical analysis.
Drug application.
Drugs were applied by gravity-driven superfusion of the brain slice in the ACSF (∼2 ml/min). Solution flow into the recording chamber (1 ml volume) was controlled with a three-way stopcock. Some compounds were also included in the internal solution of the patch pipette for intracellular application (see below, Drugs).
Live-cell imaging of superoxide formation in brain slices
Coronal brain slices containing the CeLC were prepared as in the electrophysiology experiments. Slices were incubated in oxygenated ACSF for 1–3 h for stabilization. Immediately before imaging, the brain slice was incubated in a redox-sensitive, mitochondrial-specific fluorescent dye (MitoSOX, Invitrogen) for 30 min. The reduced form of MitoSOX does not show fluorescence and penetrates into the cells and sequesters in the mitochondria. MitoSOX fluoresces red when oxidized by superoxide produced by mitochondria. For live-cell imaging, one slice was placed in a perfusion/imaging chamber on an inverted epifluorescence microscope (confocal system Zeiss LSM 510 META with Zeiss Axiovert 200M) and continuously perfused with oxygenated ACSF (2 ml/min; 32°C). In some experiments, focal electrical stimulation of presumed PB afferents at low frequencies (1 Hz) and high frequencies (100 Hz) was used (see above, Patch-clamp recording). Scanning parameters, pinhole diameter, and laser intensity were optimized to minimize photobleaching and other photodynamic artifacts while maintaining high-image quality (resolution and signal-to-noise ratio). The viewing field was placed over the target area of the brain slice (CeLC), and confocal images with red fluorescence were photographed every minute. The time lapse confocal image acquisition was controlled using the LSM 510 META workstation software (version 3.2) equipped with a “physiology package.” Image analysis and quantification of regions of interest within each slice (shown as pseudocolor) was performed “off-line” using the software package MetaMorph (version 6.2; Molecular Devices). Background fluorescence was determined from areas containing unlabeled regions under control conditions. Background-subtracted pixel intensity in the target area was averaged for each brain slice and measured every minute.
Behavioral tests
Spinal reflexes.
Thresholds of hindlimb withdrawal reflexes evoked by mechanical stimulation of the knee joint were measured as described previously (Neugebauer et al., 2007). Mechanical stimuli of continuously increasing intensity were applied to the knee joint using a calibrated forceps equipped with a force transducer; its output signal was amplified and displayed in grams on a liquid crystal display screen (Han et al., 2005a; Neugebauer et al., 2007; Fu and Neugebauer, 2008; Ji et al., 2010). Three consecutive measurements (2 min intervals) were made under each experimental condition (predrug, drug) and averaged. Withdrawal threshold was defined as the minimum stimulus intensity that evoked a withdrawal reflex.
Vocalizations.
Audible and ultrasonic vocalizations were recorded and analyzed as described previously (Han et al., 2005a; Neugebauer et al., 2007; Fu and Neugebauer, 2008; Ji et al., 2010). The experimental setup (U.S. Patent 7,213,538) included a custom-designed recording chamber, a condenser microphone (audible range, 20 Hz to 16 kHz) connected to a preamplifier, an ultrasound detector (25 ± 4 kHz), filter and amplifier (UltraVox four-channel system; Noldus Information Technology), and data acquisition software (UltraVox 2.0; Noldus Information Technology), which automatically monitored the occurrence of vocalizations within user-defined frequencies and recorded the number and duration of digitized events (vocalizations). The computerized recording system was set to suppress nonrelevant audible sounds (background noise) and ignore ultrasounds outside the defined frequency range. Animals were placed in the recording chamber for acclimation 1 h before the vocalization measurements.
Brief (15 s) innocuous (500 g/30 mm2) and noxious (2000 g/30 mm2) mechanical stimuli were applied to the knee using a calibrated forceps (see above, Spinal reflexes). For colorectal distension (CRD), an inflatable latex balloon (5 cm) connected to a sphygmomanometer was inserted into the distal colon. Innocuous (20 mmHg), moderate (40 mmHg), and noxious (60 and 80 mmHg) intraluminal pressures (15 s each) were achieved by inflating the balloon (Ji and Neugebauer, 2010). The total duration of vocalizations (arithmetic sum of the duration of individual events) was recorded for 1 min, starting with the onset of the mechanical stimulus. Audible and ultrasonic vocalizations reflect supraspinally organized nocifensive and affective responses to aversive stimuli (Neugebauer et al., 2007). The results of two to three consecutive measurements (2 min intervals) were averaged for each experimental condition (predrug, drug).
Drug application by microdialysis in awake behaving animals.
As described in detail previously (Neugebauer et al., 2007; Fu and Neugebauer, 2008; Han et al., 2010; Ji et al., 2010), a guide cannula was implanted stereotaxically the day before behavioral measurements using a stereotaxic apparatus (Kopf Instruments). The animal was anesthetized with pentobarbital sodium (Nembutal; 50 mg/kg, i.p.), and a small unilateral craniotomy was performed at the sutura frontoparietalis level. The guide cannula was implanted on the dorsal margin of the CeA in the right hemisphere using the following coordinates (in mm): 2.0 caudal to bregma; 4.0 lateral to midline; depth, 7.5. In some experiments a guide cannula was implanted into the striatum as a placement control using the following stereotaxic coordinates (in mm): 2.0 caudal to bregma; 4.0 lateral to midline; depth of tip, 6.0. The cannula was fixed to the skull with dental acrylic (Plastics One). Antibiotic ointment was applied to the exposed tissue to prevent infection. On the day of the experiment, a microdialysis probe (CMA11; CMA/Microdialysis ; membrane diameter, 250 μm; membrane length, 1 mm; 8 kDa cutoff, sufficient for the application of small molecules used in this study) was inserted through the guide cannula so that the probe protruded by 1 mm. The probe was connected to a Harvard infusion pump and perfused with ACSF containing the following (in mm): 125.0 NaCl, 2.6 KCl, 2.5 NaH2PO4, 1.3 CaCl2, 0.9 MgCl2, 21.0 NaHCO3, and 3.5 glucose; oxygenated and equilibrated to pH 7.4 at 5 μl/min. Before each drug application, ACSF was pumped through the fiber for at least 1 h to establish equilibrium in the tissue. Drugs were dissolved in ACSF on the day of the experiment at a concentration 100-fold that predicted to be needed based on data from our previous microdialysis (Li and Neugebauer, 2004; Han and Neugebauer, 2005), in vitro studies (Neugebauer et al., 2003; Fu et al., 2008), and data in the literature (Lee et al., 2007; Schwartz et al., 2008; Schwartz et al., 2009). Drug concentration in the tissue is at least 100 times lower than in the microdialysis probe as a result of the concentration gradient across the dialysis membrane and diffusion in the tissue (Ji and Neugebauer, 2007; Han et al., 2010; Ji et al., 2010). Numbers in the text refer to drug concentrations in the microdialysis fiber.
Histological verification of drug administration sites.
At the end of a behavioral experiment, the animal was killed by decapitation using a guillotine (Harvard Apparatus decapitator). This method of killing is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association and approved by the IACUC of the University of Texas Medical Branch. The brain was removed and submerged in 10% formalin. Tissues were stored in 20% sucrose before they were frozen sectioned at 50 μm. Sections were stained with Neutral Red, mounted on gel-coated slides, and coverslipped. Positions of the microdialysis fibers were identified under the microscope (Fu and Neugebauer, 2008) and plotted on standard diagrams (from Paxinos and Watson, 1998).
Drugs
The following compounds were used in this study: (S)-3,5-dihydroxyphenylglycine (DHPG, mGluR1/5 agonist); 2-chloro-5-hydroxyphenyl-glycine (CHPG, mGluR5 agonist); (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385, mGluR1 antagonist); 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP, mGluR5 antagonist); 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (tempol, ROS scavenger, superoxide dismutase mimetic); (9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester (KT5720, PKA inhibitor); 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene (U0126, MEK1/2 inhibitor); 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene (U0124, inactive structural analog of U0126); these were purchased from Tocris Bioscience Cookson. Phenyl-N-t-butyl nitrone (PBN, ROS scavenger); tert-butyl hydroperoxide (tBOOH, ROS donor); Nω-nitro-l-arginine methyl ester (l-NAME; neuronal nitric oxide synthase [nNOS] inhibitor); phorbol-12-myristate-13-acetate (PMA, phorbol ester, PKC activator); guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S, nonhydorlyzable GDP analog, G protein inhibitor); these were purchased from Sigma. IP3 and xestospongin C (XeC, blocker of IP3-mediated Ca2+ release) were purchased from A.G. Scientific.
Stock solutions of DHPG, MTEP, PBN, tempol, tBOOH, GDP-β-S, IP3, and l-NAME were prepared with water. LY367385 was dissolved in NaOH (3%). DMSO was used for stock solutions of PMA, XeC (30% DMSO in deionized water), GF109203X (50%), U0124, U0126, and KT5720 (70%). Drugs were dissolved in ACSF to their final concentrations on the day of the experiment. The dilution factor was 1:10,000 for NaOH and 1:1000 (PMA, XeC) or 1:10,000 (U0124, U0126, KT5720, GF109203X) for DMSO. ACSF served as vehicle control in all experiments. PBN, tempol, l-NAME, GDP-β-S, IP3, XeC, U0126, and KT5720 were also applied intracellularly through the patch pipette. The pH of the internal solution was adjusted to 7.2–7.3 and osmolarity to 280 mOsm/kg. Selectivity and target concentrations have been established in the literature for mGluRs (Varney and Gereau, 2002; Lesage, 2004; Lea and Faden, 2006; Neugebauer, 2007; Pernia-Andrade et al., 2009), ROS (Klann, 1998; Hu et al., 2006; Schwartz et al., 2009; Lee et al., 2010), nitric oxide synthase (NOS) (Mallozzi et al., 2007), XeC (Steinmann et al., 2008), and kinase inhibitors (Cabell and Audesirk, 1993; Favata et al., 1998; Fu et al., 2008; Kolber et al., 2010).
Statistical analysis
All averaged values are given as the mean ± SE. Statistical significance was accepted at the level of p < 0.05. GraphPad Prism 3.0 software was used for all statistical analyses. For multiple comparisons, one-way ANOVA or two-way ANOVA was used with appropriate posttests as indicated in the text and figure legends (Dunnett's multiple-comparison test was used to compare all sets of data to a control value; Newman–Keuls multiple-comparison posttest was used to compare all pairs of data using GraphPad Prism 3.0 software).
Results
Group I mGluRs act presynaptically to facilitate synaptic transmission in CeLC neurons (Neugebauer et al., 2003), but they may have additional postsynaptic effects because higher (micromolar) concentrations of group I mGluR agonists (DHPG and CHPG) affected membrane properties (slope conductance) of these neurons (Neugebauer et al., 2003). The present study explored direct actions of group I mGluRs on CeLC neurons to test the hypothesis that mitochondrial ROS are downstream signaling molecules in these cells. Therefore, patch-clamp analysis focused on action potential firing as a measure of neuronal excitability and output function rather than on synaptic transmission. Still, all CeLC neurons included in this study responded with monosynaptic EPSCs to electrical stimulation of the parabrachial input (PB-CeLC synapse) as in our previous studies (Neugebauer et al., 2003; Bird et al., 2005; Han et al., 2005b; Fu and Neugebauer, 2008). In some experiments, synaptically evoked action potentials in response to electrical stimulation of the PB input were also measured.
Group I mGluR5 increases excitability of CeLC neurons
In a majority of CeLC neurons (31 of 45), DHPG (10 μm, applied by superfusion of the brain slices) increased the input–output function of neuronal excitability significantly (F(1,420) = 37.52, p < 0.001, main effect of drug, two-way ANOVA) (Fig. 1a,b). Whole-cell patch recordings were made in coronal brain slices from the right hemisphere as in our previous studies, because accumulating evidence suggests that pain-related amygdala functions are lateralized to the right hemisphere (Carrasquillo and Gereau, 2007, 2008; Ji and Neugebauer, 2009). Action potentials were generated by direct intracellular current injections (500 ms) of increasing magnitude (up to 300 pA in 50 pA steps). DHPG also increased the input resistance of these neurons significantly (predrug, 141 ± 12 MΩ; DHPG, 179 ± 17 MΩ; n = 12; p < 0.05, paired t test). In a separate set of experiments, DHPG induced an inward current (25.86 ± 6.43 pA) in eight of eight CeLC neurons in the presence of TTX (1 μm), indicating a direct membrane effect. Superfusion of DHPG onto the brain slices had no significant effect in the presence of GDP-β-S (1 mm), a nonhydorlyzable GDP analog that was included in the patch pipette to inhibit G protein-mediated intracellular effects (F(1,42) = 0.84, n = 4; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 1c,d).
Because DHPG can activate mGluR1 and mGluR5, we examined which receptor subtype mediates the facilitatory effects. An mGluR5 antagonist (MTEP, 1 μm, applied by superfusion) decreased the facilitatory effect of DHPG (10 μm) on neuronal excitability significantly (F(1,70) = 20.14, n = 6 neurons; p < 0.0001, main effect of drug, two-way ANOVA) (Fig. 1e,f). In contrast, an mGluR1 antagonist LY367385 (10 μm; applied by superfusion) had no significant effect on DHPG-induced increases of excitability (F(1,56) = 1.95, n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 1g,h). Superfusion of MTEP and LY367385 onto the brain slices had no significant effect on action potential firing in the absence of DHPG (MTEP, F(1,42) = 0.59, n = 4 neurons, p > 0.05, main effect of drug, two-way ANOVA, see Fig. 9a; LY367385, F(1,70) = 0.01, n = 6 neurons, p > 0.05, main effect of drug, two-way ANOVA, Fig. 9b).
Figure 9.
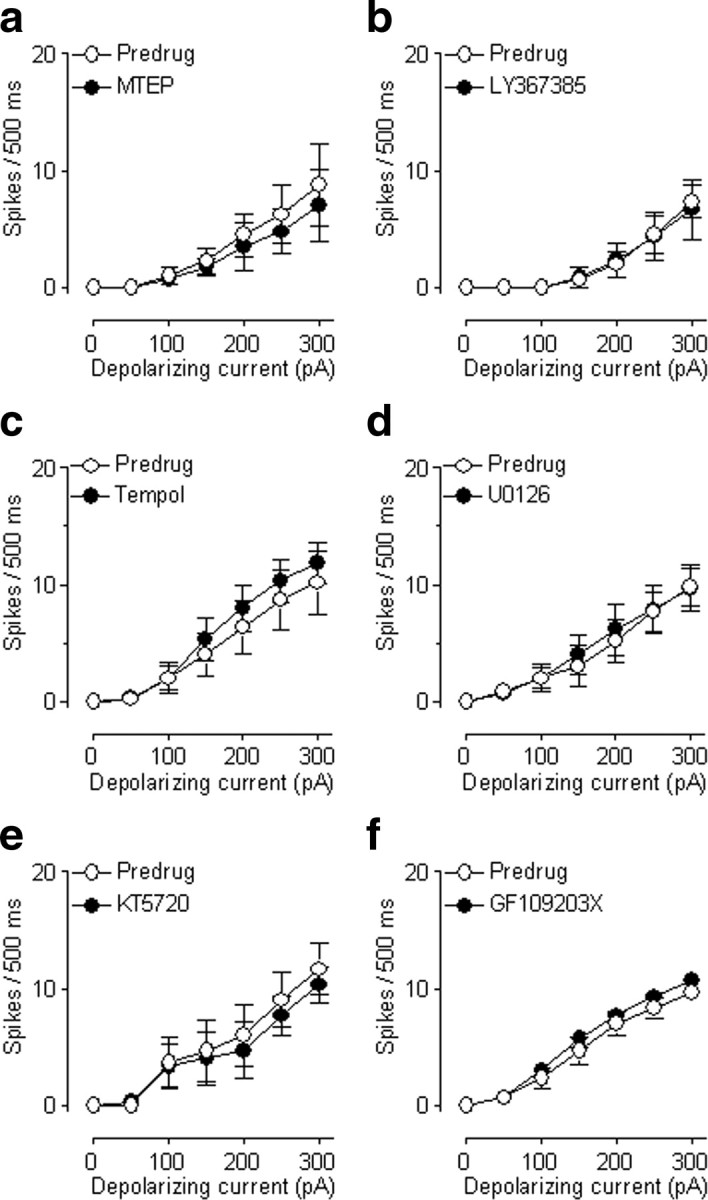
Antagonists and inhibitors by themselves have no effect on neuronal excitability. a, MTEP administered alone (1 μm, 15 min) had no significant effect on input–output functions of neuronal excitability (n = 4 neurons; p > 0.05, main effect of drug, two-way ANOVA). Action potentials were generated in CeLC neurons by intracellular current injections of increasing magnitude (see Fig. 1). Symbols show mean (± SE) number of spikes per 500 ms. b, LY367385 (10 μm) had no significant effect on action potential firing in the absence of DHPG (n = 6 neurons; p > 0.05, main effect of drug, two-way ANOVA). c, Application of tempol without DHPG had no significant effect (n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA). d, U0126 (500 nm) alone had no significant effect on action potential firing (n = 6 neurons, p > 0.05, main effect of drug, two-way ANOVA). e, KT5720 (200 nm) did not affect action potential firing in the absence of DHPG (n = 3 neurons, p > 0.05, main effect of drug, two-way ANOVA). GF109203X (1 μm) alone had no significant effect (n = 3 neurons, p > 0.05, main effect of drug, two-way ANOVA). a–f, All drugs were applied by superfusion of the brain slices. See Results for details of the statistical analysis.
mGluR5 effects on neuronal excitability require intracellular ROS
DHPG (10 μm, applied by superfusion) had no significant effects on neuronal excitability when a ROS scavenger (PBN, 1 mm) was applied intracellularly through the patch pipette (F(1,42) = 0.00, n = 4 neurons; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 2a,b). PBN can inhibit the formation of different types of ROS, including superoxide, hydrogen peroxide, and hydroxyl radical (Kotake, 1999). Therefore, we also tested a superoxide dismutase mimetic (tempol) that selectively scavenges superoxide (Tal, 1996). In the presence of intracellularly applied tempol (5 mm), superfusion of DHPG onto the brain slices had no significant effect on action potential firing (F(1,56) = 0.07, n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 2c,d). The data suggest that superoxide is the type of ROS that mediates the effects of mGluR5 on neuronal excitability.
Figure 2.
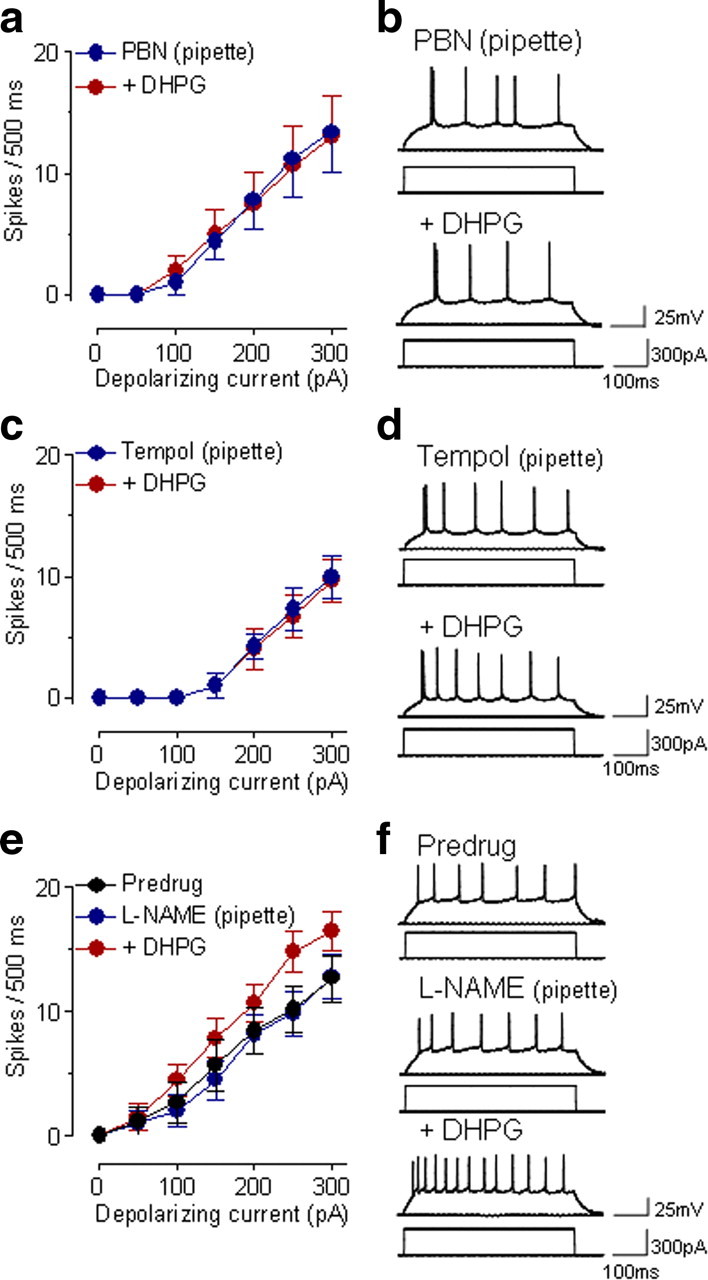
ROS scavengers, but not a NOS inhibitor, block group I mGluR effects. a, In the presence of an intracellularly applied ROS scavenger (PBN, 1 mm, included in the patch pipette) DHPG (10 μm) had no significant effect on action potential firing rate (n = 4 neurons; p > 0.05, main effect of drug, two-way ANOVA). Symbols show mean ± SE (see Fig. 1). b, Individual example of action potentials generated in a CeLC neuron by intracellular current injections (300 pA, 500 ms) before and during DHPG (10 μm) while PBN was injected into the cell through the patch pipette. c, Intracellular application of a superoxide dismutase mimetic (tempol, 5 mm in the patch pipette) also prevented any significant effects of DHPG (n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA). d, Individual example of a CeLC neuron recorded with tempol in the patch pipette. e, Intracellular application of l-NAME (400 μm) did not block the significant effect of DHPG on neuronal excitability (n = 6 neurons; p < 0.01, main effect of drug, two-way ANOVA). Action potential firing was measured immediately after whole-cell configuration was obtained, again 10 min after rupturing the membrane, and during application of DHPG (10 μm, for 10 min). f, Individual example of a CeLC neuron. a–f, DHPG was superfused onto the brain slices. See Results for details of the statistical analysis.
In these experiments it was impossible to determine whether DHPG alone had any effect, because the ROS scavengers (PBN or tempol) were included in the pipette. Therefore, in some experiments (n = 6 neurons) PBN (1 mm) was applied by superfusion after it was established that DHPG (10 μm) had a significant excitatory effect on the input–output function of neuronal excitability in these cells (F(1,70) = 22.71, p < 0.0001, main effect of drug, two-way ANOVA). PBN inhibited the effect of DHPG significantly (F(1,70) = 8.94, p < 0.01, main effect of drug, two-way ANOVA).
Mitochondrial superoxide is generated as a metabolic byproduct of the electron transport chain and oxidative phosphorylation, but some evidence suggests that NOS can also generate superoxide (Kishida and Klann, 2007). Therefore, we used a well established NOS inhibitor (l-NAME) to determine the possible involvement of NOS (Mallozzi et al., 2007). Intracellular application of l-NAME (400 μm) through the patch pipette did not significantly affect excitability when comparing action potential firing immediately after obtaining whole-cell configuration and 10 min after “break-in” (F(1,70) = 0.19, n = 6 neurons; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 2e,f). In the presence of l-NAME, DHPG (10 μm, applied by superfusion) still increased neuronal excitability significantly (F(1,70) = 11.37, n = 6 neurons; p < 0.01, main effect of drug, two-way ANOVA) (Fig. 2e,f), arguing against a major role of NOS in group I mGluR-induced effects on CeLC neurons.
Next, we used an mGluR5 agonist (CHPG) to confirm the link between mGluR5 and ROS. Superfusion of CHPG (10 μm) onto the brain slices significantly increased input–output functions of neuronal excitability (F(1,84) = 16.27, n = 7 neurons; p < 0.0001, main effect of drug, two-way ANOVA) (Fig. 3a,b). When PBN was included in the patch pipette, CHPG (10 μm, applied by superfusion) had no significant effect (F(1,56) = 0.17, n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 3c,d).
Figure 3.
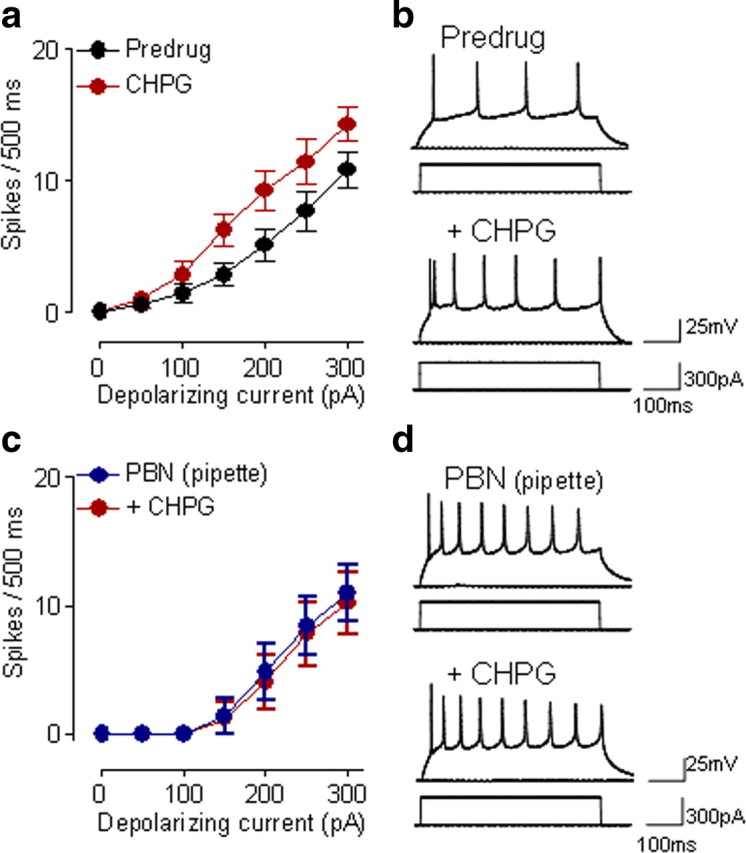
A ROS scavenger blocks mGluR5 agonist effects. a, CHPG (10 μm, 10–15 min) increased input–output function of neuronal excitability of CeLC neurons (n = 7) significantly (p < 0.0001, main effect of drug, two-way ANOVA), thus mimicking the effect of DHPG (see Fig. 1a,b). Symbols show mean ± SE (see Fig. 1). b, Individual example of a CeLC neuron tested with CHPG. c, Intracellular application of PBN (1 mm in the patch pipette) prevented significant effects of CHPG on neuronal excitability (n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA). d, Individual example of CeLC neuron recorded with PBN included in the patch pipette. a–d, CHPG was superfused onto the brain slices. See Results for details of the statistical analysis.
Group I mGluRs increase mitochondrial superoxide formation
The results so far showed that the facilitatory effects of mGluR5 on neuronal excitability require ROS, particularly superoxide, the predominant ROS in mitochondria. Therefore, we sought to determine whether mitochondrial superoxide formation is induced by group I mGluR activation by using quantitative fluorescence live-cell imaging in brain slices. Brain slices containing the CeLC were incubated with a fluorescent dye (MitoSOX, 1.25 μm; 30 min) that is specific for mitochondrial superoxide (Robinson et al., 2008). MitoSOX is rapidly and selectively targeted to the mitochondria where it is readily oxidized by superoxide, but not by other ROS- or NO-generating systems. Oxidized MitoSOX fluoresces red upon binding to mitochondrial nucleic acids (Liu et al., 2007; Robinson et al., 2008). DHPG (10 μm, 15 min) increased fluorescence intensity significantly in the CeLC (n = 6 slices) compared with ACSF-treated controls (n = 6 slices; F(1,262) = 2406.51; p < 0.0001, main effect of drug, two-way ANOVA comparing DHPG-treated and control slices for a time period of 28 min) (Fig. 4a,c). The effect of DHPG showed reversibility upon washout. The DHPG-induced increase of superoxide-related fluorescence was completely inhibited by a superoxide dismutase mimetic (tempol, 1 mm, 10 min; n = 3 slices; F(1,140) = 291.65; p < 0.0001, main effect of drug, two-way ANOVA comparing tempol-treated and DHPG-treated slices for a time period of 20 min) (Fig. 4a,d). The data suggest that group I mGluRs increase mitochondrial superoxide formation in CeLC neurons.
Figure 4.
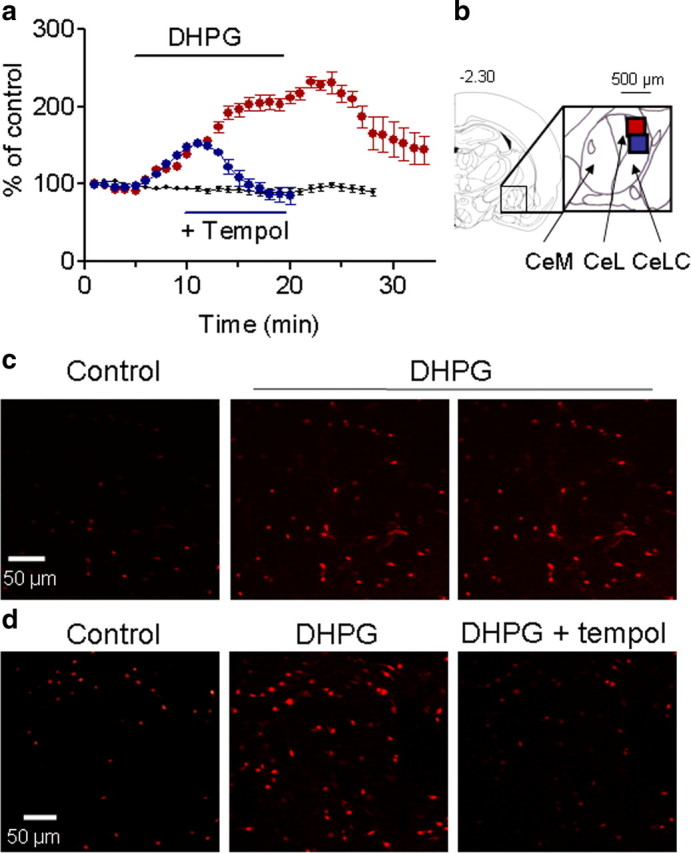
Group I mGluRs increase mitochondrial superoxide in CeLC neurons. Fluorescence generated by oxidation of a mitochondrial superoxide specific dye (MitoSOX) was measured in live brain slices containing the CeLC (see Materials and Methods for quantitative imaging analysis). a, Time course data. After incubation in MitoSOX (1.25 μm in ACSF; 30 min), slices were superfused with DHPG alone (10 μm, 15 min; n = 6 slices; red symbols) or with DHPG followed 5 min later by coapplication of a superoxide dismutase mimetic (tempol, 1 mm, 10 min; n = 3 slices; blue symbols). Control slices (n = 6) were superfused with ASCF only (black symbols). DHPG increased fluorescence intensity significantly compared with control slices (p < 0.0001, main effect of drug, two-way ANOVA). Tempol decreased the DHPG effect significantly compared with DHPG-treated slices (p < 0.0001, main effect of drug, two-way ANOVA). b, Coronal brain section 2.30 mm posterior to bregma; diagram adapted from Paxinos and Watson, 1998. Inset shows the central nucleus with its medial (CeM), lateral (CeL), and laterocapsular (CeLC) subdivisions and the area of the photographs in c (red square) and d (blue square). The image analysis system (see Materials and Methods), measured superoxide-related fluorescence levels in individual cells every minute. Values measured in each slice were averaged. Symbols and error bars show mean ± SE of intensity levels averaged for the slices in each of the three experimental groups normalized to predrug control values (set to 100%). c, d, Individual fluorescence images in slices treated with DHPG alone (c) or with DHPG followed by coapplication of tempol (d). Photographs were taken at 5 (control), 10, and 20 min (see a). See Results for details of the statistical analysis.
Synaptically evoked mitochondrial superoxide formation
Next, we addressed the question of whether mitochondrial ROS is also formed in response to synaptic activation of the parabrachial input that carries nociceptive information to the CeLC (Gauriau and Bernard, 2002; Neugebauer et al., 2004). Using quantitative fluorescence live-cell imaging of superoxide formation in the CeLC we found that high-frequency stimulation (three trains of five stimuli at 100Hz; n = 5 brain slices), but not low-frequency synaptic stimulation (1 Hz for 5 min; n = 5 brain slices), significantly increased superoxide-related fluorescence [p < 0.05, compared with prestimulation control, paired t test (Fig. 5c); individual examples are shown in Figure 5b]. The data suggest that synaptic activation of the PB input to the CeLC can increase ROS formation.
Figure 5.
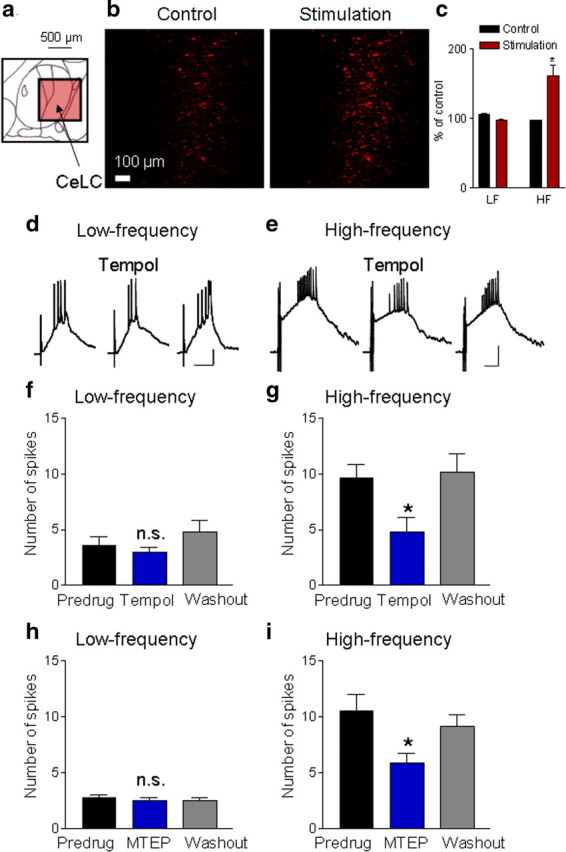
Synaptically evoked increase of mitochondrial superoxide and modulation of synaptic activity by mGluR5 and ROS. a, Diagram (adapted from Paxinos and Watson, 1998) shows the central nucleus with its subdivisions including the CeLC (see Fig. 4b) and the area of the photographs in b (red square). b, Individual images of fluorescence generated by oxidation of a mitochondrial superoxide specific dye (MitoSOX) in live brain slices containing the CeLC (see Fig. 4 and Materials and Methods for quantitative imaging analysis). Photographs were taken before (control) and 10 min after high-frequency stimulation (three trains, five stimuli at 100 Hz; see Materials and Methods). c, Bar histograms show averaged fluorescence activity for 10 min before (control) and 10 min after low-frequency (LF, 1 Hz, 5 min; n = 5 slices) or high-frequency (HF, n = 5 slices) electrical stimulation of PB input (see Materials and Methods). Values are normalized to the first minute of prestimulation control values; *p < 0.05 (compared with prestimulation control, paired test). b, c, High-frequency stimulation increased mitochondrial superoxide fluorescence activity. d, e, Current-clamp recordings of synaptically evoked depolarizations and action potentials before, during, and after superfusion of tempol (1 mm, 10 min) onto the brain slice. Tempol had little effect on synaptic responses evoked by low-frequency (0.033 Hz) stimulation (d) but inhibited responses to high-frequency stimulation (e, 5 stimuli at 100 Hz) of PB-input. Scale bars, 100 ms, 15 mV. f, g, Tempol (1 mm, applied by superfusion) had no significant effect on action potential firing evoked by low-frequency synaptic stimulation (f, n = 5 neurons), but inhibited synaptic activity evoked by high-frequency stimulation (g, n = 5 neurons). h, i, MTEP (1 μm, applied by superfusion) had no significant effect on spike firing evoked by low-frequency stimulation (h, n = 4 neurons), but inhibited synaptic activity evoked but high-frequency stimulation (i, n = 4 neurons). f–i, Bar histograms show means ± SE of the number of synaptically evoked spikes per stimulus before (predrug), during (10 min), and after (washout for 10 min) drug application; n.s., not significant; *p < 0.05 (compared with predrug; Dunnett's multiple-comparison tests).
Synaptically evoked activity requires mGluR5 and ROS
To determine the contribution of the mGluR5- IP3-ROS signaling cascade to synaptically evoked activity, we measured the effect of MTEP (mGluR5 antagonist) and tempol (superoxide dismutase mimetic) on action potentials evoked by synaptic stimulation of the parabrachial input. Superfusion of tempol (1 mm, 10 min) had no effect on low-frequency synaptic stimulation (n = 5 neurons) (Fig. 5d,f), but inhibited action potentials generated by high-frequency stimulation significantly (n = 5 neurons; p < 0.05, Dunnett's multiple-comparison test) (Fig. 5e,g). MTEP (1 μm, 10 min) also inhibited synaptic activity evoked by high-frequency stimulation significantly (n = 4 neurons; p < 0.05, Dunnett's multiple-comparison test) (Fig. 5h) without significantly affecting spike activity evoked by low-frequency stimulation (n = 4 neurons) (Fig. 5i). The data suggest activity-dependent involvement of mGluR5 and ROS in the synaptic activation of CeLC neurons.
IP3, but not PKC, links group I mGluRs to ROS signaling
Group I mGluRs couple to IP3 formation and PKC activation (Varney and Gereau, 2002; Lesage, 2004; Neugebauer, 2007). IP3-mediated calcium release has been linked to mitochondrial ROS production (Hajnoczky et al., 2006; Wu et al., 2007). To determine the involvement of IP3 in group I mGluR-induced increases of neuronal excitability, we used XeC, a selective inhibitor of IP3-induced calcium release (Oka et al., 2002; Steinmann et al., 2008). Intracellular application of XeC (1 μm in the patch pipette) prevented any significant facilitatory effect of DHPG (10 μm, applied by superfusion; F(1,70) = 0.01, n = 6 neurons; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 6a,b). To link IP3 and ROS signaling we determined the effect of a ROS scavenger on IP3 receptor activation with IP3 (Ding et al., 2010). Intracellular application of IP3 (1 mm in patch pipette) increased neuronal excitability significantly (F(1,56) = 41.78, n = 5 neurons; p < 0.0001, main effect of drug, two-way ANOVA) (Fig. 6c,d) when comparing action potential firing immediately after obtaining whole-cell configuration and 10 min after rupturing the membrane. Superfusion of tempol (5 mm) onto the brain slices inhibited the facilitatory effect of IP3 significantly (F(1,56) = 18.41, n = 5 neurons; p < 0.0001, main effect of drug, two-way ANOVA) (Fig. 6c,d). Tempol (5 mm, applied by superfusion) had no significant effect on action potential firing in the absence of DHPG (F(1,56) = 1.08, n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 9c).
Figure 6.
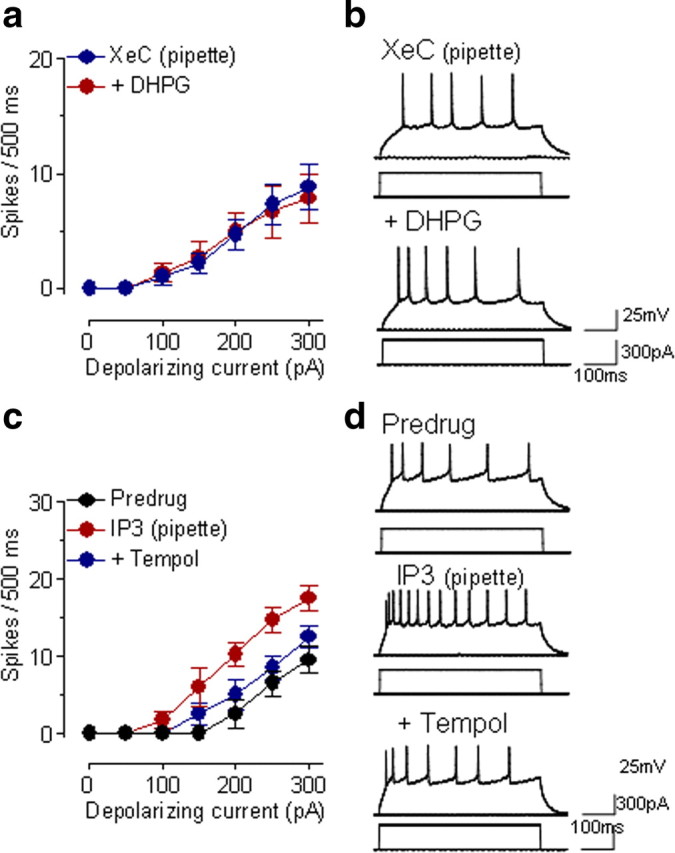
IP3 links group I mGluRs to ROS. a, In the presence of an intracellularly applied IP3 receptor blocker (XeC, 1 μm, included in the patch pipette) superfusion of DHPG (10 μm) onto the brain slices had no significant effect on neuronal excitability (n = 6 neurons; p > 0.05, main effect of drug, two-way ANOVA). Symbols show mean ± SE (see Fig. 1). b, Individual example of action potentials generated in a CeLC neuron by intracellular current injections (300 pA, 500 ms) before and during DHPG (10 μm, applied by superfusion) while XeC was injected into the cell. c, Intracellular application of IP3 (1 mm in the patch pipette) increased action potential firing significantly (n = 5 neurons; p < 0.0001, main effect of drug, two-way ANOVA). Superfusion of tempol (5 mm) onto the brain slices inhibited the facilitatory effect of IP3 significantly (n = 5 neurons; p < 0.0001, main effect of drug, two-way ANOVA). Action potential firing was measured immediately after whole-cell configuration was obtained, 10 min after rupturing the membrane, and during application of tempol for 10 min. d, Individual example of a CeLC neuron. See Results for details of the statistical analysis.
Next we determined whether ROS signaling is downstream of PKC, because group I mGluRs couple not only to IP3 but also to PKC activation. A commonly used phorbol ester (PMA, 1 μm, applied by superfusion) had mixed effects in CeLC neurons. In the presence of intracellularly applied tempol (5 mm), PMA increased action potential firing in eight neurons (F(1,98) = 13.65; p < 0.001, main effect of drug, two-way ANOVA) (Fig. 7a,b), but decreased firing rate in seven neurons (F(1,84) = 19.94; p < 0.0001, main effect of drug, two-way ANOVA) (Fig. 7c,d). PMA alone had excitatory effects in seven neurons (F(1,84) = 11.13; p < 0.01, main effect of drug, two-way ANOVA) (Fig. 7e), but decreased firing rate in five neurons (F(1,56) = 14.44; p < 0.001, main effect of drug, two-way ANOVA) (Fig. 7f). The results argue against the involvement of ROS, because the pattern of excitatory and inhibitory effects of PMA persisted in the presence of intracellularly applied tempol. The data are consistent with an mGluR5- IP3-ROS signaling cascade that does not involve PKC.
Figure 7.
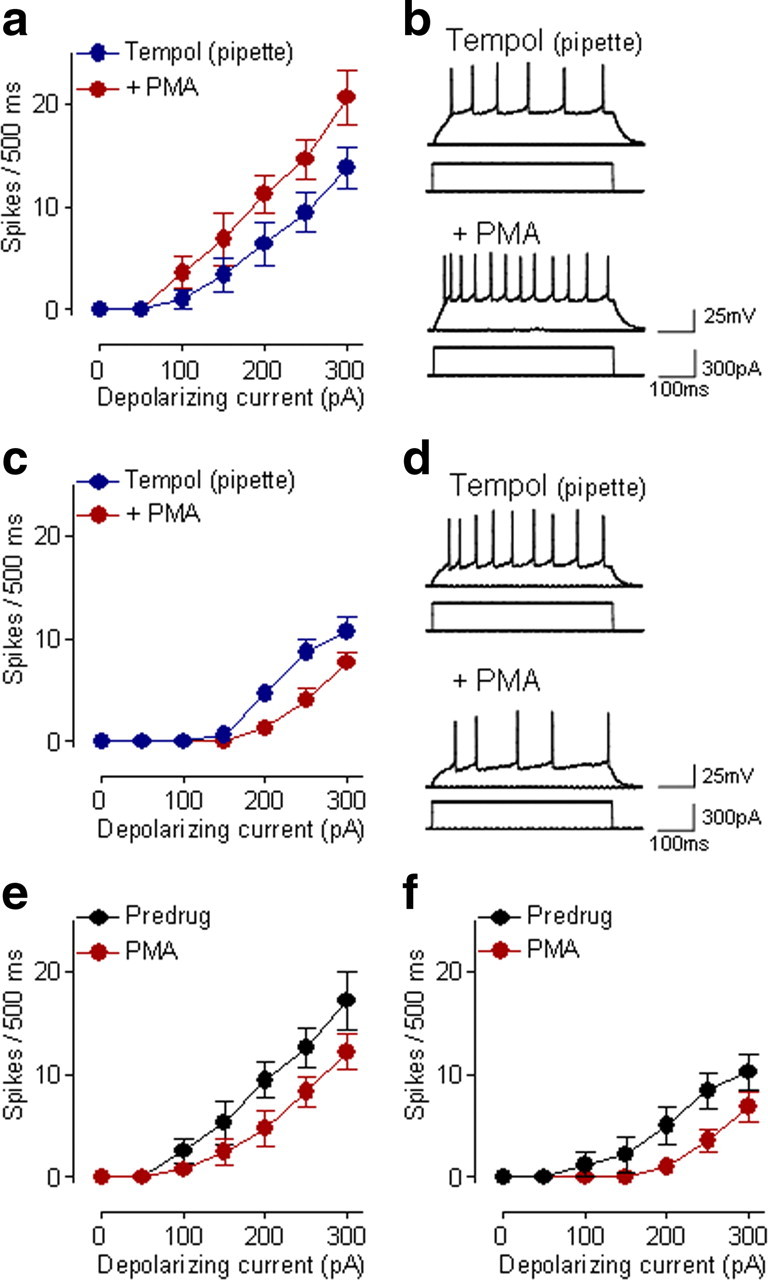
Effects of PKC activation do not involve ROS. a, b, A phorbol ester (PMA, 1 μm, superfused onto the brain slice) increased neuronal excitability of CeLC neurons significantly (n = 8 neurons; p < 0.001, main effect of drug, two-way ANOVA) while tempol (5 mm) was applied intracellularly through the patch pipette. a, Action potential firing was compared before and during PMA (10 min) in the presence of tempol. Symbols show mean ± SE (see Fig. 1). b, Individual example of action potentials generated in a CeLC neuron by intracellular current injections (300 pA, 500 ms) before and during PMA (10 μm, applied by superfusion) while tempol was injected into the cell. c, d, In another set of CeLC neurons, PMA (1 μm, superfused onto the brain slices) inhibited action potential firing of CeLC neurons significantly (n = 7 neurons; p < 0.0001, main effect of drug, two-way ANOVA) while tempol (5 mm) was applied intracellularly through the patch pipette. Analysis and display as in a and b. e, PMA alone (1 μm, superfused onto the brain slice) increased action potential firing in seven neurons (p < 0.01, main effect of drug, two-way ANOVA). f, In another sample of five neurons, superfusion of PMA alone inhibited action potential firing (p < 0.001, main effect of drug, two-way ANOVA). See Results for details of the statistical analysis.
ERK and PKA, but not PKC, are downstream targets of ROS
Next, we sought to determine the effector mechanism of ROS. ERK is required for superoxide-induced synaptic potentiation in the hippocampus (Huddleston et al., 2008). Group I mGluRs also can activate ERK (Karim et al., 2001; Giles et al., 2007; Kolber et al., 2010), but the mechanism is not clearly understood. Therefore, we tested the hypothesis that ERK acts downstream of ROS in the novel mGluR5- IP3-ROS-ERK signaling pathway to increase excitability of CeLC neurons. A ROS donor (tBOOH, 100 μm, applied by superfusion) increased action potential firing of CeLC neurons significantly (F(1,294) = 21.42, n = 22 neurons; p < 0.0001, main effect of drug, two-way ANOVA) (Fig. 8a). A similar significant effect was observed when tBOOH was included in the patch pipette for intracellular application (F(1,84) = 14.57, n = 7 neurons; p < 0.001, main effect of drug, two-way ANOVA).
Figure 8.
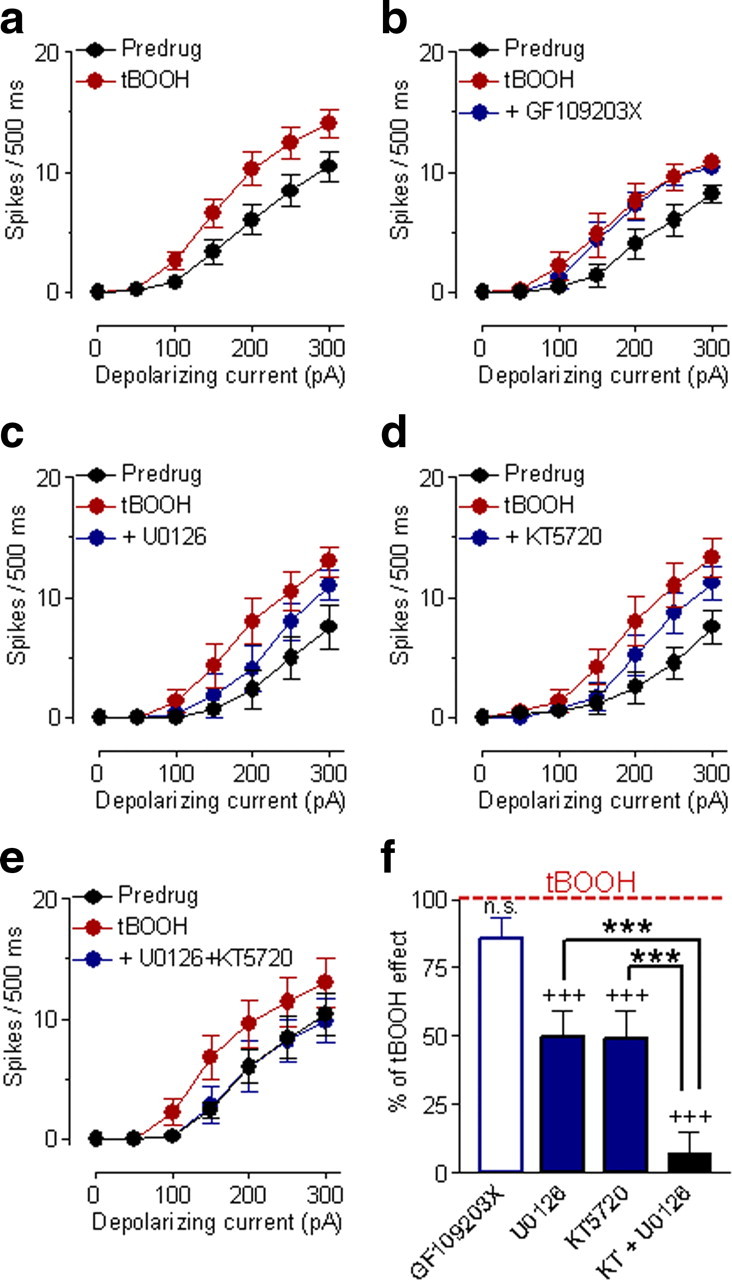
ERK and PKA, but not PKC, are downstream targets of ROS to increase excitability. a, Superfusion of a ROS donor (tBOOH, 100 μm) onto the brain slices increased action potential firing generated by intracellular current injections (300 pA, 500 ms) significantly (n = 22 CeLC neurons; p < 0.0001, two-way ANOVA). Symbols show mean ± SE (see Fig. 1). b, Coapplication of a PKC inhibitor (GF109203X, 1 μm) had no significant effect (n = 5 neurons; p > 0.05, two-way ANOVA). c, An ERK inhibitor (U0126, 500 nm) coapplied with tBOOH decreased, but did not completely block, the excitatory effect of tBOOH (n = 6 neurons; p < 0.05, two-way ANOVA). d, A PKA inhibitor (KT5720, 200 nm) also decreased, but did not completely block, the effect of tBOOH (n = 6 neurons; p < 0.05, main effect of drug, two-way ANOVA). e, Coapplication of U0126 and KT5720 (n = 5 neurons) completely blocked the effect of tBOOH (n = 5 neurons; p < 0.01, main effect of drug, two-way ANOVA). f, Comparison of the effects of different inhibitors. The effects of U0126 and KT5720 applied alone or together produced significant inhibition of the tBOOH effect. Coapplication of U0126 and KT5720 (KT) (n = 5 neurons) produced significantly greater inhibition of tBOOH effects than either inhibitor alone. Inhibition was calculated as follows: (inhibitor − predrug)/(tBOOH − predrug) × 100. Bar histograms show means ± SE; n.s., not significant, +++p < 0.001 (compared with tBOOH); ***p < 0.001 (compared with coapplication of U0126 and KT5720; Newman–Keuls multiple-comparison test). a–f, All drugs were applied by superfusion of the brain slices.
Coapplication of a PKC inhibitor (GF109203X, 1 μm, applied by superfusion) did not change the excitatory effect of tBOOH (100 μm, applied by superfusion; F(1,56) = 0.42, n = 5 neurons; p > 0.05, main effect of drug, two-way ANOVA) (Fig. 8b,f). Inhibition of ERK with U0126 (500 nm, applied by superfusion) decreased the excitatory effect of tBOOH significantly without completely blocking it (F(1,70) = 6.27, n = 6 neurons; p < 0.05, main effect of drug, two-way ANOVA) (Fig. 8c,f). An inactive structural analog of U0126 (U0124, 1 μm) had no significant effect (n = 5 neurons). Also, Inhibition of ERK did not completely block the behavioral effect of DHPG in the CeLC in a recent study (Kolber et al., 2010). PKA, but not PKC, has emerged as another important signaling molecule in pain-related amygdala function and acts via a mechanism that does not require ERK (Fu et al., 2008). Group I mGluRs can activate PKA in expression systems (Joly et al., 1995). A PKA inhibitor (KT5720, 200 nm, applied by superfusion) decreased, but did not completely block, the effect of tBOOH (100 μm, applied by superfusion; F(1,70) = 5.18, n = 6 neurons; p < 0.05, main effect of drug, two-way ANOVA) (Fig. 8d,f).
Therefore, we tested whether the combined application of KT5720 and U0126 produced greater inhibition than that by each drug alone. Coapplication of KT5720 and U0126 completely blocked the effect of tBOOH (F(1,56) = 8.45, n = 5 neurons; p < 0.001, main effect of drug, two-way ANOVA) (Fig. 8e,f). The inhibitory effect of the combined application of KT5720 and U0126 was significantly greater than that of each inhibitor alone (p < 0.01, Newman–Keuls multiple-comparison test (Fig. 8f). Drugs were applied by superfusion onto the brain slice. Appropriate concentrations of the inhibitors were selected based on data in the literature (see Materials and Methods). At the concentrations used in this study, inhibitors of MEK1/2, PKA, or PKC by themselves had no effect on neuronal excitability [U0126: 500 nm, F(1,70) = 0.11, n = 6 neurons, p > 0.05, main effect of drug, two-way ANOVA (Fig. 9d); KT5720: 200 nm, F(1,28) = 0.45, n = 3 neurons, p > 0.05, main effect of drug, two-way ANOVA (Fig. 9e); GF109203X, 1 μm, F(1,28) = 2.92, n = 3 neurons, p > 0.05, main effect of drug, two-way ANOVA (Fig. 9f)]. The finding is consistent with the results of our previous study (Fu et al., 2008) that showed a lack of effect of these compounds on basal synaptic transmission in the CeLC in brain slices from naive animals. In summary, the results of the kinase inhibitor experiments suggest that both ERK and PKA are required for the full effect of ROS on neuronal excitability.
Activation of mGluR5, but not mGluR1, increases nocifensive and affective behaviors through an IP3 and ROS-dependent mechanism
To determine the behavioral significance of mGluR5- IP3-ROS signaling, we analyzed hindlimb withdrawal reflexes and audible and ultrasonic vocalizations (see Materials and Methods). Withdrawal reflexes are spinally organized nocifensive responses that can be modulated by descending systems such as from the amygdala, whereas audible and ultrasonic vocalizations reflect supraspinally organized nocifensive and affective behaviors, respectively (Neugebauer et al., 2007, 2009). Our previous study suggested differential contributions of mGluR1 and mGluR5 to the processing of somatosensory (mGluR5 only) and visceral (mGluR1 and mGluR5) nociceptive information in CeLC neurons (Ji and Neugebauer, 2010). Therefore, we measured vocalizations to brief (15 s) noxious stimulation of somatic (compression of the knee joint) and visceral (colorectal distension or CRD) tissues (Fig. 10). Stereotaxic administration of DHPG (100 μm, concentration in microdialysis probe; 15 min) into the CeA increased vocalizations evoked by knee joint compression (Fig. 10a,c,e,g) and by CRD (Fig. 10b,d,f,h). The facilitatory effects of DHPG were blocked by coapplication of MTEP (100 μm, 15 min; n = 9 rats, knee; n = 6 rats, CRD) (Fig. 10a,b), but not by LY367385 (1 mm, 15 min; n = 10 rats, knee; n = 6 rats, CRD) (Fig. 10c,d). Significance of drug effects was determined with Newman–Keuls multiple-comparison tests (Fig. 10 shows results). A higher concentration of LY367385 (1 mm) was used to exclude the possibility that insufficient drug levels were achieved in the tissue (100 μm was tested in preliminary experiments and had no effect). Figure 10 shows the results for ultrasonic vocalizations only, because no difference of drug effects was found on audible and ultrasonic vocalizations. In the absence of DHPG, mGluR5 or mGluR1 antagonists had no significant effects (p > 0.05, paired t test) on vocalizations to knee joint stimulation (MTEP, 93.9 ± 4.8% of predrug, n = 5 rats; LY367385, 101.7 ± 7.0%, n = 5 rats) or to CRD (MTEP, 103.9 ± 8.1% of predrug, n = 5 rats; LY367385, 105.4 ± 7.2%, n = 5 rats).
Figure 10.
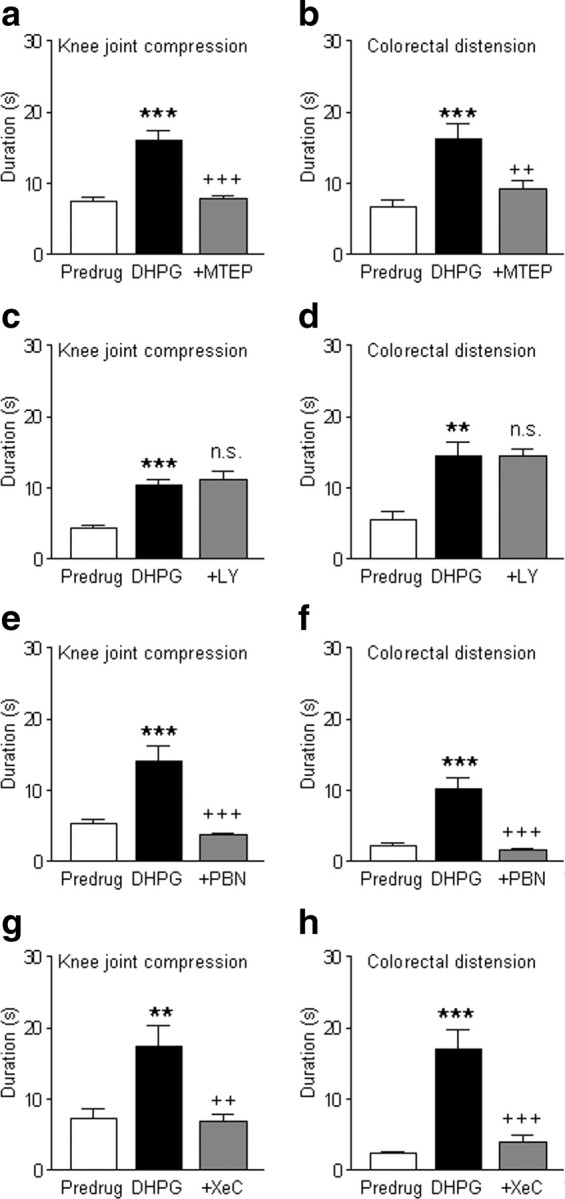
Activation of mGluR5- IP3-ROS signaling in the CeA increases vocalizations. Vocalizations in the ultrasonic range (25 ± 4 kHz; see Materials and Methods) were evoked by brief (15 s) noxious stimuli (a, c, e, g, compression of the knee joint, 2000 g/30 mm2; b, d, f, h, colorectal distension, 60 mmHg). Duration of vocalizations was measured for 2 min following the onset of the stimulus. At least two measurements were made and averaged for each animal before (predrug, in ACSF) and during each drug application. a, b, Administration of DHPG (100 μm, concentration in microdialysis probe; 15 min) into the CeA increased the duration of vocalizations. Coapplication of MTEP (100 μm; 15 min) blocked the effect (knee compression, n = 9 rats; CRD, n = 6 rats). c, d, Coapplication of LY367385 (1 mm) did not change the effect of DHPG (knee compression, n = 10 rats; CRD, n = 6 rats). e, f, Coapplication of PBN (100 mm) blocked the facilitatory effects of DHPG (knee compression, n = 6 rats; CRD, n = 6 rats). g, h, Coapplication of XeC (100 μm) also blocked the effect of DHPG (knee compression, n = 5 rats; CRD, n = 5 rats). Bar histograms in a–h show means ± SE, **p < 0.01, ***p < 0.001 (compared with predrug in ACSF); ++p < 0.01, +++p < 0.001 (compared with DHPG); n.s., not significant (compared with DHPG; Newman–Keuls multiple-comparison test).
The analysis of spinal reflexes showed similar results (Fig. 11). Stereotaxic administration of DHPG (100 μm, concentration in microdialysis probe; 15 min) into the CeA decreased the hindlimb withdrawal threshold, and this facilitatory effect was reversed by MTEP (100 μm, 15 min; n = 6 rats) (Fig. 11a), but not by LY367385 (1 mm, 15 min; n = 6 rats) (Fig. 11c). Significance of drug effects was determined with the Newman–Keuls multiple-comparison test (Fig. 11 shows results). Application of the antagonists (MTEP, n = 5 rats; LY367385, n = 5 rats) without DHPG had no significant effect as determined with paired t tests (Fig. 11b,d). The data suggest that the facilitatory effects of group I mGluRs on spinally and supraspinally organized behaviors are mediated by mGluR5 rather than mGluR1 under normal conditions in the absence of tissue injury.
Figure 11.
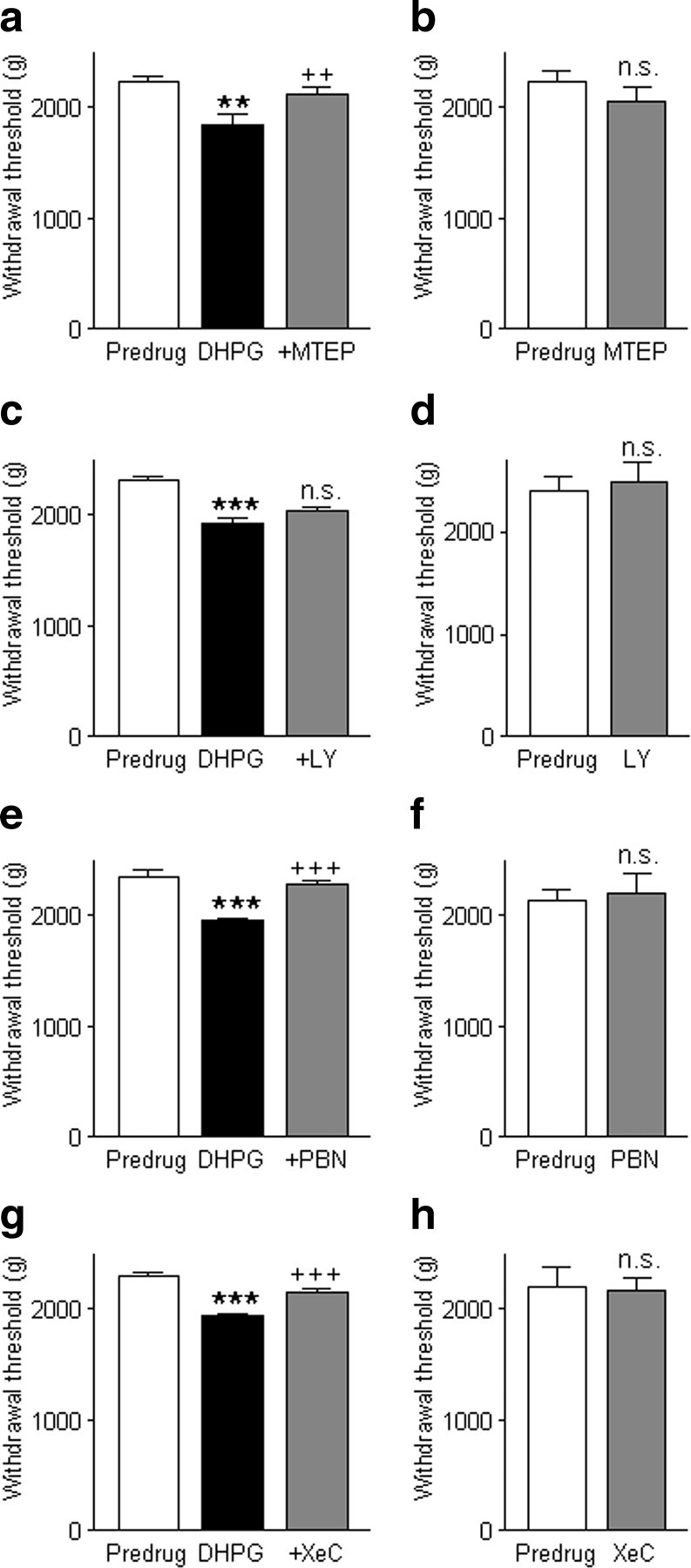
Activation of mGluR5- IP3-ROS signaling in the CeA increases spinal reflexes. Hindlimb withdrawal thresholds were measured by compressing the knee joint with increasing intensities using a calibrated forceps (see Materials and Methods). Three measurements were made and averaged in each animal before (predrug, in ACSF) and during each drug application. a, Stereotaxic administration of DHPG (100 μm, concentration in microdialysis probe; 15 min) into the CeA decreased the hindlimb withdrawal threshold. Coapplication of MTEP (100 μm, 15 min; n = 6 rats) reversed this facilitatory effect. b, MTEP (100 μm) alone had no significant effect (n = 5 rats). c, LY367385 (1 mm, 15 min; n = 6 rats) did not inhibit the effect of DHPG. d, LY367385 (1 mm) had no significant effect by itself (n = 5 rats). e, PBN (100 mm; n = 6 rats) reversed the facilitatory effect of DHPG. f, PBN (100 mm) alone had no significant effect (n = 5 rats). g, XeC (100 μm; n = 5 rats) also blocked the effect of DHPG. h, XeC (100 μm) had no significant effect in the absence of DHPG (n = 5 rats). Bar histograms in a–h show means ± SE, **p < 0.01, ***p < 0.001 (compared with predrug in ACSF); ++p < 0.01, +++p < 0.001 (compared with DHPG); n.s., not significant (compared with DHPG in a, c, e, and g, Newman–Keuls multiple-comparison test; compared with predrug in b, d, f, and h, paired t test).
The facilitatory effects of DHPG (100 μm) on vocalizations to noxious stimuli were blocked significantly (p < 0.001, Newman–Keuls multiple-comparison tests) by a ROS scavenger (PBN, 100 mm; knee joint compression, n = 6 rats; CRD, n = 6 rats) (Fig. 10e,f), confirming the involvement of ROS. PBN had no significant effect on its own (p > 0.05, paired t test; knee, 106.1 ± 5.5% of predrug, n = 5 rats; CRD, 93.2 ± 7.0%, n = 5 rats). Inhibition of IP3 receptor function with xestospongin C (100 μm) also blocked the facilitatory effects of DHPG on vocalizations significantly (p < 0.01–0.001, Newman–Keuls multiple-comparison tests; knee compression, n = 5 rats; CRD, n = 5 rats) (Fig. 10g,h). XeC alone had no significant effect on vocalizations (p > 0.05, paired t test; knee, 103.3 ± 4.6% of predrug, n = 5 rats; CRD, 94.3 ± 9.9%, n = 5 rats). No difference was found between drug effects on audible and ultrasonic vocalizations.
The facilitatory effects of DHPG on spinal reflexes (Fig. 11) were inhibited by PBN (100 mm; n = 6 rats) (Fig. 11e) and XeC (100 μm; n = 5 rats) (Fig. 11g). Significance of drug effects was determined with the Newman–Keuls multiple-comparison test (Fig. 11 shows results). Application of PBN (n = 5 rats) or XeC (n = 5 rats) without DHPG had no significant effect as determined with paired t tests (Fig. 11f,h). The lack of effects of antagonists and inhibitors on baseline spinal reflexes argues against the contribution of any motor deficits to the inhibition of DHPG-induced behaviors by blockers of the mGluR5- IP3-ROS signaling pathway. Concentrations were selected based on data in the literature (see Materials and Methods), data from slice experiments (this study), and preliminary concentration–response experiments that determined the maximum concentration that by itself had no effect on vocalizations.
As a placement control for drug spread, we performed off-site applications into the neighboring striatum (1.5 mm away from the CeLC) in some experiments (n = 5 rats). Because the inhibitors of the mGluR5- IP3-ROS-ERK and PKA pathway had no effect on their own, we used the mGluR1/5 agonist DHPG (100 μm). DHPG had no significant effect (p > 0.05, paired t test) on vocalizations evoked by noxious knee joint compression (107.75 ± 10.01% of predrug control, n = 4 rats) or by CRD (94.95 ± 17.23%, n = 4 rats) and on hindlimb withdrawal thresholds (101.94 ± 5.23%, n = 4 rats). The behavioral data are consistent with the electrophysiological results showing that IP3 and ROS mediate the facilitatory effects of mGluR5 in the CeA.
Discussion
ROS play a critical role in cytotoxicity and oxidative stress (Maher and Schubert, 2000; Chinopoulos and Adam-Vizi, 2006) but have also emerged as important signaling molecules in physiological plasticity (Klann, 1998; Hu et al., 2006; Kishida and Klann, 2007). More recent evidence suggests that ROS such as superoxide and hydrogen peroxide also contribute to peripheral (Twining et al., 2004; Keeble et al., 2009) and spinal (Kim et al., 2004; Wang et al., 2004; Gao et al., 2007; Lee et al., 2007; Schwartz et al., 2008, 2009; Kim et al., 2009; Lee et al., 2010) mechanisms of inflammatory and neuropathic pain. However, mechanisms of ROS activation and downstream effectors related to pain remain to be determined.
The key finding of the present study is that ROS, specifically mitochondrial superoxide, link mGluR5 to ERK and PKA through an IP3-dependent mechanism that does not require PKC and NO and does not involve mGluR1. The second novelty is that this signaling cascade can produce pain-like behaviors by increasing the excitability and output of neurons in the amygdala (CeLC), a brain area that is concerned with emotional-affective aspects of behavior and pain (Neugebauer et al., 2004, 2009; Maren, 2005; Phelps and Ledoux, 2005; Seymour and Dolan, 2008).
The rationale for focusing on group I mGluRs as activators of ROS and on ERK and PKA as effectors was as follows. Pain-related synaptic plasticity and central sensitization of CeLC neurons depend critically on group I mGluRs (Neugebauer et al., 2003; Li and Neugebauer, 2004). Antagonists for mGluR1 and mGluR5 largely reversed electrophysiological changes of CeLC neurons in the arthritis pain model (Neugebauer et al., 2003; Li and Neugebauer, 2004) and also normalized nocifensive and affective responses in arthritic and neuropathic pain models (Han and Neugebauer, 2005; Ansah et al., 2010). Function of mGluR1 showed a more pronounced change than that of mGluR5 in the arthritis pain model. Disruption of mGluR5 function in the CeLC pharmacologically or with a conditional knock-out approach reversed and prevented, respectively, formalin-induced mechanical hypersensitivity (Kolber et al., 2010). Conversely, activation of group I mGluRs in the CeLC under normal conditions increased neuronal activity and synaptic transmission through a mechanism that mainly involved mGluR5 (Neugebauer et al., 2003; Li and Neugebauer, 2004; Ji and Neugebauer, 2010). Activation of mGluR5 in the CeLC also produced nocifensive behaviors in animals without tissue injury (Kolber et al., 2010). Interestingly, DHPG in the CeA induced no or negligible avoidance behavior in normal animals (Ansah et al., 2009), but enhanced fear conditioning when injected into the basolateral amygdala (Rudy and Matus-Amat, 2009). Possible regional differences of group I mGluR function related to pain and fear remain to be determined. Off-site drug applications into the adjacent striatum as placement controls in our previous studies (for mGluR1 and mGluR5 antagonists, see Han and Neugebauer, 2005; for MEK1/2, PKA, and PKC inhibitors, see Fu et al., 2008) and in the present study show that the spread of drugs applied by microdialysis does not exceed 0.5–1 mm. Even more importantly, a drug that can inhibit activity of neurons in the basolateral amygdala when administered into that nucleus has no effect when administered by microdialysis into the CeA (Ji et al., 2010). These findings suggest that the pharmacological manipulations of mGluR5- IP3-ROS signaling targeted the CeA rather than the basolateral nucleus, although effects on adjacent structures cannot be excluded entirely in the in vivo approach.
Group I mGluRs typically couple to IP3 formation and PKC activation via Gq/11 protein (Varney and Gereau, 2002; Lesage, 2004; Neugebauer, 2007). They can also activate PKA, at least in expression systems (Joly et al., 1995), and have been linked to ERK activation in the spinal cord (Karim et al., 2001; Giles et al., 2007) and amygdala (Kolber et al., 2010). ERK (Carrasquillo and Gereau, 2007; Fu et al., 2008) and PKA (Bird et al., 2005; Han et al., 2005b; Fu and Neugebauer, 2008) appear to play more important roles than PKC (Fu et al., 2008) in pain-related amygdala plasticity and amygdala-mediated pain behaviors. It is not clear how group I mGluRs couple to the activation of ERK and PKA.
Here, we tested the novel hypothesis that ROS serves as an important link, because IP3-mediated calcium release increases ROS production (Hajnoczky et al., 2006; Wu et al., 2007) and ROS can activate protein kinases probably through redox modification (Kishida and Klann, 2007; Huddleston et al., 2008). Indeed, the results of a recent study from our group suggest that the facilitatory effects of group I mGluRs on nociceptive processing in CeLC neurons involve ROS (Ji and Neugebauer, 2010), although the in vivo approach did not allow us to determine whether mGluRs and ROS are linked in the same cell or through indirect mechanisms such as presynaptic to postsynaptic signaling. The present study used intracellular injections of signaling blockers and measured excitability changes to link mGluR5, but not mGluR1, to ROS activation in the same cell.
The differential effects of mGluR1 and mGluR5 antagonists on neuronal excitability and behavior argue against nonselective drug effects. LY367385 is a potent and selective mGluR1 antagonist that does not interact with other mGluR subtypes at concentrations up to 100 μm (Kingston et al., 2002). LY367385 had no significant effect at concentrations of 10 μm in slices and up to 1 mm in the microdialysis fiber, which further confirms the appropriateness of the factor 100 to estimate tissue concentration (see Materials and Methods). MTEP is a more selective mGluR5 antagonist than the commonly used compound MPEP and has fewer off-target effects. Concentrations used in our study (1 μm in slices, 100 μm in microdialysis probe) are well within the concentration range (<10 μm) that is highly selective for mGluR5 (Lea and Faden, 2006).
Importantly, mGluR5-dependent ROS activation required IP3 but not PKC activation. ROS scavengers did not block the effect of PKC activation with a phorbol ester. Unexpectedly, PKC activation produced mixed excitatory and inhibitory effects, which may be explained by known interactions between group I mGluRs and PKC. On the one hand, PKC is an important signaling molecule for group I mGluR functions (Varney and Gereau, 2002; Conn, 2003; Lesage, 2004). On the other hand, PKC can desensitize these receptors (Gereau and Heinemann, 1998). The mixed effects of PKC activation in CeLC neurons may explain why we have not yet found evidence for the involvement of PKC in pain-related neuroplasticity in this region and in amygdala-mediated pain modulation (Bird et al., 2005; Han et al., 2005b; Fu and Neugebauer, 2008; Fu et al., 2008). Nitric oxide (NO), another important signaling molecule in pain mechanisms (Freire et al., 2009), is also not involved in the mGluR5- IP3-ROS pathway. Superoxide and NO have been shown to act independently in the spinal cord to generate hyperalgesia in a neuropathic pain model (Kim et al., 2009).
A novelty of this study is the demonstration that ROS plays an important role in pain mechanisms in the brain. Evidence for the involvement of ROS in group I mGluR signaling comes from live-cell imaging of mitochondrial superoxide production in the CeLC, but is also based on the pharmacological effects of ROS scavengers. PBN is the prototype of “spin-trapping” nitrones that can inhibit the formation of different types of ROS such as superoxide, hydrogen peroxide, hydroxyl radical, and peroxynitrates (Kotake, 1999). The cellular levels of ROS are carefully controlled by detoxifying enzymes such as the superoxide dismutases (Hidalgo and Donoso, 2008). Tempol is a potent nontoxic superoxide dismutase mimetic that converts superoxide radical to hydrogen peroxide, which is further metabolized into molecular oxygen and water (Tal, 1996). Tempol has been used to determine the role of superoxide in pain-related neuroplasticity (Schwartz et al., 2008, 2009; Lee et al., 2010). The inhibitory effects of tempol and live-cell imaging of mitochondrial ROS production suggest that superoxide plays an important role in the mGluR5- IP3-ROS cascade. This is consistent with circumstantial evidence that superoxide is necessary for long-term potentiation whereas other forms of ROS, such as hydrogen peroxide, may have a negative impact (Knapp and Klann, 2002).
Possible effector mechanisms of ROS include oxidative modification or phosphorylation of protein kinases such as PKA and PKC (Hidalgo and Donoso, 2008) and calcium release receptors of the ryanodine type that can link ROS to ERK activation (Kemmerling et al., 2007). ERK has emerged as a key signaling molecule in pain-related amygdala functions (Carrasquillo and Gereau, 2007; Fu et al., 2008; Kolber et al., 2010). Importantly, ERK activation does not appear to account fully for all effects of mGluR5-induced signaling in the CeLC. ERK inhibition did not completely block all pronociceptive effects of mGluR5 activation in a recent study (Kolber et al., 2010) or the mGluR5-induced increase of neuronal excitability in the present study. However, simultaneous inhibition of ERK and PKA completely blocked the effect of DHPG (Fig. 8). The additive effect may argue against a simple serial arrangement of PKA and ERK signaling, because blocking either molecule would be expected to produce the full effect in this model. We also showed previously that PKA activation in the CeLC with forskolin is not inhibited by an ERK inhibitor (Fu et al., 2008), suggesting some degree of independence of PKA and ERK signaling in the CeLC.
The ionic mechanisms of mGluR5- IP3-ROS-ERK/PKA-induced excitability changes remain to be determined. Kv4.2 has been identified as a key target for ERK to increase excitability of hippocampal cells (Yuan et al., 2002) and spinal neurons (Hu et al., 2003). Although Kv4.2 has phosphorylation sites for ERK, PKA, and PKC (Birnbaum et al., 2004), it was suggested that PKA and PKC did not modulate channel function directly but acted as upstream activators of ERK (Yuan et al., 2002; Hu et al., 2003). As mentioned before, results from our previous study in the CeLC argue against ERK acting downstream of PKA (Fu et al., 2008). PKA has also been linked to the modulation of other ion channels such as Kv3 to regulate excitability of CeLC neurons (Fu and Neugebauer, 2008). mGluR5- IP3-ROS-ERK/PKA signaling is likely to target multiple ionic mechanisms. Additive effects of ERK and PKA inhibitors in this study are consistent with a parallel arrangement and separate targets. Their analysis was beyond the scope of this study.
In summary, novel findings of this study include the important role of ROS in the modulation of neuronal excitability of amygdala neurons and amygdala-mediated behavior, a novel mGluR5- IP3-ROS-ERK/PKA signaling mechanism that explains how group I mGluRs couple to ERK and PKA, and ROS as a critical activator of ERK and PKA to increase neuronal excitability and pain behavior. Increased nocifensive and affective responses as a consequence of altered brain function in the absence of tissue injury may have important implications for pain disorders with little or no clearly defined tissue pathology, such as irritable bowel syndrome, fibromyalgia, and others (Gebhart, 2004).
Footnotes
Research related to this study was supported by National Institute of Neurological Disorders and Stroke Grants NS-38261 and NS-11255. We thank Drs. Jin-Mo Chung and William D. Willis, Jr., University of Texas Medical Branch, Galveston, TX, for critical reading of and valuable comments on the manuscript.
References
- Ansah OB, Gonçalves L, Almeida A, Pertovaara A. Enhanced pronociception by amygdaloid group I metabotropic glutamate receptors in nerve-injured animals. Exp Neurol. 2009;216:66–74. doi: 10.1016/j.expneurol.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Ansah OB, Bourbia N, Gonçalves L, Almeida A, Pertovaara A. Influence of amygdaloid glutamatergic receptors on sensory and emotional pain-related behavior in the neuropathic rat. Behav Brain Res. 2010;209:174–178. doi: 10.1016/j.bbr.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol. 2005;564:907–921. doi: 10.1113/jphysiol.2005.084780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Cabell L, Audesirk G. Effects of selective inhibition of protein kinase C, cyclic AMP–dependentdependent protein kinase, and Ca(2+)-calmodulin-dependent protein kinase on neurite development in cultured rat hippocampal neurons. Int J Dev Neurosci. 1993;11:357–368. doi: 10.1016/0736-5748(93)90007-z. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Hemispheric lateralization of a molecular signal for pain modulation in the amygdala. Mol Pain. 2008;4:24. doi: 10.1186/1744-8069-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology: novel aspects of an enduring theme. FEBS J. 2006;273:433–450. doi: 10.1111/j.1742-4658.2005.05103.x. [DOI] [PubMed] [Google Scholar]
- Chung JM. The role of reactive oxygen species (ROS) in persistent pain. Mol Interv. 2004;4:248–250. doi: 10.1124/mi.4.5.3. [DOI] [PubMed] [Google Scholar]
- Conn PJ. Physiological roles and therapeutic potential of metabotropic glutamate receptors. Ann NY Acad Sci. 2003;1003:12–21. doi: 10.1196/annals.1300.002. [DOI] [PubMed] [Google Scholar]
- Ding Z, Rossi AM, Riley AM, Rahman T, Potter BV, Taylor CW. Binding of inositol 1,4,5-trisphosphate (IP3) and adenophostin A to the N-terminal region of the IP3 receptor: thermodynamic analysis using fluorescence polarization with a novel IP3 receptor ligand. Mol Pharmacol. 2010;77:995–1004. doi: 10.1124/mol.109.062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Freire MA, Guimarães JS, Leal WG, Pereira A. Pain modulation by nitric oxide in the spinal cord. Front Neurosci. 2009;3:175–181. doi: 10.3389/neuro.01.024.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Neugebauer V. Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci. 2008;28:3861–3876. doi: 10.1523/JNEUROSCI.0227-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Han J, Ishola T, Scerbo M, Adwanikar H, Ramsey C, Neugebauer V. PKA and ERK, but not PKC, in the amygdala contribute to pain-related synaptic plasticity and behavior. Mol Pain. 2008;4:26–46. doi: 10.1186/1744-8069-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Kim HK, Chung JM, Chung K. Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain. Pain. 2007;131:262–271. doi: 10.1016/j.pain.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauriau C, Bernard JF. Pain pathways and parabrachial circuits in the rat. Exp Physiol. 2002;87:251–258. doi: 10.1113/eph8702357. [DOI] [PubMed] [Google Scholar]
- Gebhart GF. Descending modulation of pain. Neurosci Biobehav Rev. 2004;27:729–737. doi: 10.1016/j.neubiorev.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Gereau RW, 4th, Heinemann SF. Role of protein kinase C phosphorylation in rapid desensitization of metabotropic glutamate receptor 5. Neuron. 1998;20:143–151. doi: 10.1016/s0896-6273(00)80442-0. [DOI] [PubMed] [Google Scholar]
- Giles PA, Trezise DJ, King AE. Differential activation of protein kinases in the dorsal horn in vitro of normal and inflamed rats by group I metabotropic glutamate receptor subtypes. Neuropharmacology. 2007;53:58–70. doi: 10.1016/j.neuropharm.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Csordás G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006;40:553–560. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Neugebauer V. mGluR1 and mGluR5 antagonists in the amygdala inhibit different components of audible and ultrasonic vocalizations in a model of arthritic pain. Pain. 2005;113:211–222. doi: 10.1016/j.pain.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Han JS, Bird GC, Li W, Jones J, Neugebauer V. Computerized analysis of audible and ultrasonic vocalizations of rats as a standardized measure of pain-related behavior. J Neurosci Methods. 2005a;141:261–269. doi: 10.1016/j.jneumeth.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Han JS, Li W, Neugebauer V. Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci. 2005b;25:10717–10728. doi: 10.1523/JNEUROSCI.4112-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Adwanikar H, Li Z, Ji G, Neugebauer V. Facilitation of synaptic transmission and pain responses by CGRP in the amygdala of normal rats. Mol Pain. 2010;6:10–23. doi: 10.1186/1744-8069-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan EA, Magnuson DJ, Thunstedt GM, Gray TS. Corticotropin releasing factor neurons are innervated by calcitonin gene-related peptide terminals in the rat central amygdaloid nucleus. Brain Res Bull. 1994;33:529–534. doi: 10.1016/0361-9230(94)90079-5. [DOI] [PubMed] [Google Scholar]
- Hidalgo C, Donoso P. Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:1275–1312. doi: 10.1089/ars.2007.1886. [DOI] [PubMed] [Google Scholar]
- Hu D, Serrano F, Oury TD, Klann E. Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2006;26:3933–3941. doi: 10.1523/JNEUROSCI.5566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Glauner KS, Gereau RW., 4th ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. I. Modulation of A-type K+ currents. J Neurophysiol. 2003;90:1671–1679. doi: 10.1152/jn.00340.2003. [DOI] [PubMed] [Google Scholar]
- Huddleston AT, Tang W, Takeshima H, Hamilton SL, Klann E. Superoxide-induced potentiation in the hippocampus requires activation of ryanodine receptor type 3 and ERK. J Neurophysiol. 2008;99:1565–1571. doi: 10.1152/jn.00659.2007. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Differential effects of CRF1 and CRF2 receptor antagonists on pain-related sensitization of neurons in the central nucleus of the amygdala. J Neurophysiol. 2007;97:3893–3904. doi: 10.1152/jn.00135.2007. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Hemispheric lateralization of pain processing by amygdala neurons. J Neurophysiol. 2009;102:2253–2264. doi: 10.1152/jn.00166.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Reactive oxygen species are involved in group I mGluR-mediated facilitation of nociceptive processing in amygdala neurons. J Neurophysiol. 2010;104:218–229. doi: 10.1152/jn.00223.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Sun H, Fu Y, Li Z, Pais-Vieira M, Galhardo V, Neugebauer V. Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. J Neurosci. 2010;30:5451–5464. doi: 10.1523/JNEUROSCI.0225-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly C, Gomeza J, Brabet I, Curry K, Bockaert J, Pin JP. Molecular, functional, and pharmacological characterization of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J Neurosci. 1995;15:3970–3981. doi: 10.1523/JNEUROSCI.15-05-03970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeble JE, Bodkin JV, Liang L, Wodarski R, Davies M, Fernandes ES, Coelho Cde F, Russell F, Graepel R, Muscara MN, Malcangio M, Brain SD. Hydrogen peroxide is a novel mediator of inflammatory hyperalgesia, acting via transient receptor potential vanilloid 1-dependent and independent mechanisms. Pain. 2009;141:135–142. doi: 10.1016/j.pain.2008.10.025. [DOI] [PubMed] [Google Scholar]
- Kemmerling U, Muñoz P, Müller M, Sánchez G, Aylwin ML, Klann E, Carrasco MA, Hidalgo C. Calcium release by ryanodine receptors mediates hydrogen peroxide-induced activation of ERK and CREB phosphorylation in N2a cells and hippocampal neurons. Cell Calcium. 2007;41:491–502. doi: 10.1016/j.ceca.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Kim HK, Park SK, Zhou JL, Taglialatela G, Chung K, Coggeshall RE, Chung JM. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain. 2004;111:116–124. doi: 10.1016/j.pain.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Kim HY, Wang J, Lu Y, Chung JM, Chung K. Superoxide signaling in pain is independent of nitric oxide signaling. Neuroreport. 2009;20:1424–1428. doi: 10.1097/WNR.0b013e328330f68b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston AE, Griffey K, Johnson MP, Chamberlain MJ, Kelly G, Tomlinson R, Wright RA, Johnson BG, Schoepp DD, Harris JR, Clark BP, Baker RS, Tizzano JT. Inhibition of group I metabotropic glutamate receptor responses in vivo in rats by a new generation of carboxyphenylglycine-like amino acid antagonists. Neurosci Lett. 2002;330:127–130. doi: 10.1016/s0304-3940(02)00751-6. [DOI] [PubMed] [Google Scholar]
- Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2007;9:233–244. doi: 10.1089/ars.2007.9.ft-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? J Neurosci Res. 2002;70:1–7. doi: 10.1002/jnr.10371. [DOI] [PubMed] [Google Scholar]
- Kolber BJ, Montana MC, Carrasquillo Y, Xu J, Heinemann SF, Muglia LJ, Gereau RW., 4th Activation of metabotropic glutamate receptor 5 in the amygdala modulates pain-like behavior. J Neurosci. 2010;30:8203–8213. doi: 10.1523/JNEUROSCI.1216-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake Y. Pharmacologic properties of phenyl N-tert-butylnitrone. Antioxid Redox Signal. 1999;1:481–499. doi: 10.1089/ars.1999.1.4-481. [DOI] [PubMed] [Google Scholar]
- Lea PM, 4th, Faden AI. Metabotropic glutamate receptor subtype 5 antagonists MPEP and MTEP. CNS Drug Rev. 2006;12:149–166. doi: 10.1111/j.1527-3458.2006.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Kim HK, Kim JH, Chung K, Chung JM. The role of reactive oxygen species in capsaicin-induced mechanical hyperalgesia and in the activities of dorsal horn neurons. Pain. 2007;133:9–17. doi: 10.1016/j.pain.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Chung K, Chung JM. Involvement of reactive oxygen species in long-term potentiation in the spinal cord dorsal horn. J Neurophysiol. 2010;103:382–391. doi: 10.1152/jn.90906.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage ASJ. Role of group I metabotropic glutamate receptors mGlu1 and mGlu5 in nociceptive signalling. Curr Neuropharmacol. 2004;2:363–393. [Google Scholar]
- Li W, Neugebauer V. Differential roles of mGluR1 and mGluR5 in brief and prolonged nociceptive processing in central amygdala neurons. J Neurophysiol. 2004;91:13–24. doi: 10.1152/jn.00485.2003. [DOI] [PubMed] [Google Scholar]
- Liu T, Hannafon B, Gill L, Kelly W, Benbrook D. Flex-Hets differentially induce apoptosis in cancer over normal cells by directly targeting mitochondria. Mol Cancer Ther. 2007;6:1814–1822. doi: 10.1158/1535-7163.MCT-06-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P, Schubert D. Signaling by reactive oxygen species in the nervous system. Cell Mol Life Sci. 2000;57:1287–1305. doi: 10.1007/PL00000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallozzi C, Martire A, Domenici MR, Metere A, Popoli P, Di Stasi AM. l-NAME reverses quinolinic acid-induced toxicity in rat corticostriatal slices: Involvement of src family kinases. J Neurosci Res. 2007;85:2770–2777. doi: 10.1002/jnr.21178. [DOI] [PubMed] [Google Scholar]
- Maren S. Synaptic mechanisms of associative memory in the amygdala. Neuron. 2005;47:783–786. doi: 10.1016/j.neuron.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Minami M. Neuronal mechanisms for pain-induced aversion behavioral studies using a conditioned place aversion test. Int Rev Neurobiol. 2009;85:135–144. doi: 10.1016/S0074-7742(09)85010-1. [DOI] [PubMed] [Google Scholar]
- Myers B, Greenwood-Van Meerveld B. Divergent effects of amygdala glucocorticoid and mineralocorticoid receptors in the regulation of visceral and somatic pain. Am J Physiol Gastrointest Liver Physiol. 2010;298:G295–G303. doi: 10.1152/ajpgi.00298.2009. [DOI] [PubMed] [Google Scholar]
- Myers B, Dittmeyer K, Greenwood-Van Meerveld B. Involvement of amygdaloid corticosterone in altered visceral and somatic sensation. Behav Brain Res. 2007;181:163–167. doi: 10.1016/j.bbr.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Neugebauer V. Glutamate receptor ligands. Handb Exp Pharmacol. 2007;177:217–249. doi: 10.1007/978-3-540-33823-9_8. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Bhave G, Gereau RW., 4th Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci. 2003;23:52–63. doi: 10.1523/JNEUROSCI.23-01-00052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10:221–234. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Han JS, Adwanikar H, Fu Y, Ji G. Techniques for assessing knee joint pain in arthritis. Mol Pain. 2007;3:8–20. doi: 10.1186/1744-8069-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Galhardo V, Maione S, Mackey SC. Forebrain pain mechanisms. Brain Res Rev. 2009;60:226–242. doi: 10.1016/j.brainresrev.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Sato K, Hori M, Ozaki H, Karaki H. Xestospongin C, a novel blocker of IP3 receptor, attenuates the increase in cytosolic calcium level and degranulation that is induced by antigen in RBL-2H3 mast cells. Br J Pharmacol. 2002;135:1959–1966. doi: 10.1038/sj.bjp.0704662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pedersen LH, Scheel-Krüger J, Blackburn-Munro G. Amygdala GABA-A receptor involvement in mediating sensory-discriminative and affective-motivational pain responses in a rat model of peripheral nerve injury. Pain. 2007;127:17–26. doi: 10.1016/j.pain.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Pernía-Andrade AJ, Kato A, Witschi R, Nyilas R, Katona I, Freund TF, Watanabe M, Filitz J, Koppert W, Schüttler J, Ji G, Neugebauer V, Marsicano G, Lutz B, Vanegas H, Zeilhofer HU. Spinal endocannabinoids and CB1 receptors mediate C-fiber-induced heterosynaptic pain sensitization. Science. 2009;325:760–764. doi: 10.1126/science.1171870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc. 2008;3:941–947. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Matus-Amat P. DHPG activation of group 1 mGluRs in BLA enhances fear conditioning. Learn Mem. 2009;16:421–425. doi: 10.1101/lm.1444909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaber JS, Sternini C, Brecha NC, Rogers WT, Card JP. Neurons containing calcitonin gene-related peptide in the parabrachial nucleus project to the central nucleus of the amygdala. J Comp Neurol. 1988;270:416–426. doi: 10.1002/cne.902700310. [DOI] [PubMed] [Google Scholar]
- Schwartz ES, Lee I, Chung K, Chung JM. Oxidative stress in the spinal cord is an important contributor in capsaicin-induced mechanical secondary hyperalgesia in mice. Pain. 2008;138:514–524. doi: 10.1016/j.pain.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz ES, Kim HY, Wang J, Lee I, Klann E, Chung JM, Chung K. Persistent pain is dependent on spinal mitochondrial antioxidant levels. J Neurosci. 2009;29:159–168. doi: 10.1523/JNEUROSCI.3792-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour B, Dolan R. Emotion, decision making, and the amygdala. Neuron. 2008;58:662–671. doi: 10.1016/j.neuron.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Steinmann C, Landsverk ML, Barral JM, Boehning D. Requirement of inositol 1,4,5-trisphosphate receptors for tumor-mediated lymphocyte apoptosis. J Biol Chem. 2008;283:13506–13509. doi: 10.1074/jbc.C800029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal M. A novel antioxidant alleviates heat hyperalgesia in rats with an experimental painful peripheral neuropathy. Neuroreport. 1996;7:1382–1384. doi: 10.1097/00001756-199605310-00010. [DOI] [PubMed] [Google Scholar]
- Twining CM, Sloane EM, Milligan ED, Chacur M, Martin D, Poole S, Marsh H, Maier SF, Watkins LR. Peri-sciatic proinflammatory cytokines, reactive oxygen species, and complement induce mirror-image neuropathic pain in rats. Pain. 2004;110:299–309. doi: 10.1016/j.pain.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Varney MA, Gereau RW., 4th Metabotropic glutamate receptor involvement in models of acute and persistent pain: prospects for the development of novel analgesics. Curr Drug Targets CNS Neurol Disord. 2002;1:283–296. doi: 10.2174/1568007023339300. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004;309:869–878. doi: 10.1124/jpet.103.064154. [DOI] [PubMed] [Google Scholar]
- Wu J, Holstein JD, Upadhyay G, Lin DT, Conway S, Muller E, Lechleiter JD. Purinergic receptor-stimulated IP3-mediated Ca2+ release enhances neuroprotection by increasing astrocyte mitochondrial metabolism during aging. J Neurosci. 2007;27:6510–6520. doi: 10.1523/JNEUROSCI.1256-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]