

Abstract
Chronic morphine treatment resulting in the alteration of postsynaptic levels of AMPA receptors, thereby modulating synaptic strength, has been reported. However, the mechanism underlying such drug-induced synaptic modification has not been resolved. By monitoring the GluR1 trafficking in primary hippocampal neurons using the pHluorin-GluR1 imaging and biotinylation studies, we observed that prolonged morphine exposure significantly induced loss of synaptic and extrasynaptic GluR1 by internalization. The morphine-induced GluR1 endocytosis was independent of neural network activities or NMDA receptor activities, as neither blocking the sodium channels with tetrodotoxin nor NMDA receptors with dl-APV altered the effects of morphine. Instead, morphine-induced GluR1 endocytosis is attributed to a change in the phosphorylation state of the GluR1 at Ser845 as morphine significantly decreased the dephosphorylation of GluR1 at this site. Such changes in Ser845 phosphorylation required morphine-induced activation of calcineurin, based on the observations that a calcineurin inhibitor, FK506, completely abrogated the dephosphorylation, and morphine treatment led to an increase in calcineurin enzymatic activity, even in the presence of dl-APV. Importantly, pretreatment with FK506 and overexpression of the GluR1 mutants, S845D (phospho-mimic) or S845A (phospho-blocking) attenuated the morphine-induced GluR1 endocytosis. Therefore, the calcineurin-mediated GluR1-S845 dephosphorylation is critical for the morphine-induced changes in the postsynaptic AMPA receptor level. Together, these findings reveal a novel molecular mechanism for opioid-induced neuronal adaptation and/or synaptic impairment.
Introduction
Drug addiction is related to experience-based behaviors such as the pursuit of rewards and cues linking drugs. Hence, it is considered a pathological form of learning and memory (Kelley, 2004; Hyman, 2005), which involves reorganization and/or strengthening of synaptic connections in specific neural circuits. In the classical addiction model, the dopaminergic system, including the ventral tegmental area (VTA) and nucleus accumbens, is a well known reward circuitry important for addiction (Di Chiara and Imperato, 1988). However, the hippocampus, a vital brain region for contextual learning and memory consolidation, has recently been found to regulate the rewarding effects of abused drugs (Fan et al., 1999; Vorel et al., 2001). Furthermore, hippocampal glutamatergic neurons directly project to the nucleus accumbens (Floresco et al., 2001) and can modulate activity of dopaminergic neurons in the VTA (Legault et al., 2000). Therefore, the hippocampus is believed to play an important role in drug addiction, and the exploration of hippocampal neuroadaptions induced by addictive drugs will be critical in the eventual understanding of the mechanisms involved in drug addiction (Koob and Volkow, 2010; Morón and Green, 2010).
Previous reports demonstrate that chronic morphine exposure not only decreases dendritic branching and spine density, but also alters both AMPA receptor-mediated responses and synaptic plasticity in the hippocampus (Robinson and Kolb, 1999; Pu et al., 2002; Liao et al., 2005). AMPA receptors, which belong to ionotropic glutamate receptors, mediate the majority of excitatory synaptic transmissions in the CNS and they are tetramers consisting of different combinations of four subunits (GluR1 to GluR4) (Hollmann and Heinemann, 1994). In response to neuronal activity, AMPA receptors undergo dynamic trafficking between plasma membrane and cytosolic compartments. Alterations of these dynamic processes lead to a relocation of AMPA receptors that is crucially involved in the expression of synaptic plasticity, such as long-term potentiation or depression (Malinow and Malenka, 2002). Hence, abused drugs including morphine are hypothesized to affect the trafficking of AMPA receptors that might underlie these neuroadaptations. Our previous study demonstrated that chronic treatment with morphine diminished the frequency and amplitude of miniature excitatory synaptic currents (mEPSCs) in culture hippocampal neurons, indicating a reduction in synaptic AMPA receptors (Liao et al., 2005). Nonetheless, no direct evidence has yet shown that morphine treatment can regulate the trafficking of AMPA receptors through internalization. The signaling pathway(s) involved also remain unclear.
In this study, we sought to determine the cellular mechanism underlying morphine-induced AMPA receptor internalization. By using superecliptic pHluorin-tagged GluR1 and DsRed in live-cell imaging techniques, we recorded temporal translocation of GluR1 from surface membrane to the inside of the cell following morphine treatment. Based on our biochemical experiments, this loss of surface-expressed AMPA receptors is due to receptor internalization. Similar morphine treatment led to an increase in calcineurin activity and the dephosphorylation of GluR1 at Ser845 (S845), which was subsequently found to be required for the drug-induced GluR1 internalization. Therefore, the calcineurin-dependent dephosphorylation of AMPA receptors provides a novel mechanism for morphine-induced neuroadaptive alterations in synaptic strength.
Materials and Methods
Hippocampal cultures and transfection.
Dissociated neuronal cultures were prepared from hippocampi of Sprague Dawley rats (Harlan) at postnatal days 1 and 2 and maintained as previously described (Ghosh and Greenberg, 1995; Liao et al., 1999). For live-cell imaging experiments, neurons were plated at a density of 1 × 106 cells per dish onto a 35 mm glass-bottom Petri dish coated with poly-l-lysine (thickness of glass coverslip, 0.08 mm) (Lin et al., 2004). For all other biochemical studies, 3 × 106 cells were plated onto a poly-l-lysine-treated 60 mm Petri dish. From the day of plating, cultured neurons were counted as DIV1 (day 1 in vitro). Neurons at DIV5–DIV9 were transfected with various plasmids by standard calcium phosphate coprecipitation method (Ghosh and Greenberg, 1995) and used for experiments between DIV18–DIV24.
Construction of recombinant receptors.
Superecliptic pHluorin was a gift from Dr. Gero Miesenböck (University of Oxford, Oxford, UK). The pHluorin is a pH-sensitive mutant of green fluorescent protein (GFP), which emits strong fluorescence on cell surface but becomes nonfluorescent inside the cells (Miesenböck et al., 1998; Ashby et al., 2004b). Its coding sequence was amplified by PCR (QuikChange protocol; Stratagene) and inserted into the N terminus of rat GluR1 after a signal peptide in pRK5 vector. The tagging position with pHluorin in GluR1 was exactly the same as that with enhanced GFP, which has been shown to have no effect on the receptor function (Shi et al., 1999; Lin e al., 2004). A point mutant of GluR1 at Ser845 was also generated by PCR.
Live-cell confocal imaging and data analysis.
Images were acquired by a 63× oil-immersion objective of a Leica DMIRE2 fluorescence microscope, connected to a BD CARVII confocal imager and a Hamamatsu EM CCD camera (Joiner et al., 2009). To maintain neuronal viability during image capturing, transfected hippocampal cultures were placed in a temperature-controlled chamber installed on the microscope (heated at 37°C) with continuous 5% CO2 infusion (Leica Microsystems). Cells were then immediately returned to a humidified 5% CO2 incubator at 37°C for prolonged drug treatment. All transfected cells, except those with abnormal morphologies, were randomly photographed. The x,y coordinates of individual neurons on the stage fitted with x,y translation were recorded by the IPLab4 software program (BD Biosciences) so that the same neuron could be located and imaged at different time points during the drug treatment.
All confocal images were taken as stacks (≤ 25 z planes) at 0.4 μm intervals and then merged into one single image before further analysis with IPLab4 software, as described previously (Joiner et al., 2009). The averaged pHluorin-GluR1 fluorescence of the cells was based on the quantification of the pHluorin-GluR1 intensity of each individual neuron in the field of view. To obtain these data, total area of the cell from the entire image was selected by an autosegmentation function in the IPLab4 program.
In the case of measuring averaged pHluorin-GluR1 fluorescence on specific regions such as spines and dendrites, the region of interest was manually highlighted with Segment Tools. All fluorescence measurements represented raw data with background subtractions of the averaged blank field intensities. Dendritic spine was defined as a dendritic protrusion containing a rounded head (Murai et al., 2002; Liao et al., 2005). Spine density was calculated by the number of spines normalized against the dendritic length (100 μm) in DsRed images. All data are expressed as mean ± SEM of the indicated number of experiments. Statistical significance was determined using t test for a comparison between two groups and ANOVA for a comparison between multiple groups (SPSS 13.0); a value of p < 0.05 was considered statistically significant.
Biotinylation.
To label all surface proteins, after washing with ice-cold PBS/Ca2+/Mg2+ (10 mm phosphate buffer, 2.7 mm KCl, 137 mm NaCl, 1 mm CaCl2, 0.5 mm MgCl2, pH 7.4), 3-week-old hippocampal neurons in 60 mm dishes were incubated with sulfo-NHS-SS-biotin (300 μg/ml; Pierce) for 30 min at 4°C to biotinylate cell surface proteins, as described previously (Mammen et al., 1997; Lin et al., 2000). The unbound biotin was washed away by PBS/Ca2+/Mg2+ containing 0.1% BSA at 4°C. The biotinylated cells were then incubated with the original growth media with or without 10 μm morphine (in the presence or absence of inhibitors if necessary) and returned to the 5% CO2 incubator at 37°C for various time periods for receptor internalization. Receptor trafficking was stopped by rapidly cooling the cells at 4°C. Biotinylated proteins remaining on the cell surface were stripped by glutathione (150 mm glutathione, 150 mm NaCl, pH 8.75), but internalized receptors were protected and still contained biotin. Subsequently, 50 mm iodoacetamide in PBS/Ca2+/Mg2+ was used to neutralize glutathione. Cells were then immediately solubilized in extraction buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 30 mm NaF, 1 mm sodium orthovanadate and complete protease inhibitor mixture; Roche). After centrifugation at 13,000 × g for 5 min, supernatants with equal amounts of total proteins were agitated with streptavidin-agarose beads (Pierce) at 4°C overnight. Finally, the biotinylated receptors in the pull-down complexes, which should represent internalized receptors, were eluted by 2× Laemmli buffer and resolved by SDS-PAGE. Antibody against GluR1 (AB1504; Millipore Bioscience Research Reagents) was used to detect internalized receptors in the pull-down complexes and total receptor expressions in cell extracts.
Immunoprecipitation and immunoblotting.
Three-week-old hippocampal neurons in 60 mm dishes were treated with 10 μm morphine and/or 2 mm dbcAMP (Sigma) for indicated time periods. If necessary, various inhibitors were applied before agonist treatment, including FK506 (1 μm for 45 min; Calbiochem), tetrodotoxin (TTX; 1 μm for 1 h; Sigma), and APV (100 μm for 1 h; Sigma). Cells were then lysed in the extraction buffer as shown above, followed by a centrifugation at 13,000 × g for 5 min. Approximately 400–500 μg of total protein was immunoprecipitated with 2 μg of anti-GluR1 antibody (Millipore Bioscience Research Reagents) at 4°C overnight. After a 3 h incubation with protein A-Sepharose, samples were washed three times with the extraction buffer without sodium deoxycholate. Proteins were then separated by SDS-PAGE and immunoblotted with antibodies against phospho-GluR1-S845 (Millipore Bioscience Research Reagents), GluR1, and PSD-95 (sc-32290; Santa Cruz Biotechnology).
In vitro phosphatase assay.
To measure phosphatase activity of calcineurin, 32P-labeled RII peptide substrate (Sigma) corresponding to 19 residues in the regulatory subunit of type II cAMP-dependent protein kinase was prepared as described previously (Fruman et al., 1996). After drug treatments, hippocampal neurons were lysed in hypotonic lysis buffer (50 mm Tris, pH 7.5; 1 mm EDTA; 0.1 mm EGTA; 0.5 mm DTT; 50 μg/ml PMSF; 10 μg/ml leupeptin. and 10 μg/ml aprotinin). Cell extracts (6–8 μg) were used for the phosphatase assay. Okadaic acid (OA) was added to the assay buffer (50 mm Tris, pH 7.5, 100 mm NaCl, 0.5 mm DTT, 100 μg/ml BSA, 0.1 mm CaCl2) in all experiments. Since OA can inhibit the activities of Ca2+-independent phosphatases, including phosphatases 1 and 2A, by >90%, but has no significant effect on calcineurin, calcineurin activity can be measured clearly under the assay condition containing OA (Fruman et al., 1996). Reaction mixtures for the phosphatase assay contained 20 μl of cell lysates, 20 μl of assay buffer, 100 nm OA, and 20 μl of 15 μm 32P-labeled RII peptide substrate. The mixtures were incubated at 30°C for 10 min. Next, the reactions were terminated by adding 20 μl of 50% trichloroacetic acid and 80 μl of BSA (6 mg/ml). The samples were incubated on ice for 10 min, followed by centrifuging at 13,000 × g for a further 10 min at 4°C. The amount of 32Pi released from RII peptide in the supernatant was measured by liquid scintillation. Blank samples containing only assay buffer were used. To determine the specific activity of the substrate, counts per minute in 20 μl of 15 μm substrate was measured. Net counts per minute for samples was calculated by subtracting counts per minute measured in the blanks from the counts per minute in the samples. The amount of 32Pi released (pmol) in each sample was calculated by dividing the net counts per minute for the sample by the specific activity of the substrate. Final phosphatase activity was expressed as percentage of the control without drug treatment.
Results
Chronic exposure to morphine caused loss of both synaptic and extrasynaptic GluR1 subunits
Prolonged treatment of the hippocampal neurons with morphine not only induces collapse of preexisting spines, but also decreases frequency and amplitude of mEPSCs, suggesting a reduction in the postsynaptic AMPA receptors (Liao et al., 2005). However, no study has shown the temporal dynamics of AMPA receptor trafficking in response to morphine. To measure the dynamics of morphine-regulated AMPA receptor trafficking, cultured hippocampal neurons at DIV5–DIV9 were cotransfected with plasmids encoding superecliptic pHluorin-GluR1 and DsRed. The pHluorin-GluR1 was used to visualize surface-expressed AMPA receptors (Joiner et al., 2009; Ashby et al., 2004a,b), whereas the soluble red fluorescence protein DsRed was used to label dendritic spines. At DIV18–DIV24, a live-imaging system was used to take images of same-labeled neuron before and after 10 μm morphine exposure at various time points (3 h, 1 d, and 3 d) (Fig. 1). Upon morphine exposure, a gradual decrease in fluorescence signal of the pHluorin-GluR1 was observed (Fig. 1A). The average fluorescence of pHluorin-GluR1 from the entire image of morphine-treated neurons significantly decreased by 12.8% at 3 h, 21.6% at 1 d, and 33.9% at 3 d after the drug application (relative to before morphine treatment) (Fig. 1B, top), implicating a loss of surface-expressed GluR1 by morphine. In contrast, the fluorescence intensity of pHluorin-GluR1 in untreated neurons remained constant or even slightly increased during the 3 d of observation, indicating that the decrease in green fluorescence by morphine was not due to a photobleaching effect (Fig. 1A,B). Also, no significant change in overall protein expression of AMPA receptors was observed during drug treatment (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material in supplemental data). Interestingly, quantitative analysis on individual spines and the adjacent dendrites in Figure 1B, middle and bottom, showed that treatment with morphine for 3 h clearly diminished the surface fluorescence of pHluorin-GluR1 on spines but not at dendritic shafts. Therefore, the loss of synaptic GluR1 by morphine should occur before the loss of extrasynaptic GluR1. Consistent with our previous studies (Liao et al., 2005), the density of DsRed-labeled spines was substantially reduced after 1 and 3 d of morphine treatment (before, 63.4 ± 4.47 spines/100 μm; 1 d, 43.3 ± 4.02 spines/100 μm; 3 d, 37.2 ± 3.28 spines/100 μm) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). These findings provide direct live-imaging evidence that chronic treatment with morphine results in two distinct processes, the loss of surface-expressed AMPA receptors and the loss of spines.
Figure 1.
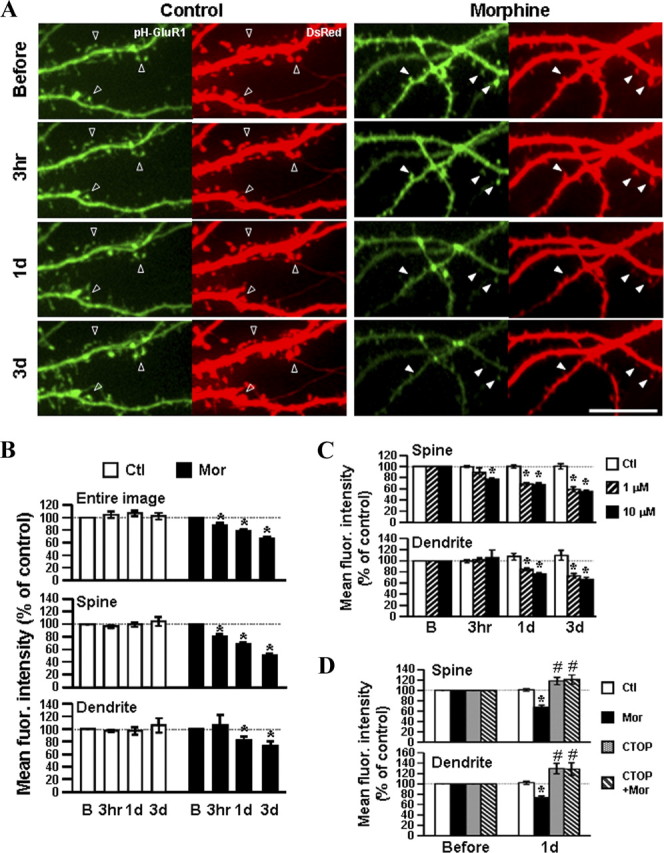
Chronic treatment with morphine causes loss of synaptic and extrasynaptic surface pHluorin-GluR1 subunits. A, Examples of confocal images of primary hippocampal neurons expressing pHluorin-GluR1 (pH-GluR1) and DsRed (Left, untreated control neurons; Right, morphine-treated neurons). The transfected cells were imaged before and after exposure to 10 μm morphine for various durations (3 h, 1 d, and 3 d). Open arrowheads denote that both fluorescence intensities remained constant or even increased under untreated control conditions. Solid arrowheads denote that morphine progressively decreased the fluorescence intensity of pHluorin-GluR1 and the density of DsRed-labeled dendritic spines. Scale bar, 10 μm. B, Mean pHluorin-GluR1 fluorescence from different regions (the entire images, spines, and dendrites) of untreated (Ctl; open bars; n = 16) or morphine-treated neurons (Mor; black bars; n = 16). Morphine significantly decreased the amount of surface-expressed pHluorin-GluR1 on both spines and dendrites in a time-dependent manner. C, Neurons expressing pHluorin-GluR1 and DsRed were treated with morphine at two different concentrations (1 and 10 μm; no drug as the control; n = 10 in each group) for various times (before, 3 h, 1 d, and 3 d). D, Neurons expressing the two proteins were exposed to no drug (Ctl), 10 μm morphine, 10 μm CTOP, or morphine with CTOP for 1 d (n = 12 in each group). Changes in averaged pHluorin-GluR1 fluorescence on spines (upper) and dendrites (lower) were normalized with data before treatments and are shown as percentages. *, Significant decrease compared with the value before treatment. #, Significant increase (p < 0.05). In each group, a minimum of five dendrites from a single neuron were analyzed. Error bars represent ±SEM. Confocal images of neurons in C and D are shown in supplemental Figure S2 (available at www.jneurosci.org as supplemental material). fluor., Fluorescence.
The effect of morphine on AMPA receptor trafficking was mediated through μ-opioid receptor
To investigate the specificity of morphine's effect on AMPA receptor trafficking, a reduced dosage of morphine (1 μm) was tested with the selective μ-opioid receptor (MOR) antagonist d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-ThrNH2 (CTOP) (Fig. 1C,D and supplemental Fig. S2, available at www.jneurosci.org as supplemental material). In contrast to the higher concentration of morphine (10 μm), 1 μm morphine did not significantly reduce the fluorescence signal of synaptic pHluorin-GluR1 at the 3 h time point (Fig. 1C, top). Nevertheless, this lower concentration of morphine did induce a comparable reduction in fluorescence of the receptor at both spines and dendrites at longer time points (1 and 3 d) (Fig. 1C and supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). Hence, a higher concentration of morphine was likely to induce a more rapid loss of the synaptic AMPA receptors. More importantly, this downregulated effect of morphine was attenuated by CTOP (10 μm), indicating the participation of MOR in morphine-modulated surface-expressed GluR1 (Fig. 1D and supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). It should be noted that both CTOP alone and CTOP with morphine enhanced the surface delivery of GluR1 at spines and dendrites due to increased fluorescence intensity in both regions (Fig. 1D and supplemental Fig. S2B, available at www.jneurosci.org as supplemental material), but this did not alter the receptor protein expressions (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). These results imply that the constitutive activity of MOR and/or endogenous opioids could regulate the distribution of AMPA receptors on cell surface.
Morphine treatment triggered internalization of GluR1
The loss of the AMPA receptors on the cell surface could be a result of either an increase in receptor internalization or a decrease in surface recruitment of the receptor. To directly examine whether morphine exposure mediates endocytosis of the AMPA receptors, surface proteins including GluR1 in the live hippocampal cultures were biotinylated before morphine treatment. After stripping, only internalized receptors would be protected and still contain biotin (see Materials and Methods). As shown in Figure 2, A and B, upon application of morphine (10 μm), the amount of biotinylated GluR1 subunits progressively increased by twofold to threefold over the control (at 12–24 h), indicating a time-dependent increase in internalized GluR1 subunits. However, there was no significant difference in the amount of the internalized NMDA receptor between control and morphine-treated neurons (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), suggesting that morphine specifically affected trafficking of GluR1 subunits. In addition, this morphine-induced GluR1 internalization was blocked in the presence of CTOP (10 μm) (Fig. 2C,D). Collectively, these data indicate that morphine-induced GluR1 internalization is mediated by MOR.
Figure 2.

Surface biotinylation assay demonstrates a significant increase in GluR1 internalization upon morphine treatment. A, Surface biotinylated hippocampal neurons were treated with no drug (Ctl) or 10 μm morphine for various durations (3, 6, 12, and 24 h). After cleavage of extracellular biotin, internalized GluR1 subunits were precipitated with immobilized streptavidin beads and detected with an anti-GluR1 antibody by Western blotting (top) (see Materials and Methods). Total GluR1 subunits from whole-cell lysates were also detected for a comparison (bottom). B, Densitometric quantifications of Western blots in A on internalized GluR1 (normalized to the untreated control level; n = 4). C, Neurons were treated with no drug, 10 μm morphine, 10 μm CTOP, or morphine with CTOP for 24 h. Top, Internalized GluR1 subunits were isolated by using streptavidin precipitation. Bottom, Total GluR1 subunits from whole-cell lysates were loaded for comparison. D, The amount of internalized GluR1 subunits was normalized to the untreated control (n = 3). *p < 0.05, compared with the untreated control. Error bars represent ± SEM.
Morphine-regulated GluR1 trafficking is not caused by alteration of neural circuitry
Opioid agonists have been shown to inhibit presynaptic release of transmitters such as GABA through activation of a voltage-dependent potassium channels. This results in a change in neural circuitry (Williams et al., 2001), which may indirectly contribute to the regulation of AMPA receptor trafficking. To test this possibility, a blockade of action potentials was achieved by using the sodium channel blocker TTX. As shown in Figure 3A, hippocampal neurons expressing pHluorin-GluR1 and DsRed were imaged before and 1 d after the addition of 10 μm morphine, 1 μm TTX, TTX with morphine, or no drug (control). TTX was applied to cultured neurons 1 h before addition of morphine and was present during the course of experiment. Although incubation with TTX alone did not change fluorescence intensity of pHluorin-GluR1 from the entire images, the fluorescence intensity was significantly elevated at dendritic spines and diminished at dendritic shafts (entire, 107 ± 6%; spine, 117 ± 5%; dendrite, 88 ± 5%) (Fig. 3A,B). This suggests that TTX can cause a translocation of GluR1 subunits from dendritic shafts to dendritic spines. Despite TTX-induced redistribution of GluR1 on surface membrane, application of both TTX and morphine resulted in a significant reduction on the fluorescence of pHluorin-GluR1 at spines and dendrites (entire, 76 ± 3%; spine, 70 ± 4%; dendrite, 60 ± 4%) (Fig. 3A,B). In addition, biotinylation studies demonstrated that morphine still enhanced internalization of GluR1 even in the presence of TTX (Fig. 3C). These findings indicate that morphine-evoked loss of synaptic and extrasynaptic GluR1 via the receptor internalization is not caused by indirect alteration in neural activity.
Figure 3.
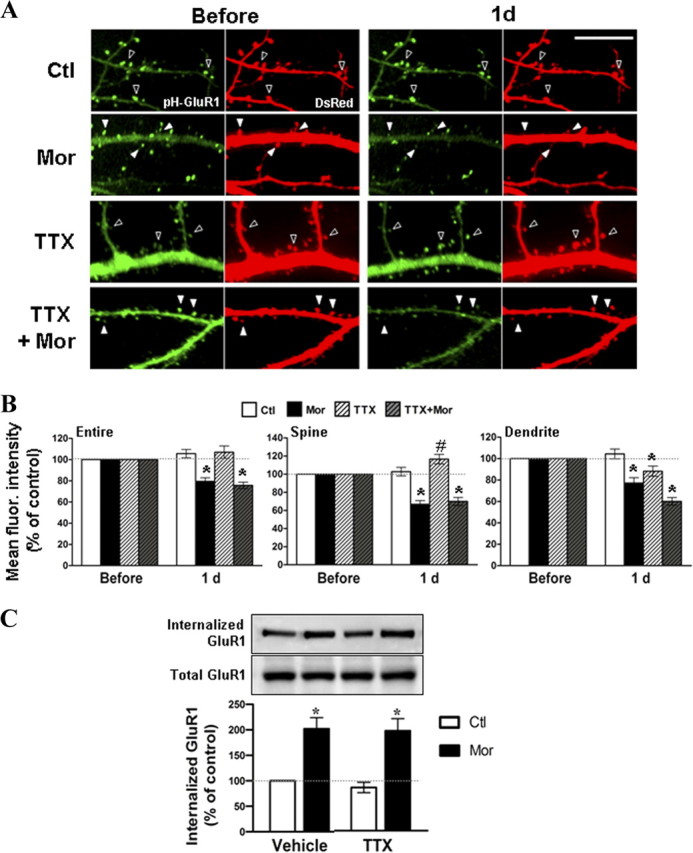
Morphine-regulated GluR1 trafficking is not due to alterations of neural networks. A, Time-lapse images were taken on hippocampal neurons coexpressing pHluorin-GluR1 (pH-GluR1) and DsRed before and 1 d after various treatments [untreated control (Ctl), 10 μm morphine (Mor), 1 μm TTX, TTX with morphine]. Open arrowheads indicate that both fluorescence intensities remained constant or even increased under untreated control conditions. Solid arrowheads indicate that morphine progressively decreased the fluorescence intensity of pHluorin-GluR1 and the density of DsRed-labeled dendritic spines. Scale bar, 10 μm. B, Mean fluorescence (fluor.) intensities of pHluorin-GluR1 from different regions of neurons (entire images, spines, and dendritic shafts) were normalized to data before treatments. *, Significant decrease comparing with the value before treatment; #, Significant increase (p < 0.05, n = 15 in each group). C, Surface biotinylated neurons were preincubated with no drug (vehicle) or 1 μm TTX for 1 h, and then were exposed to morphine (10 μm; 1 d). Top immunoblots, Internalized GluR1 subunits were isolated by using streptavidin precipitation and detected with an anti-GluR1 antibody. Bottom immunoblots, Total GluR1 subunits from whole-cell lysates were measured for comparison. Densitometric quantifications of Western blots on the internalized GluR1 are shown in the bottom (normalized to the control without any drug treatment; *p < 0.05; n = 3). Error bars represent ± SEM.
Morphine decreased phosphorylation of GluR1 at Ser845 and its interaction to PSD-95
AMPA receptor trafficking and synaptic plasticity can be mediated by receptor phosphorylation at cytoplasmic C termini (Malinow and Malenka, 2002). To investigate the molecular mechanism by which morphine regulates GluR1 internalization, we hypothesized that morphine can affect the phosphorylation status of GluR1. Because activation of Gi-coupled MOR can inhibit adenylyl cyclase, decrease intracellular cAMP level, and inactivate PKA (Duman et al., 1988; Ueda et al., 1988), we examined the effect of morphine on the phosphorylation status of GluR1 at S845, a substrate of cAMP-dependent kinase (PKA) (Esteban et al., 2003). Cultured hippocampal neurons were incubated with 10 μm morphine in the absence or presence of 10 μm CTOP. The GluR1 complex was immunoprecipitated with an anti-GluR1 antibody followed by the Western blotting detection with the phosphorylated GluR1-S845 and PSD-95 antibodies (Fig. 4). The amount of GluR1 phosphorylation at S845 was significantly decreased along the duration of the morphine treatment from 3 h to 1 d, whereas the total amount of GluR1 was unchanged (Fig. 4A,B), indicating that morphine treatment either inhibited the phosphorylation of GluR1-S845 or enhanced the dephosphorylation of the receptor subunit. Meanwhile, the amount of PSD-95 in the receptor complex also gradually decreased when compared with the untreated neurons (Fig. 4A,B), suggesting the dissociation of GluR1 subunits from the postsynaptic density (PSD) of dendritic spines and subsequent translocation. In contrast, cotreatment with CTOP reversed these effects (Fig. 4C–E), further confirming the important role of MOR in the effects of morphine. Altogether, the morphine-decreased phosphorylation of GluR1-S845 and morphine-induced dissociation from PSD-95 indicate a probable mechanism for the opioid agonist to regulate the AMPA receptor trafficking. Therefore, to detect obvious GluR1 dephosphorylation, hippocampal neurons were exposed to 10 μm morphine for 1 d in all subsequent experiments.
Figure 4.

Morphine decreases the phosphorylation of GluR1 at Ser845 and inhibits the interaction between GluR1 and PSD-95 through μ-opioid receptor. A, Neurons were treated with no drug (Ctl) or 10 μm morphine for various durations (3 h, 6 h, and 1 d). The cell lysates were immunoprecipitated with an anti-GluR1 antibody. The amount of phosphorylated GluR1 at Ser845 (Phospho-S845), PSD95 proteins, and total GluR1 subunits in the immunoprecipitation complex were detected with appropriate antibodies. Total GluR1 from whole-cell extracts (Input) was also determined. B, The Western blots in A were quantified with densitometry and normalized to the untreated control (n = 5). Note that the amount of PSD95 immunoprecipitated by the anti-GluR1 antibody significantly decreases at 6 h and 1 d after addition of morphine, indicating a dissociation between the postsynaptic density and GluR1 subunits upon drug treatment. C, Neurons were treated with no drug (Ctl) or 10 μm morphine for 6 h and 1 d in the absence (vehicle) or presence of 10 μm CTOP. The cell lysates were immunoprecipitated with an anti-GluR1 antibody. The amount of GluR1–S845 phosphorylation (Phospho-S845), PSD95, and total GluR1 in the immunoprecipitation complex was detected. Total GluR1 subunits from whole-cell lysates (Input) were also determined. D, E, Densitometric quantifications of Western blots in C on the amount of phospho-S845 GluR1 and PSD-95 in the receptor complex, respectively (normalized to the untreated control; n = 3). *p < 0.05 relative to the control. Error bars represent ± SEM.
Morphine regulated GluR-1 phosphorylation in a calcineurin-dependent manner
The phosphorylation status of GluR1-S845 depends on the balance between PKA and calcineurin/protein phosphatases 1 (Dell'Acqua et al., 2006). We therefore examined whether morphine-inhibited phosphorylation of GluR1-S845 was induced by suppressing the cAMP/PKA signaling pathway. Hippocampal neurons were preincubated with 10 μm morphine for 3 d, followed by stimulation with a PKA activator, dibutyryl-cAMP (dbcAMP), for 5 and 15 min (2 mm) (Fig. 5). In response to dbcAMP, the phosphorylation of GluR1-S845 was significantly increased at 5 min and further elevated at 15 min, indicating that dbcAMP-induced PKA activation stimulated GluR1-S845 phosphorylation in a time-dependent manner (Fig. 5). However, pretreatment with morphine caused complete and partial inhibitions of GluR1-S845 phosphorylation induced by dbcAMP at 5 min and 15 min, respectively (Fig. 5). The complete inhibition on the effect of dbcAMP at 5 min suggests that, in the presence of morphine, the dephosphorylation exceeds the phosphorylation of GluR1 by PKA. The partial restoration by longer incubation with dbcAMP implicates that the phosphorylation of GluR1-S845 by PKA may overcome the phosphatase activity that morphine may regulate.
Figure 5.

Morphine-induced PKA inhibition is not a dominant factor mediating GluR1-S845 dephosphorylation. A, Hippocampal neurons were pretreated with no drug (vehicle; left) or morphine at 10 μm (right) for 3 d, followed by stimulation with 2 mm dbcAMP (a cAMP analog to activate PKA) for 0, 5, and 15 min. Cell lysates were immunoblotted using antibodies against phospho-GluR1-S845 (top) and GluR1 (bottom). B, Densitometric quantifications of Western blots in A on the level of phosphorylated GluR1-S845 (normalized to the controls without dbcAMP stimulation, 0 min). *, Significant increase in GluR1-S845 phosphorylation by dbcAMP; #, Significant decrease in the effect of dbcAMP by morphine pretreatment (p < 0.05; n = 4). Error bars represent ± SEM. Note that pretreatment with morphine elicits a clear but not complete reduction in the dbcAMP-induced GluR1-S845 phosphorylation.
Due to the kinetics of morphine's effect on the dbcAMP-induced GluR1-S845 phosphorylation, we hypothesized that morphine treatment might promote dephosphorylation of GluR1-S845 through the activation of phosphatases. Thus, we examined the role of calcineurin with its specific inhibitor FK506 (Lieberman and Mody, 1994). A concentration of FK506 at 1 μm was shown to have a significant inhibition on calcineurin in neuronal cells (Asai et al., 1999). Figure 6, A and B, shows the level of GluR1-S845 phosphorylation in untreated and morphine-treated hippocampal cultures pretreated with FK506 (1 μm; 45 min). Compared with untreated control, exposure to morphine alone led to a significant decrease in GluR1-S845 phosphorylation. In contrast, the pretreatment with FK506 completely attenuated the effect of morphine (Fig. 6A,B), suggesting that morphine-mediated dephosphorylation of GluR1-S845 is dependent on calcineurin. To further support this conclusion, we performed an in vitro phosphatase assay to determine whether morphine exposure can increase the activation of calcineurin. Hippocampal neurons were treated with 10 μm morphine for various durations before cell lysis (Fig. 6C). To establish an assay condition in which the activity of calcineurin can be examined clearly, okadaic acid (100 nm), an inhibitor for phosphatase 1 and 2A, was included in all assay buffers (as described in Materials and Methods) (Fruman et al., 1996; Ranta et al., 2008). As shown in Figure 6C, phosphatase activity markedly increased 30 min after morphine application and persisted during 1 d of morphine treatment (30 min, 147 ± 11%; 1 h, 141 ± 9%; 3 h, 140 ± 5%; 6 h, 142 ± 3%; 1 d, 160 ± 14%) (Fig. 6C). However, this effect of morphine was eliminated, when FK506 (1 μm) was added to the assay buffers (30 min, 100 ± 7%; 1 h, 110 ± 5%; 3 h, 100 ± 8%; 6 h, 98 ± 12%; 1 d, 106 ± 6%) (Fig. 6C), indicating that the increase in phosphatase activity due to morphine is primarily activation of calcineurin. This morphine-induced calcineurin activation was totally suppressed by cotreatment with CTOP as well (Fig. 7A). Together, these results suggest that morphine acts via MOR to increase the activity of calcineurin, which in turn promotes dephosphorylation of GluR1-S845.
Figure 6.

Calcineurin plays a crucial role in mediating morphine-induced GluR1-S845 dephosphorylation. A, A complete elimination of morphine-induced GluR1-S845 dephosphorylation by a calcineurin inhibitor FK506. Hippocampal neurons were incubated in the absence (left) or presence (right) of 1 μm FK506 for 45 min, followed by exposure to morphine (10 μm; 1 d). Cell lysates were immunoprecipitated by an anti-GluR1 antibody and the pull-down complexes were immunoblotted using antibodies against phospho-GluR1-S845 (top) and GluR1 (bottom). B, Densitometric quantifications of Western blots in A on the level of phosphorylated GluR1-S845 (normalized to the control without any drug treatment). *, Significant decrease in GluR1-S845 phosphorylation upon morphine treatment (p < 0.05; n = 5). Error bars represent ± SEM. C, A time-dependent activation of calcineurin by morphine treatment. Hippocampal neurons were treated with 10 μm morphine for various durations (0, 0.5, 1, 3, 6, and 24 h) and harvested for preparation of cell lysates. Phosphatase activity in each sample was measured in the absence (solid line) and presence (dotted line) of 1 μm FK506. The phosphatase activity was expressed as percentage of the control by dividing each value for the phosphatase activity (pmol/mg) of morphine-treated cells by that of untreated cells. Assays were performed in duplicate, and the data shown are means ± SEM from four independent experiments. *, Significant increase in phosphatase activity compared with the control (p < 0.05).
Figure 7.

The effect of CTOP, dl-APV, and TTX on morphine-induced calcineurin activation and GluR1-S845 dephosphorylation. A, Hippocampal neurons were incubated with no drug (vehicle), CTOP (10 μm), dl-APV (100 μm), or TTX (1 μm) for 45 min, followed by exposure to 10 μm morphine (Mor) for 1 and 3 h. Calcineurin activity in cell lysates was measured. Assays were performed in duplicate, and the data shown are means ± SEM from three to four independent experiments. *, Significant increase in phosphatase activity compared with the control without any drug treatment (p < 0.05). B, C, Neurons were pretreated with no drug, 100 μm dl-APV (B), or 1 μm TTX (C) for 1 h and then were exposed to morphine (10 μm) for 1 d. Cell lysates were immunoprecipitated by an anti-GluR1 antibody and the pull-down complexes were immunoblotted using antibodies against phospho-GluR1-S845 (top) and GluR1 (middle). Total GluR1 from whole-cell extracts (Input) was also detected (bottom). Bar charts in B and C show densitometric quantifications of Western blots on the level of phosphorylated GluR1-S845 (normalized to the control without any drug treatment). *, Significant decrease in GluR1-S845 phosphorylation upon morphine treatment (p < 0.05; n = 4). Error bars represent ± SEM.
Morphine-induced calcineurin activation and GluR1-S845 dephosphorylation is independent of NMDA receptor activation
Calcineurin is a Ca2+-dependent protein phosphatase (Klee et al., 1979) and has been shown to be activated by Ca2+ influx through NMDA receptors (Li et al., 2002). Because morphine is reported to alter protein expression and functional property of NMDA receptors in vivo (Yang et al., 2000; Inoue et al., 2003; Ma et al., 2007), which may change the intracellular concentration of Ca2+, we evaluated the participation of NMDA receptor in morphine-regulated calcineurin activity and GluR1-S845 dephosphorylation. dl-APV (100 μm), a NMDA receptor antagonist, was applied 1 h before treatment with morphine. We found that morphine still increased calcineurin activity and decreased GluR1-S845 phosphorylation in neurons pretreated with dl-APV when compared with neurons without drug pretreatment (Fig. 7A,B). Therefore, the contribution of NMDA receptors to the morphine's effect is excluded. In agreement with the data in Figure 3, application of TTX did not affect the morphine-induced calcineurin activation (Fig. 7A) or GluR1-S845 dephosphorylation (Fig. 7C), further verifying that the calcineurin-dependent GluR1-S845 dephosphorylation is not an indirect consequence of altered neural network by morphine.
Calcineurin plays an important role in morphine-regulated GluR1 trafficking
To explore the participation of calcineurin in morphine-regulated GluR1 endocytosis, surface-biotinylated hippocampal cultures were incubated with FK506 (1 μm) for 45 min before morphine exposure (10 μm; 1 d) (Fig. 8A). Compared with the untreated control, the 2.5-fold increase of the GluR1 internalization by morphine was completely blocked in the presence of FK506 (Fig. 8B), suggesting an essential role of calcineurin in morphine-induced GluR1 endocytosis. To verify the involvement of calcineurin in morphine-induced loss of surface-expressed GluR1, hippocampal neurons overexpressing pHluorin-GluR1 and DsRed were imaged before and after treatment with no drug (control), morphine, FK506, or FK506 with morphine (Fig. 8C,D). Upon morphine treatment alone (10 μm for 1 d), the averaged fluorescence of pHluorin-GluR1 from the entire image, spines, and dendrites was significantly reduced (Fig. 8C,D), revealing the removal of synaptic and extrasynaptic GluR1. In contrast, pretreatment with FK506 (1 μm, 45 min) attenuated the morphine-induced decrease in pHluorin-GluR1 on spines and dendrites (Fig. 8C,D). These data demonstrated that morphine regulates AMPA receptor trafficking primarily through a calcineurin-dependent pathway.
Figure 8.
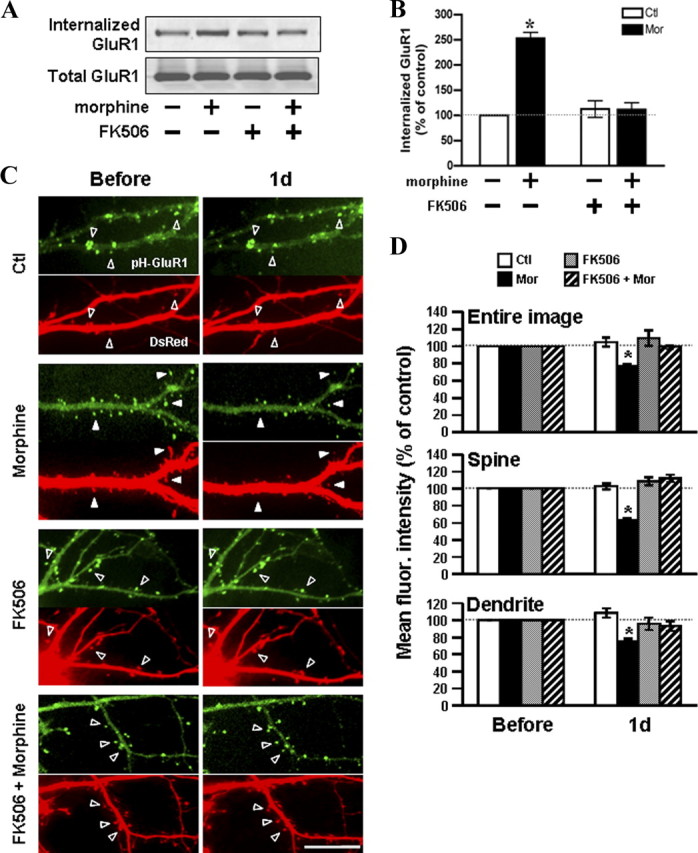
Inhibition of calcineurin blocks the morphine-regulated trafficking of GluR1. A, Surface biotinylated hippocampal neurons were incubated in the absence (left) or presence (right) of 1 μm FK506 for 45 min, followed by treatment with morphine (10 μm; 1 d). Top, The internalized GluR1 subunits were precipitated with immobilized streptavidin and detected with an anti-GluR1 antibody by Western blotting. Bottom, Total GluR1 subunits from whole-cell lysates were detected for a comparison. B, Densitometric quantifications of Western blots in A on the internalized GluR1 (normalized to the control without any drug treatment; p < 0.05; n = 3). C, Neurons coexpressing pHluorin-GluR1 and DsRed were imaged before (left) and 1 d (right) after treatment with no drug, 10 μm morphine, 1 μm FK506, or FK506 with morphine. Open arrowheads denote no change in pHluorin-GluR1 fluorescence and spine morphology. Solid arrowheads denote a decrease in the pHluorin-GluR1 fluorescence and spine morphology by morphine. Scale bar, 10 μm. D, Mean fluorescence (fluor.) intensities of pHluorin-GluR1 from different regions of neurons (the entire image, spines, and dendritic shafts) were normalized to data before treatments in the four groups of experiments [no drug (Ctl), morphine (Mor), FK506, or FK506 with morphine]. *p < 0.05, compared with data before treatment. At least five dendrites of single neurons were analyzed (n = 15 in each group). Error bars represent ± SEM.
Dephosphorylation of GluR1-S845 was essential for morphine-induced GluR1 internalization
To directly test the involvement of calcineurin-dependent GluR1-S845 dephosphorylation in the regulation of GluR1 trafficking by morphine, we generated a GluR1 mutant, in which the Ser845 residue was mutated to a negatively charged aspartic acid (S845D) to mimic continuous phosphorylation of AMPA receptors (Boehm et al., 2006). Hippocampal cultures overexpressing either GFP-GluR1 wild-type or GFP-GluR1-S845D mutant were subjected to biotinylation before morphine treatment (10 μm for 1 d) (Fig. 9A,B). Internalized recombinant receptors (∼127 kDa) were detected by an anti-GFP antibody, and internalized endogenous GluR1 (∼100 kDa) was determined by an anti-GluR1 antibody (Fig. 9A). Equal protein amounts of both recombinant and endogenous receptors from each sample were also validated by the anti-GluR1 antibody (Fig. 9A). Without drug treatment, we found a trend that the basal amount of internalized GFP-GluR1-S845D mutant was slightly less than that of internalized GFP-GluR1 wild-type, but this did not reach statistical significance (wild-type, 100%; S845D, 90 ± 7.8%) (Fig. 9B). Morphine treatment enhanced internalization of GFP-GluR1 wild-type, but not the GFP-GluR1-S845D mutant (wild-type/morphine, 174 ± 12.1%; S845D/morphine, 87 ± 12.3%) (Fig. 9A,B). This suggests that the dephosphorylation of GluR1-S845 is important for morphine to regulate the AMPA receptor endocytosis. The effect of morphine on endogenous GluR1 was used as a positive control, in which morphine was able to induce a significant internalization of endogenous GluR1 regardless of whether the cells were transfected with GFP-GluR1 wild-type or S845D mutant (Fig. 9A,B). Nevertheless, in the presence of the receptor mutant, the endogenous GluR1 was internalized by morphine to a lesser extent than that in the presence of wild-type (from 178 ± 10.2% to 144 ± 10.3%) (Fig. 9B), implying that some endogenous GluR1 might form a functional receptor unit with the recombinant receptor.
Figure 9.
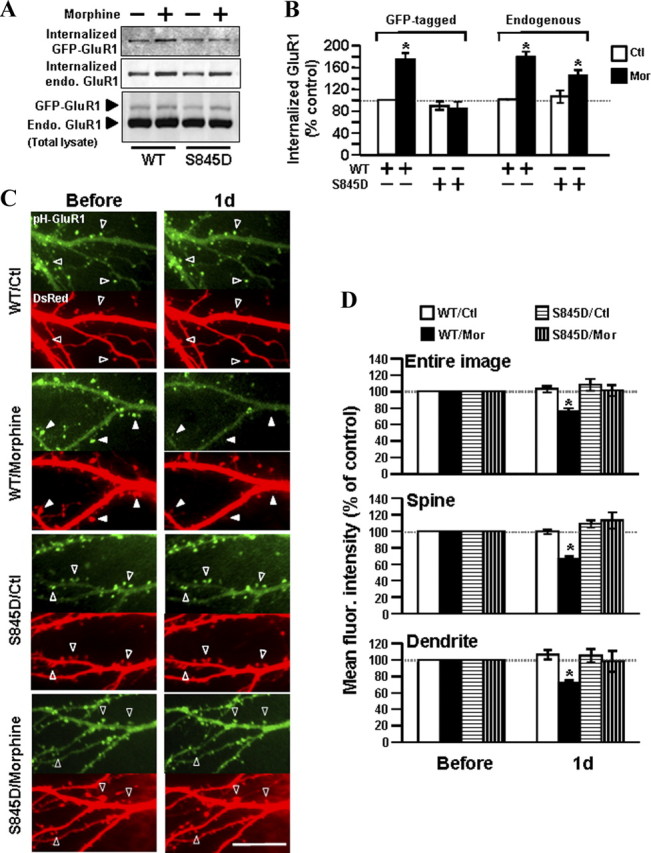
Morphine-induced GluR1 internalization requires dephosphorylation of the receptor at Ser845. A, Hippocampal neurons were transfected with either GFP-GluR1 wild-type (WT; left) or GFP-GluR1-S845D (right). The transfected cells were subjected to surface biotinylation, followed by morphine treatment (10 μm for 1 d). All internalized receptors were isolated by streptavidin precipitation. Top, Internalized exogenous GFP-GluR1 was detected with an anti-GFP antibody. Middle, Internalized endogenous (endo.) GluR1 was detected with an anti-GluR1 antibody. Bottom, Total protein expressions of exogenous and endogenous GluR1 subunits from whole-cell lysates were detected for comparison. In neurons expressing GFP-GluR1-WT (left), morphine induced internalization of both exogenous GFP-GluR1-WT and endogenous GluR1. In neurons expressing GFP-GluR1-S845D (right), morphine induced internalization of endogenous GluR1 but not exogenous GFP-GluR1 mutant. B, Quantification of Western blots in A on the internalized receptor [normalized to the untreated control (Ctl); *p < 0.05; n = 3]. C, Neurons coexpressing DsRed and pHluorin-GluR1-WT (top) or pHluorin-GluR1-S845D (bottom) were imaged before (left) and after morphine (Mor) treatment (right, 10 μm for 1 d). Open arrowheads denote no change in pHluorin-GluR1 and spine morphology. Solid arrowheads denote loss of surface pHluorin-GluR1 (green fluorescence) and shrinkage of spines (DsRed fluorescence) by morphine. Scale bar, 10 μm. D, Averaged fluorescence (fluor.) of pHluorin-GluR1 from different regions of neurons (entire image, spines, and dendritic shafts) were normalized to values before treatment in the four groups of experiments (WT/Ctl, overexpression of pHluorin-GluR1 WT with no drug; WT/Mor, pHluorin-GluR1 WT with morphine; S845D/Ctl, pHluorin-GluR1-S845D with no drug; S845D/Mor, pHluorin-GluR1-S845D with morphine). *p < 0.05, compared with data before treatment. At least five dendrites of single neurons were analyzed (n = 13 in each group). Error bars represent ± SEM.
To further confirm the above biochemical studies, live-cell imaging experiments were also performed using both pHluorin-GluR1-S845D and pHluorin-GluR1-S845A. The pHluorin-GluR1-S845A is a phospho-blocking mutant carrying a substitution of Ser845 with neutral amino acid, alanine (S845A). It is expected that both mutants are unable to be further dephosphorylated (Man et al., 2007). Therefore, hippocampal neurons were cotransfected with DsRed and either pHluorin-GluR1 wild-type, pHluorin-GluR1-S845D, or pHluorin-GluR1-S845A. The transfected cells subsequently were imaged before and 1 d after morphine treatment (10 μm) (Figs. 9C,D, 10). No obvious morphological difference was observed among cells overexpressing wild-type and mutant receptors (Figs. 9C, 10A). This is similar to the findings of earlier studies that mimicking GluR1 phosphorylation or dephosphorylation at Ser845 does not significantly change surface or synaptic GluR1 levels (Lee et al., 2003; Kessels et al., 2009). In the pHluorin-GluR1 wild-type labeled cells, morphine significantly decreased the averaged green fluorescence from the entire images, spines, and dendrites (Figs. 9C,D, 10). In contrast, morphine did not alter the green fluorescence intensity when the cells were overexpressed with either pHluorin-GluR1-S845D or pHluorin-GluR1-S845A (Figs. 9C,D, 10), demonstrating that morphine-induced AMPA receptor internalization can be blocked by both phospho-mimic S845D and S845A phospho-blocking mutants. Since both mutants can provide a constant phosphorylation status of GluR1 and they are effective in suppressing morphine-induced GluR1 trafficking, this suggests that a change in GluR1 phosphorylation by morphine is required for the regulation of receptor trafficking. It is possible that the dephosphorylation itself may send a downstream signal transduction to trigger or modulate AMPA receptor internalization.
Figure 10.
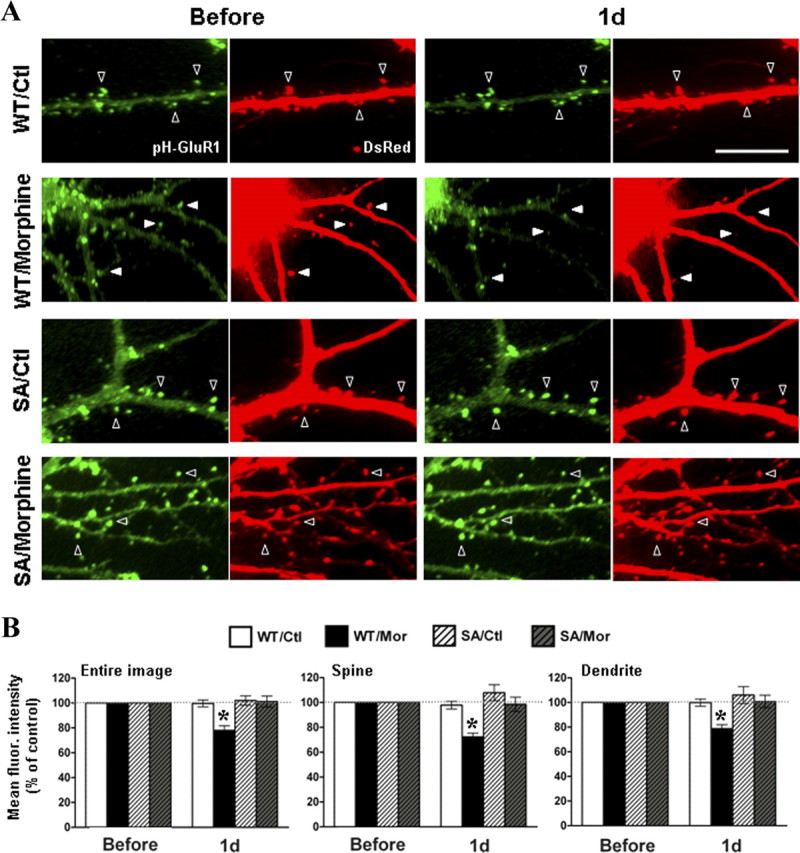
Overexpression of pHluorin-GluR1-S845A mutant blocked the morphine-regulated AMPA receptors internalization. A, Neurons coexpressing DsRed and pHluorin-GluR1-WT (top) or pHluorin-GluR1-S845A (bottom) were imaged before (left) and after (right) morphine treatment (10 μm for 1 d). Open arrowheads denote no change in pHluorin-GluR1 and spine morphology. Solid arrowheads denote loss of surface pHluorin-GluR1 (green fluorescence) and shrinkage of spines (DsRed fluorescence) by morphine. Scale bar, 10 μm. B, Averaged fluorescence of pHluorin-GluR1 from different regions of neurons (entire image, spines, and dendritic shafts) were normalized to values before treatment in the four groups of experiments (WT/Ctl, overexpression of pHluorin-GluR1-WT with no drug; WT/Mor, pHluorin-GluR1-WT with morphine; S845A/Ctl, pHluorin-GluR1-S845A with no drug; S845A/Mor, pHluorin-GluR1-S845A with morphine). *p < 0.05, comparing with data before treatment. At least five dendrites of single neurons were analyzed (n = 10 in each group). Error bars represent ± SEM.
Discussion
Morphine-induced internalization of AMPA receptors
Earlier electrophysiological studies on hippocampal cultures have demonstrated that chronic exposure to morphine decreases the amplitude of mEPSCs (Liao et al., 2005, 2007), which can be attributed to a removal of preexisting AMPA receptor, an inhibition in synaptic delivery of the receptor, and/or a suppression of the receptor channel opening. In the current study, time-lapse imaging and biochemical experiments provided direct evidence that morphine treatment induced a time-dependent loss of preexisting GluR1 at both spines and dendrites, likely through receptor internalization (Figs. 1, 2). However, the receptor internalization may not be the exclusive mechanism, as rats administrated with morphine have a marked reduction of long-term potentiation at hippocampi (Pu et al., 2002), which suggests a decrease in the reinsertion rate of synaptic AMPA receptors. Therefore, it is plausible that morphine-induced loss of postsynaptic GluR1 is a combination of increased internalization and decreased synaptic delivery (Ehlers, 2000; Man et al., 2007). Moreover, it should be noted that the loss of surface pHluorin-GluR1 could be detected by live-imaging 3 h after morphine treatment (Fig. 1), but an increase in endogenous internalized GluR1 appeared only after 6 h, as shown in the biochemical experiments (Fig. 2). This time discrepancy may be due to different sensitivity of the two analysis techniques. The live imaging is based on detection of overexpressed recombinant receptors in a single neuron, whereas the biochemical study measures endogenous receptors in a population of neurons. Hence, the imaging technique may have higher sensitivity to detect subtle changes in the receptor movement at an earlier time point.
Morphine is well known to inhibit the presynaptic release of GABA through activation of inwardly rectifying potassium channels in GABAergic neurons of hippocampus, leading to a disinhibition of neural circuits (Wimpey and Chavkin, 1991). Since TTX pretreatment, which prevents depolarization, cannot attenuate morphine-induced GluR1 endocytosis and loss of surface-expressed GluR1 (Fig. 3), the morphine effect is not due to an alteration of the neural network. Notably, CTOP cotreatment can completely abrogate the morphine-induced AMPA receptor trafficking (Figs. 1, 2). In addition, previous studies have found that MORs predominantly express in postsynaptic membrane (Arvidsson et al., 1995; Liao et al., 2005) and mainly colocalized with postsynaptic AMPA receptors (Liao et al., 2005). All of these observations suggest that morphine likely acts on postsynaptic MORs to regulate AMPA receptor trafficking.
Morphine-induced calcineurin-dependent dephosphorylation of GluR1 at Ser845 and GluR1 endocytosis
We found that prolonged application of morphine promotes the dephosphorylation of GluR1 at Ser845 and reduces the amount of PSD-95 in the immunoprecipitated GluR1 complex (Fig. 4), indicating the detachment of GluR1 from the PSD. Importantly, the prevention of GluR1-S845 dephosphorylation by either GluR1-S845D or GluR1-S845A mutant clearly suppresses the morphine-induced GluR1 endocytosis (Figs. 9, 10). These observations are consistent with the prevalent view that the phosphorylation of AMPA receptors can regulate the receptor trafficking by affecting the interactions between the receptors' cytoplasmic tails and postsynaptic scaffolding proteins, especially PDZ-domain containing proteins such as SAP97, PSD-95, GRIP/ABP, and PICK (Dong et al., 1997; Leonard et al., 1998; Xia et al., 1999; Shepherd and Huganir, 2007). As our results suggest that an alteration of phosphorylation status of GluR1-S845 by calcineurin-induced dephosphorylation is important for morphine-regulated GluR1 trafficking (Figs. 9, 10), this implies that the induction of dephosphorylation can facilitate the dissociation of the GluR1 cytoplasmic tails and postsynaptic scaffolding proteins. Furthermore, since AMPA receptors are believed to cluster in PSD of synapses via the binding of stargazin or related transmembrane AMPA receptor binding proteins (TARPs) to PSD-95 (Nicoll et al., 2006), it will be of interest to determine whether GluR1-S845 dephosphorylation by morphine affects the interaction between AMPA receptors and TARPs or TARPs and PSD-95, and how the dephosphorylation suppresses this interaction.
The net level of GluR1-S845 phosphorylation depends on the opposing activities of PKA and phosphatases such as calcineurin (Barria et al., 1997; Beattie et al., 2000). Since postsynaptic protein A kinase-anchoring protein 150 (AKAP150) can scaffold PKA and calcineurin at different binding sites and is recruited to the complexes of SAP-97/PSD-95 and GluR1, AKAP150 acts as a link to position these intracellular enzymes adjacent to the surface receptors. The physical proximity of these proteins in turn allows PKA and calcineurin to regulate GluR1-S845 phosphorylation in a highly coordinated manner (Colledge et al., 2000, Dell'Acqua et al., 2006). In this study, 3 d of morphine treatment only elicited a transient reduction on GluR1-S845 phosphorylation by dbcAMP (Fig. 5), suggesting that morphine-induced PKA inhibition alone is insufficient to reduce GluR1-S845 phosphorylation level. Conversely, FK506 pretreatment completely reversed the morphine-induced GluR1-S845 dephosphorylation and morphine is able to increase calcineurin activity (Fig. 6), indicating that activated calcineurin appears to be the dominant regulator of GluR1-S845 phosphorylation when the primary hippocampal neurons were treated with morphine.
Recently, several in vivo studies have found that discontinuous treatment of animals with morphine increases GluR1-S845 phosphorylation, in contrast to our findings (Billa et al., 2009, 2010). This difference may be related to different experimental models, particularly the method of morphine application. They used a 12 h period without morphine injections before animal dissection, potentially leading to a decrease in morphine level by metabolism and elimination. The decreased morphine level may cause drug withdrawal that in turn increases adenylate cyclase activity, cAMP level, and PKA activity (Sharma et al., 1975). Once PKA activity overwhelms calcineurin activity, GluR1-S845 phosphorylation level could then be increased. In contrast, our study used a continuous incubation of hippocampal cultures with morphine, in which PKA was inhibited, thus favoring the action of activated calcineurin (Figs. 5, 6). As a result, there was dephosphorylation of GluR1-S845. Furthermore, our in vitro primary culture studies were not affected by the circuitry synaptic activities, as shown in our TTX and APV studies, which might not be case in the in vivo studies if the animals treated with morphine were undergoing withdrawal. Hence, cultured hippocampal neurons can provide an experimental model to directly study any neuromodulation induced by morphine itself or μ-opioid receptor.
Calcineurin is a Ca2+-dependent protein phosphatase (Klee et al., 1979) and it plays a pivotal role in morphine-regulated GluR1-S845 dephosphorylation at Ser845 and receptor endocytosis (Figs. 6, 8). The key issue is how calcineurin is stimulated or where Ca2+ comes from upon morphine treatment. Although the precise mechanism underlying morphine-activated calcineurin in our study is still unclear, at least we narrow down the pathway to one that NMDA receptors are not involved with, as dl-APV has no effect on morphine-increased calcineurin activity (Fig. 7). Apart from NMDA receptors, other glutamate receptors such as Gq-coupled metabotropic glutamate receptors (mGluRs) and the related intracellular messengers such as phospholipids-Ca2+-PKC are believed to regulate morphine-induced signal transduction in vitro and tolerance development in vivo (Fundytus and Coderre, 1996; Narita et al., 2005; Chu et al., 2010). Therefore, mGluRs or/and phospholipid-Ca2+-PKC may be the potential signal pathways mediating the calcineurin activation. Considering a recent report that Ca2+-permeable AMPA receptor is absent from perisynaptic sites in GluR1-S845A mutant mice (He et al., 2009), and our data that overexpression of pHluorin-GluR1-S845A causes the attenuation of morphine-regulated GluR1 trafficking (Fig. 10), Ca2+-permeable AMPA receptor may be another potential source of Ca2+ activating calcineurin.
Implications and conclusion
The present study directly demonstrates that chronic treatment with morphine induces GluR1 endocytosis, resulting in a loss of GluR1 at excitatory synapses of hippocampi (Figs. 1, 2). This morphine effect may represent a key step in an addiction model proposed by Lüsher and Bellone (2008). In this model, chronic exposure to addictive drugs such as cocaine induces an initial synaptic plasticity in which the amount of synaptic AMPA receptors in nucleus accumbens initially decreases (Thomas et al., 2001). However, later withdrawal of the drug increases synaptic insertion of the receptors, even exceeding the baseline. Intriguingly, Ca2+-impermeable-AMPA receptors are gradually replaced by Ca2+-permeable ones during the late withdrawal (Conrad et al., 2008). Such exchange for Ca2+-permeable AMPA receptors is considered to conduct synaptic currents more readily and to contribute long-lasting alterations in synaptic strength that implies the relapse of addiction. This multiple-step addiction model emphasizes that the addictive drug-evoked synaptic plasticity indeed is a hierarchical organization: an initial induction of synaptic plasticity is important for developing subsequent plasticity (Lüsher and Bellone, 2008). Therefore, chronic exposure to morphine-induced removal of synaptic GluR1 is probably the initial induction of plasticity underlying the drug addiction. Moreover, the present study points out the calcineurin-dependent signal pathway in morphine-regulated GluR1 trafficking. Likewise, an earlier report demonstrated the impairment of the reinforcing effect of morphine in memory-deficient mice overexpressing calcineurin (Biala et al., 2005), further suggesting the key role of calcineurin in morphine-induced neuroadaptations.
The current study increases the knowledge on how opioids induce neuroadaptation. Chronic treatment with morphine triggers loss of both synaptic and extrasynaptic AMPA receptors through internalization. This effect of morphine requires calcineurin-mediated dephosphorylation of GluR1 at Ser845. The intervention of this calcineurin-dependent signal pathway may provide a new approach for the prevention and/or treatment of opiate addiction and the drug-induced developmental deficits.
Footnotes
This work was supported by grants from the National Institutes of Health (DA000564 and DA011806 to H.H.L and P.Y.L., DA020582 and K02-DA025048 to D.L., K05-DA00513 to P.Y.L.) and a grant from the American Health Assistance Foundation (to D.L.). We are grateful to Dr. Gero Miesenböck for kindly providing the pHluorin plasmid. We also thank Paul Higgins for preparing neuronal cultures and Steph Porter for helpful discussion.
References
- Arvidsson U, Riedl M, Chakrabarti S, Lee JH, Nakano AH, Dado RJ, Loh HH, Law PY, Wessendorf MW, Elde R. Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J Neurosci. 1995;15:3328–3341. doi: 10.1523/JNEUROSCI.15-05-03328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai A, Qiu J, Narita Y, Chi S, Saito N, Shinoura N, Hamada H, Kuchino Y, Kirino T. High level calcineurin activity predisposes neuronal cells to apoptosis. J Biol Chem. 1999;274:34450–34458. doi: 10.1074/jbc.274.48.34450. [DOI] [PubMed] [Google Scholar]
- Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMAPRs. J Neurosci. 2004a;24:5172–5176. doi: 10.1523/JNEUROSCI.1042-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, Ibaraki K, Henley JM. It's green outside: tracking cell surface proteins with pH-sensitive GFP. Trends Neurosci. 2004b;27:257–261. doi: 10.1016/j.tins.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the -amino-3-hydroxy-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem. 1997;272:32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;12:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Biala G, Betancur C, Mansuy IM, Giros B. The reinforcing effects of chronic d-amphetamine and morphine are impaired in a line of memory-deficient mice overexpressing calcineurin. Eur J Neurosci. 2005;21:3089–3096. doi: 10.1111/j.1460-9568.2005.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billa SK, Sinha N, Rudrabhatla SR, Morón JA. Extinction of morphine-dependent conditioned behavior is associated with increased phosphorylation of the GluR1 subunit of AMPA receptors at hippocampal synapses. Eur J Neurosci. 2009;29:55–64. doi: 10.1111/j.1460-9568.2008.06560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billa SK, Liu J, Bjorklund NL, Sinha N, Fu Y, Shinnick-Gallagher P, Morón JA. Increased insertion of glutamate receptor 2-lacking alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors at hippocampal synapses upon repeated morphine administration. Mol Pharmacol. 2010;77:874–883. doi: 10.1124/mol.109.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Chu J, Zheng H, Zhang Y, Loh HH, Law PY. Agonist-dependent mu-opioid receptor signaling can lead to heterologous desensitization. Cell Signal. 2010;22:684–696. doi: 10.1016/j.cellsig.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES, Gomez LL. Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur J Cell Biol. 2006;85:627–633. doi: 10.1016/j.ejcb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interact with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- Duman RS, Tallman JF, Nestler EJ. Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Ther. 1988;246:1033–1039. [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPAR receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Fan GH, Wang LZ, Qiu HC, Ma L, Pei G. Inhibition of calcium/calmodulin-dependent protein kinase II in rat hippocampus attenuates morphine tolerance and dependence. Mol Pharmacol. 1999;56:39–45. doi: 10.1124/mol.56.1.39. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Blaha CD, Yang CR, Phillips AG. Modulation of hippocampal and amygdalar-evoked activity of nucleus accumbens neurons by dopamine: cellular mechanisms of input selection. J Neurosci. 2001;21:2851–2860. doi: 10.1523/JNEUROSCI.21-08-02851.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Pai SY, Klee CB, Burakoff SJ, Bierer BE. Measurement of calcineurin phosphatase activity in cell extracts. Methods. 1996;9:146–154. doi: 10.1006/meth.1996.0020. [DOI] [PubMed] [Google Scholar]
- Fundytus ME, Coderre TJ. Chronic inhibition of intracellular Ca2+ release or protein kinase C activation significantly reduces the development of morphine dependence. Eur J Pharmacol. 1996;300:173–181. doi: 10.1016/0014-2999(95)00871-3. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee HK. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc Natl Acad Sci U S A. 2009;106:20033–20038. doi: 10.1073/pnas.0910338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–1422. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- Inoue M, Mishina M, Ueda H. Locus-specific rescue of GluRepsilon1 NMDA receptors in mutant mice identifies the brain regions important for morphine tolerance and dependence. J Neurosci. 2003;23:6529–6536. doi: 10.1523/JNEUROSCI.23-16-06529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner ML, Lisé MF, Yuen EY, Kam AY, Zhang M, Hall DD, Malik ZA, Qian H, Chen Y, Ulrich JD, Burette AC, Weinberg RJ, Law PY, El-Husseini A, Yan Z, Hell JW. Assembly of a beta(2)-adrenergic receptor-GluR1 signalling complex for localized cAMP signalling. EMBO J. 2010;29:482–495. doi: 10.1038/emboj.2009.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and memory mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Kopec CD, Klein ME, Malinow R. Roles of stargazin and phosphorylation in the control of AMPA receptor subcellular distribution. Nat Neurosci. 2009;12:888–896. doi: 10.1038/nn.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee CB, Crouch TH, Krinks MH. Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci U S A. 1979;76:6270–6273. doi: 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Legault M, Rompré PP, Wise RA. Chemical stimulation of the ventral hippocampus elevates nucleus accumbens dopamine by activating dopaminergic neurons of the ventral tegmental area. J Neurosci. 2000;20:1635–1642. doi: 10.1523/JNEUROSCI.20-04-01635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxyl-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- Li ST, Kato K, Tomizawa K, Matsushita M, Moriwaki A, Matsui H, Mikoshiba K. Calcineurin plays different roles in group II metabotropic glutamate receptor- and NMDA receptor-dependent long-term depression. J Neurosci. 2002;22:5034–5041. doi: 10.1523/JNEUROSCI.22-12-05034.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- Liao D, Lin H, Law PY, Loh HH. Mu-opioid receptors modulate the stability of dendritic spines. Proc Natl Acad Sci U S A. 2005;102:1725–1730. doi: 10.1073/pnas.0406797102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Grigoriants OO, Wang W, Wiens K, Loh HH, Law PY. Distinct effects of individual opioids on the morphology of spines depend upon the internalization of mu opioid receptors. Mol Cell Neuorsci. 2007;35:456–469. doi: 10.1016/j.mcn.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature. 1994;369:235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- Lin H, Huganir R, Liao D. Temporal dynamics of NMDA receptor-induced changes in spine morphology and AMPA receptor recruitment to spines. Biochem Biophys Res Commun. 2004;316:501–511. doi: 10.1016/j.bbrc.2004.02.086. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Bellone C. Cocaine-evoked synaptic plasticity: a key to addiction? Nat Neurosci. 2008;11:737–738. doi: 10.1038/nn0708-737. [DOI] [PubMed] [Google Scholar]
- Ma YY, Chu NN, Guo CY, Han JS, Cui CL. NR2B-containing NMDA receptor is required for morphine-but not stress-induced reinstatement. Exp Neurol. 2007;203:309–319. doi: 10.1016/j.expneurol.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Huganir RL, O'Brien RJ. Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J Neurosci. 1997;17:7351–7358. doi: 10.1523/JNEUROSCI.17-19-07351.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Morón JA, Green TA. Exploring the molecular basis of addiction: drug-induced neuroadaptations. Neuropsychopharmacology. 2010;35:337–338. doi: 10.1038/npp.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat Neurosci. 2002;6:153–160. doi: 10.1038/nn994. [DOI] [PubMed] [Google Scholar]
- Narita M, Suzuki M, Narita K, Niikura A, Nakamura M, Miyatake T, Aoki Y, Yajima Y, Suzuki T. Involvement of spinal metabotropic glutamate receptor 5 in the development of tolerance to morphine-induced antinociception. J Neurochem. 2005;94:1297–1305. doi: 10.1111/j.1471-4159.2005.03296.x. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- Pu L, Bao GB, Xu NJ, Ma L, Pei G. Hippocampal long-term potentiation is reduced by chronic opiate treatment and can be restored by re-exposure to opiates. J Neurosci. 2002;22:1914–1921. doi: 10.1523/JNEUROSCI.22-05-01914.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranta F, Düfer M, Stork B, Wesselborg S, Drews G, Häring HU, Lang F, Ullrich S. Regulation of calcineurin activity in insulin-secreting cells: stimulation by Hsp90 during glucocorticoid-induced apoptosis. Cell Signal. 2008;20:1780–1786. doi: 10.1016/j.cellsig.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Morphine alters the structure of neurons in the nucleus accumbens and neocortex of rats. Synapse. 1999;33:160–162. doi: 10.1002/(SICI)1098-2396(199908)33:2<160::AID-SYN6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci U S A. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Ueda H, Harada H, Nozaki M, Katada T, Ui M, Satoh M, Takagi H. Reconstitution of rat brain mu opioid receptors with purified guanine nucleotide-binding regulatory proteins, Gi and Go. Proc Natl Acad Sci U S A. 1988;85:7013–7017. doi: 10.1073/pnas.85.18.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL. Relapse to cocaine seeking after hippocampal theta burst stimulation. Science. 2001;292:1175–1178. doi: 10.1126/science.1058043. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Wimpey TL, Chavkin C. Opioids activate both an inward rectifier and a novel voltage-gated potassium conductance in the hippocampal formation. Neuron. 1991;6:281–289. doi: 10.1016/0896-6273(91)90363-5. [DOI] [PubMed] [Google Scholar]
- Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- Yang SN, Yang JM, Wu JN, Kao YH, Hsieh WY, Chao CC, Tao PL. Prenatal exposure to morphine alters kinetic properties of NMDA receptor-mediated synaptic currents in the hippocampus of rat offspring. Hippocampus. 2000;10:654–662. doi: 10.1002/1098-1063(2000)10:6<654::AID-HIPO1003>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]