

Abstract
Water is an inhospitable environment for protein hydrogen bonds because it is polarizable and capable of forming competitive hydrogen bonds. In contrast, the apolar core of a biological membrane seems like an ideal environment for hydrogen bonds, and it has long been assumed that hydrogen bonding should be a powerful force driving membrane protein folding. Nevertheless, while backbone hydrogen bonds may be much stronger in membrane proteins, experimental measurements indicate that side chain hydrogen bond strengths are not strikingly different in membrane and water soluble proteins. How is this possible? I argue that that model compounds in apolar solvents do not adequately describe the system because the protein itself is ignored. The protein chain provides a rich source of competitive hydrogen bonds and a polarizable environment that can weaken hydrogen bonds. Thus, just like water soluble proteins, evolution can drive the creation of potent hydrogen bonds in membrane proteins where necessary, but mitigating forces in their environment must still be overcome.
Many residues in the hydrocarbon-spanning region of membrane proteins are polar [1] and they are often engaged in hydrogen bonding interactions between transmembrane helices. Indeed almost all transmembrane helices have at least one interhelical hydrogen bond [2]. There is no doubt that hydrogen bonding interactions can play key roles in structure stabilization [3-7][8,9]. Polar side chains are likely sites of disease causing mutations [10] and mutations to polar side chains can lead to inappropriate interactions that cause disease [11-15]. Nevertheless, quantitative measures of hydrogen bond strengths to date suggest that the stabilization afforded by side chain hydrogen bonds is usually not much different than is seen in water soluble proteins. Moreover, the fraction of apparently unsatisfied hydrogen bond donors and acceptors is similar in both classes of proteins [16]. As discussed here, many factors can reduce hydrogen bond strength in membrane proteins.
How strong can hydrogen bonds be?
The energy of an amide hydrogen bond in vacuo is arguably the upper limit of its free energy since the entropic contribution is presumably unfavorable and anything more polarizable than a vacuum will reduce the contribution of the electrostatic interactions [17]. Quantum mechanical calculations of amide hydrogen bond strengths using a formamide-formaledehyde model suggest that an amide hydrogen bond has an in vacuo energy of 6.6 kcal/mol[17]. Calculations for an N-methylacetamide dimer also agree that the energy of the hydrogen bond in a vacuum is about 6.6 kcal/mol [18]. Experimentally determined vapor phase enthalpies of hydrogen bonds between neutral species range from 3-6 kcal/mol[19]. Thus, it appears that the upper end for neutral hydrogen bonds is around 6 kcal/mol, but could be even higher in special cases [20], particularly charge stabilized hydrogen bonds [19]. Clearly, there is potential for hydrogen bonds to be a powerful stabilizing force.
Hydrogen bond energies will be reduced from the optimal values by a number of factors. First, hydrogen bond strength is very sensitive to geometry [17,21] and geometry may not be optimized. Second, solvent polarizability decreases hydrogen bond strength, which is dominated by electrostatics [17]. Third, the entropy cost for fixing the donors and acceptors decreases the free energy of the interaction. Finally, the ability of the solvent to participate in hydrogen bonds can reduce the net energy difference. For example, in water (W) the making and breaking of a backbone hydrogen bond can be written as follows [22]:
Thus, a simple hydrogen bond inventory approach implies no net change in the number of hydrogen bonds. If all hydrogen bonds were equal (and they are certainly not), the net contribution of a hydrogen bond should be zero. While the hydrogen bond inventory concept is overly simplistic [23], it is clear that for the formation of a protein hydrogen bond to be energetically favorable, it must be more favorable than solvation of the broken hydrogen bond by water.
How much more stable can a protein hydrogen bond be compared to water solvation? It seems that a good place to look is in functional binding sites or enzymes where there may be strong evolutionary pressure to optimize particular hydrogen bonds. Indeed, Shan & Herschlag argue that an optimized neutral hydrogen bond in an enzyme active site could be around 9 kcal/mol more stable than an alternative hydrogen bond to water [24]. A Tyr to Phe mutation in the active site of ketosteroid isomerase reduces transition state stabilization by 6.3 kcal/mol [25]. Thus, even in water solution, optimized hydrogen bonds can be very important contributors to intermolecular complexes.
In an apolar solvent like the center of a membrane bilayer that has a low dielectric constant and no competitive hydrogen bonding potential, hydrogen bond contributions could be even stronger. As discussed below, however, most hydrogen bonds in both water soluble and membrane proteins seem to be far from these optimized limits.
How strong are backbone hydrogen bonds?
Early model compound studies argued that backbone hydrogen bonds could not be stabilizing in aqueous solution. Classic experiments by Klotz and Franzen[26], who studied the aggregation of N-methylacetamide in polar and apolar solvents, found that association is unfavorable by 3.1 kcal/mol in water. Their conclusion from this work was that “…in aqueous solution, interpeptide hydrogen bonds cannot contribute significantly to the stabilization of macromolecular organization, except perhaps in a few regions with very low local dielectric…” It is not possible, however, to directly compare an intermolecular reaction to the intramolecular reaction that occurs in protein folding, where there is potential for dramatically enhanced effective concentrations[19]. Eberhardt and Raines studied the ability of different solvents to donate hydrogen bonds to model solutes. Secondary amide solvents, which mimic the backbone, were much weaker hydrogen bond donors than water even at concentrations higher than 10 M [27]. Their results imply that backbone hydrogen bonds cannot be stabilizing.
In spite of these model compound studies, other experimental work in proteins and peptides suggests a net favorable contribution of backbone hydrogen bonds. Calorimetric measurements of the enthalpy helix-coil transitions report a net ethalpic contribution of 0.9-1.0 kcal/mol [28,29]. Elegant “backbone mutagenesis” studies by Kelly and coworkers found that a hydrogen bond in a β-sheet contributed a favorable 1.3 kcal/mol and one in an α-helix contributed 1.4 kcal/mol [30,31].
Backbone hydrogen bonds can have variable strength, however. Recent work from the Kelly lab showed that the polarity of the environment around a backbone hydrogen bond can alter its free energy by 1.2 kcal/mol [32]. Moreover, backbone hydrogen bond lengths vary significantly as a function of the environment polarity [33,16].
There is no comparable experimental data for membrane proteins, but presumably the net free energy of backbone hydrogen bonds must be significantly larger in the apolar core. White and Wimley argue based on the free energy of partitioning hydrogen bonded and non-hydrogen bonded amides into apolar solvents that the contribution could be as high as 4-5 kcal/mol [34]. Calculations based on the N-methylacetamide model system also suggest a free energy of 5 kcal/mol [18]. As discussed below, however, naked hydrogen bonds in model compounds surrounded by an apolar solvent may not be appropriate and lead to an overestimate of the hydrogen bond contribution. Nevertheless, the fact that secondary structure persists in denatured membrane proteins [35-38] suggests that backbone hydrogen bonds in the membrane are more stable than in water solution where isolated helices are usually unstable. Moreover, backbone hydrogen bonds in transmembrane helices are shorter and more regular on average than backbone hydrogen bonds in water soluble protein helices[33,16] [39]. Frommel and co-workers made the interesting observation that backbone hydrogen bonds in membrane proteins have a higher level of bifurcation between the i, i+3 and i, i+4 residues [33]. It is unclear how the environment creates this alteration or how the bifurcation might influence helix stability.
How strong are side chain hydrogen bonds in water soluble proteins?
Hydrogen bonding contributions in water soluble proteins have been probed extensively through mutagenesis experiments. Mutation of a hydrogen bonding residue not only deletes the hydrogen bond, but can also alter the hydrophobicity of the side chain, the conformational entropy cost of folding, and the packing of the side chain in the protein. Efforts have been made to carefully account for these effects [40-42]. Pace, however, provides a rather elegant solution to this problem which is outlined in Fig. 1 [42]. He examined the effects on the free energy of folding for 52 Tyr to Phe and 40 Thr to Val mutants. For the Thr to Val mutants, the unfolding free energy was reduced by 1.0 ± 1.0 kcal/mol when the Thr was hydrogen bonded in the folded protein and only 0.0 ± 0.7 kcal/mol when it was not hydrogen bonded. Similarly, for the Tyr to Phe mutants, stability was lowered by 1.4 ± 0.9 kcal/mol when the Tyr was hydrogen bonded and only 0.2 ± 0.4 kcal/mol when it was not. Thus, on average, hydrogen bonds were worth an extra 1.2 ± 1.0 kcal/mol for the Tyr to Phe mutants and 1.0 ± 1.2 kcal/mol for the Thr to Val mutants. The beauty of this analysis is that the mutations being compared are identical except that some are hydrogen bonded and some are not, so the extra correction factors should be the same (on average). These results argue that hydrogen bonds typically stabilize a water soluble protein by about 1 ± 1 kcal/mol. The range is large, however, partly due to intrinsic errors in such complex experiments, but also because some hydrogen bonds are stronger than others [42].
Figure 1. Method for estimating of the average net hydrogen bond free energy.

Pace collected unfolding free energy data for Tyr to Phe mutants and Thr to Val mutations in which the wild-type side chain was either hydrogen bonded or not hydrogen bonded. The analysis is illustrated for Tyr to Phe mutations. A mutation from Tyr to Phe will break the hydrogen bond, reflected in ΔGHB. It may also alter other contributions of the side chain such as hydrophobicity, conformational entropy and packing, reflected in ΔGSC(Tyr) and ΔGSC(Phe) for the Tyr and Phe side chains, respectively. All other contributions to folding that are not involved in the mutation are lumped into ΔGother. For the hydrogen bonded side chains, the difference in unfolding free energies, ΔGu(1) and ΔGu(2), reflects both ΔGHB and the difference in other side chain contributions. For the non-hydrogen bonded side chains, the difference in unfolding free energies, ΔGu(3) and ΔGu(4), reflects only the difference in the other side chain contributions. By subtracting the average unfolding free energy changes for the mutants in the two classes (hydrogen bonded and not), the average other side chain contributions cancel, leaving the average hydrogen bond contribution.
Another way to assess the strength of a hydrogen bond in the folded protein is to perform double mutant cycle analysis [43]. A double mutant cycle is illustrated in Fig. 2. The basic idea is that single point mutants will not only delete the hydrogen bonding interaction between two side chains, but will also affect any other interaction the side chain makes in the folded or unfolded states of the protein. By subtracting the energetic effects of a double mutant, the effects of these other interactions can be cancelled out. An advantage of the double mutant cycle is that it does not require assumptions about desolvation free energies or conformational entropy changes that are needed for the single mutants. On the other hand, the double mutant cycle is a complex experiment fraught with uncertainties. In particular it requires three different unfolding experiments, each requiring some level of interpretation. Moreover, the approach cannot account for structural changes or new interactions in the single mutants that are not present in the double mutant.
Figure 2. Double mutant cycle analysis of hydrogen bond interactions.
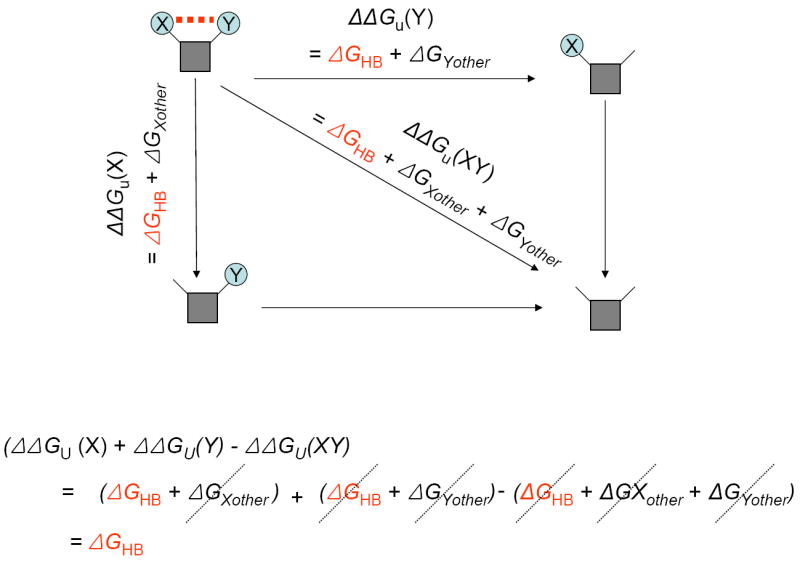
In this example residue X and residue Y form a hydrogen bond. The change in unfolding free energy upon mutation of residue Y, ΔΔGu(Y), will reflect the hydrogen bond contribution, ΔGHB, and any other interactions made by residue Y in the folded or unfolded state, ΔGYother. Similarly, the change in unfolding free energy upon mutation of residue X, ΔΔGu(X), will reflect the hydrogen bond contribution, ΔGHB, and any other interactions made by residue X in the folded or unfolded state, ΔGXother. The change in unfolding free energy upon mutation of both residue X and Y, ΔΔGu(XY), will reflect the hydrogen bond contribution, ΔGHB, and any other interactions made by residues X and Y in the folded or unfolded state, ΔGXother + ΔGYother. As illustrated below the cycle, adding the single mutant effects and subtracting the double mutant effects results in cancellation of the other contributions and leaves the hydrogen bond free energy. The key and rather bold assumption is that the effects of the mutants are additive and independent.
In a search of the literature, we found a total of 15 double mutant cycle analyses of hydrogen bond contributions to protein folding, which are listed in Table I. Ignoring the outlier at an unfavorable 2.6 kcal/mol, the remaining 14 interaction free energies made average favorable contribution of 0.5 ± 0.7 kcal/mol, which is in the range found in Pace’s single mutant analysis [42].
Table I.
Interaction free energies for hydrogen bonded residue pairs measured by double mutant cycle analysis
| PROTEIN | RESIDUE PAIR | INTERACTION FREE ENERGY | REFERENCE |
|---|---|---|---|
| apoflavodoxin | H34/ Y47 | -1.32 ± 0.01 | [64] |
| apoflavodoxin | H34/ Y47 | -0.66 ± 0.01 | [64] |
| apoflavodoxin | H34/ F7 | -0.47 ± 0.03 | [64] |
| apoflavodoxin | D96/ N128 | -0.19 ± 0.09 | [65] |
| apoflavodoxin | H34/ F7 | 0.08 ± 0.03 | [64] |
| apoflavodoxin | D96/ N128 | 0.3 ± 0.1 | [65] |
| ketosteroid isomerase | Y14/ D99 | -1.3 ± 0.2 | [66] |
| ketosteroid isomerase | Y55/ D99 | 0.6 ± 0.1 | [66] |
| ketosteroid isomerase | Y30/ D99 | 2.7 ± 0.3 | [66] |
| lambda repressor | D14/ S77 | -1.53 | [67] |
| lambda repressor | D14/ S77 | -1.24 | [67] |
| lambda repressor | D14/ R17 | -0.81 | [67] |
| lambda repressor | D14/ R17 | -0.52 | [67] |
| lambda repressor | R17/ S77 | -0.24 | [67] |
| lambda repressor | R17/ S77 | 0.05 | [67] |
Double mutant cycles have also been performed on hydrogen bonds in functional binding sites. As noted above, these hydrogen bonds have the potential to be particularly strong because of evolutionary pressure to optimize binding interactions, but it again appears that very strong hydrogen bonds are not the norm. Fersht et al. measured hydrogen bonds to tyrosyl-tRNA-synthetase substrates and found that neutral hydrogen bonding interactions contribute in the range of 0.5-1.5 kcal/mol. A thorough analysis of a hydrogen bonding network between β-lactamase and a peptide inhibitor found that individual pairs of hydrogen bonding residue pairs contribute only 0.3 kcal/mol [44].
How strong are side chain hydrogen bonds in membrane proteins?
The results from soluble proteins suggest that even in a water solution, hydrogen bonds in a protein are generally stabilizing by about 1 kcal/mol, and with evolutionary pressure, can be much stronger indeed. Moreover, there is little disagreement that an apolar environment can be dramatically stabilizing. For example, dimerization of N-methylacetamide is about 4 kcal/mol stronger more favorable in carbon tetrachloride than in water. Consistent with these measurements, calculations of Ben-Tal et al. suggest the energy of an amide hydrogen bond is reduced from 6.6 kcal/mol in a vacuum to 5.3 kcal/mol in an alkane solvent and 1.3 kcal/mol in water[18].
Experimental results in membrane proteins tell a different story, however. Quantitative free energy measurements of regular hydrogen bonds in membrane proteins are summarized in Table II. Both Bill Degrado and Don Engelman’s groups found that the introduction of polar residues into a hydrophobic transmembrane helix could drive trimerization of the helix [4,7,45-47]. Degrado’s group put these results on a quantitative basis by measuring the difference in trimerization free energy for a series of polar and nonpolar substitutions using equilibrium sedimentation in detergent solution [48]. They found that the strongest effects relative to Ala were 1.8 kcal/mol per monomer for an Asp side chain. Fleming’s group probed the contribution of hydrogen bonding to the dimerization of a β-barrel membrane protein, OMPLA, and found a modest contribution of 0.5 kcal/mol per monomer [49]. In glycophorin A, a Thr makes a hydrogen bond to the backbone of a neighboring dimer and contributes approximately 0.5 kcal/mol per side chain [50]. A caveat for these observations is that they were made in detergent solution. As a result, there might be a significant change in the solvation free energy of the mutant side chains that could lead to an underestimation of the net contribution in an apolar environment. Hristova and co-workers, however, measured the contribution of a Glu side chain to the dimerization of an FGF receptor TM helix in natural bilayers and found a value of 0.7 kcal/mol. In unpublished results, we have re-examined the contribution of the glycophorin A Thr hydrogen bond in POPC bilayers and find a value of 1 kcal/mol per monomer. To summarize, the hydrogen bonding interactions measured by single site mutants in membrane proteins all average 1.0 ± 0.5 kcal/mol. This is essentially the same as what is seen in soluble proteins.
Table II. Experimentally measured energetic consequences of single mutations in hydrogen bonded residues in membrane proteins.
We performed double mutant cycle analysis of the interaction free energies for eight different hydrogen bonds in bacteriorhodopsin [16]. The interaction free energies ranged from an unfavorable 0.4 kcal/mol to a favorable 1.7 kcal/mol, with an average of a favorable 0.6 kcal/mol. Again, this is very similar to the results of double mutant cycle analyses in soluble proteins discussed above.
The similar contribution of hydrogen bonding in both soluble and membrane protein is also suggested by the finding that the fraction of unsatisfied hydrogen bonds is similar in the two protein classes[16]. Fleming and Rose have pointed out that many of these apparently unsatisfied hydrogen bonds are simply errors or could be satisfied by small adjustments to the structure [51]. On the other hand, one could also argue that the fraction of observed unsatisfied hydrogen bonds is an underestimate, since crystal structures are a static snapshot and do not report the fraction of the time a particular hydrogen bond is broken. In either case, soluble and membrane proteins do not seem to be strikingly different by this measure.
How can membrane protein hydrogen bonds be so weak?
One of the primary arguments that hydrogen bonds should be strong in the hydrocarbon core of a bilayer is that the solvent cannot compete for hydrogen bonds in the folded state of the protein. But that does not mean that membrane proteins do not experience competitive hydrogen bonding potential as the protein itself is a plentiful source of alternative hydrogen bonding partners[49]. For example, the unfolded state of helical membrane proteins is thought to consist of separated transmembrane helices, in which the secondary structure remains largely intact [37,52,53]. Most polar residues in a helix can hydrogen bond back to the backbone [54]. Moreover, there are other polar side chain partners in the unfolded protein. Thus, in the unfolded state, there is considerable local hydrogen bonding potential that can compete with alternative hydrogen bonds in the folded protein. Indeed it is possible that these alternative hydrogen bonds are more potent than hydrogen bonds to water because they are intramolecular. As illustrated in Fig. 3, for a hydrogen bond to be net stabilizing, it must be more stable than the alternative hydrogen bonds. For most polar side chains in a helix, a backbone competitor is always readily available.
Figure 3. Native hydrogen bonds must compete with alternative hydrogen bonds.
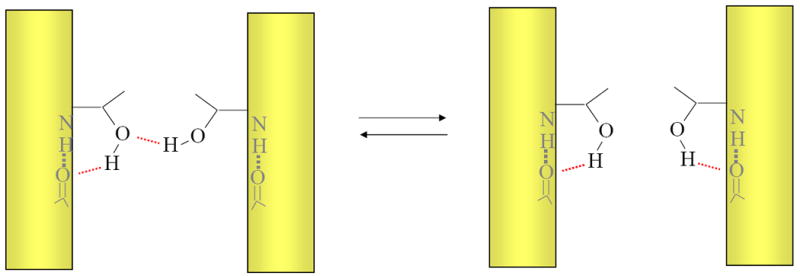
The figure illustrates a hypothetical hydrogen bond made between two threonine side chains on the left must compete with an alternative hydrogen bond. The alternative hydrogen bond could be made in either the folded or unfolded state because transmembrane helical structure can be maintained in unfolded membrane proteins. For the hydrogen bond on the left to be net stabilizing, it must be stronger than the hydrogen bond on the right.
A second argument that hydrogen bonds should be strong in the membrane is that the environment has a low dielectric, which should strengthen electrostatic interactions. No doubt an apolar solvent can strengthen naked hydrogen bonds relative to a polar solvent, but hydrogen bonding is a local interaction that usually occurs buried in a protein environment –a much higher dielectric medium than pure alkanes. Moreover, the interior of membrane proteins often contains considerable water that can form competitive hydrogen bonds. Indeed, the similar polarity in the interior of soluble and membrane proteins argues that hydrogen bond contributions should not be much different in the two protein classes [1,55].
As noted above, hydrogen bonds can be strong if there is evolutionary pressure driving their optimization. It seems reasonable to suppose that the most potent evolutionary pressure for optimizing hydrogen bonds would only occur for functional reasons rather than to stabilize the protein fold. Indeed most polar side chains in membrane proteins seem to be important for function and so there may be strong pressure to interact strongly with substrates rather than with other polar groups in the protein.
Glycine and CαH⋯O hydrogen bonds
Glycine residues are common in TM helix interfaces [9,56-58] and close apposition of glycine residues in transmembrane helix oligomers can allow for the formation of potential CαH⋯O hydrogen bonds [59]. Ab initio quantum mechanics calculations suggest that these special hydrogen bonds could be half as strong as regular hydrogen bonds[60]. Arkin and co-workers measured the change in stretching frequency of hydrogen bonded CαH bond upon dimerization of glycophorin A [61]. The results suggest that this particular interaction could contribute a favorable 0.9 kcal/mol to the dimer. While the magnitude of the estimate is uncertain because it requires extrapolation from much stronger hydrogen bonds, it is apparently a favorable interaction. How can these hydrogen bonds be significant contributors to stability when regular hydrogen bonding appears to be so weak? A major advantage of glycine is that there is no flexibility to form alternative hydrogen bonds like polar side chains. Thus, the lack of competition could make glycine a particular potent partner in these non-traditional hydrogen bonds. Consistent with this view, we investigated the strength of a CαH⋯O hydrogen bond in which the donor oxygen is contributed by a Thr side chain and found that it was not stabilizing [62].
Conclusion
While it appears that the average side chain hydrogen bonding interaction seen in both soluble and membrane proteins are fairly modest contributors to stability, the importance of hydrogen bonds should not be discounted. The strongest hydrogen bonds seen so far in membrane proteins contribute about 2 kcal/mol to protein stability, which are significant interactions that could mean the difference between life and death. Morever, multiple weak interactions can sum to a significant stabilizing influence. An excellent recent example is the network of CαH⋯O that appear to stabilize the helical hairpin formed by the influenza hemagglutinin fusion peptide [63]. Moreover, there is little doubt that hydrogen bonds can be extremely strong if evolutionary pressure favors it [24,25].
Acknowledgments
The author would like to thank Nick Pace for helpful discussions and comments and members of the Bowie lab for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rees DC, DeAntonio L, Eisenberg D. Hydrophobic organization of membrane proteins. Science. 1989;245:510–513. doi: 10.1126/science.2667138. [DOI] [PubMed] [Google Scholar]
- 2.Adamian L, Liang J. Interhelical hydrogen bonds and spatial motifs in membrane proteins: polar clamps and serine zippers. Proteins. 2002;47:209–218. doi: 10.1002/prot.10071. [DOI] [PubMed] [Google Scholar]
- 3.Choma C, Gratkowski H, Lear JD, DeGrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7:161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 4.Gratkowski H, Lear JD, DeGrado WF. Polar side chains drive the association of model transmembrane peptides. Proc Natl Acad Sci USA. 2001;98:880–885. doi: 10.1073/pnas.98.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tatko CD, Nanda V, Lear JD, Degrado WF. Polar networks control oligomeric assembly in membranes. J Am Chem Soc. 2006;128:4170–4171. doi: 10.1021/ja055561a. [DOI] [PubMed] [Google Scholar]
- 6.Dawson JP, Weinger JS, Engelman DM. Motifs of serine and threonine can drive association of transmembrane helices. J Mol Biol. 2002;316:799–805. doi: 10.1006/jmbi.2001.5353. [DOI] [PubMed] [Google Scholar]
- 7.Zhou FX, Cocco MJ, Russ WP, Brunger AT, Engelman DM. Interhelical hydrogen bonding drives strong interactions in membrane proteins. Nat Struct Biol. 2000;7:154–160. doi: 10.1038/72430. [DOI] [PubMed] [Google Scholar]
- 8.Call ME, Wucherpfennig KW. Molecular mechanisms for the assembly of the T cell receptor-CD3 complex. Mol Immunol. 2004;40:1295–1305. doi: 10.1016/j.molimm.2003.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr Opin Struct Biol. 2004;14:465–479. doi: 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Partridge AW, Therien AG, Deber CM. Missense mutations in transmembrane domains of proteins: phenotypic propensity of polar residues for human disease. Proteins. 2004;54:648–656. doi: 10.1002/prot.10611. [DOI] [PubMed] [Google Scholar]
- 11.Li E, You M, Hristova K. FGFR3 dimer stabilization due to a single amino acid pathogenic mutation. J Mol Biol. 2006;356:600–612. doi: 10.1016/j.jmb.2005.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith SO, Smith CS, Bormann BJ. Strong hydrogen bonding interactions involving a buried glutamic acid in the transmembrane sequence of the neu/erbB-2 receptor. Nat Struct Mol Biol. 1996;3:252–258. doi: 10.1038/nsb0396-252. [DOI] [PubMed] [Google Scholar]
- 13.Weiner DB, Liu J, Greene MI. A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature. 1989;339:230–231. doi: 10.1038/339230a0. [DOI] [PubMed] [Google Scholar]
- 14.Choi MY, Cardarelli L, Therien AG, Deber CM. Non-native interhelical hydrogen bonds in the cystic fibrosis transmembrane conductance regulator domain modulated by polar mutations. Biochemistry. 2004;43:8077–8083. doi: 10.1021/bi0494525. [DOI] [PubMed] [Google Scholar]
- 15.Partridge AW, Therien AG, Deber CM. Polar mutations in membrane proteins as a biophysical basis for disease. Biopolymers. 2002;66:350–358. doi: 10.1002/bip.10313. [DOI] [PubMed] [Google Scholar]
- 16.Joh NH, Min A, Faham S, Whitelegge JP, Yang D, Woods VL, Bowie JU. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell JBO, Price SL. The nature of the N &bond; H…O&dbond;C hydrogen bond: An intermolecular perturbation theory study of the formamide/formaldehyde complex. Journal of Computational Chemistry. 1990;11:1217–1233. [Google Scholar]
- 18.Ben-Tal N, Sitkoff D, Topol IA, Yang A, Burt SK, Honig B. Free Energy of Amide Hydrogen Bond Formation in Vacuum, in Water, and in Liquid Alkane Solution. The Journal of Physical Chemistry B. 1997;101:450–457. [Google Scholar]
- 19.Rose GD, Wolfenden R. Hydrogen Bonding, Hydrophobicity, Packing, and Protein Folding. Annu Rev Biophys Biomol Struct. 1993;22:381–415. doi: 10.1146/annurev.bb.22.060193.002121. [DOI] [PubMed] [Google Scholar]
- 20.Tsemekhman K, Goldschmidt L, Eisenberg D, Baker D. Cooperative hydrogen bonding in amyloid formation. Protein Sci. 2007;16:761–764. doi: 10.1110/ps.062609607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheiner S, Hillenbrand EA. Modification of pK values caused by change in H-bond geometry. Proc Natl Acad Sci U S A. 1985;82:2741–2745. doi: 10.1073/pnas.82.9.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MM, Winter G. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:235–238. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 23.Baldwin RL. In search of the energetic role of peptide hydrogen bonds. J Biol Chem. 2003;278:17581–17588. doi: 10.1074/jbc.X200009200. [DOI] [PubMed] [Google Scholar]
- 24.Shan SO, Herschlag D. Hydrogen bonding in enzymatic catalysis: analysis of energetic contributions. Meth Enzymol. 1999;308:246–276. doi: 10.1016/s0076-6879(99)08013-1. [DOI] [PubMed] [Google Scholar]
- 25.Kraut DA, Sigala PA, Fenn TD, Herschlag D. Dissecting the paradoxical effects of hydrogen bond mutations in the ketosteroid isomerase oxyanion hole. Proc Natl Acad Sci USA. 2010;107:1960–1965. doi: 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klotz IM, Franzen JS. Hydrogen Bonds between Model Peptide Groups in Solution. Journal of the American Chemical Society. 1962;84:3461–3466. [Google Scholar]
- 27.Eberhardt ES, Raines RT. Amide-Amide and Amide-Water Hydrogen Bonds: Implications for Protein Folding and Stability. Journal of the American Chemical Society. 1994;116:2149–2150. doi: 10.1021/ja00084a067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez MM, Chin D, Baldwin RL, Makhatadze GI. The enthalpy of the alanine peptide helix measured by isothermal titration calorimetry using metal-binding to induce helix formation. Proc Natl Acad Sci U S A. 2002;99:1298–1302. doi: 10.1073/pnas.032665199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goch G, Maciejczyk M, Oleszczuk M, Stachowiak D, Malicka J, Bierzyński A. Experimental investigation of initial steps of helix propagation in model peptides. Biochemistry. 2003;42:6840–6847. doi: 10.1021/bi027339d. [DOI] [PubMed] [Google Scholar]
- 30.Fu Y, Gao J, Bieschke J, Dendle MA, Kelly JW. Amide-to-E-olefin versus amide-to-ester backbone H-bond perturbations: Evaluating the O-O repulsion for extracting H-bond energies. J Am Chem Soc. 2006;128:15948–15949. doi: 10.1021/ja065303t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao J, Kelly JW. Toward quantification of protein backbone-backbone hydrogen bonding energies: An energetic analysis of an amide-to-ester mutation in an alpha-helix within a protein. Protein Sci. 2008;17:1096–1101. doi: 10.1110/ps.083439708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J, Bosco DA, Powers ET, Kelly JW. Localized thermodynamic coupling between hydrogen bonding and microenvironment polarity substantially stabilizes proteins. Nat Struct Mol Biol. 2009;16:684–690. doi: 10.1038/nsmb.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hildebrand PW, Preissner R, Frömmel C. Structural features of transmembrane helices. FEBS Lett. 2004;559:145–151. doi: 10.1016/S0014-5793(04)00061-4. [DOI] [PubMed] [Google Scholar]
- 34.White SH, Wimley WC. MEMBRANE PROTEIN FOLDING AND STABILITY: Physical Principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 35.Lau FW, Bowie JU. A method for assessing the stability of a membrane protein. Biochemistry. 1997;36:5884–5892. doi: 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- 36.Riley ML, Wallace BA, Flitsch SL, Booth PJ. Slow alpha helix formation during folding of a membrane protein. Biochemistry. 1997;36:192–196. doi: 10.1021/bi962199r. [DOI] [PubMed] [Google Scholar]
- 37.Engelman DM, Chen Y, Chin C, Curran A, Dixon AM, Dupuy AD, Lee AS, Lehnert U, Matthews EE, Reshetnyak YK, et al. Membrane protein folding: beyond the two stage model. FEBS Letters. 2003;555:122–125. doi: 10.1016/s0014-5793(03)01106-2. [DOI] [PubMed] [Google Scholar]
- 38.Haltia T, Freire E. Forces and factors that contribute to the structural stability of membrane proteins. Biochim Biophys Acta. 1995;1241:295–322. doi: 10.1016/0304-4157(94)00161-6. [DOI] [PubMed] [Google Scholar]
- 39.Page RC, Kim S, Cross TA. Transmembrane helix uniformity examined by spectral mapping of torsion angles. Structure. 2008;16:787–797. doi: 10.1016/j.str.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takano K, Yamagata Y, Funahashi J, Hioki Y, Kuramitsu S, Yutani K. Contribution of intra- and intermolecular hydrogen bonds to the conformational stability of human lysozyme(,) Biochemistry. 1999;38:12698–12708. doi: 10.1021/bi9910169. [DOI] [PubMed] [Google Scholar]
- 41.Myers JK, Pace CN. Hydrogen bonding stabilizes globular proteins. Biophys J. 1996;71:2033–2039. doi: 10.1016/S0006-3495(96)79401-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takano K, Scholtz JM, Sacchettini JC, Pace CN. The contribution of polar group burial to protein stability is strongly context-dependent. J Biol Chem. 2003;278:31790–31795. doi: 10.1074/jbc.M304177200. [DOI] [PubMed] [Google Scholar]
- 43.Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 44.Albeck S, Unger R, Schreiber G. Evaluation of direct and cooperative contributions towards the strength of buried hydrogen bonds and salt bridges. J Mol Biol. 2000;298:503–520. doi: 10.1006/jmbi.2000.3656. [DOI] [PubMed] [Google Scholar]
- 45.Zhou FX, Merianos HJ, Brunger AT, Engelman DM. Polar residues drive association of polyleucine transmembrane helices. Proc Natl Acad Sci USA. 2001;98:2250–2255. doi: 10.1073/pnas.041593698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choma C, Gratkowski H, Lear JD, DeGrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7:161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 47.Bowie JU. Understanding membrane protein structure by design. Nat Struct Biol. 2000;7:91–94. doi: 10.1038/72454. [DOI] [PubMed] [Google Scholar]
- 48.Gratkowski H, Lear JD, DeGrado WF. Polar side chains drive the association of model transmembrane peptides. Proc Natl Acad Sci USA. 2001;98:880–885. doi: 10.1073/pnas.98.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanley AM, Fleming KG. The role of a hydrogen bonding network in the transmembrane beta-barrel OMPLA. J Mol Biol. 2007;370:912–924. doi: 10.1016/j.jmb.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 50.Fleming KG, Engelman DM. Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc Natl Acad Sci U S A. 2001;98:14340–14344. doi: 10.1073/pnas.251367498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fleming PJ, Rose GD. Do all backbone polar groups in proteins form hydrogen bonds? Protein Sci. 2005;14:1911–1917. doi: 10.1110/ps.051454805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 53.Hong H, Joh NH, Bowie JU, Tamm LK. Methods for measuring the thermodynamic stability of membrane proteins. Meth Enzymol. 2009;455:213–236. doi: 10.1016/S0076-6879(08)04208-0. [DOI] [PubMed] [Google Scholar]
- 54.Chamberlain AK, Bowie JU. Analysis of side-chain rotamers in transmembrane proteins. Biophys J. 2004;87:3460–3469. doi: 10.1529/biophysj.104.044024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adamian L, Nanda V, DeGrado WF, Liang J. Empirical lipid propensities of amino acid residues in multispan alpha helical membrane proteins. Proteins. 2005;59:496–509. doi: 10.1002/prot.20456. [DOI] [PubMed] [Google Scholar]
- 56.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 57.Walters RFS, DeGrado WF. Helix-packing motifs in membrane proteins. Proc Natl Acad Sci USA. 2006;103:13658–13663. doi: 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S, Jeon T, Oberai A, Yang D, Schmidt JJ, Bowie JU. Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc Natl Acad Sci USA. 2005;102:14278–14283. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Senes A, Ubarretxena-Belandia I, Engelman DM. The Calpha ---H…O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vargas R, Garza J, Dixon DA, Hay BP. How Strong Is the Cα–H⋯OC Hydrogen Bond? Journal of the American Chemical Society. 2000;122:4750–4755. [Google Scholar]
- 61.Arbely E, Arkin IT. Experimental measurement of the strength of a C alpha-H…O bond in a lipid bilayer. J Am Chem Soc. 2004;126:5362–5363. doi: 10.1021/ja049826h. [DOI] [PubMed] [Google Scholar]
- 62.Yohannan S, Faham S, Yang D, Grosfeld D, Chamberlain AK, Bowie JU. A C alpha-H…O hydrogen bond in a membrane protein is not stabilizing. J Am Chem Soc. 2004;126:2284–2285. doi: 10.1021/ja0317574. [DOI] [PubMed] [Google Scholar]
- 63.Lorieau JL, Louis JM, Bax A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc Natl Acad Sci USA. 2010;107:11341–11346. doi: 10.1073/pnas.1006142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fernández-Recio J, Romero A, Sancho J. Energetics of a hydrogen bond (charged and neutral) and of a cation-pi interaction in apoflavodoxin. J Mol Biol. 1999;290:319–330. doi: 10.1006/jmbi.1999.2863. [DOI] [PubMed] [Google Scholar]
- 65.Campos LA, Cuesta-López S, López-Llano J, Falo F, Sancho J. A double-deletion method to quantifying incremental binding energies in proteins from experiment: example of a destabilizing hydrogen bonding pair. Biophys J. 2005;88:1311–1321. doi: 10.1529/biophysj.104.050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jang DS, Cha HJ, Cha S, Hong BH, Ha N, Lee JY, Oh B, Lee H, Choi KY. Structural double-mutant cycle analysis of a hydrogen bond network in ketosteroid isomerase from Pseudomonas putida biotype B. Biochem J. 2004;382:967–973. doi: 10.1042/BJ20031871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marqusee S, Sauer RT. Contributions of a hydrogen bond/salt bridge network to the stability of secondary and tertiary structure in lambda repressor. Protein Sci. 1994;3:2217–2225. doi: 10.1002/pro.5560031207. [DOI] [PMC free article] [PubMed] [Google Scholar]