

Abstract
We report the synthesis and preliminary characterization of “clickable” inhibitors of human transglutaminase 2 (TG2). These inhibitors possess the 3-halo-4,5-dihydroisoxazole warhead along with bio-orthogonal groups such as azide or alkyne moieties that enable subsequent covalent modification with fluorophores. Their mechanism for inhibition of TG2 is based on halide displacement, resulting in the formation of a stable imino thioether. Inhibition assays against recombinant human TG2 revealed that some of the clickable inhibitors prepared in this study have comparable specificity as benchmark dihydroisoxazole inhibitors reported earlier. At low micromolar concentrations they completely inhibited transiently activated TG2 in a WI-38 fibroblast scratch assay, and could subsequently be used to visualize the active enzyme in situ. The potential use of these inhibitors to probe the role of TG2 in celiac sprue as well as other diseases is discussed.
Mammalian transglutaminases play important roles in many biological processes such as blood clotting, wound healing, extracellular matrix formation, and cell adhesion and motility (Akimov and Belkin, 2001; Fesus et al., 1987; Iismaa et al., 2009; Lorand, 2007; Lorand and Graham, 2003; Piacentini et al., 1991; Zemskov et al., 2006). Transglutaminase 2 (TG2), a ubiquitous member of this enzyme family, is found in intracellular as well as extracellular environments of many organs. In the presence of calcium and the absence of GTP or GDP, TG2 cross-links selected -glutaminyl and -lysine residues on proteins, leading to the formation of proteolytically resistant covalent bonds. This catalytically active form of TG2 exists in an extended (“open”) conformation (Pinkas et al., 2007). In contrast, in the absence of calcium and the presence of GTP or GDP, TG2 assumes an inactive (“closed”) form (Liu et al., 2002), and functions as a G protein in the phospholipase C signal transduction cascade of the -adrenergic receptor (Nakaoka et al., 1994; Singh et al., 1995). TG2 activity is also subject to regulation by local redox conditions (Stamnaes et al., 2010). Under oxidizing conditions, the formation of an intramolecular disulfide bond inactivates the enzyme; activity is reversibly restored under reducing conditions. Given the ubiquitous nature of TG2 and its ability to form covalent “scars”, its catalytic activity must be tightly regulated in mammals. However, the physiological signals that interconvert these active and inactive forms of the enzyme remain largely unknown. For example, whereas the majority of extracellular TG2 in the mouse small intestine is inactive, it can be rapidly activated at the villus tips by administration of poly(I:C), a ligand of the TLR-3 innate immune receptor (Siegel et al., 2008). Chemical probes of in vivo TG2 biology should therefore be of considerable use in resolving some of these fundamental mysteries associated with allosteric TG2 regulation.
From a translational standpoint, TG2 is implicated in the pathogenesis of a number of unrelated disorders including neurological diseases such as Huntington’s, Alzheimer’s and Parkinson’s disease, certain types of cancers and renal diseases, and celiac sprue (Ruan and Johnson, 2007; Shweke et al., 2008; Verma and Mehta, 2007). For example, in celiac sprue, peptides derived from dietary gluten are deamidated by TG2 to enhance their affinity towards the disease-associated class II major histocompatibility complexes, which in turn activates inflammatory Th1 cells (Sollid and Lundin, 2009). Therefore, TG2 may be a suitable target for celiac sprue therapy. However, absent knowledge of the location, timing and mechanism of TG2 activation in the celiac small intestine, this hypothesis cannot be rationally tested. Again, chemical probes could provide answers to these questions. Last but not least, small molecule TG2 inhibitors could also serve as drug leads and eventually as drug candidates for one or more of the above diseases (Siegel and Khosla, 2007).
Many classes of irreversible, active site inhibitors of human TG2 have been reported thus far (Case et al., 2005; Choi et al., 2005; Freund et al., 1994; Halim et al., 2007; Hausch et al., 2003; Keillor, 2005; Pardin et al., 2006; Pardin et al., 2008; Schaertl et al.). One example is the 3-bromo-4,5-dihydroisoxazoles (DHI) family of compounds (Castelhano et al., 1988; Choi et al., 2005; Watts et al., 2006). Guided by the excellent tolerance of these molecules in acute and chronic administration studies in rodents (Choi et al., 2005; Yuan et al., 2007), here we have used the DHI scaffold to develop a class of covalent TG2 probes that bind to active but not inactive TG2 and can therefore be used to visualize the enzyme via a selective (bioorthogonal) chemical reaction (Figure 1). Analogous strategies have been used to modify cell surface glycans and proteins (Chin et al., 2002; Deiters and Schultz, 2005; Wang and Schultz, 2005), to profile protein glycosylation (Agard and Bertozzi, 2009), and for the detection of lipid-modified proteins (Hang et al., 2007). Recently, bioorthogonal chemical reporters have also been used in living animals as noninvasive imaging tools (Laughlin et al., 2008; Laughlin and Bertozzi, 2009; Prescher and Bertozzi, 2005).
Figure 1.

Labeling and visualization of catalytically active (but not catalytically inactive) transglutaminase 2 with alkynyl-DHI inhibitors. Similar labeling strategy can be employed using azido-DHI inhibitors.
RESULTS
Design and Synthesis of Azido- and Alkynyl-DHI inhibitors
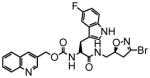
In this study we describe the synthesis and properties of 3-bromo-4,5-dihydroisoxazole inhibitors of human TG2 that are conjugated to alkyne or azide functional groups. We incorporated alkyne and azide groups because of their bioorthogonality (Sletten and Bertozzi, 2009). Figure 2 describes the structure and synthesis of a representative DHI inhibitor. Both azide and alkyne groups were introduced as substitutions on the carbobenzyloxy (Cbz) moiety, because previous studies had highlighted the advantage of an aromatic group at this position (Watts et al., 2006). To identify a clickable inhibitor with optimal activity, we also varied the amino acid moiety of our DHI inhibitors using side-chains that had previously been found to be beneficial. The compounds synthesized and evaluated in this study are summarized in Table 1; all of them were made in good yield and purity (Supporting Information; Schemes S1-S2). Four previously synthesized DHI inhibitors, 13–16, were chosen as reference compounds in this study; their TG2 inhibitory activities vary over a 8-fold range (Table 1).
Figure 2.

General synthesis of azido- and alkynyl-DHI inhibitors. Supported by Figure S1.
Table 1.
TG2 inhibitory activity of enantiopure azido- and alkynyl-DHI compounds. Compounds 17 and 18 have a stereochemically altered DHI moiety, and are therefore considerably less potent than their diastereomeric analogs, 11 and 12, respectively.
| Cmpd | K I(μM) | kinh (min−1) | kinh/KI(mM−1 min−1) | Cmpd | K I(μM) | kinh (min−1) | kinh/KI(mM−1 min−1) |
|---|---|---|---|---|---|---|---|
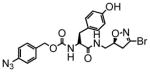 3 |
280 | 1.63 | 5.8 |
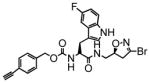 11 |
7.0 | 0.13 | 19 |
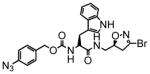 4 |
15 | 0.23 | 15 |
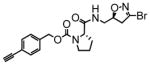 12 |
1.8 | 0.052 | 30 |
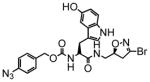 5 |
100 | 0.82 | 8.0 |
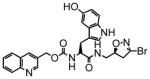 13 |
41 | 0.51 | 12 |
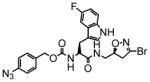 6 |
19 | 0.31 | 16 |
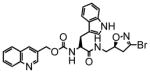 14 |
47 | 0.53 | 11 |
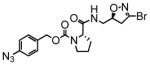 7 |
9.2 | 0.12 | 13 |
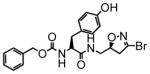 15 |
22 | 0.13 | 6.1 |
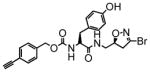 8 |
4.2 | 0.016 | 4.0 |
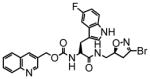 16 |
19 | 0.73 | 38 |
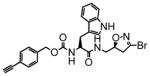 9 |
13 | 0.15 | 11 |
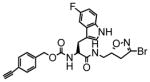 17 |
51 | 0.21 | 4.2 |
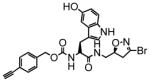 10 |
12 | 0.14 | 8.0 |
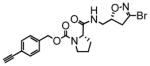 18 |
38 | 0.27 | 7.2 |
Inhibition of Recombinant Human TG2 by Azido- and Alkynyl-DHI inhibitors
The potencies of alkynyl- and azido-DHI inhibitors 1–12 were evaluated in a kinetic assay using recombinant human TG2. In this assay, TG2-catalyzed deamidation of the dipeptide substrate Cbz-Gln-Gly is monitored under steady-state conditions. Analogous to the parameters of the Michaelis-Menten equation, the first order rate constant kinh is a measure of the reactivity of the DHI warhead with the active site sulfhydryl of TG2, whereas Ki estimates the reversible binding affinity of the inhibitor to the active site. The bimolecular rate constant kinh/Ki can be thought of as the specificity constant of TG2 for the inhibitor.
As summarized in Table 1, inhibitors harboring tryptophan analogs are more potent than those with tyrosine as the amino acid residue. Amongst tryptophan analogs, the 5-fluorotryptophan analogs have the highest specificity constants. The relative trends are similar between the azido-DHI and alkynyl-DHI inhibitor series. The most active alkynyl- and azido-DHI inhibitors (12 and 6, respectively) have specificity constants that are within 1.3 and 2.3-fold, respectively, of the most potent DHI inhibitor 16 identified to date. Together, these results encouraged us to evaluate some of these compounds in a cell culture assay for activated TG2.
It is noteworthy that the azido-DHI inhibitors prepared in this study have moderately higher TG2 specificity (kinh/Ki) than their alkynyl-DHI counterparts (Table 1). However, this benefit is primarily derived from higher reactivity towards the active site cysteine residue of TG2; the reversible binding affinity (Ki) of the alkynyl-DHI inhibitors is actually higher. Although the reason for this inverse correlation between affinity and specificity remains to be understood, in subsequent studies we placed greater emphasis on biological evaluations of the most promising alkynyl-DHI inhibitors.
Inhibition of TG2 in a Tissue Culture Wounding Model
To evaluate the activity of the clickable TG2 inhibitors identified above in a representative biological assay, we used a tissue culture wounding model. TG2 plays a role in wound healing in mammals by cross-linking extracellular matrix proteins after a wound has been inflicted. Recently, we demonstrated that, when a confluent monolayer of WI-38 fibroblast cells is scratched, TG2 is rapidly and transiently activated in the zone surrounding the “wound” (Siegel et al., 2008). This activated TG2 enzyme can be visualized using 5-biotinyl pentylamine (5BP), which presumably cross-links onto glutamine side-chains of fibronectin and other proteins along the scratch border that are recognized as TG2 substrates (Figure 3). Here we have exploited this assay to compare the relative activities of TG2 inhibitors in a biological system.
Figure 3.

Titration of 11 against active TG2 in a fibroblast scratch assay. TG2 activity was visualized in situ after scratching a confluent WI-38 monolayer with a small pipette tip. Significant TG2 activity was detected around the wound in the presence of 300 μM 5BP and vehicle (DMSO) (a-c). Clear inhibition of active TG2 was observed in the presence of 6.25 μM 11 (g-i), and complete inhibition was observed at or above 12.5 μM 11 (j-o). In this assay, active TG2 was detected by exposing fixed cultures to streptavidin Alexa fluor 555 (a, d, g, j, m). The scratch geometry is conveniently visualized by co-staining with polyclonal anti-fibronectin antibody, followed by a secondary antibody conjugated Alexa fluor 488 (b, e, h, k, n). Overlays of the left and middle images are in the right column (c, f, i, l, o). Scale bar represents 200 μm and applies to all panels.
To quantify inhibitor potency, selected inhibitors were added to scratched WI-38 cultures at incremental two-fold dilutions. The lowest inhibitor concentration at which complete inhibition of transiently activated TG2 was observed was recorded as the minimum inhibitory concentration (MIC) of that inhibitor. For example, the effect of increasing concentrations of 11 on the extent of 5BP cross-linking around WI-38 scratches is shown in Figure 3; from this data, its MIC was estimated to lie between 6.25–12.5 μM. The MIC values of different inhibitors are compared in Table 2 (see also Figures S2 and S3 in Supporting Information). Whereas the most potent inhibitors (e.g., 6, 11, 14 and 16) completely blocked TG2 activity around the wound at 3.1–6.25 μM, the less potent inhibitor 15 (kinh/KI= 6.1 mM−1 min−1) only showed partial inhibition at 25 μM.
Table 2.
Titration of DHI inhibitors against TG2 in a fibroblast wound model. Each value is a representative of 3 or more scratches. Supported by Figure S2 and S3.
| Cmpd | MIC(μM) |
|---|---|
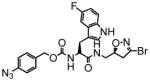 6 |
[6.25–12.5] |
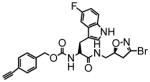 11 |
[6.25–12.5] |
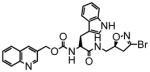 14 |
[6.25–12.5] |
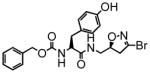 15 |
[50–75] |
 16 |
[3.1–6.25] |
The above cell culture measurements verified that the biological potencies of the most active azido-DHI inhibitor (6) and the most active alkynyl-DHI inhibitors (11 and 12) were comparable to the benchmark DHI inhibitor of human TG2 (16) identified to date (Watts et al., 2006). This in turn encouraged us to develop procedures for visualizing inhibitor-bound TG2 in biological samples, as described below. Future improvements to these compounds will focus on improving their water solubility and rendering them resistant to chymotrypsin-catalyzed hydrolysis. Both properties would be necessary for chronic oral administration in animal studies directed at elucidating TG2 regulatory mechanisms in the small intestine, and can be achieved by incorporating alternative amino acid residues or isosteres in place of 5-fluorotryptophan in compounds 6 and 11.
Labeling and Visualization of Catalytically Active TG2
In principle, the alkyne and azide groups incorporated into DHI inhibitors can be used as handles to selectively visualize the location of TG2 protein that is transiently activated in a biological sample. To test this hypothesis, alkynyl inhibited TG2 was chemoselectively ligated to an azide-tagged biotin or fluorophore molecule via a Cu1-catalyzed alkyne-azide [3+2] cycloaddition reaction (Figure 1). Initial experiments were performed with recombinant TG2 protein activated with Ca+2 or inhibited with GTP. The proteins were resolved by gel electrophoresis, and visualized by Western blotting using neutravidin-linked horseradish peroxidase. Catalytically active TG2 treated with 12 can be efficiently conjugated with a suitable azide partner molecule, and subsequently visualized in a dose-dependent manner (Figure 4, lanes 1–4). The limit of detection of active TG2 by this Western blotting method was below 1 g. GTP-bound, catalytically inactive TG2 was not labeled, and therefore not visualized (Figure 4, lanes 5–8).
Figure 4.

Visualization of recombinant TG2 with alkynyl-DHI inhibitor 12. Recombinant TG2 was inhibited with 12 in the presence of CaCl2 (5 mM) or GTP (500 μM) and MgCl2 (1 mM). The resulting mixture was reacted with biotin-azide, resolved by SDS-PAGE, and detected by Western blotting with neutravidin-HRP. Lane 1:TG2 (0.2μg) + Ca2+ + 12, lane 2:TG2 (0.4μg) + Ca2+ + 12, lane 3:TG2 (1μg) + Ca2+ + 12, lane 4:TG2 (4μg) + Ca2+ + 12, lane 5:TG2 (0.4μg) + GTP/MgCl2 + 12, lane 6:TG2 (1μg) + GTP/MgCl2 + 12, lane 7: TG2 (1μg) + GTP/MgCl2 + 12, lane 8: TG2 (4μg)+GTP/MgCl2 + 12.
To investigate the utility of this method for visualizing catalytically active TG2 in biological systems, we performed fluorescence microscopy on scratched WI-38 fibroblast cultures that had been incubated with two representative alkynyl-DHI inhibitors, 11 and 12. In both cases, cells were extensively washed to remove excess inhibitor, fixed with paraformaldehyde, permeabilized with 0.1% Triton X-100, washed again, and subjected to the [3+2] cycloaddition reaction with biotin azide TG2 (active + inactive). The biotin conjugate was visualized by exposing fixed cultures to streptavidin conjugated with Alexa fluor 555. The scratch was visualized with rabbit anti-fibronectin antibody, followed by exposure to an Alexa fluor 488-conjugated goat anti-rabbit IgG secondary antibody.
Analogous to experiments with 5BP (see, for example, Figure 3), a strong red fluorescence signal was observed around the scratch in samples treated with inhibitors and reacted with biotin azide (Figure 5 and Figure S4, Supporting Information), corresponding to catalytically active TG2. As negative controls, DMSO vehicle and two substantially weaker inhibitors, 17 and 18 (Table 1), were evaluated. These compounds, which are diastereomeric analogs of 11 and 12, respectively, were used because earlier work had shown that TG2 specificity was strongly dependent upon the stereochemistry of the chiral center in the dihydroisoxazole ring. Scratches treated with 17 and 18 resulted into no red fluorescence around the scratch (Figure 5 and Figure S4, Supporting Information).
Figure 5.

Visualization of catalytically active TG2 in a scratched WI-38 fibroblast culture. TG2 activity was visualized after scratching confluent WI-38 monolayers in the presence of 50 μM 11 (a-f) or 17 (g-i) for 1 h. Cells were then fixed, permeabilized, reacted with biotin azide followed by strepatavidin-Alexa fluor 555 conjugates (red), and imaged by epifluorescence microscopy through a 10x objective. (a, d, g) Rabbit anti-fibronectin antibody, followed by a secondary antibody labeled with Alexa fluor 488, were used to visualize fibronectin on the WI-38 monolayers. (b, e, h) Catalytically active TG2 was visualized by coupling the alkynyl-DHI inhibitor to biotin azide. (c, f, i) Overlays of the left and middle image pairs are shown in the right column. Scale bar represents 200 μm and applies to all panels. Supported by Figure S4 and S5.
Two other observations were made during the course of these studies with WI-38 fibroblasts. First, the signal-to-noise was significantly superior in scratches reacted with biotin azide as compared to rhodamine azide (Figure S5). The reason for this difference was not investigated. Second, 5BP yields a distinctly different labeling pattern around the fibroblast scratch than the clickable inhibitor. For example, whereas, the overlap between red and green signals is excellent in Figure 3a, the two colors are proximal but clearly discernable in Figure 5e. Presumably, this is due to subtle differences in the distribution of catalytically active TG2 (i.e., the target of the clickable inhibitor) and its substrates (principally fibronectin, which is the target of 5BP) in the vicinity of the scratch.
DISCUSSION
Human transglutaminase 2 (TG2) catalyzes transamidation and deamidation reactions that are believed to contribute to a variety of physiological processes as well as to the pathogenesis of several diseases. Therefore, understanding the mechanisms responsible for spatio-temporal regulation of TG2 activity could have important implications for human biology and disease. However, because the protein is abundantly expressed in most organs and tissues, most mechanisms of interest are allosteric in nature, and conventional molecular biology approaches are not applicable. Consequently, new tools are needed to visualize rapid, tissue-specific transitions of TG2 from a catalytically inactive to an active state (or vice versa). Because TG2 is localized in multiple intracellular compartments as well as the extracellular environment, the ideal tools should also be able to resolve the sub-cellular location of the target protein. Here we have described a new class of small molecule reagents that are well suited for such a task. As shown, our clickable TG2 inhibitors can visualize the active enzyme in a manner that is only limited by the resolution of optical microscopy. Specifically, we have shown that compounds 11 and 12 can be used to visualize the burst of TG2 catalytic activity that rapidly appears at the scratch boundary of a WI-38 fibroblast cell culture system. Although the fibroblast scratch assay mimics a pathological, rather than a physiological state, its utility for interrogating the allosteric transition of TG2 from an inactive to an active state has been established (Nicholas et al., 2003; Siegel et al., 2008). Our method has two principal advantages over previous studies that visualized transiently activated TG2 around scratched cells using amine substrates such as 5BP. First, whereas the use of amines only enables detection of TG2 substrates (principally fibronectin) that have undergone modification by catalytically active enzyme, our new method enables visualization of the activated enzyme itself. As illustrated by the differences between the labeling patterns around scratches in Figures 3 and 5, these differences may prove significant in further investigations of TG2 biology, especially in situations where sub-cellular resolution of the active enzyme is desired. Second, because the specificity of transglutaminases for their electrophilic (glutamine-bearing) substrates is considerably higher than for nucleophilic (amine-bearing) substrates, the development of isozyme-specific clickable DHI inhibitors should be feasible.
Pending verification of their in vivo stability, several important biological and medical problems could be addressed with clickable DHI inhibitors such as 11 and 12. In metastatic cancers, TG2 is believed to play a crucial role in cell migration, and has been shown to localize primarily at the leading edge of migrating cells (Antonyak et al., 2009). However, the relative importance of its catalytic versus signaling activity is unclear. For example, the catalytically inactive C277S mutant of TG2 is indistinguishable from the wild-type protein with respect to cell motility(Balklava et al., 2002). Visualizing the precise location(s) of catalytically active TG2 over the course of a nascent and metastasizing tumor could shed important light on the role of protein cross-linking in cancer. In celiac sprue, the pathogenic role of catalytic TG2 is well established; however, the precise location at which catalytically active TG2 encounters immunotoxic gluten peptides remains a topic of controversy. Again, small molecule probes that can highlight the distribution of active enzyme in normal versus inflamed intestinal mucosa could be invaluable. More fundamentally, biochemical and structural biological investigations have demonstrated that TG2 undergoes allosteric changes between at least three distinct conformational states, as summarized in the scheme below:
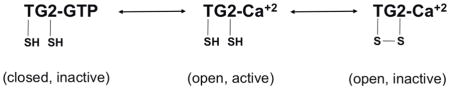
Tools that can distinguish between the Ca+2-bound active state versus the GTP-bound or disulfide bonded inactive states could greatly facilitate a deeper understanding of the dynamics of this multifunctional protein in biological systems.
In conclusion, we also note that our lead clickable inhibitors reported here could be further improved with respect to several characteristics. First, they have relatively low solubility in aqueous buffers. The efficiency of labeling TG2-bound inhibitors via [3+2] cycloaddition could also be improved by varying the spacer length between the alkyne and the benzyl groups. Last but not least, the reactivity of these inhibitors with other mammalian transglutaminases could be further engineered in order to develop probes with orthogonal specificity for individual isozymes of this protein family.
SIGNIFICANCE
We have developed non-radioactive chemical probes for detecting and imaging catalytically active transglutaminase 2 (TG2) in mammalian cells and tissues. A series of electrophilic dihydroisoxazole compounds have been engineered with alkyne and azide groups that can be further modified by “click chemistry” (Cu1-catalyzed alkynyl-azide [3+2] cycloaddition). These compounds have high specificity for the TG2 active site, and undergo an irreversible reaction with the catalytically active cysteine residue. The biological utility of these mechanism-based irreversible inhibitors is demonstrated by their ability to visualize transiently activated TG2 in a fibroblast scratch assay. These probes should be broadly useful in elucidating mechanisms responsible for allosteric regulation of this multifunctional protein in cell biology, mammalian development, as well as a variety of human diseases.
Experimental Procedures
Cell Culture and Antibodies
WI-38 fibroblasts were grown in minimum essential media (Gibco, 11095) containing L-glutamine, Earle’s salts, non-essential amino acids (Gibco, 11140), sodium pyruvate (Gibco, 11360), 10% fetal bovine serum (FBS) (Atlanta Biologicals), and 100 U/ml penicillin + 100 μg/ml Streptomycin (Gibco, 15140). Cells were typically grown to 80–90% confluency before treating with trypsin-EDTA (Gibco 15400) and splitting. All cells were grown in a humidified incubator at 37 °C, 5% CO2. The antibodies used were anti-fibronectin antibody produced in Rabbit (Sigma, F3648), Alexa fluor 488 goat anti-rabbit IgG (H-L) antibody (Invitrogen, A-11008), and Alexa fluor 555 labeled streptavidin (Invitrogen, S-21381).
Synthesis of alkynyl- and azido-DHI inhibitors
Compounds 3–18 were synthesized as previously reported(Watts et al., 2006). Briefly, 4-ethynylbenzyl alcohol was reacted with p-nitrophenyl choloroformate in methylene chloride and N-methylmorpholine to give a carbonate of 4-ethynyl benzyl and p-nitrophenol after purification by flash chromatography. The carbonate was reacted with the methyl ester of the amino acid in DMF and N-methylmorpholine to give 4-ethynyl benzyl carbamate protected amino acid methyl ester. Methyl ester hydrolysis, followed by EDCI coupling of resulting acid to the (S)-isomer of the halo-dihydroisoxazole, gave the indicated compound as a final product after flash chromatography purification. The azido-DHI compounds were synthesized in similar fashion.
TG2 inhibition assays
Recombinant human TG2 was expressed in E. coli and purified to >90% homogeneity as described previously(Choi et al., 2005). The potency of individual DHI inhibitors was assayed in a reaction mixture containing 200 mM MOPS (pH 7.2), 5 mM CaCl2, 1 mM EDTA, 10 mM -ketoglutarate, 18 U/ml glutamate dehydrogenase, 0.4 mM NADH, 3.3% (v/v) DMSO, 0.5 μM TG2, 10, 20, and 30 mM Cbz-Gln-Gly (KM = 6.28 mM, kcat. = 37 min−1). The concentration of each inhibitor was varied from 0.001 to 0.3 mM. The reaction was initiated by adding TG2, and the consumption of NADH was monitored by UV spectroscopy (340 nm, ε = 6220 cm−1 M−1). Kinetic parameters were obtained by plotting the reaction progress curves against theoretical equations for irreversible enzyme inhibition.
Labeling and visualization of TG2 with clickable inhibitors
TG2 protein was subjected to the probe labeling in 50 μl volume reaction containing a final concentration of 2 mM of 12, and 5 mM CaCl2 or 500 mM GTP/1 mM MgCl2 in 100 mM Tris HCl, pH 7.2, for 1h at RT. Then, 2 mM of biotin azide was added, 1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich) dissolved in water, 0.2 mM tris[(1-benzyl-1H-1,2,3-triazole-4-yl)methyl amine (TBTA, Sigma-Aldrich) dissolved in DMSO/tert-butanol (20%/80%) and 1 mM CuSO4 in PBS. The reaction was quenched with 2X loading buffer. Samples were resolved by SDS-PAGE using 4–20% Tris-glycine gels, and transferred onto a nitrocellulose membrane. The membrane was blocked with PBS, 0.1% Tween-20 (PBST), and 5% non-fat dried milk for 1 h at RT. The membrane was washed three times with PBST (5 min each), and incubated with neutravidin horseradish peroxidase (Invitrogen, 1:200 in 5% non-fat dried milk) for 1 h at RT. The membrane was washed again with PBST three times (10 min each) and developed according to manufacturer’s recommendation (Amersham Biosciences).
Detection of TG2 activity in fibroblast scratch assays using 5-biotinyl pentylamine (5BP)
WI-38 fibroblasts were plated in an 8-well chamber glass slide at 2 × 105 cells/well and grown for 3 days with medium change every 2 days. Small scratches were made in the monolayer with 0.1–10 μl pipette tip, and the cells were incubated with 5BP for 1 h at 37°C, 5% CO2. To capture the highest TG2 activity around the wound, we performed titration assays using 50, 100, 150 and 300 μM 5BP; the best results were obtained at 300 μM 5BP. To quantify TG2 inhibition in cultured cells, inhibitors 3–18 were added at the indicated concentrations to the culture media for 10–15 min. An equal amount of vehicle (DMSO) was added to the media of control cells at 37 °C. Then, 5BP was added at a final concentration 300 μM, and incubated for an additional 1 h at 37 °C, 5% CO2. The cells were washed three times with PBS and fixed with −20 °C methanol for 10 min. Fixed cells were washed again with PBS twice for 5 min, blocked with 1% BSA in PBS for 15 min at room temperature, and washed twice more with PBS. Anti-fibronectin antibody produced in Rabbit (F3648, Sigma-Aldrich) was diluted 1:500 in blocking buffer and incubated for 1 h at RT. Cells were washed three times with PBS and incubated with Alexa fluor 488 goat anti rabbit IgG (H+L) (A-11034, Invitrogen) and Alexa fluor 555 streptavidin conjugate in blocking buffer for 1 h at RT. After three further washes with PBS, 300 μl PBS was added onto each sample before visualization via fluorescence microscopy. Note that Factor XIII, another ubiquitous member of the transglutaminase family, is not expressed in WI-38 fibroblasts (data not shown).
Detection of TG2 activity in fibroblast scratch assays using clickable inhibitors
Following WI-38 fibroblast cell growth and scratching as above, compounds 11–12 were added to living cells for 10–15 min at 50 μM final concentration at 37 °C, 5% CO2. Control cells were incubated with 300 μM 5-BP and vehicle (DMSO). Cells were washed three times with PBS to remove excess compounds and fixed with 4% paraformaldehyde (PFA) for 10 min at RT. Cells were then permeabilized with PBS/0.1% Triton X-100 for 1–2 min at RT, washed extensively with PBS, and subjected to the [3+2] cycloaddition reaction at RT for 1 h in 175 μl solution containing 1 mM biotin azide, 1 mM TCEP dissolved in water, and 1 mM CuSO4 in PBS. The labeled samples were rinsed extensively with PBS, and blocked in PBS/5% BSA for 45 min at RT. To visualize the fibronectin protein, samples were treated with rabbit anti-fibronectin for 1 h at RT in PBS/5%BSA, then washed three times with PBS, and further stained with the goat anti-Rabbit Alexa fluor 488 conjugated secondary antibody for 45 min in PBS/5% BSA at RT. After three further washes, 300 μl of PBS was added onto the cells for visualization via microscopy. Fluorescent images were captured in an upright Zeiss microscope.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NIH (R01 DK063158) to C.K. L.D. is a recipient of a postdoctoral fellowship from “Fonds de Recherche sur la Nature et les Technologies” (Canada).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agard NJ, Bertozzi CR. Chemical Approaches To Perturb, Profile, and Perceive Glycans. Acc Chem Res. 2009;42:788–797. doi: 10.1021/ar800267j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimov SS, Belkin AM. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fibronectin. Blood. 2001;98:1567–1576. doi: 10.1182/blood.v98.5.1567. [DOI] [PubMed] [Google Scholar]
- Antonyak MA, Li B, Regan AD, Feng QY, Dusaban SS, Cerione RA. Tissue Transglutaminase Is an Essential Participant in the Epidermal Growth Factor-stimulated Signaling Pathway Leading to Cancer Cell Migration and Invasion. J Biol Chem. 2009;284:17914–17925. doi: 10.1074/jbc.M109.013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balklava Z, Verderio E, Collighan R, Gross S, Adams J, Griffin M. Analysis of tissue transglutaminase function in the migration of swiss 3T3 fibroblasts - The active-state conformation of the enzyme does not affect cell motility but is important for its secretion. J Biol Chem. 2002;277:16567–16575. doi: 10.1074/jbc.M109836200. [DOI] [PubMed] [Google Scholar]
- Case A, Ni J, Yeh LA, Stein RL. Development of a mechanism-based assay for tissue transglutaminase - results of a high-throughput screen and discovery of inhibitors. Anal Biochem. 2005;338:237–244. doi: 10.1016/j.ab.2004.09.047. [DOI] [PubMed] [Google Scholar]
- Castelhano AL, Billedeau R, Pliura DH, Bonaventura BJ, Krantz A. Synthesis, Chemistry, And Absolute-Configuration Of Novel Transglutaminase Inhibitors Containing A 3-Halo-4,5-Dihydroisoxazole. Bioorg Chem. 1988;16:335–340. [Google Scholar]
- Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG. Addition of p-azido-L-phenylaianine to the genetic code of Escherichia coli. J Am Chem Soc. 2002;124:9026–9027. doi: 10.1021/ja027007w. [DOI] [PubMed] [Google Scholar]
- Choi K, Siegel M, Piper JL, Yuan L, Cho E, Strnad P, Omary B, Rich KM, Khosla C. Chemistry and biology of dihydroisoxazole derivatives: Selective inhibitors of human transglutaminase 2. Chem Biol. 2005;12:469–475. doi: 10.1016/j.chembiol.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Deiters A, Schultz PG. In vivo incorporation of an alkyne into proteins in Escherichia coli. Bioorg Med Chem Lett. 2005;15:1521–1524. doi: 10.1016/j.bmcl.2004.12.065. [DOI] [PubMed] [Google Scholar]
- Fesus L, Thomazy V, Falus A. Induction And Activation Of Tissue Transglutaminase During Programmed Cell-Death. FEBS Lett. 1987;224:104–108. doi: 10.1016/0014-5793(87)80430-1. [DOI] [PubMed] [Google Scholar]
- Freund KF, Doshi KP, Gaul SL, Claremon DA, Remy DC, Baldwin JJ, Pitzenberger SM, Stern AM. Transglutaminase Inhibition By 2-[(2-Oxopropyl)Thio]Imidazolium Derivatives - Mechanism Of Factor Xiiia Inactivation. Biochemistry. 1994;33:10109–10119. doi: 10.1021/bi00199a039. [DOI] [PubMed] [Google Scholar]
- Halim D, Caron K, Keillor JW. Synthesis and evaluation of peptidic maleimides as transglutaminase inhibitors. Bioorg Med Chem Lett. 2007;17:305–308. doi: 10.1016/j.bmcl.2006.10.061. [DOI] [PubMed] [Google Scholar]
- Hang HC, Geutjes EJ, Grotenbreg G, Pollington AM, Bijlmakers MJ, Ploegh HL. Chemical probes for the rapid detection of fatty-acylated proteins in mammalian cells. J Am Chem Soc. 2007;129:2744. doi: 10.1021/ja0685001. [DOI] [PubMed] [Google Scholar]
- Hausch F, Halttunen T, Maki M, Khosla C. Design, synthesis, and evaluation of gluten peptide analogs as selective inhibitors of human tissue transglutaminase. Chem Biol. 2003;10:225–231. doi: 10.1016/s1074-5521(03)00045-0. [DOI] [PubMed] [Google Scholar]
- Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev. 2009;89:991–1023. doi: 10.1152/physrev.00044.2008. [DOI] [PubMed] [Google Scholar]
- Keillor JW. Tissue transglutaminase inhibition. Chem Biol. 2005;12:410–412. doi: 10.1016/j.chembiol.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. In vivo imaging of membrane-associated glycans in developing zebrafish. Science. 2008;320:664–667. doi: 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin ST, Bertozzi CR. In Vivo Imaging of Caenorhabditis elegans Glycans. ACS Chem Biol. 2009;4 doi: 10.1021/cb900254y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SP, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci U S A. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorand L. Crosslinks in blood: transglutaminase and beyond. FASEB J. 2007;21:1627–1632. doi: 10.1096/fj.07-0602ufm. [DOI] [PubMed] [Google Scholar]
- Lorand L, Graham RM. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- Nakaoka H, Perez DM, Baek KJ, Das T, Husain A, Misono K, Im MJ, Graham RM. G(H) - A Gtp-Binding Protein With Transglutaminase Activity And Receptor Signaling Function. Science. 1994;264:1593–1596. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- Nicholas B, Smethurst P, Verderio E, Jones R, Griffin M. Cross-linking of cellular proteins by tissue transglutaminase during necrotic cell death: a mechanism for maintaining tissue integrity. Biochem J. 2003;371:413–422. doi: 10.1042/BJ20021949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardin C, Gillet S, Keillor JW. Synthesis and evaluation of peptidic irreversible inhibitors of tissue transglutaminase. Bioorg Med Chem. 2006;14:8379–8385. doi: 10.1016/j.bmc.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Pardin C, Pelletier JN, Lubell WD, Keillor JW. Cinnamoyl inhibitors of tissue transglutaminase. J Org Chem. 2008;73:5766–5775. doi: 10.1021/jo8004843. [DOI] [PubMed] [Google Scholar]
- Piacentini M, Autuori F, Dini L, Farrace MG, Ghibelli L, Piredda L, Fesus L. Tissue Transglutaminase Is Specifically Expressed In Neonatal Rat-Liver Cells Undergoing Apoptosis Upon Epidermal Growth Factor-Stimulation. Cell Tissue Res. 1991;263:227–235. doi: 10.1007/BF00318764. [DOI] [PubMed] [Google Scholar]
- Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:2788–2796. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescher JA, Bertozzi CR. Chemistry in living systems. Nat Chem Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- Ruan QM, Johnson GVW. Transglutaminase 2 in neurodegenerative disorders. Frontiers in Bioscience. 2007;12:891–904. doi: 10.2741/2111. [DOI] [PubMed] [Google Scholar]
- Schaertl S, Prime M, Wityak J, Dominguez C, Munoz-Sanjuan I, Pacifici RE, Courtney S, Scheel A, Macdonald D. A Profiling Platform for the Characterization of Transglutaminase 2 (TG2) Inhibitors. J Biomol Screen. 15:478–487. doi: 10.1177/1087057110366035. [DOI] [PubMed] [Google Scholar]
- Shweke N, Boulos N, Jouanneau C, Vandermeersch S, Melino G, Dussaule JC, Chatziantoniou C, Ronco P, Boffa JJ. Tissue transglutaminase contributes to interstitial renal fibrosis by favoring accumulation of fibrillar collagen through TGF-beta activation and cell infiltration. Am J Pathol. 2008;173:631–642. doi: 10.2353/ajpath.2008.080025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol Ther. 2007;115:232–245. doi: 10.1016/j.pharmthera.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel M, Strnad P, Watts RE, Choi KH, Jabri B, Omary MB, Khosla C. Extracellular Transglutaminase 2 Is Catalytically Inactive, but Is Transiently Activated upon Tissue Injury. PLoS One. 2008;3 doi: 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh US, Erickson JW, Cerione RA. Identification And Biochemical-Characterization Of An 80 Kilodalton Gtp-Binding Transglutaminase From Rabbit Liver Nuclei. Biochemistry. 1995;34:15863–15871. doi: 10.1021/bi00048a032. [DOI] [PubMed] [Google Scholar]
- Sletten EM, Bertozzi CR. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew Chem, Int Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollid LM, Lundin KEA. Diagnosis and treatment of celiac disease. Mucosal Immunol. 2009;2:3–7. doi: 10.1038/mi.2008.74. [DOI] [PubMed] [Google Scholar]
- Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010 doi: 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Mehta K. Tissue transglutaminase-mediated chemoresistance in cancer cells. Drug Resistance Updates. 2007;10:144–151. doi: 10.1016/j.drup.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Wang L, Schultz PG. Expanding the genetic code. Angew Chem, Int Ed. 2005;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- Watts RE, Siegel M, Khosla C. Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J Med Chem. 2006;49:7493–7501. doi: 10.1021/jm060839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Siegel M, Choi K, Khosla C, Miller CR, Jackson EN, Piwnica-Worms D, Rich KM. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene. 2007;26:2563–2573. doi: 10.1038/sj.onc.1210048. [DOI] [PubMed] [Google Scholar]
- Zemskov EA, Janiak A, Hang J, Waghray A, Belkin AM. The role of tissue transglutaminase in cell-matrix interactions. Frontiers in Bioscience. 2006;11:1057–1076. doi: 10.2741/1863. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.