

Abstract
Please cite this paper as: Easterbrook et al. (2011) Immunization with 1976 swine H1N1‐ or 2009 pandemic H1N1‐inactivated vaccines protects mice from a lethal 1918 influenza infection. Influenza and Other Respiratory Viruses DOI: 10.1111/j.1750‐2659.2010.00191.x.
Background Zoonotic infections with H1N1 influenza viruses that evolved initially from the 1918 virus (1918) and adapted to swine threatened a pandemic in 1976 (1976 swH1N1) and a novel reassortant H1N1 virus caused a pandemic in 2009–2010 (2009 pH1N1). Epidemiological and laboratory animal studies show that protection from severe 2009 pH1N1 infection is conferred by vaccination or prior infection with 1976 swH1N1 or 1918.
Objectives Our aim was to demonstrate cross‐protection by immunization with 2009 pH1N1 or 1976 swH1N1 vaccines following a lethal challenge with 1918. Further, the mechanisms of cross‐protective antibody responses were evaluated.
Methods Mice were immunized with 1976 swH1N1, 2009 pH1N1, 2009 seasonal trivalent, or 1918 vaccines and challenged with 1918. Cross‐reactive antibody responses were assessed and protection monitored by survival, weight loss, and pathology in mice.
Results and Conclusions Vaccination with the 1976 swH1N1 or 2009 pH1N1 vaccines protected mice from a lethal challenge with 1918, and these mice lost no weight and had significantly reduced viral load and pathology in the lungs. Protection was likely due to cross‐reactive antibodies detected by microneutralization assay. Our data suggest that the general population may be protected from a future 1918‐like pandemic because of prior infection or immunization with 1976 swH1N1 or 2009 pH1N1. Also, influenza protection studies generally focus on cross‐reactive hemagglutination‐inhibiting antibodies; while hemagglutinin is the primary surface antigen, this fails to account for other influenza viral antigens. Neutralizing antibody may be a better correlate of human protection against pathogenic influenza strains and should be considered for vaccine efficacy.
Keywords: 1918 influenza, 1976 influenza, 2009 pandemic H1N1, cross‐protection, microneutralization
Introduction
During 1918–1919, an influenza pandemic was responsible for the deaths of up to 50 million people worldwide, killing more people than any influenza pandemic in recorded history. 1 , 2 Approximately 30% of the global population was clinically infected during the 1918 pandemic and survivors developed specific antibody responses. 3 In the last decade, genomic RNA was isolated from several patients who died from influenza in 1918, and using plasmid‐based reverse genetics, the complete 1918 H1N1 influenza virus has been reconstructed. 4 The fully reconstructed 1918 virus is lethal to mice and macaques following a low dose inoculation 4 , 5 and although rigorous biocontainment precautions are used when handling this virus, there has been some concern that this reconstructed 1918 virus could be re‐introduced into the population, causing another devastating 1918‐like pandemic.
The origin of the 1918 influenza virus is unresolved, but the genome of the virus is very avian‐like. 6 , 7 It is likely that humans subsequently transmitted the 1918 virus to swine 8 and the North American classical swine H1N1 viruses have circulated in pigs since that time, causing occasional zoonotic infections in humans, such as the 1976 outbreak at Fort Dix in New Jersey. 9 No prior contact with pigs and human‐to‐human transmission was documented for more than 200 recruits at Fort Dix, thus inciting fear of a 1918‐like outbreak in a population with little immunity to H1N1 influenza. 10 Scientific and government organizations supported a preventative measure to produce enough vaccine to protect the population, and more than 40 million people were vaccinated against 1976 swine H1N1 (1976 swH1N1). 11
After the 1957 H2N2 influenza pandemic, 1918‐derived seasonal H1N1 influenza strains were no longer isolated, only to return to human circulation in 1977. 12 These H1N1 viruses continued to circulate in humans and became a more mild seasonal strain by accumulating mutations in antigenic sites 13 and acquiring other mutations, such as a truncated PB1‐F2. The novel H1N1 of the pandemic of 2009–2010 (2009 pH1N1) derives its hemagglutinin (HA) from the North American swine triple reassortant H1N2 virus and its neuraminidase (NA) from an evolutionarily, independently emerged Eurasian avian‐like swine H1N1 virus. 14 An estimated 61 million people have been infected globally, and millions have been vaccinated against 2009 pH1N1. 15
Seroepidemiologic studies have demonstrated pre‐existing immunity in the population, primarily in people over 60 years old, against the 2009 pH1N1. 16 , 17 , 18 , 19 The origin of this immunity is likely due to previous exposures and/or vaccinations with cross‐reactive viruses, such as pre‐1957 H1N1 viruses or the 1976 swH1N1 vaccine. Adults who received at least 1 dose of the 1976 vaccine have high levels of cross‐reactive antibodies to 2009 pH1N1. 17 Vaccination with the current seasonal trivalent influenza vaccine (2009 TIV) offers little or no protection against 2009 pH1N1 infection. 16 , 20 Animal studies have further supported the human epidemiologic data, demonstrating protection against a lethal dose of 2009 pH1N1 by prior infection or vaccination with the 1918 or 1976 swine H1N1, while only partial protection with recent seasonal H1N1 viruses or vaccines. 21 , 22 , 23 Additionally, a recent study has demonstrated that 2009 pH1N1 vaccination protects mice against a lethal 1918 infection. 24 Structural and antigenic analyses have shown high similarity of the HA of 1918 and 2009 pH1N1. 22 , 23 , 25 Whether vaccination with the closely related 1976 swH1N1 or 2009 pH1N1 vaccines confers protection against infection with the 1918 virus and the role of HA and NA in cross‐protection were therefore examined in the studies described in this report. Mice were vaccinated with 1976 swH1N1, 2009 pH1N1, or 2009 TIV vaccines and then challenged with a lethal dose of 1918 virus. We demonstrated protection against weight loss, which correlated with significantly reduced pulmonary pathology and virus titers in the lungs of mice vaccinated with the inactivated 1976 and 2009 pH1N1 preparations. Protection correlated with cross‐reactive microneutralization (MN) antibody titers, which likely involves both HA to reduce virus attachment and entry and NA to reduce the release of virus from cells and thus virus spread. 26 These data suggest that either prior infection or vaccination with 1976 swH1N1 or 2009 pH1N1 may protect the population from a possible future 1918 or 1918‐like pandemic. Further, the protection conferred by NA, not only HA, should be considered in immunologic and cross‐protection analyses.
Materials and methods
Virus
The fully reconstructed 1918 H1N1 influenza virus was previously prepared using a standard reverse genetics system. 4 Virus and infectious samples were handled under biosafety level 3 (BSL3) enhanced laboratory conditions in accordance with the select agent guidelines of the National Institutes of Health and the Centers for Disease Control and Prevention.
Vaccines
The vaccine strain used for the 1976 swH1N1 vaccine was the original X‐53 reassortant, comprised of HA and NA from A/New Jersey/11/76 and the remaining segments from A/PR/8/34 for high yield. 27 The fully reconstructed South Carolina/1/18 H1N1 4 was used for the 1918 vaccine. Both of these viruses were grown in 10‐day‐old eggs, and allantoic fluid was harvested and clarified by low‐speed centrifugation. Virus was inactivated using β‐propiolactone (BPL; Sigma, St Louis, MO, USA) and concentrated by ultracentrifugation at 100 000 g for 2 h. Inactivated virus was purified using a 30% sucrose cushion at 100 000 g for 2 h and pelleted by ultracentrifugation at 100 000 g for 2 h. Total protein was quantified using the Bradford BCA assay (Pierce, Rockford, IL, USA), and the proportion of HA and NA of total protein was determined by Coomassie blue staining. The commercial monovalent pandemic 2009 H1N1 subunit vaccine, which is derived from BPL‐inactivated A/California/7/2009 H1N1 (Novartis, Cambridge, MA, USA), and the trivalent seasonal subunit vaccine comprised of BPL‐inactivated A/Brisbane/59/2007 (H1N1), A/Uruguay/716/2007 (H3N2), and B/Brisbane/60/2008 (Novartis) were provided by Dr Matt Memoli, NIAID/NIH (Bethesda, MD, USA).
Homology analysis
Sequences for HA and NA from all four influenza vaccine strains were downloaded from GenBank were aligned and compared using the LaserGene Megalign program (DNAStar, Inc., Madison, WI, USA).
Mouse experiments
Groups of 6‐ to 8‐week‐old female BALB/c mice (Jackson Labs, Bar Harbor, ME, USA) were lightly anaesthetized with isofluorane supplemented with O2 (1·5 l/min) and immunized with 1·5 μg HA (H1) of the inactivated virus vaccine in 50‐μl total volume (1/10 commercial human dose) by intramuscular injection in the hind leg. Two weeks later, mice were boosted with the same amount of vaccine. Four weeks after the initial vaccination, mice were anaesthetized as described previously and challenged intranasally with 10× LD50 (2·5 × 103 pfu) of 1918 virus in 50 μl DMEM. Survival and body weight were monitored for 14 days, and mice were humanely euthanized if more than 25% body weight was lost. Lungs were collected for viral titration (n = 3 per vaccine group) and pathologic examination (n = 2 per vaccine group) at 2 and 4 dpi. Virus titers were measured from 10% (w/v) lung suspensions by plaque assay after homogenization in sterile L15 media. All experimental work was performed in an enhanced ABSL3 laboratory at the NIH, following approval of animal safety protocols by the NIH Animal Care and Use Committee.
Hemagglutination inhibition assay
Sera were collected 1–2 days before and 28 days after vaccination from all mice. Sera were treated with receptor destroying enzyme (RDE; Denka Seiken, Tokyo, Japan), and the hemagglutination inhibition (HI) assay was performed as described previously. 22 Data are presented as the reciprocal geometric mean titers (GMT) of the highest serum dilution completely inhibiting turkey red blood cell agglutination by 8 HA units of the appropriate homologous virus or 1918.
Microneutralization assay
For determination of MN titers, serially diluted serum samples were incubated with 50× TCID50 of 1918 in 50 μl for 1 h and 2·5 × 104 MDCK cells were added and incubated overnight. The inoculum was removed, and cells were incubated for 72 h in DMEM supplemented with 2 μg/ml TPCK‐trypsin. MN titers are reported as the reciprocal of the final dilution that neutralizes virus, as defined by a negative HA result at a 1:1 dilution, using turkey RBCs.
Neuraminidase inhibition (NI) assay
To measure NA‐inhibiting antibody titers against NA, a reassortant virus containing the appropriate N1, an avian H6, and the remaining segments from A/Puerto Rico/8/34 (H1N1) (PR8) was created using plasmid‐based reverse genetics, as described previously 4 and inactivated with BPL. BPL inactivation did not impact NA activity. The NI assay was performed as described previously. 28 , 29 Briefly, serial dilutions of heat‐inactivated sera were transferred to 96‐well plates coated with fetuin. Virus diluted in PBS with Ca/Mg to a standard NA activity level was then added to the wells and the mixture incubated overnight at 37°C. The plates were washed and incubated for 2 h at room temperature with peanut agglutinin conjugated to peroxidase. The plates were washed before the addition of O‐phenylenediamine dihydrochloride, and the reaction with this substrate stopped after 10 min by the addition of 1 N H2SO4. Absorbance was read at 490 nm on a plate reader. The NA inhibition titer was defined as the inverse of the greatest dilution that gave at least 50% inhibition of NA activity.
Histopathological and immunohistochemical analyses
Following 24‐h fixation in 10% formaldehyde, inflated lung samples were embedded in paraffin, cut into 5 μm sections, and mounted on positively charged slides (American HistoLabs, Gaithersburg, MD, USA). Influenza virus antigen distribution was evaluated by immunohistochemistry using the manufacturer’s protocol (Invitrogen Corporation, Carlsbad, CA, USA) as described previously. 30 A single pathologist reviewed the histopathology and immunohistochemistry in a blinded fashion.
Statistical analyses
The percent weight loss and lung virus titers were assessed using one‐way analyses of variances (anova) with one between group variable (dpi or vaccine treatment; Graph Pad Prism, La Jolla, CA, USA). Specific comparisons to mock‐vaccinated mice were carried out using the student’s t‐test. Mean differences were considered statistically significant if P < 0·05.
Results
Mice were protected from lethal infection with 1918 by vaccination with the 1976 and 2009 pH1N1 vaccines
To determine whether vaccines using viruses that have evolved from the parental 1918 strain provide protection against a lethal 1918 infection, mice were immunized with commercial 2009 pH1N1 or 2009 TIV subunit vaccines or 1976 or 1918 H1N1 whole virus vaccines (see Materials and methods) and subsequently infected with the 1918 virus. All of the mice that were immunized with the 1976, 2009 pH1N1, or 1918 vaccines (5/5 per group) survived challenge with a lethal dose (10× LD50) of 1918 (Figure 1A) and did not lose weight during infection (Figure 1B; P > 0·05). Immunization with the 2009 TIV resulted in 80% (4/5) protection against death following infection with 1918; however, all of these mice lost an average of 20·2 ± 1·8% of body weight, which was significantly more than 1918, 1976, or 2009 pH1N1‐vaccinated mice (Figure 1A,B; anova: F 3,59 = 18·5, P < 0·0001). Mice that were mock‐vaccinated with PBS (0/5) did not survive (Figure 1A,B).
Figure 1.
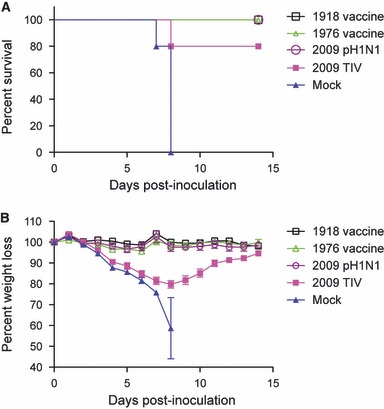
Mice administered the 1976 or 2009 pH1N1 survived a lethal 1918 influenza infection. Mice were immunized with the 1976, 2009 pH1N1, 2009 trivalent influenza vaccine, 1918 vaccine or vehicle alone and were monitored daily for survival (a) and weight loss (b) for 14 days.
Immunization with 1976 and 2009 pH1N1 vaccines reduced the amount of infectious virus in the lungs of mice infected with 1918
Mice immunized with the 1976 vaccine had about 2 logs less infectious virus in the lungs at both 2 and 4 dpi with 1918 than mock‐vaccinated mice (Figure 2; anova: F 3,11 = 8·3, P = 0·008). Mice that received the 2009 pH1N1 vaccine had almost 3 logs lower viral titers in the lungs during peak infection with 1918, at 2 dpi, than mock‐vaccinated mice (Figure 2; t = 2·9, P = 0·04, d.f. = 4). Mice that received the 1918 vaccine had no detectable virus in the lungs after challenge with the homologous virus (Figure 2). Administration of the 2009 TIV did not reduce viral titers in the lungs at either time point during infection with 1918 when compared with mock‐vaccinated mice (Figure 2).
Figure 2.
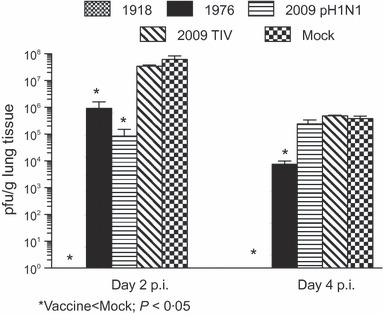
Mice immunized with the 1976 or 2009 pH1N1 vaccines had reduced virus titers in the lungs following challenge with 1918 than mice that received the 2009 trivalent influenza vaccine or vehicle alone. Mice were vaccinated, and the amount of infectious virus in the lungs following challenge with 1918 was measured by plaque assay.
Vaccination with 1976 and p2009 H1N1 protected mice from severe pathology caused by 1918 infection
The histopathology and influenza viral antigen distribution of lung tissue in mice challenged with the 1918 virus were characterized by analyzing formalin‐fixed, paraffin‐embedded stained sections. Consistent with previous studies, 4 mock‐vaccinated mice that were subsequently challenged with the 1918 virus showed pathology consisting of a marked, transmural necrotizing bronchiolitis and multifocal alveolitis. This pattern was present at 2 dpi but was more prominent at 4 dpi (Figure 3A–D). Correlating to the marked histopathological changes, abundant influenza viral antigen was observed in both bronchiolar and alveolar epithelium (Figure 3B,D). Mice vaccinated with the 2009 TIV and subsequently challenged with 1918 showed a histopathological pattern very similar to mock‐vaccinated mice (Figure 3E,F), with transmural necrotizing bronchiolitis and multifocal alveolitis and abundant influenza viral antigen in both bronchiolar and alveolar epithelial cells. In contrast, mice vaccinated with either the 2009 pH1N1 (Figure 3G,H) or 1976 (Figure 3I,J) vaccines and subsequently challenged with the 1918 virus showed a histopathological pattern of focal bronchiolitis with luminal necrotic debris and only rare, focal alveolitis. Influenza viral antigen was noted in luminal necrotic debris with bronchioli but not in alveolar epithelial cells. In contrast, mice vaccinated with the homologous 1918 vaccine before challenge showed no histopathological changes in the lung and no evidence of influenza viral antigen by immunohistochemistry (Figure 3K,L).
Figure 3.
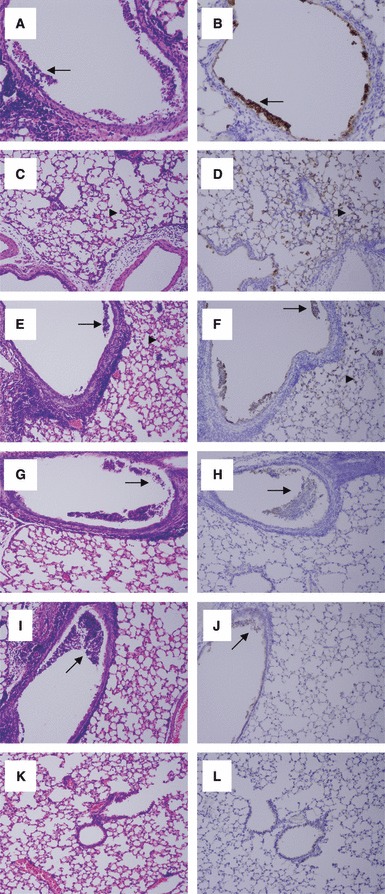
Pulmonary pathology caused by infection with 1918 was reduced in mice vaccinated with 1976 or 2009 pH1N1 vaccines. Photomicrographs of hematoxylin‐ and eosin‐stained tissue sections and immunohistochemically stained sections to detect influenza viral antigen from mice infected with different influenza virus constructs at day 4 post‐challenge with 1918. Viral antigen is stained red‐brown on a hematoxylin‐stained background. Original magnifications, ×100. (A–D) Sections from a vehicle‐alone vaccinated animal subsequently challenged with the 1918 virus showing (A) marked transmural necrotizing bronchiolitis (arrows) and (C) multifocal alveolitis (arrowhead). Abundant viral antigen was noted in (B) bronchiolar epithelium (arrow) and (D) alveolar lining cells (arrowhead). (E, F) Sections from a 2009 trivalent influenza vaccine‐vaccinated animal subsequently challenged with the 1918 virus showing (E) marked transmural necrotizing bronchiolitis (arrows) and multifocal alveolitis (arrowhead). Abundant viral antigen (F) was noted in bronchiolar epithelium (arrow) and alveolar lining cells (arrowhead). (G, H) Sections from a 2009 pH1N1‐vaccinated animal subsequently challenged with the 1918 virus showing (G) focal bronchiolitis with luminal necrotic debris (arrow). Viral antigen (H) was noted in the necrotic debris within the bronchiolar lumen (arrow). (I, J) Sections from a 1976‐vaccinated mouse subsequently challenged with the 1918 virus showing (I) focal bronchiolitis with luminal necrotic debris (arrow). Viral antigen (J) was noted in the necrotic debris within the bronchiolar lumen (arrow). (K, L) Sections from a 1918‐vaccinated animal subsequently challenged with the homologous virus showing (K) no pathological changes. No viral antigen (L) was noted.
Sera from mice immunized with the 1976 or 2009 pH1N1 vaccines neutralized 1918
Vaccination resulted in serum HI GMT >40 to each homologous virus and was similar among vaccination groups (75–150 GMT; data not shown), demonstrating both a sufficient and comparable immunization success. Sera from mice immunized with the 1976 or 2009 pH1N1 vaccines had detectable HI GMTs against 1918, though reduced 15‐ to 21‐fold when compared to the homologous virus (Table 1). Sera from mice vaccinated with the 2009 TIV had no inhibition of HA activity of the 1918 virus (Table 1).
Table 1.
Microneutralization (MA) of 1918 and inhibition of its HA and neuraminidase activity following immunization with 1918, 1976, 2009 pH1N1, and 2009 TIV vaccines
| Vaccine | MN: 1918 | NI: 1918 | HI: SC/18 |
|---|---|---|---|
| 1918 | 400 ± 49 | 120·0 ± 19·1 | 107 ± 13 |
| 1976 | 179 ± 34 | 13·3 ± 2·1 | 6·5 ± 1·9 |
| 2009 pH1N1 | 165 ± 56 | <10 | 7·2 ± 1·9 |
| 2009 TIV | <10 | <10 | <1 |
| Vehicle | <10 | <10 | <1 |
Mice were immunized with 1·5 μg HA and boosted 2 weeks later, and sera were collected 28 days after the initial vaccination.
HA, hemagglutinin; NI, neuraminidase inhibition; HI, hemagglutination inhibition; TIV, trivalent influenza vaccine.
NA‐inhibiting (NI) antibody titers were measured against a virus expressing the NA of the 1918 strain, using an unrelated HA reassortant virus to avoid interference by HA‐specific antibodies. As expected, inhibition of 1918 NA activity was highest for the 1918 vaccinated mice, with an NI GMT of 120·0 ± 19·1 (Table 1). Although reduced, 60% (6/10) of mice immunized with the 1976 vaccine had detectable NI titers against 1918, averaging 13·3 ± 2·1, whereas mice that received the 2009 pH1N1, 2009 TIV, or vehicle alone did not have detectable NI GMT against 1918 (Table 1). All mice vaccinated with 1976 had detectable NI titers, averaging 112·5 ± 19·2, against the appropriate H6N1 virus with the homologous NA, whereas only 40% of mice immunized with the commercial inactivated 2009 pH1N1 vaccine had detectable NI titers against the homologous virus. Vaccination with the commercial 2009 TIV did not elicit any detectable antibodies against NA of the homologous strain, A/Brisbane/59/2007 (H1N1).
Cross‐reactivity of antibodies elicited by vaccination with 1976, 2009 pH1N1, 2009 TIV, or 1918 inactivated vaccines or vehicle alone with the 1918 virus was measured by MN. Sera from mice administered the 1918 vaccine had the highest MN titers against 1918, 400 ± 49 (Table 1). In contrast to HI and NI titers, substantial heterologous activity was observed by MN; mice immunized with 1976 or 2009 pH1N1 vaccines had MN titers of 179 ± 34 and 165 ± 56, respectively (Table 1). Administration of the 2009 TIV vaccine did not elicit detectable cross‐neutralization titers (Table 1).
Discussion
Antigenic sites of HA that are targeted by antibodies to neutralize influenza viruses have been defined previously by Brownlee and Fodor, 31 using H1 residue numbering, as the strain‐specific epitopes Sa (141–142, 170–174, 176–181) and Sb (201–212), as well as more conserved epitopes Ca1 (183–187, 219–222, 252–254), Ca2 (154–160, 238–239), and Cb (87–92). Comparison of the amino acid sequences of these epitopes in A/South Carolina/1/1918 (H1N1) (1918) with the 1976 swH1N1 vaccine strain, A/New Jersey/11/76 (H1N1) (1976), the 2009 pH1N1, A/California/07/2009 (H1N1), and the 2009 TIV H1N1 component, A/Brisbane/59/2007 (H1N1), shows 86·5%, 80·8%, and 53·8% homology, respectively. Human seasonal H1N1 viruses have been undergoing rapid antigenic drift from 1918–1957 and again since the late 1970s. 32 The seasonal H1N1 viruses have also gained two highly conserved glycosylation sites, whereas the pandemic viruses do not have any glycosylation sites that can mask antigenic regions and further reduce binding of pre‐existing cross‐reactive antibodies. 23 This suggests that HA‐specific immunity, primarily in antigenic regions of HA, that is induced by the 1976 and 2009 pH1N1 vaccines would provide greater protection against 1918 challenge than seasonal TIV.
Antigenic regions of NA, specifically N1, have not been defined, but the total sequence homology to 1918 is similar among the H1N1 viruses examined: 87·4% for 1976 swH1N1, 87·2% for 2009 pH1N1, and 86·6% for the seasonal A/Brisbane/59/2007. Future research should be conducted to better define regions of influenza NA antigenicity. The 2009 pH1N1 received the NA from a Eurasian swine H1N1 virus and the seasonal H1N1 virus has undergone substantial antigenic drift, so one could hypothesize that the antigenic regions of the NA of the 2009 pandemic and seasonal H1N1 viruses would be less homologous to the antigenic regions of the 1918 NA than the NA of 1976 swH1N1 that was derived from 1918.
Mice immunized with the 1976 swH1N1 or commercial human 2009 pH1N1 vaccines survived a lethal challenge with 1918, lost no weight, and had 2–3 logs less virus in the lungs early in infection when compared to mock‐vaccinated mice. This was comparable to recently reported protection from a lethal infection with 1918 observed in mice immunized with an inactivated 2009 pH1N1 reassortant virus. 24 Similarly, laboratory and epidemiological data support cross‐protection between 2009 pH1N1 and 1918. 17 , 18 , 19 , 21 , 22 , 23 Infection induces T‐cell‐mediated responses (e.g. CD8+ and CD4+ T‐cell responses), in addition to humoral responses, which may contribute to observations of greater protection against challenge with a non‐homologous, but closely related, virus than protection following vaccination with inactivated influenza viruses that primarily induces humoral immunity. 33 , 34
In contrast to 1976 and 2009 pH1N1 vaccination, immunization with the 2009 seasonal TIV offered only partial protection from a lethal 1918 infection. This is consistent with other human and animal studies that demonstrate little protection against a challenge with 2009 pH1N1 or 1918 after infection or immunization with seasonal H1N1 viruses or vaccines. 16 , 20 , 21 , 22 , 23 , 24 , 35 The protection from death for some mice vaccinated with the 2009 TIV may be a result of antibodies to conserved epitopes of HA or matrix protein (M2), 36 or the possible minor induction of IFN‐γ secreting CD4+ T cells or CD8+ T cells. In humans, the H1N1 antibody response is likely boosted or primed by seasonal influenza infection or vaccination, but does not offer complete protection because of high variability in the antigenic regions of HA and NA.
Mice immunized with 1976 swH1N1 or 2009 pH1N1 vaccines induced MN titers (>150) against 1918, whereas administration of the 2009 TIV or vehicle alone did not neutralize against 1918. Protection against viral replication, weight loss, and death was evident in mice that had measurable MN titers against the 1918 virus, but no detectable or very low (<40 GMT) HI or NI titers, supporting MN as a means to evaluate vaccine efficacy. Such protective antibodies may be detected by MN assay, but not HI or NI assays, because they bind to HA epitopes that are not closely associated with the receptor‐binding domain, have specificity for conserved epitopes on antigens other than HA, or have low affinity for HA or NA.The laboratory‐produced whole virus vaccines were made as similarly as possible to the commercial vaccines, including the use of vaccine strains when available (1976 swH1N1) and inactivation with β‐propiolactone, but HA quantification was performed by densitometry to determine the percentage of HA of total protein in the laboratory vaccines and by single radial immunodiffusion (SRID) analysis for the commercial vaccines, so discrepancies in the amount of HA administered may be possible.
Virus neutralization was due primarily to cross‐reactive antibodies to HA that were elicited by vaccination. Neuraminidase, though to a lesser extent than HA, also may contribute to cross‐protection among related influenza viruses. Recombinant NA protein, in the absence of other influenza virus proteins, can induce NA‐specific antibodies, reduce the replication of both homologous and heterovariant virus, and suppress weight loss in mice by reducing virus release from cells. 26 Additionally, high levels of population immunity to the 1957 pandemic N2, as well as the N2 of subsequent seasonal strains, may have lessened the severity of the 1968 pandemic in which the HA, but not NA, underwent reassortment. 37 In our experiments, mice that received the 1976 vaccine had lower titers of virus in the lungs than mock‐vaccinated mice throughout infection, whereas mice that received the 2009 pH1N1 vaccine had less virus in the lungs that was limited to early infection at 2 dpi. It is possible that this may be because of a reduced ability of the virus to be released from cells and spread efficiently by cross‐reactive NA‐inhibiting antibodies elicited by vaccination with the 1976 vaccine that were not evident following 2009 pH1N1 vaccination. The 2009 pH1N1 and 2009 seasonal TIV vaccines were commercially derived and elicited low or no antibody responses to the NA of the appropriate homologous virus. This likely reflects steps in the manufacturing process that do not retain a stable immunogenic form of NA, while NA activity itself was maintained. The role of NA in cross‐protection is often overlooked 38 and should be recognized in vaccine development and evaluation of population immunity. Future studies will examine the role of NA in protection against influenza infection and whether NI titers from a laboratory‐produced 2009 pH1N1 vaccine that possesses an immunogenic NA can inhibit the activity of the 1918 NA.
Mice infected with a lethal dose of 1918 have necrotizing bronchitis and bronchiolitis and moderate to severe alveolitis that is composed of neutrophils, lymphocytes, and macrophages, with accompanying edema and hemorrhage and viral antigen is distributed throughout the lungs. 39 , 40 Mock and 2009 TIV‐vaccinated mice demonstrated similar pathology following infection with 1918, while immunization with 2009 pH1N1 or 1976 vaccines significantly reduced the extent of pathology and the amount of viral antigen in the lungs. These data correlated with survival data and the amount of infectious virus in the lungs as determined by plaque assay, thus reinforcing that the amount of 1918 virus in the lungs directly correlates with pathology.
The 1976 vaccination campaign may have helped protect some of the normally high‐risk older population during the 2009 pH1N1 outbreak, as well as any potential future exposure to a 1918‐like H1N1. Similarly, exposure to the 2009 pH1N1 and the associated vaccination campaign also may contribute to protection against an accidental release or zoonotic re‐introduction of 1918 or 1918‐like influenza. Vaccination is an effective method of protection against influenza infection and the associated morbidity and mortality, but vaccine development should optimize the range and duration of protection. Vaccines should not only elicit antibodies against HA but also induce other protective responses, such as antibodies against other surface antigens, including NA and possibly M2, as well as innate, antiviral, and T‐cell responses. Thus, neutralization of influenza infection may be a better correlate of protection for vaccines than the more limited and currently accepted measurement of the ability to inhibit HA activity, as expressed by HI titers.
Acknowledgements
We thank the Comparative Medicine Branch (NIH/NIAID) for their assistance with animal studies. This work was supported by the Intramural Research Program of the NIH and the NIAID.
References
- 1. Taubenberger JK, Morens DM. 1918 Influenza: the mother of all pandemics. Emerg Infect Dis 2006; 12:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kilbourne ED. Influenza pandemics of the 20th century. Emerg Infect Dis 2006; 12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frost W. Statistics of influenza morbidity. Public Health Rep 1920; 35:584–597. [Google Scholar]
- 4. Tumpey TM, Basler CF, Aguilar PV et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 2005; 310:77–80. [DOI] [PubMed] [Google Scholar]
- 5. Kobasa DS, Jones M, Shinya K et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 2007; 445:319–323. [DOI] [PubMed] [Google Scholar]
- 6. Rabadan R, Levine AJ, Robins H. Comparison of avian and human influenza A viruses reveals a mutational bias on the viral genomes. J Virol 2006; 80:11887–11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG. Characterization of the 1918 influenza virus polymerase genes. Nature 2005; 437:889–893. [DOI] [PubMed] [Google Scholar]
- 8. Taubenberger JK, Reid AH, Janczewski TA, Fanning TG. Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos Trans R Soc Lond B Biol Sci 2001; 356:1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans: a review of the literature. Clin Infect Dis 2007; 44:1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gaydos JC, Top FH Jr, Hodder RA, Russell PK. Swine influenza a outbreak, Fort Dix, New Jersey, 1976. Emerg Infect Dis 2006; 12:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kilbourne ED. National immunization for pandemic influenza. Hosp Pract 1976; 11:15–16. [DOI] [PubMed] [Google Scholar]
- 12. Nakajima K, Desselberger U, Palese P. Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950. Nature 1978; 274:334–339. [DOI] [PubMed] [Google Scholar]
- 13. Hay AJ, Gregory V, Douglas AR, Lin YP. The evolution of human influenza viruses. Philos Trans R Soc Lond B Biol Sci 2001; 356:1861–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morens DM, Taubenberger JK, Fauci AS. The persistent legacy of the 1918 influenza virus. N Engl J Med 2009; 361:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Centers for Disease Control and Prevention . FluView: a weekly influenza surveillance report. Available at: http://www.cdc.gov/flu/weekly/2010.
- 16. Centers for Disease Control and Prevention . Serum cross‐reactive antibody response to a novel influenza A (H1N1) virus after vaccination with seasonal influenza vaccine. MMWR Morb Mortal Wkly Rep 2009; 58:521–524. [PubMed] [Google Scholar]
- 17. Hancock K, Veguilla V, Lu X et al. Cross‐reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N Engl J Med 2009; 361:1945–1952. [DOI] [PubMed] [Google Scholar]
- 18. Itoh Y, Shinya K, Kiso M et al. In vitro and in vivo characterization of new swine‐origin H1N1 influenza viruses. Nature 2009; 460:1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dawood FS, Jain S, Finelli L et al. Emergence of a novel swine‐origin influenza A (H1N1) virus in humans. N Engl J Med 2009; 360:2605–2615. [DOI] [PubMed] [Google Scholar]
- 20. Garcia‐Garcia L, Valdespino‐Gomez JL, Lazcano‐Ponce E et al. Partial protection of seasonal trivalent inactivated vaccine against novel pandemic influenza A/H1N1 2009: case–control study in Mexico City. BMJ 2009; 339:b3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manicassamy B, Medina RA, Hai R et al. Protection of mice against lethal challenge with 2009 H1N1 influenza A virus by 1918‐like and classical swine H1N1 based vaccines. PLoS Pathog 2010; 6:e1000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kash JC, Qi L, Dugan VG et al. Prior infection with classical swine H1N1 influenza viruses is associated with protective immunity to the 2009 pandemic H1N1 virus. Influenza Other Respi Viruses 2010; 4:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei CJ, Boyington JC, Dai K et al. Cross‐neutralization of 1918 and 2009 influenza viruses: role of glycans in viral evolution and vaccine design. Sci Transl Med 2010; 2:24ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Medina RA, Manicassamy S, Stertz S et al. Pandemic 2009 H1N1 vaccine protects against 1918 Spanish influenza virus. Nat Commun 2010; 1:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE Jr, Wilson IA. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 2010; 328:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kilbourne ED, Pokorny BA, Johansson B, Brett I, Milev Y, Matthews JT. Protection of mice with recombinant influenza virus neuraminidase. J Infect Dis 2004; 189:459–461. [DOI] [PubMed] [Google Scholar]
- 27. Palese P, Ritchey MB, Schulman JL, Kilbourne ED. Genetic composition of a high‐yielding influenza A virus recombinant: a vaccine strain against “Swine” influenza. Science 1976; 194:334–335. [DOI] [PubMed] [Google Scholar]
- 28. Cate TR, Rayford Y, Nino D et al. A high dosage influenza vaccine induced significantly more neuraminidase antibody than standard vaccine among elderly subjects. Vaccine 2010; 28:2076–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lambre CR, Terzidis H, Greffard A, Webster RG. Measurement of anti‐influenza neuraminidase antibody using a peroxidase‐linked lectin and microtitre plates coated with natural substrates. J Immunol Methods 1990; 135:49–57. [DOI] [PubMed] [Google Scholar]
- 30. Memoli MJ, Tumpey TM, Jagger BW et al. An early ‘classical’ swine H1N1 influenza virus shows similar pathogenicity to the 1918 pandemic virus in ferrets and mice. Virology 2009; 393:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brownlee GG, Fodor E. The predicted antigenicity of the haemagglutinin of the 1918 Spanish influenza pandemic suggests an avian origin. Philos Trans R Soc Lond B Biol Sci 2001; 356:1871–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nelson MI, Viboud C, Simonsen L et al. Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog 2008; 4:e1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quan FS, Compans RW, Nguyen HH, Kang SM. Induction of heterosubtypic immunity to influenza virus by intranasal immunization. J Virol 2008; 82:1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thomas PG, Keating R, Hulse‐Post DJ, Doherty PC. Cell‐mediated protection in influenza infection. Emerg Infect Dis 2006; 12:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tumpey TM, Garcia‐Sastre A, Taubenberger JK et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 2005; 79:14933–14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tompkins SM, Zhao ZS, Lo CY et al. Matrix protein 2 vaccination and protection against influenza viruses, including subtype H5N1. Emerg Infect Dis 2007; 13:426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kilbourne ED. Influenza pandemics in perspective. JAMA 1977; 237:1225–1228. [PubMed] [Google Scholar]
- 38. Johansson BE, Brett IC. Changing perspective on immunization against influenza. Vaccine 2007; 25:3062–3065. [DOI] [PubMed] [Google Scholar]
- 39. Tumpey TM, Garcia‐Sastre A, Taubenberger JK, Palese P, Swayne DE, Basler CF. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc Natl Acad Sci 2004; 101:3166–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qi L, Kash JC, Dugan VG et al. Role of sialic acid binding specificity of the 1918 influenza virus hemagglutinin protein in virulence and pathogenesis for mice. J Virol 2009; 83:3754–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]