

Abstract
Homeostatic synaptic plasticity adjusts the strength of synapses during global changes in neural activity, thereby stabilizing the overall activity of neural networks. Suppression of synaptic activity increases synaptic strength by inducing synthesis of retinoic acid (RA), which activates postsynaptic synthesis of AMPA-type glutamate receptors (AMPARs) in dendrites and promotes synaptic insertion of newly synthesized AMPARs. Here, we show that fragile X mental retardation protein (FMRP), an RNA-binding protein that regulates dendritic protein synthesis, is essential for increases in synaptic strength induced by RA or by blockade of neural activity in the mouse hippocampus. Although activity-dependent RA synthesis is maintained in Fmr1 knock-out neurons, RA-dependent dendritic translation of GluR1-type AMPA receptors is impaired. Intriguingly, FMRP is only required for the form of homeostatic plasticity that is dependent on both RA signaling and local protein synthesis. Postsynaptic expression of wild-type or mutant FMRP(I304N) in knock-out neurons reduced the total, surface, and synaptic levels of AMPARs, implying a role for FMRP in regulating AMPAR abundance. Expression of FMRP lacking the RGG box RNA-binding domain had no effect on AMPAR levels. Importantly, postsynaptic expression of wild-type FMRP, but not FMRP(I304N) or FMRPΔRGG, restored synaptic scaling when expressed in knock-out neurons. Together, these findings identify an unanticipated role for FMRP in regulating homeostatic synaptic plasticity downstream of RA. Our results raise the possibility that at least some of the symptoms of fragile X syndrome reflect impaired homeostatic plasticity and impaired RA signaling.
Introduction
Homeostatic synaptic plasticity, working in concert with Hebbian-type synaptic plasticity, refines neuronal connectivity during development and contributes to network stability (Davis and Bezprozvanny, 2001; Turrigiano and Nelson, 2004; Kaneko et al., 2008). One well studied form of homeostatic plasticity, called synaptic scaling, is induced by long-term blockade of neuronal firing and synaptic transmission and is manifest as new synthesis and insertion of AMPA-type glutamate receptors (AMPARs) (Ju et al., 2004; Thiagarajan et al., 2005; Sutton et al., 2006; Aoto et al., 2008).
We recently reported a critical role for all-trans retinoic acid (RA) in the induction of the synaptic scaling form of homeostatic plasticity (Aoto et al., 2008). Inhibition of action potential firing with tetrodotoxin (TTX), along with blockade of NMDA receptors with aminophosphonovalerate (APV), stimulates synthesis of RA in neurons. RA alone is both necessary and sufficient to induce local translation and synaptic scaling, placing RA into a key role in regulating synaptic strength (Aoto et al., 2008). The effect of RA is mediated by dendritically localized retinoic acid receptor RARα, which inhibits protein translation through direct binding to specific target mRNAs (Poon and Chen, 2008). Addition of RA reverses the RARα-dependent repression of translation of target mRNAs (Maghsoodi et al., 2008; Poon and Chen, 2008), and acute knockdown of RARα completely blocks synaptic scaling (Aoto et al., 2008). One of the RARα targets is the mRNA encoding GluR1, an AMPA receptor subunit (Poon and Chen, 2008). TTX + APV treatment or direct RA application leads to the local translation of GluR1 receptors in dendrites and the insertion of GluR1 homotetramers at the synapse, increasing synaptic strength (Aoto et al., 2008).
Fragile X mental retardation protein (FMRP), encoded by the Fmr1 gene, is another dendritically localized RNA-binding protein. Absence of FMRP in human patients causes fragile X syndrome, the most common inherited form of mental retardation. FMRP knock-out (KO) mice exhibit normal baseline synaptic transmission but have altered spine morphology (Comery et al., 1997; Irwin et al., 2000), impairments in certain forms of long-term potentiation (LTP) (Li et al., 2002; Larson et al., 2005), and exaggerated metabotropic glutamate receptor (mGluR)-dependent long-term depression (LTD) (Huber et al., 2002). FMRP is associated with both translationally repressed messenger ribonucleoprotein particles and actively translating polyribosomes (Corbin et al., 1997; Zalfa et al., 2003) and is believed to specifically bind to mRNAs and regulate their translation (Laggerbauer et al., 2001; Li et al., 2001; Bassell and Warren, 2008). Consistent with this notion, dysregulated translation and elevated basal protein synthesis are found in Fmr1 knock-out neurons (Dolen et al., 2007; Muddashetty et al., 2007). However, whether FMRP is involved in translational regulation during homeostatic plasticity is unknown.
Here we report that FMRP is required postsynaptically for the form of synaptic scaling that is mediated by RA. Although RA synthesis is normal in Fmr1 knock-out neurons, RA-induced local translation of specific mRNAs is impaired. As a consequence, activity blockade or RA treatment fails to increase synaptic strength in the absence of FMRP. Our data reveal an unanticipated role for FMRP in homeostatic synaptic plasticity and RA signaling.
Materials and Methods
DNA constructs.
The 3xRARE–EGFP reporter construct is as described (Aoto et al., 2008). Briefly, three copies of the retinoic acid response element were placed upstream of a TK promoter driving enhanced green fluorescent protein (EGFP). All FMRP constructs used were the full-length isoform 1 (Ashley et al., 1993). For coimmunoprecipitation (Co-IP) experiments, FMRP was tagged with FLAG at the N terminus, RARα with Myc at the N terminus, and FXR1 with Myc at the C terminus. The lentiviral transfer vector JHUG was derived from the original L307 vector. The internal ribosomal entry site sequence downstream of a ubiquitin promoter in L307 was deleted and replaced with a multiple cloning site (MCS) followed by the EGFP coding sequence. Mouse FMRP and FMRP(I304N) coding sequences were then inserted into the MCS. The RGG box [amino acids RRGDGRRRGGGGRGQGGRGRGGGFKGN (as described by Darnell et al., 2005a)] was removed using PCR deletion.
Antibodies.
The following mouse monoclonal primary antibodies were used in this study: actin, FMRP, GluR1 N terminus, GluR2, and RARα (Millipore Corporation), PSD-95 (Affinity Bioreagents), NR1 (BD Biosciences Pharmingen), Arc (Santa Cruz Biotechnology), FLAG (Sigma), and Myc (Roche). The following rabbit polyclonal primary antibodies were used: GluR1 (Millipore Corporation), EF2 and phospho-EF2 (Thr56) (Cell Signaling Technology), Stargazin and Myc (Abcam), and MAP1b 750 (a generous gift from Dr. Itzhak Fischer, Drexel University, Philadelphia, PA).
Drugs and chemicals.
The following drugs and chemicals were purchased from Sigma-Aldrich: all-trans retinoic acid, actinomycin D, cycloheximide, picrotoxin, philanthotoxin-433 (PhTx), and 4-(diethylamino)-benzaldehyde (DEAB). Tetrodotoxin was purchased from Tocris Biosciences and d-APV from Thermo Fisher Scientific.
Mice.
Wild-type (WT) and Fmr1 knock-out mice in the FVB background were obtained from The Jackson Laboratory.
Cell cultures and drug treatment.
Primary hippocampal cultures were prepared from mice at postnatal day 0 (P0) or P1 and maintained in serum-free Neurobasal medium supplemented with B-27 and Glutamax (Invitrogen) for 2 weeks in vitro (Nam and Chen, 2005). Hippocampal slice cultures were prepared from P6 or P7 animals and maintained in Neurobasal-A medium supplemented with horse serum (Hyclone), insulin (Sigma), and Glutamax (Aoto et al., 2008). Stock solutions of all-trans RA in DMSO were freshly made immediately before treatment, and the final concentration of DMSO in culture media was 0.05% or lower. Twenty-four hour treatment of 1 μm TTX and 100 μm APV was used to induce synaptic scaling in dissociated cultures, and 36 h treatment of 10 μm TTX and 1 mm APV was used to induce synaptic scaling in slice cultures. Four hour treatment of 2 μm RA was used to induce synaptic scaling in slice cultures, and 30 min treatment of 1 μm RA followed by 1 h of washout was used to induce synaptic scaling in dissociated cultures. When indicated, 100 μm cycloheximide or 0.5 μg/ml actinomycin D was applied for 30 min before RA treatment and remained in the media during RA treatment. To induce RA-independent synaptic scaling, 48 h treatment of 1 μm TTX in dissociated culture or 60 h treatment of 10 μm TTX in slice culture was used. DEAB at 10 μm was applied when indicated.
Retinoic acid response element assay.
Dissociated cultures used for retinoic acid response element (RARE) imaging were transfected using Lipofectamine 2000 (Invitrogen) with a protocol described previously (Aoto et al., 2008) and were fixed with 4% paraformaldehyde (15 min, room temperature) and washed with PBS before mounting. Images were acquired and quantified as described previously (Nam and Chen, 2005) using an Olympus FV1000 BX61WI laser-scanning confocal microscope.
Lentivirus production and infection of slices and dissociated neurons.
Lentivirus was produced and purified as described previously (Aoto et al., 2008). Briefly, human embryonic kidney 293T (HEK293T) cells were transfected using calcium phosphate with the transfer vector and three helper plasmids. After 48 h, supernatants were pooled, spun at 25,000 rpm through a sucrose cushion for 1.5 h, and resuspended in PBS. Virus was injected into the CA1 region of slices on the day of cutting. For imaging, slices at 6–7 d in vitro (DIV) were fixed overnight in 4% paraformaldehyde at 4°C. Slices were washed in PBS, mounted, and imaged as described above. To infect dissociated cells, purified virus was applied to the culture media overnight and washed out the following day. Neurons were infected at 7 DIV and lysates were harvested at 13 DIV to mimic the expression time seen in slice cultures.
Electrophysiology.
Patch-clamp recordings from the CA1 region of slice cultures were made at room temperature from 5–7 DIV slices with a 4–6 MΩ patch pipette filled with an internal solution containing the following (in mm): 140 CsCl, 2 MgCl2, 5 EGTA, 10 HEPES, 0.3 Na3-GTP, and 4 Na2-ATP, pH 7.35. Slices were continuously superfused with external solution containing the following (in mm): 120 NaCl, 26 NaHCO3, 2.5 KCl, 11 glucose, 2.5 CaCl2, 1.3 MgSO4, and 1.0 NaH2PO4. Tetrodotoxin (1 μm) and picrotoxin (100 μm) were included in the external saline, along with 5 μm philanthotoxin when indicated. Cells were held at −60 mV. Miniature responses were analyzed with Mini Analysis Program (Synaptosoft).
Surface biotinylation assay.
Cultured hippocampal cells were washed with cold PBS/Mg2+/Ca2+, and surface proteins were biotinylated with 1 mg/ml Ez-link sulfo-NHS-SS-biotin (Pierce) in PBS/Mg2+/Ca2+ for 25 min on ice. Cells were washed with 0.1 m glycine in ice-cold PBS/Mg2+/Ca2+ to stop additional biotinylation of the surface proteins. After additional washes with ice-cold PBS, cells were collected and solubilized in lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.25% Na-deoxycholate, 1% NP-40, 1 mm EDTA, 0.1% SDS, and protease inhibitor cocktail). Lysates were centrifuged to remove cell debris and nuclei at 14,000 rpm for 20 min, and supernatants were rotated with Ultralink-immobilized streptavidin beads (Thermo Fisher Scientific) for 2 h at 4°C to bind biotinylated proteins. Beads were then pelleted and washed four times with lysis buffer. Biotinylated surface proteins were eluted with denaturing buffer at 65°C. Surface-expressed AMPA receptors were detected by Western blot analysis.
Synaptoneurosome preparation.
Whole hippocampi or cultured hippocampal slices were gently homogenized in a solution containing 33% sucrose, 10 mm HEPES, 0.5 mm EGTA, pH 7.4, and protease inhibitors. Nuclei and other debris were pelleted at 2000 × g for 5 min at 4°C, and the supernatant was filtered through three layers of 100 μm pore nylon mesh (Millipore Corporation) and a 5 μm pore polyvinylidene difluoride (PVDF) syringe filter (Millipore Corporation). The filtrate was then centrifuged for 10 min at 10,000 × g at 4°C, and the supernatant was removed. The synaptoneurosome-containing pellet was then resuspended in lysis buffer (see above).
Spine morphology assay.
Neurons were transfected as described above at 12–13 DIV with pSUPER, a plasmid that expresses high levels of EGFP. Cells were then treated, fixed, and imaged as described above. Two to three secondary branches per cell were analyzed for spine density and spine length using Matlab software; length was determined by measuring the distance from the dendritic shaft to the spine tip.
Quantitative PCR.
RNA from cultured slices or synaptoneurosomes was isolated using the Aurum Total RNA Mini kit (Bio-Rad). Equal amounts of RNA from each sample were reverse transcribed using SuperScriptII transcriptase and random hexamer primers (Invitrogen) according to the protocol of the manufacturer. Quantitative PCR (qPCR) was performed using Sybr Green supermix (Bio-Rad) on an iQ5 thermal cycler (Bio-Rad). Cycle threshold values obtained from triplicate technical replicates for each sample were averaged, and relative abundance was determined using a dilution curve. Expression levels for all genes were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). GluR1, GluR2, and GAPDH primer sequences were adapted for mouse from those used by Dijk et al. (2004). All primers were tested for specificity and efficiency using melting curve and dilution curve analysis. Primer sequences are available in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
35S metabolic labeling and immunoprecipitation.
Culture media was removed from dissociated neurons at 13–14 DIV, and cells were washed twice and maintained in DMEM lacking methionine and cysteine (Invitrogen). RA or DMSO was added along with EasyTag Express 35S protein labeling mix (PerkinElmer Life and Analytical Sciences). After 2 h, media were removed and cells were washed twice with ice-cold PBS. Immunoprecipitation was performed as described by Muddashetty et al. (2007). Briefly, cells were lysed with rotating at 4° in lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% NP-40, and 0.5% Na-deoxycholate). Debris was pelleted with centrifugation, and supernatants were rotated for 1 h at 4° with the appropriate antibody. Protein G beads (Invitrogen) were added and rotated with lysates overnight at 4°. Beads were washed one time in lysis buffer, three times in wash buffer 2 (50 mm Tris-HCl, pH 7.4, 300 mm NaCl, 0.1% NP-40, and 0.05% Na-deoxycholate), three times in wash buffer 3 (50 mm Tris-HCl, pH 7.4, 0.1% NP-40, and 0.05% Na-deoxycholate), and in cold PBS. Samples were eluted in SDS sample buffer, loaded on polyacrylamide gels, and transferred to PVDF membranes. The appropriately sized band (identified by Western blot) was cut out from the membrane and analyzed for incorporated radioactivity using liquid scintillation counting. Background counts per minute (determined by analyzing a similar-sized band cut out from an unstained region of each lane) were subtracted from the AMPAR counts per minute values. Duplicate technical replicates were averaged for each sample.
Co-IP.
HEK293T cells were transfected using calcium phosphate with equal amounts of each construct, as indicated. At 24 h after transfection, cells were washed and resuspended in PBS, pelleted, and lysed with rotating for 30 min at 4° (50 mm Tris, pH 7.4, 150 mm NaCl, 5 mm EDTA, 10% glycerol, 0.5% NP-40, and protease inhibitors). Samples were spun to pellet nuclei, and lysate was precleared with Protein-G beads (Invitrogen). Cleared lysates were rotated at 4° with antibody for 4 h and then with beads overnight. Beads were washed three times in lysis buffer, and bound protein was eluted in SDS sample buffer.
Statistical analysis.
Single-factor ANOVA was used for statistical analysis unless otherwise stated. Values are presented as mean ± SEM in the figures.
Results
FMRP is required for TTX + APV-induced synaptic scaling
To directly investigate a possible role for FMRP in homeostatic plasticity, we examined the effect of activity blockade on synaptic transmission in cultured hippocampal slices from Fmr1 knock-out mice. Although 24 h of TTX + APV is sufficient to induce homeostatic plasticity in dissociated neurons, 36 h of treatment is required for robust scaling in slice culture (Aoto et al., 2008). TTX + APV treatment increased the amplitude of miniature EPSC (mEPSC) events in slices obtained from wild-type mice (Fig. 1A,B). In contrast, TTX + APV had no effect on mEPSCs in slices from Fmr1 knock-out mice (Fig. 1A,B), indicating that loss of FMRP causes a defect in synaptic scaling. Consistent with previous reports (Braun and Segal, 2000), the baseline amplitude and frequency of mEPSC events was not different between wild-type and knock-out slices (Fig. 1B) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Neither genotype showed a change in the frequency of mEPSCs after treatment (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Figure 1.

FMRP is required for TTX + APV-induced synaptic scaling. A, Representative mEPSC traces from wild-type and Fmr1 knock-out (untreated and TTX + APV treated) neurons in hippocampal slice culture. Calibration: 10 pA, 40 ms. B, Cumulative distribution of mEPSC amplitudes from WT and KO neurons treated with 36 h of TTX + APV (p < 0.001, Kolmogorov–Smirnov test). Inset, Quantification of average mEPSC amplitude (n = 28–34; ***p < 0.001). C, Representative blots for biotinylation of surface AMPARs in primary cultured neurons after 24 h of TTX + APV treatment. IB, Immunoblot. D, Quantification of C. Surface band intensity was normalized to input, and all groups were compared with WT untreated (n = 4–6; *p < 0.05). Error bars represent SEM.
During synaptic scaling induced by TTX + APV, the AMPA receptor subunit GluR1 is synthesized locally in dendrites, and homomeric GluR1 AMPA receptors are inserted into the synaptic membrane, thereby increasing the strength of the synapse (Ju et al., 2004; Sutton et al., 2006; Aoto et al., 2008). Consistent with this process, activity blockade with TTX + APV caused a significant increase in the levels of surface GluR1, but not GluR2 protein, in wild-type neurons (Fig. 1C,D). However, TTX + APV treatment failed to increase the surface levels of either GluR1 or GluR2 protein in neurons from Fmr1 knock-out mice (Fig. 1C,D). This result corroborates the impairment in homeostatic plasticity seen with electrophysiology and indicates that FMRP is required for synaptic scaling upstream of the insertion of new GluR1 receptors into the plasma membrane. Dendritic GluR1 and GluR2 mRNA levels are normal in Fmr1 knock-out neurons (Muddashetty et al., 2007), and the basal levels of GluR1 and GluR2 protein in both whole hippocampal lysate and synaptoneurosomes were not different between wild-type and knock-out mice (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material). In addition, we saw no difference in the levels of RARα protein (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material), which is also required for synaptic scaling (Aoto et al., 2008).
RA synthesis is normal in Fmr1 knock-out neurons
Because RA synthesis is both necessary and sufficient for synaptic scaling (Aoto et al., 2008), we tested whether impaired synaptic scaling in Fmr1 knock-out neurons is attributable to altered RA synthesis, using a genetic reporter system (Aoto et al., 2008). Dissociated hippocampal neurons from wild-type or Fmr1 knock-out mice were transfected with a plasmid containing multiple copies of an RARE driving transcription of GFP (Fig. 2A). RARα is not only a translational regulator but also a transcription factor that binds to RARE sequences in the presence of RA and promotes transcription of GFP from the reporter plasmid. Thus, the GFP intensity in transfected neurons serves as a readout of RA levels in those neurons. Using this system, we found that TTX + APV treatment caused a significant increase in the intensity of GFP fluorescence in both wild-type and knock-out neurons (Fig. 2B,C), demonstrating that FMRP is not required for the stimulation of RA synthesis in response to activity blockade.
Figure 2.

FMRP is not required for RA synthesis but is specifically required for RA-induced local translation-dependent synaptic scaling. A, Schematic of the 3xDR5–RARE–GFP reporter construct. B, Representative images of RARE–GFP reporter expression in WT and KO neurons with and without 24 h TTX + APV treatment. Scale bar, 10 μm. C, Quantification of B (n = 16–18; *p < 0.05). D, Representative traces and quantification of mEPSC amplitude in WT and KO neurons after 4 h DMSO or RA treatment (n = 31–33; ***p < 0.001). Philanthotoxin-433 was used to block GluR2-lacking AMPA receptor-mediated responses in the WT–RA group (n = 22). Calibration: 10 pA, 40 ms. E, Effect of transcription inhibitor actinomycin D and translation inhibitor cycloheximide on RA-induced synaptic scaling in WT neurons (n = 22–27; ***p < 0.001). Error bars represent SEM.
RA-dependent scaling requires FMRP and new protein translation
Because RA synthesis is maintained in Fmr1 knock-out neurons, we wondered whether direct application of RA is still capable of upscaling synaptic strength in these neurons. In cultured hippocampal slices from wild-type mice, 4 h of RA treatment caused a significant increase in mEPSC amplitude without affecting event frequency (Fig. 2D) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The increase in mEPSC amplitude in wild-type neurons was fully reversed by PhTx, a blocker of GluR2-lacking AMPA receptors (Fig. 2D), indicating that the increase in synaptic strength after RA treatment is caused by insertion of homomeric GluR1 receptors at the synapse. In addition, RA-induced upscaling in wild-type slices was unaffected by transcription blockers but required de novo translation of preexisting mRNAs (Fig. 2E). This is similar to previous findings demonstrating increased local translation of specific proteins during TTX + APV or RA-mediated synaptic scaling (Aoto et al., 2008). Importantly, RA treatment did not increase synaptic strength in slices from Fmr1 knock-out mice (Fig. 2D). Thus, FMRP is required for synaptic scaling induced by TTX + APV or RA and acts downstream of RA.
RA treatment does not affect synaptic AMPAR mRNA levels or spine morphology
FMRP is known to play a role in the activity-dependent dendritic trafficking of specific mRNAs (Dictenberg et al., 2008). We wondered whether RA induces the movement of mRNAs toward synapses and, if so, whether FMRP is required for this process. Synaptoneurosomes were collected from wild-type or knock-out cultured hippocampal slices treated with DMSO or RA, and total RNA was isolated from these preparations. qPCR showed no differences in AMPAR mRNA levels between wild-type and knock-out synaptoneurosomes (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). Also, no effect of RA on mRNA levels was found in either genotype (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). This implies that AMPAR mRNA trafficking does not play a significant role in synaptic scaling and that FMRP has no obvious effect on the synaptic localization of these mRNAs.
Neurons from FMRP knock-out mice have altered dendritic spine morphology, showing an increased spine length and a larger proportion of immature spines (Comery et al., 1997; Nimchinsky et al., 2001; Grossman et al., 2006). Because changes in spine morphology are known to accompany changes in synapse strength (Muller et al., 2000; Matsuzaki et al., 2004), we asked whether TTX + APV or RA treatment affects spine morphology and whether this might account for impaired homeostatic plasticity in FMRP knock-out animals. Analysis of GFP-expressing wild-type and knock-out neurons treated with TTX + APV or RA found no changes in spine density between genotypes or between treatments (Fig. 3A,B), confirming our physiology results showing no change in mEPSC frequency during homeostatic plasticity (supplemental Figs. 1A, 2, available at www.jneurosci.org as supplemental material). Consistent with the literature, we found an increased average spine length in knock-out neurons compared with wild-type, but neither TTX + APV nor RA treatment had any effect on spine length in either genotype (Fig. 3A,C). Although this does not rule out that subtle spine shape changes may occur during synaptic scaling, we find no obvious link between the FMRP knock-out altered spine phenotype and the inability of these neurons to increase their synaptic strength after TTX + APV or RA treatment.
Figure 3.

RA does not affect spine morphology in WT or KO neurons. A, Sample images of GFP-expressing WT or KO neurons treated with TTX + APV or RA. Scale bar, 5 μm. B, C, Quantification of spine density and spine length in WT and KO neurons treated with 24 h of TTX + APV or 30 min (plus 1 h washout) of RA (n = 9–10 cells per group, 2–3 branches per cell). For spine density, p > 0.5. For spine length *p < 0.05, **p < 0.01. Error bars represent SEM.
RA and FMRP are not involved in the slow transcription-dependent form of synaptic scaling
Activity blockade with TTX and APV, applied for 24 h in dissociated neurons or 36 h in slice culture, is only one of several manipulations that can be used to induce homeostatic plasticity. A similar magnitude of increase in mEPSC amplitude is seen after long-term treatment with TTX only [48 h in dissociated neurons (Turrigiano et al., 1998) or 60 h in slice culture (our results)]. Different from the rapid GluR1-dependent upscaling induced by TTX + APV, upscaling produced by TTX alone is mediated by an increase in GluR1/GluR2 heteromeric receptors and is transcription dependent (Wierenga et al., 2005; Ibata et al., 2008). We wondered whether FMRP is also necessary for this slower, transcription-dependent scaling induced by TTX alone. First, we confirmed that, in our hands, 36 h of TTX-alone treatment in wild-type slice cultures is insufficient to induce synaptic scaling (supplemental Fig. 4A,B, available at www.jneurosci.org as supplemental material). Intriguingly, however, we found that long-term (60 h) TTX treatment induced synaptic upscaling of mEPSC amplitudes even in the absence of FMRP (Fig. 4A) (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). The increase in synaptic strength was not reversed by philanthotoxin, confirming that the change in mEPSC amplitude is caused by the insertion of GluR2-containing receptors (Fig. 4A).
Figure 4.
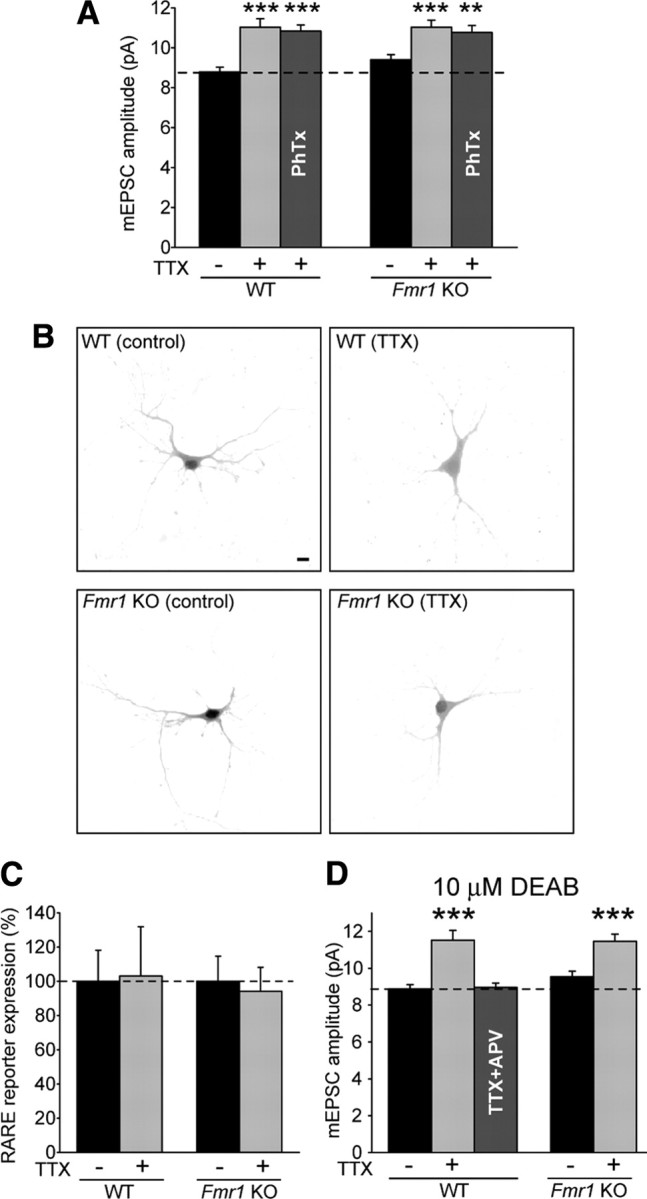
Neither FMRP nor RA is required for TTX-alone-induced synaptic scaling. A, Sixty hours of TTX induced synaptic scaling in WT and KO neurons. PhTx was used to block GluR2-lacking AMPA receptor-mediated responses (n = 18–39; **p < 0.01; ***p < 0.001). B, Representative images of RARE–GFP reporter expression in WT and KO neurons with and without 48 h of TTX treatment. Scale bar, 10 μm. C, Quantification of B (n = 22–28). D, Effect of the RA synthesis blocker DEAB on 60 h of TTX-induced synaptic scaling and 36 h of TTX + APV-induced scaling (n = 21–27; ***p < 0.001). Error bars represent SEM.
The specific involvement of FMRP in TTX + APV- and RA-induced synaptic scaling but not in TTX-induced scaling suggests that RA may not be involved in the slow, transcription-dependent form of homeostatic plasticity. Indeed, when we used the RARE reporter to measure RA synthesis after 48 h of TTX treatment in dissociated neurons, we found no increase in GFP fluorescence in either wild-type or knock-out neurons (Fig. 4B,C), indicating no change in RA levels. Moreover, blocking RA synthesis with DEAB, an inhibitor of retinal dehydrogenase (an enzyme in the RA synthesis pathway), blocked TTX + APV-induced scaling but did not prevent synaptic scaling induced by long-term TTX-alone treatment in wild-type or Fmr1 knock-out slices (Fig. 4D) (supplemental Fig. 4D, available at www.jneurosci.org as supplemental material). Thus, FMRP and RA synthesis are both specifically required for the form of synaptic scaling that is induced by TTX + APV and accomplished via local translation.
Translation of RARα target mRNAs requires FMRP
To probe the mechanism by which FMRP acts downstream of RA in synaptic scaling, we examined the local synthesis of synaptic proteins in dendrites in response to RA. We isolated total lysates and synaptoneurosomes from wild-type and Fmr1 knock-out hippocampal slices treated with DMSO or RA and examined the levels of synaptic proteins by Western blotting. We found that, in synaptoneurosomes from wild-type slices, RA significantly increased the levels of GluR1, GluR2, and eEF2 proteins (Fig. 5A,C). This effect was blocked by cycloheximide, indicating that the change is dependent on new protein translation (Fig. 5D). RA also marginally increased the levels of FMRP, but this was not statistically significant (Fig. 5C). RA had no effect on the levels of other synaptic proteins examined, including phosphorylated eEF2, PSD-95, Stargazin, or NR1 (Fig. 5A,C).
Figure 5.
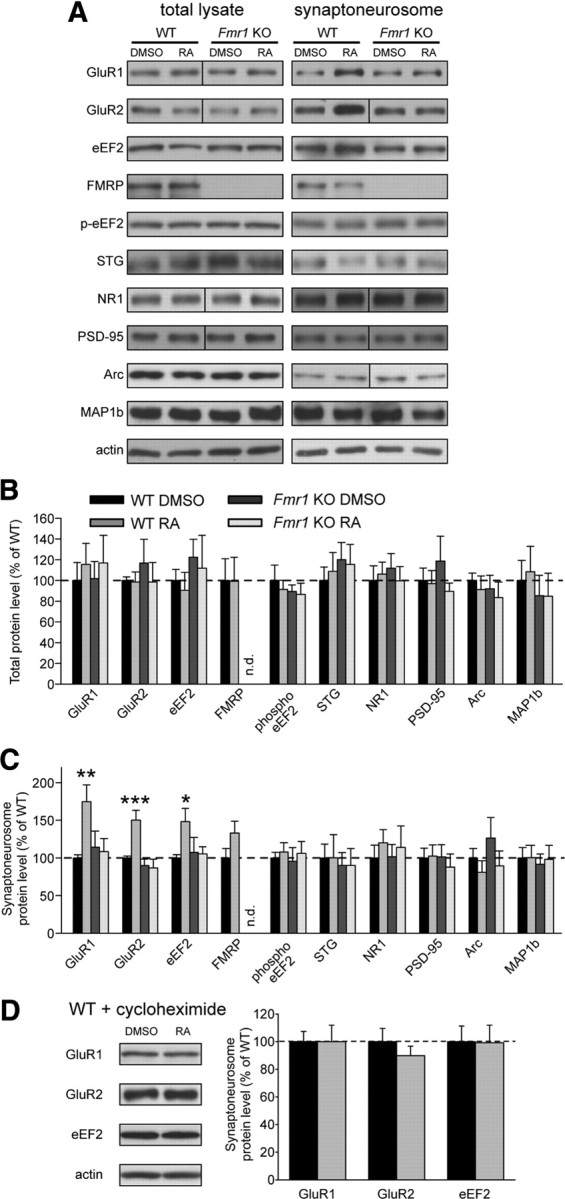
FMRP is required for RA-induced local translation of specific synaptic proteins. A, Representative blots of synaptic proteins from whole lysates and synaptoneurosome (SNS) fractions of WT and KO hippocampal slices, treated with 4 h of DMSO or RA. A vertical line indicates the removal (for ease of viewing) of extra lanes between WT and KO lanes. B, C, Quantification of synaptic proteins in the total lysate (B) and synaptoneurosome fraction (C) from treated hippocampal slices. Band intensities were normalized to actin (n.d., not detected; STG, Stargazin; n = 5–16; *p < 0.05; **p < 0.01; ***p < 0.001). D, Representative blots and quantification of proteins from the synaptic fraction of WT slices pretreated with the translation blocker cycloheximide before DMSO or RA treatment (n = 5). Error bars represent SEM.
Strikingly, RA treatment failed to elicit changes in any synaptic protein in synaptoneurosomes from Fmr1 knock-out slices (Fig. 5A,C), indicating that FMRP is indeed required for RA-stimulated increases in synaptic protein levels. In contrast to synaptoneurosomes, we observed no RA-dependent changes in the abundance of any protein in whole-cell lysates from either wild-type or Fmr1 knock-out slices (Fig. 5A,B), consistent with the notion that RA-induced translation in wild-type slices is a local phenomenon, occurring in dendrites near synapses.
We also examined the effect of RA on the synaptic levels of two verified FMRP target proteins, MAP1b and Arc. Although some groups have reported increased baseline MAP1b levels in FMRP knock-out animals at some (but not all) developmental stages (Lu et al., 2004; Hou et al., 2006), others have seen decreased levels in knock-out tissue (Chen et al., 2003; Wei et al., 2007). We saw no detectable differences in MAP1b levels between wild-type and knock-out slices at this developmental stage and no effect of RA on MAP1b levels (Fig. 5A–C). The immediate early gene Arc, which promotes internalization of AMPARs, is not only thought to be regulated by FMRP (Zalfa et al., 2003) but is also known to play a role in synaptic scaling (Shepherd et al., 2006). We saw no change in Arc levels after RA treatment and no baseline differences in protein level between wild-type and knock-out slices (Fig. 5A–C). It should be noted, however, that Arc has been implicated only in the form of scaling induced by long-term TTX-alone treatment (Shepherd et al., 2006) and has not been examined in the context of TTX + APV-induced scaling.
RA-induced GluR1 translation is dependent on FMRP
Although our analysis of specific proteins after RA treatment showed an increase in synaptic AMPAR levels that was dependent on both FMRP and new protein translation (Fig. 5), we wanted to more directly assay the effect of RA on the translation of new AMPA receptors. 35S-labeled methionine and cysteine were added to wild-type and knock-out neurons along with DMSO or RA. Dissociated cultures were used for these experiments to ensure a rapid and complete penetration of the labeling mix to all cells. After 2 h of treatment, cell lysates were collected and radioimmunoprecipitation was used to analyze synthesis of GluR1 and GluR2 proteins (Muddashetty et al., 2007).
First, total cell lysate samples were subjected to gel electrophoresis and autoradiography to verify effective labeling. No obvious effect of RA on global translation was seen in either wild-type or knock-out neurons, consistent with our observation that RA only affects the local translation of specific proteins (Fig. 6A). Consistent with previous reports (Dolen et al., 2007), we did observe a slight increase in overall 35S incorporation in knock-out neurons compared with wild-type (Fig. 6A), indicating globally elevated baseline translation in knock-out cells.
Figure 6.

Metabolic labeling shows increased GluR1 translation with RA treatment, and FMRP and RARα do not interact directly. A, Autoradiography of total protein lysate from dissociated neurons treated with 2 h of DMSO or RA in the presence of 35S-labeled amino acids. B, Quantification of incorporated radioactivity using liquid scintillation counting after immunoprecipitation of AMPARs. Each genotype was normalized separately to its DMSO group (n = 5–8; **p < 0.01). C, Representative blots for biotinylation of surface AMPARs in primary cultured neurons after RA treatment. IB, Immunoblot. D, Quantification of surface GluR1 and GluR2 protein levels after RA treatment. Surface band intensity was normalized to input, and all groups were compared with WT DMSO (n = 3–9; *p < 0.05). E, Attempted coimmunoprecipitation of FMRP and RARα. Tagged constructs were expressed in HEK293T cells as indicated, and immunoprecipitation (IP) of either FMRP (anti-FLAG) or RARα (anti-Myc) was performed (Input: 10% of total). Although pull down of FMRP was able to coimmunoprecipitate the known binding partner FXR1 (positive control), no interaction was seen between FMRP and RARα. Error bars represent SEM.
Immunoprecipitation of GluR1 and quantification of 35S incorporation showed a significant increase in radiolabeled GluR1 after RA treatment in wild-type, but not knock-out, neurons (Fig. 6B). This demonstrates both that RA induces translation of GluR1 and that FMRP is required for this translation to occur. We saw no increase in radiolabeled GluR2 protein after RA treatment (Fig. 6B), although we saw an increase in GluR2 protein in synaptoneurosomes from RA-treated slice cultures (Fig. 5). Although we cannot rule out the possibility that the constitutive somatic translation of GluR2 in the absence of RA masks induced translation of GluR2 in neuronal dendrites, this may also reflect a difference between dissociated and slice preparations or may imply that the increased GluR2 seen at the synapse in slices is attributable to altered trafficking or degradation of existing protein, not new protein translation.
Indeed, the concurrent increase of both GluR1 and GluR2 proteins seen in RA-treated wild-type synaptoneurosomes (Fig. 5) was somewhat surprising, because the increase in mEPSC amplitude that follows TTX + APV or RA treatment is attributable to synaptic insertion of GluR2-lacking AMPA receptors (Fig. 2D) (Aoto et al., 2008). Surface biotinylation confirmed that RA treatment only increased GluR1, but not GluR2, protein levels on the cell surface (Fig. 6C,D), indicating that the increased GluR2 protein seen in synaptoneurosomes after RA treatment is not reaching the surface or contributing to synaptic transmission at the time point examined.
FMRP and RARα proteins do not interact directly
Because of the previously demonstrated role for RARα in binding to GluR1 mRNA and regulating its translation (Aoto et al., 2008; Poon and Chen, 2008), we wondered whether FMRP might affect GluR1 translation by interacting directly with RARα protein. To test for a possible interaction under permissive conditions, we expressed FLAG-tagged FMRP and Myc-tagged RARα proteins in HEK293T cells and attempted coimmunoprecipitation in both directions. Although we were able to demonstrate co-IP of FMRP and FXR1 (a known binding partner), we found no evidence of direct interaction between FMRP and RARα (Fig. 6E).
Acute postsynaptic expression of FMRP in knock-out neurons rescues synaptic scaling
Is FMRP required directly for TTX + APV- and RA-induced synaptic scaling, or are the deficits seen in Fmr1 knock-out mice attributable to altered development in the absence of FMRP? To answer this question, we used lentiviral delivery to express GFP-tagged FMRP in CA1 neurons of slices obtained from Fmr1 knock-out mice and tested whether this could restore synaptic scaling. We also tested two mutant forms of FMRP in an attempt to identify which domains of the protein might be critical for the regulation of homeostatic plasticity. FMRP has two major RNA binding domains: an RGG box, which binds RNAs containing a G-quartet structure (Darnell et al., 2001), and the tandem KH domains (KH1 and KH2), which bind RNAs containing a characteristic “kissing complex” structure (Darnell et al., 2005b). To separate the functions of these two domains, we tested an FMRP construct that was missing the RGG box (FMRPΔRGG–GFP) and one containing a point mutation (I304N) in the KH2 domain [FMRP(I304N)–GFP]. The pathogenic mutation I304N does not prevent FMRP from localizing to dendrites or binding G-quartet RNAs but does inhibit binding with KH2-interacting RNAs (Darnell et al., 2005b; Zang et al., 2009). The I304N mutation also prevents the association of FMRP with actively translating polyribosomes, possibly by inhibiting homo-oligomerization of the protein (Feng et al., 1997; Laggerbauer et al., 2001; Wang et al., 2008).
We first expressed these constructs in wild-type dissociated neurons and quantified their expression levels with immunoblotting. All three constructs expressed at similar levels, and none of them altered the endogenous FMRP expression level compared with GFP-expressing cells (Fig. 7A). The exogenous expression levels were approximately equal to total endogenous FMRP levels (Fig. 7A) and were 2-fold to 2.5-fold higher than expression of the largest FMRP isoform (isoform 1) alone (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material). Neither the wild-type nor the mutant FMRP constructs altered AMPAR abundance (supplemental Fig. 5E,F, available at www.jneurosci.org as supplemental material). Additionally, overexpression of exogenous FMRP and its mutant forms did not change the basal synaptic transmission or prevent synaptic scaling in wild-type neurons in response to TTX + APV (supplemental Fig. 5B–D, available at www.jneurosci.org as supplemental material).
Figure 7.

Viral expression of FMRP in knock-out slices restores synaptic scaling. A, Representative blot and quantification of exogenous FMRP–GFP, FMRP(I304N)–GFP, and FMRPΔRGG–GFP expression compared with endogenous FMRP protein levels after 6 d of virus expression in wild-type dissociated neurons (n = 6). B, Images from the CA1 region of hippocampal slices infected with lentiviral constructs expressing GFP, FMRP–GFP, FMRP(I304N)–GFP, or FMRPΔRGG–GFP. Asterisks indicate cell body. FMRP and FMRPΔRGG exhibit a punctate expression pattern in neuronal dendrites (filled arrows), whereas GFP or FMRP(I304N) are diffusely expressed in neuronal dendrites (open arrows). Scale bar, 10 μm. C, Amplitude of mEPSC events in KO neurons expressing GFP, FMRP, FMRP(I304N), or FMRPΔRGG (n = 38–66; ***p < 0.001). D, Percentage scaling after RA treatment in neurons expressing GFP or different FMRP constructs. DMSO groups for each construct were set to 100% to account for altered baseline amplitudes (see C) (n = 18–49; ***p < 0.001). E, Percentage scaling after treatment with 36 h of TTX + APV (n = 17–20; *p < 0.05). Error bars represent SEM.
We next introduced these wild-type and mutant FMRP constructs into knock-out neurons. Consistent with the reported localization of FMRP to RNA granules (Antar et al., 2004; Aschrafi et al., 2005), we found that expression of FMRP in knock-out neurons yielded a distinct punctate pattern in dendrites, resembling that of the endogenous protein (Fig. 7B) (supplemental Fig. 6A, available at www.jneurosci.org as supplemental material). FMRPΔRGG exhibited a similar expression pattern, as has been reported (Pfeiffer and Huber, 2007). In contrast, expression of FMRP(I304N) in knock-out neurons yielded a more diffuse, less punctate expression pattern in dendrites (Fig. 7B), which is consistent with its altered function and similar to previous reports (Schrier et al., 2004; Pfeiffer and Huber, 2007).
Introduction of FMRP into knock-out neurons caused a small reduction in the baseline amplitude of mEPSCs compared with cells expressing GFP alone (Fig. 7C). No change in mEPSC frequency was observed (supplemental Fig. 6B,C, available at www.jneurosci.org as supplemental material). Similarly, FMRP(I304N) expression also reduced the baseline amplitude of mEPSCs, but FMRPΔRGG had no effect on mEPSC amplitude (Fig. 7C). These results differ somewhat from a previous report (Pfeiffer and Huber, 2007) of decreased frequency, rather than amplitude, of mini events after FMRP expression in knock-out neurons. These disparities are possibly attributable to differences in the age of slices used or in the time course or level of FMRP expression.
Our data are consistent, however, with the finding that GluR1 and GluR2 mRNA levels are elevated in polyribosomes of Fmr1 knock-out mice (Muddashetty et al., 2007), indicating overactive baseline translation of these proteins. The acute reintroduction of FMRP into Fmr1 knock-out neurons may reduce this elevated translation back down to wild-type levels, thus temporarily decreasing the amount of these proteins and, by consequence, synaptic strength. In support of this hypothesis, we found that expression of FMRP or FMRP(I304N) in dissociated neurons from knock-out animals led to a significant reduction in both GluR1 and GluR2 protein levels, although NR1 and PSD-95 levels were unchanged (Fig. 8A,B). Surface AMPAR levels, as measured by biotinylation and pull down, were reduced proportionally with total AMPAR protein levels, so that the ratio of surface to total fractions remained constant across all conditions (Fig. 8C,D). Consistent with our observation that the RGG box is required for the reduction in mEPSC amplitude caused by FMRP reintroduction, FMRPΔRGG had no effect on levels of any of the proteins measured (Fig. 8). These data suggest that FMRP does indeed regulate the abundance of AMPARs, either through direct binding to AMPAR mRNAs or through controlling the translation of other regulatory proteins.
Figure 8.

Viral expression of FMRP in knock-out neurons affects AMPAR abundance. Representative blots (A) and quantification of protein levels in lysates (B) collected from KO dissociated neurons infected with virus expressing GFP, FMRP, FMRP(I304N), or FMRPΔRGG. Band intensities were normalized to actin (n = 4–10; *p < 0.05; **p < 0.01). Representative blots (C) and quantification of surface biotinylation of AMPARs (D) in knock-out dissociated neurons infected with virus expressing GFP or different FMRP constructs. Surface band intensity was normalized to input (n = 5–7). Error bars represent SEM.
Importantly, FMRP restored the ability of Fmr1 knock-out neurons to undergo synaptic scaling after RA treatment (Fig. 7D) or TTX + APV treatment (Fig. 7E). In contrast, neither GFP alone, FMRP(I304N), nor FMRPΔRGG rescued synaptic scaling induced by RA or TTX + APV (Fig. 7D,E). This confirms that FMRP is required acutely in the postsynaptic cell for induction of the form of synaptic scaling mediated by RA/RARα and that the ability of FMRP to both repress and permit the translation of specific transcripts is critical for RA-induced local translation and synaptic scaling in neurons.
Discussion
Homeostatic plasticity, specifically synaptic scaling, maintains network stability and the coding capacity of neural circuits (Turrigiano and Nelson, 2004; Davis, 2006). It has been shown that activity blockade by TTX and APV induces a form of synaptic scaling that requires dendritic protein synthesis (Ju et al., 2004; Sutton et al., 2006), which we have shown to be mediated by RA signaling (Aoto et al., 2008). In the present study, we identify FMRP as a critical factor required for homeostatic plasticity and regulation of synaptic strength by RA. FMRP is not essential for RA production but mediates RA-induced protein synthesis and is specifically involved in the form of homeostatic plasticity that requires dendritic translation of discrete synaptic proteins. We also demonstrate that RA-dependent homeostatic plasticity in Fmr1 knock-out neurons is rescued by wild-type FMRP but not by FMRP(I304N) or FMRPΔRGG. This result indicates that FMRP regulation of protein translation mediates the induction of homeostatic plasticity triggered by RA.
The involvement of FMRP in homeostatic plasticity and in RA signaling is unexpected and raises several new questions. How do FMRP and RARα work together to regulate RA-mediated translation during homeostatic plasticity? Although we were unable to demonstrate direct binding between these two proteins, it is possible that they interact by binding to the same RNA molecules. Deciphering the functional interplay between FMRP and RARα will be a critical step toward understanding the molecular basis of FMRP-mediated translational regulation and the Fmr1 knock-out phenotype.
The ability of FMRP to function as a translational repressor has been well described (Comery et al., 1997; Laggerbauer et al., 2001; Li et al., 2001). Numerous studies have, with some success, attempted to identify mRNAs that bind directly to FMRP (Sung et al., 2000; Brown et al., 2001; Chen et al., 2003; Miyashiro et al., 2003; Zou et al., 2008), and some specific mRNA targets have been verified, including PSD-95, MAP1b, and CaMKII (Brown et al., 2001; Hou et al., 2006; Zalfa et al., 2007). Although FMRP has not been reported to directly bind GluR1 or GluR2 mRNAs (Zalfa et al., 2007), our data and those of others (Muddashetty et al., 2007; Schütt et al., 2009) do support its involvement in regulating AMPAR translation, possibly via indirect binding or regulation of secondary factors.
Our viral expression data support a model whereby elevated baseline translation of AMPARs in the absence of FMRP could partially account for the failure of Fmr1 knock-out neurons to respond to RA treatment. This elevated translation could impose a “ceiling effect,” masking or inhibiting any additional increases in translation. Reintroducing FMRP into knock-out cells lowers translation to normal levels, thus reducing AMPAR protein levels and mEPSC amplitude, allowing cells to then respond to TTX + APV or RA treatment. However, our results with I304N mutant FMRP (Fig. 7) show that simply reducing AMPAR protein levels is not sufficient to rescue plasticity. Therefore, wild-type FMRP must also participate in the activation of AMPAR translation in response to RA, which in turn leads to increased synaptic strength.
The fact that baseline AMPAR levels are unaltered in knock-out neurons does not inherently contradict our model. The effect of FMRP on AMPAR translation is intricate; our results suggest that FMRP not only represses basal translation of AMPARs but also is required for activity blockade-induced activation of AMPAR translation. Therefore, it is difficult to predict the effect that the constitutive absence of FMRP would have on total protein levels. Second, compensatory effects during development, such as altered protein degradation, could adjust the abundance of AMPARs over time in knock-out animals. This can be seen with some other validated FMRP targets, such as MAP1b (Chen et al., 2003; Lu et al., 2004; Hou et al., 2006; Wei et al., 2007) and PSD-95 (Todd et al., 2003; Schütt et al., 2009), which do not show consistently elevated levels in knock-out neurons.
We were also able to use mutant FMRP constructs to further understand the mechanism of the action of FMRP in homeostatic plasticity. FMRP lacking the RGG box RNA-binding domain failed to affect baseline AMPAR levels or to restore synaptic scaling, indicating that this domain is critical for the dynamic regulation of AMPAR levels, particularly in the context of homeostatic plasticity. This is not surprising, because the RGG box binds RNAs specifically and with high affinity (Darnell et al., 2001) and mediates the interaction of FMRP with several validated target mRNAs, including those encoding MAP1b, semaphorin 3F, and FMRP itself (Schaeffer et al., 2001; Menon and Mihailescu, 2007; Menon et al., 2008). Results with the FMRP(I304N) mutant support this model. This form of FMRP, with an intact RGG box, was still able to bind RNA and downregulate AMPAR levels, but because this mutant protein cannot enter actively translating polyribosomes, it did not restore the ability of neurons to increase AMPAR translation in response to activity blockade.
When overexpressed in wild-type neurons, none of the forms of FMRP tested had an effect on AMPAR abundance or the ability of the neurons to undergo synaptic scaling. This is not surprising, because neither FMRP(I304N) nor FMRPΔRGG is expected to act as a dominant negative in this context. FMRP(I304N) is able to heterodimerize with wild-type FMRP and subsequently be recruited to RNA granules (Laggerbauer et al., 2001; Levenga et al., 2009), and FMRPΔRGG is unable to bind G-quartet RNA and thus should not interfere with endogenous FMRP regulation of these transcripts.
We saw changes in the synaptic levels of three proteins (GluR1, GluR2, and eEF2) in response to RA. Interestingly, these three proteins are each encoded by an mRNA that binds directly to RARα through a motif in its 5′ untranslated region, which confers RARα binding ability (Poon and Chen, 2008). Previous in vitro study of the GluR1 untranslated region showed that RARα binding inhibits translation but that this inhibition is relieved during addition of RA. Our results suggest, therefore, that FMRP may be required for this RA-induced release of inhibition by RARα.
One intriguing observation is that, despite the change in GluR2 protein levels near the synapse in response to RA, we were unable to directly detect RA-induced GluR2 translation and found no evidence for trafficking of newly synthesized GluR2 protein to the cell surface or its insertion into the postsynaptic membrane. This result points to a differential regulation of GluR1 and GluR2 receptors during synaptic scaling, possibly as a result of specific trafficking or degradation of the two receptor types. In fact, this differential trafficking has already been observed, because it was shown previously that the GluR1 homomeric receptors initially inserted after activity blockade are subsequently replaced by GluR2-containing receptors (Sutton et al., 2006). The precise mechanism at work in this case requires additional investigation and may offer broader insights into the mechanism of AMPAR trafficking.
We show here that FMRP is selectively required for translation-dependent, but not transcription-dependent, synaptic scaling. This observation agrees with previous findings that different protocols used to induce homeostatic plasticity operate via distinct subcellular mechanisms (Sutton et al., 2006; Yu and Goda, 2009), similarly to what has been observed for LTP and LTD induction (Malenka and Bear, 2004). What will be critical for the future of the homeostatic plasticity field is to increase our understanding of how these different protocols correspond to various in vivo situations and how their mechanisms converge to regulate synaptic strength.
Traditionally, Hebbian-type synaptic plasticity is considered the cellular mechanism for learning and memory. As an animal model for fragile X mental retardation, Fmr1 knock-out mice have been studied extensively for defects in neuronal function and learning and memory. Fmr1 knock-out mice have impaired Hebbian-type synaptic plasticity (Huber et al., 2002; Larson et al., 2005), which may contribute to their learning deficits (Mineur et al., 2002; Yan et al., 2004; Koekkoek et al., 2005). Our study reveals an additional requirement for FMRP in homeostatic plasticity and RA-mediated translational regulation of synaptic proteins, suggesting that FMRP and its regulation of protein synthesis participate in multiple forms of activity-dependent synaptic plasticity, although seemingly through distinct mechanisms (Fig. 9). Our finding of impaired homeostatic synaptic plasticity provides a new perspective on the phenotype in Fmr1 knock-out mice and on the symptoms of human fragile X patients. It may explain, for example, the global alterations of neural activity that have been observed in Fmr1 knock-out mice and fragile X syndrome patients (Berry-Kravis, 2002; Yan et al., 2004). Moreover, although homeostatic synaptic adjustment may not be directly involved in encoding memory, its ability to influence network stability and neuronal coding capacity nonetheless could contribute significantly to the cognitive function of an organism. It is plausible that lack of homeostatic regulation destabilizes neural networks and compromises the capacity of the network to undergo Hebbian-type plasticity, which in turn may produce behavioral and learning defects in Fmr1 knock-out mice. Understanding the interplay between these different processes will provide significant additional insight into the molecular mechanisms guiding both homeostatic plasticity and fragile X syndrome.
Figure 9.

FMRP plays a critical role in multiple forms of synaptic plasticity. Synaptic activation leads to FMRP-dependent protein synthesis and the eventual removal of AMPARs from the synapse (mGluR-dependent LTD). By contrast, blockade of synaptic activity causes synthesis of new GluR1 receptors and a subsequent increase in synaptic strength, in a process that requires both FMRP and RARα.
Footnotes
The work was supported by the David and Lucile Packard Foundation, the W. M. Keck Foundation, and National Institute of Mental Health (L.C.). We thank Dr. Peng Jin (Emory University, Atlanta, GA) for providing the FMRP and FMRP(I304N) cDNAs, Dr. Thomas Südhof (Stanford University, Stanford, CA) for the original lentiviral vector, and Dr. Itzhak Fischer for the MAP1b antibody. We also thank Jason Aoto for engineering of viral transfer vectors and assistance with virus generation and purification, Sandhiya Kalyanasundaram for technical assistance, and members of the Chen laboratory for discussion and comments on this manuscript.
References
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24:2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, Chen L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 2008;60:308–320. doi: 10.1016/j.neuron.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschrafi A, Cunningham BA, Edelman GM, Vanderklish PW. The fragile X mental retardation protein and group I metabotropic glutamate receptors regulate levels of mRNA granules in brain. Proc Natl Acad Sci U S A. 2005;102:2180–2185. doi: 10.1073/pnas.0409803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley CT, Jr, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44:724–728. doi: 10.1017/s0012162201002833. [DOI] [PubMed] [Google Scholar]
- Braun K, Segal M. FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb Cortex. 2000;10:1045–1052. doi: 10.1093/cercor/10.10.1045. [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Chen L, Yun SW, Seto J, Liu W, Toth M. The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience. 2003;120:1005–1017. doi: 10.1016/s0306-4522(03)00406-8. [DOI] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin F, Bouillon M, Fortin A, Morin S, Rousseau F, Khandjian EW. The fragile X mental retardation protein is associated with poly(A)+ mRNA in actively translating polyribosomes. Hum Mol Genet. 1997;6:1465–1472. doi: 10.1093/hmg/6.9.1465. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Mostovetsky O, Darnell RB. FMRP RNA targets: identification and validation. Genes Brain Behav. 2005a;4:341–349. doi: 10.1111/j.1601-183X.2005.00144.x. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Fraser CE, Mostovetsky O, Stefani G, Jones TA, Eddy SR, Darnell RB. Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 2005b;19:903–918. doi: 10.1101/gad.1276805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I. Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol. 2001;63:847–869. doi: 10.1146/annurev.physiol.63.1.847. [DOI] [PubMed] [Google Scholar]
- Dictenberg JB, Swanger SA, Antar LN, Singer RH, Bassell GJ. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell. 2008;14:926–939. doi: 10.1016/j.devcel.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijk F, Kraal-Muller E, Kamphuis W. Ischemia-induced changes of AMPA-type glutamate receptor subunit expression pattern in the rat retina: a real-time quantitative PCR study. Invest Ophthalmol Vis Sci. 2004;45:330–341. doi: 10.1167/iovs.03-0285. [DOI] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1:109–118. doi: 10.1016/s1097-2765(00)80012-x. [DOI] [PubMed] [Google Scholar]
- Grossman AW, Elisseou NM, McKinney BC, Greenough WT. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084:158–164. doi: 10.1016/j.brainres.2006.02.044. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R, De Graaf W, Smit AE, VanderWerf F, Bakker CE, Willemsen R, Ikeda T, Kakizawa S, Onodera K, Nelson DL, Mientjes E, Joosten M, De Schutter E, Oostra BA, Ito M, De Zeeuw CI. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron. 2005;47:339–352. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Larson J, Jessen RE, Kim D, Fine AK, du Hoffmann J. Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J Neurosci. 2005;25:9460–9469. doi: 10.1523/JNEUROSCI.2638-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenga J, Buijsen RA, Rifé M, Moine H, Nelson DL, Oostra BA, Willemsen R, de Vrij FM. Ultrastructural analysis of the functional domains in FMRP using primary hippocampal mouse neurons. Neurobiol Dis. 2009;35:241–250. doi: 10.1016/j.nbd.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19:138–151. doi: 10.1006/mcne.2001.1085. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Wang H, Liang Z, Ku L, O'Donnell WT, Li W, Warren ST, Feng Y. The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci U S A. 2004;101:15201–15206. doi: 10.1073/pnas.0404995101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, Chen L. Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc Natl Acad Sci U S A. 2008;105:16015–16020. doi: 10.1073/pnas.0804801105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon L, Mihailescu MR. Interactions of the G quartet forming semaphorin 3F RNA with the RGG box domain of the fragile X protein family. Nucleic Acids Res. 2007;35:5379–5392. doi: 10.1093/nar/gkm581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon L, Mader SA, Mihailescu MR. Fragile X mental retardation protein interactions with the microtubule associated protein 1B RNA. RNA. 2008;14:1644–1655. doi: 10.1261/rna.1100708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur YS, Sluyter F, de Wit S, Oostra BA, Crusio WE. Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus. 2002;12:39–46. doi: 10.1002/hipo.10005. [DOI] [PubMed] [Google Scholar]
- Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37:417–431. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Toni N, Buchs PA. Spine changes associated with long-term potentiation. Hippocampus. 2000;10:596–604. doi: 10.1002/1098-1063(2000)10:5<596::AID-HIPO10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Nam CI, Chen L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci U S A. 2005;102:6137–6142. doi: 10.1073/pnas.0502038102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J Neurosci. 2007;27:3120–3130. doi: 10.1523/JNEUROSCI.0054-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon MM, Chen L. Retinoic acid-gated sequence-specific translational control by RARalpha. Proc Natl Acad Sci U S A. 2008;105:20303–20308. doi: 10.1073/pnas.0807740105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C, Moine H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J. 2001;20:4803–4813. doi: 10.1093/emboj/20.17.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier M, Severijnen LA, Reis S, Rife M, van't Padje S, van Cappellen G, Oostra BA, Willemsen R. Transport kinetics of FMRP containing the I304N mutation of severe fragile X syndrome in neurites of living rat PC12 cells. Exp Neurol. 2004;189:343–353. doi: 10.1016/j.expneurol.2004.05.039. [DOI] [PubMed] [Google Scholar]
- Schütt J, Falley K, Richter D, Kreienkamp HJ, Kindler S. Fragile X mental retardation protein regulates the levels of scaffold proteins and glutamate receptors in postsynaptic densities. J Biol Chem. 2009;284:25479–25487. doi: 10.1074/jbc.M109.042663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YJ, Conti J, Currie JR, Brown WT, Denman RB. RNAs that interact with the fragile X syndrome RNA binding protein FMRP. Biochem Biophys Res Commun. 2000;275:973–980. doi: 10.1006/bbrc.2000.3405. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci U S A. 2003;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wang H, Dictenberg JB, Ku L, Li W, Bassell GJ, Feng Y. Dynamic association of the fragile X mental retardation protein as a messenger ribonucleoprotein between microtubules and polyribosomes. Mol Biol Cell. 2008;19:105–114. doi: 10.1091/mbc.E07-06-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei ZX, Yi YH, Sun WW, Wang R, Su T, Bai YJ, Liao WP. Expression changes of microtubule associated protein 1B in the brain of Fmr1 knockout mice. Neurosci Bull. 2007;23:203–208. doi: 10.1007/s12264-007-0030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci. 2005;25:2895–2905. doi: 10.1523/JNEUROSCI.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP. A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav. 2004;3:337–359. doi: 10.1111/j.1601-183X.2004.00087.x. [DOI] [PubMed] [Google Scholar]
- Yu LM, Goda Y. Dendritic signalling and homeostatic adaptation. Curr Opin Neurobiol. 2009;19:327–335. doi: 10.1016/j.conb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG, Bagni C. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007;10:578–587. doi: 10.1038/nn1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang JB, Nosyreva ED, Spencer CM, Volk LJ, Musunuru K, Zhong R, Stone EF, Yuva-Paylor LA, Huber KM, Paylor R, Darnell JC, Darnell RB. A mouse model of the human Fragile X syndrome I304N mutation. PLoS Genet. 2009;5:e1000758. doi: 10.1371/journal.pgen.1000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Liu J, Zhu N, Lin J, Liang Q, Brown WT, Shen Y, Zhong N. Identification of FMRP-associated mRNAs using yeast three-hybrid system. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:769–777. doi: 10.1002/ajmg.b.30678. [DOI] [PubMed] [Google Scholar]