

Acute myocardial infarction and myocardial ischemia are leading causes of short- and long-term morbidity and mortality in the perioperative period. In part, this relates to the fact that treatment options for a myocardial infarction in surgical patients are extremely limited. For example, anti-coagulation or treatment with highly effective inhibitors of platelet aggregation may not be an option due to the risk for bleeding from the operative site. Therefore, it is not surprising that the search for new pharmacologic approaches to treat myocardial ischemia or to render the myocardium more resistant to limited oxygen availability is an area of intense investigation for perioperative scientists.1-4 In fact, the introduction of intraoperative beta-blockers for cardio-protection from ischemia was one of the most significant alterations in anesthesia practice during the past fifteen years. In line with the search for novel therapeutic approaches to render the myocardium more resistant to ischemia, a very exciting research study by Wang et al. from the research laboratory of Dr. Wei Chao from the Anesthesia Center for Critical Care Research of the Massachusetts General Hospital in Boston provides new insight into signaling pathways involving toll-like receptor (TLR)-elicited cardio-protection from ischemia.1
TLRs are named after the German word “toll” that carries the meaning of “great” or “amazing”. In fact, a team of scientists led by Dr. Christiane Nüsslein-Vollhard, PhD (Professor, Max Planck Institute, Tübingen, Germany) identified Toll as a gene critical in the embryogenesis of fruit flies by establishing the dorsal-ventral axis of Drosophila. 5,6 Their name derives from Christiane Nüsslein-Volhard’s 1985 exclamation, “Das war ja toll!”, which translates as “ that’s amazing”, in reference to the underdeveloped ventral portion of a fruit fly larva. Together with Edward B. Lewis and Eric Wieschaus, she received the Nobel Prize in Physiology or Medicine in 1995 for her discoveries concerning the genetic control of early embryonic development. Ten years following the original discovery of the Toll gene, a novel role for Toll beyond its function in embryogenesis was discovered.7 In fact, mutant flies deficient in Toll were more susceptible to fungal or bacterial infections.7 In 1997, a group of scientists from Yale University led by Ruslan Medzhitov (Professor, Yale University School of Medicine, New Haven, Connecticut) and Charles Janeway Jr. (Professor, Yale University School of Medicine, New Haven, Connecticut) (1943–2003) described a human homolog of the Drosophila Toll protein that had the resemblance of a cytoplasmatic receptor (“Toll-like”).8 In this landmark study, human Toll was identified as a type I transmembrane protein with an extracellular domain consisting of a leucine-rich repeat (LRR) domain, and a cytoplasmic domain homologous to the cytoplasmic domain of the human interleukin (IL)-1 receptor. Moreover, constitutively active mutant of human Toll transfected into human cell lines could induce the activation of nuclear factor (NF)κB and the expression of NFκB-target genes.8 As such, today’s view of “Toll-like receptors” (TLRs) has evolved towards a class of receptor proteins that play a key role in the innate immune system.9 They are single membrane-spanning, non-catalytic receptors that recognize structurally conserved molecules derived from microbes (pattern recognition receptors). For example, studies of mice deficient in TLR4 – the key TLR for the recognition of lipopolysaccharide – show hypo-responsiveness to lipopolysaccharide treatment.10 At present, 10 TLR family members have been described in human.11 They differ in their specificity for different microbial compounds. For example TLR2 can be activated by lipoteichoic acid from gram-positive bacteria, TLR4 by lipopolysaccharide from gram-negative bacteria, or TLR5 by flagellin – the filament of bacterial flagella found on most motile bacteria.12,13 Activation of TLRs involves specific signal transduction pathways, including MyD88- or Trif-dependent (MyD88-independent) pathways that culminate in the activation of NFκB, thus promoting a pro-inflammatory host program.11,14 TLR signaling has been implicated in many forms of inflammatory disorders in human. For instance, a co-segregating missense mutations (Asp299Gly and Thr399Ile) affecting the extracellular domain of the TLR4 receptor is associated with a blunted response to inhaled lipopolysaccharide in humans.15 This common Asp299Gly polymorphism of TLR4 also predicts low levels of circulating inflammatory mediators, and confers an increased risk of severe infections but carries a reduced risk of atherosclerosis in human.16
While TLR-signaling has been extensively studied in the context of innate immunity, other studies have also implicated TLR signaling in non-infectious tissue injury, such as occurs in the context of myocardial infarction. As such, the studies by Wang et al.1 build on the observation that prior administration of a small, non-lethal dose of the TLR4 agonist lipopolysaccharide results in robust cardio-protection from subsequent myocardial ischemia-reperfusion injury.17 To gain mechanistic insight towards how TLR4 activation confers cardio-protection from ischemia, the team of scientists combined very elegant pharmacologic and genetic approaches in mice treated with systemic lipopolysaccharide. For this purpose, lipopolysaccharide or vehicle treated mice were subsequently (24h later) exposed to myocardial ischemia utilizing a Langendorff apparatus, and myocardial infarct sizes and cardiac function were examined. These studies demonstrated robust cardio-protection with TLR4-agonist treatment (about 50% reduction in infarct size), that was TLR4 specific. In fact, Tlr4-/- mice were not protected by lipopolysaccharide pretreatment, while Tlr2-/- mice demonstrated similar cardio-protection as wild-type animals. Studies in mice deficient for Myd88 or Trif revealed Myd88-dependent and Trif-independent signal transduction. Consistent with previous in vitro studies from the laboratory of Dr. Chao,18 their present studies provide genetic in vivo evidence that these signaling events finally converge on inducible nitric oxide synthase and soluble guanylate cyclase. The authors have to be congratulated on putting together such a comprehensive number of genetic models that allowed their team to convincingly delineate this pathway in vivo. In contrast to studies on anesthetic preconditioning, where an in vivo knockdown of the receptor mediating the specific effects of a “cardio-protective anesthetic” has been literally impossible (at present, an “isoflurane-receptor knockout mouse” does not exist), the authors were able to target specifically the receptor critical for TLR-dependent cardio-protection, and were able to provide an extremely convincing level of evidence in support of their hypothesis via a combination of genetic and pharmacologic approaches.
In contrast to their findings, and somewhat paradoxical to the central hypothesis of their paper are studies in TLR-deficient mice exposed directly to myocardial ischemia-reperfusion injury. For example, an in situ study of myocardial ischemia-reperfusion injury exposed gene-targeted mice for Tlr4 to 60 min of coronary artery ligation, followed by 24h of reperfusion. The authors observed attenuated infarct sizes in Tlr4-deficient mice in conjunction with attenuated myocardial inflammation, as assessed by neutrophil accumulation, lipid peroxidase and complement deposition.19 Similar to these apparent discrepancies, previous studies have found a dual role of signaling pathways that result in activation of NFκB during ischemia-induced inflammation. In fact, a very elegant study from the laboratory of Michael Karin investigated the role of NFκB in acute inflammation caused by intestinal ischemia-reperfusion through selective ablation of IκB kinase (IKK)-beta, the catalytic subunit of IKK that is essential for NFκB activation.20 Ablation of IKK-beta in enterocytes prevented the systemic inflammatory response, which culminates in multiple organ dysfunction syndrome that is normally triggered by gut ischemia-reperfusion. IKK-beta removal from enterocytes, however, also resulted in severe apoptotic damage to the reperfused intestinal mucosa. Thus, these studies demonstrate a dual function of the NFκB system, which is responsible for both tissue protection and systemic inflammation. 20 Consistent with these findings, a pro-inflammatory imbalance following myocardial ischemia-reperfusion injury is attenuated in Tlr4-/- mice,19 while TLR-agonist treatment is associated with cardio-protective responses via dampening ischemia-driven cardiomyocyte apoptosis (Figure 1).17
Figure 1. Dual role of Toll-like receptor signaling in myocardial ischemia-reperfusion injury.
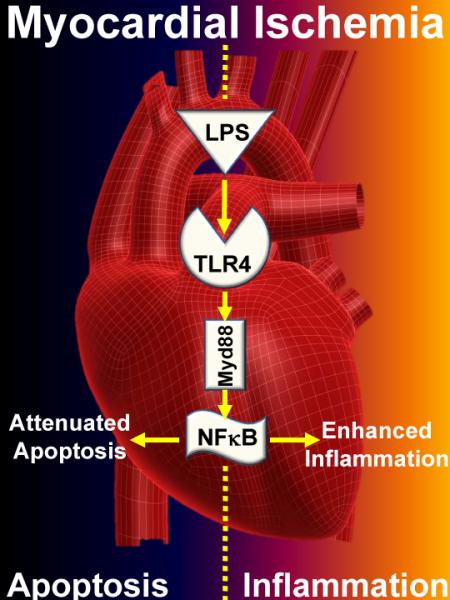
A study by Wang et al. published in the current edition of Anesthesiology provides genetic and pharmacologic evidence for a cardio-protective role of toll-like receptor signaling (TLR) during myocardial ischemia. Pre-treatment with lipopolysaccharide (LPS) provides potent protection from subsequent myocardial ischemia, involving signal transduction through Myd88, and convergence on inducible nitric oxide synthase and guanylate cyclase. As such, TLR4 dependent activation of nuclear factor (NF)κB could provide cardio-protection from ischemia by dampening programmed myocardial cell death (apoptosis). On the other hand, previous studies in knockout mice for TLR4 or Myd88 demonstrate attenuated susceptibility to myocardial ischemia-reperfusion injury. These later findings could reflect increases in myocardial inflammation during ischemia-reperfusion injury.
While ultimately the goal of the study be Wang et al is to identify novel therapeutic approaches for perioperative myocardial injury,1 some future challenges have to be overcome. The present studies were carried out in a Langendorff apparatus in the absence of inflammatory cells. It is conceivable that activation of myocardial TLRs and subsequent activation of an NFκB-elicited pro-inflammatory program could enhance inflammatory cell trafficking into the myocardium. As such, it will be an important step to test TLR4 agonists in intact models of myocardial ischemia-reperfusion injury in the presence of inflammatory cells.21,22 While other studies seek to identify ways to dampen hypoxia-elicited inflammation,23-26 the consequences of a single bolus treatment with lipopolysaccharide on myocardial inflammation following ischemia reperfusion injury needs to be clearly addressed. In addition, the therapeutic window for TLR agonist treatment has yet to be defined. While the authors can produce robust cardio-protection with a 24h pre-treatment approach, it would be highly desirable for the perioperative setting to utilize a TLR agonist at the onset of myocardial ischemia. As pointed out by the authors, additional limitations of current TLR4 agonists are their pyrogenic side effects. As such, the development of non-pyrogenic TLR agonists will be required to directly target this pathway for the perioperative treatment of myocardial ischemia reperfusion injury. However, none of these comments should distract from the enthusiasm about the studies provided by the laboratory of Dr. Chao. In our mind, the elegant experimental design, the approach of combining pharmacologic and genetic studies, the vision of characterizing molecular targets and pathways beyond the field of anesthetics, and the important therapeutic implications of their research work set the stage for future innovations in perioperative cardio-protection. No doubt, Dr. Chao and his colleagues have set the bar very high.
Acknowledgement
We would like to acknowledge Shelley A. Eltzschig, BSBA, artist, Mucosal Inflammation Program, University of Colorado Denver, USA, for the artwork included in this manuscript.
Sources of Funding, Potential Conflict of Interest: The authors have no conflict of interest. The present manuscript is supported by United States National Institutes of Health (Bethesda, Maryland) grant R01-HL0921, R01-DK083385 and R01HL098294 to HKE, and K08 HL102267 to TE.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang E, Feng Y, Zhang M, Zou L, Li Y, Buys ES, Huang P, Brouckaert P, Chao W. Toll-like receptor 4 signaling confers cardiac protection against ischemic injury via inducible nitric oxide synthase- and soluble guanylate cyclase-dependent mechanisms. Anesthesiology. 2011 doi: 10.1097/ALN.0b013e31820a4d5b. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eckle T, Kohler D, Lehmann R, El Kasmi KC, Eltzschig HK. Hypoxia-Inducible Factor-1 Is Central to Cardioprotection: A New Paradigm for Ischemic Preconditioning. Circulation. 2008;118:166–75. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 3.Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–90. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 4.Kohler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, Hart ML, Robson SC, Muller CE, Eltzschig HK. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation. 2007;116:1784–94. doi: 10.1161/CIRCULATIONAHA.107.690180. [DOI] [PubMed] [Google Scholar]
- 5.Anderson KV, Bokla L, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: The induction of polarity by the Toll gene product. Cell. 1985;42:791–8. doi: 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- 6.Anderson KV, Jurgens G, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: Genetic studies on the role of the Toll gene product. Cell. 1985;42:779–89. doi: 10.1016/0092-8674(85)90274-0. [DOI] [PubMed] [Google Scholar]
- 7.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–83. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 8.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 9.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–9. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 10.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- 11.Chao W. Toll-like receptor signaling: A critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol. 2009;296:H1–12. doi: 10.1152/ajpheart.00995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Kuhlicke J, Frick JS, Morote-Garcia JC, Rosenberger P, Eltzschig HK. Hypoxia Inducible Factor (HIF)-1 Coordinates Induction of Toll-Like Receptors TLR2 and TLR6 during Hypoxia. PLoS ONE. 2007;2:e1364. doi: 10.1371/journal.pone.0001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Y, Zou L, Si R, Nagasaka Y, Chao W. Bone marrow MyD88 signaling modulates neutrophil function and ischemic myocardial injury. Am J Physiol Cell Physiol. 2010;299:C760–9. doi: 10.1152/ajpcell.00155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–91. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 16.Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med. 2002;347:185–92. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 17.Chao W, Shen Y, Zhu X, Zhao H, Novikov M, Schmidt U, Rosenzweig A. Lipopolysaccharide improves cardiomyocyte survival and function after serum deprivation. J Biol Chem. 2005;280:21997–2005. doi: 10.1074/jbc.M413676200. [DOI] [PubMed] [Google Scholar]
- 18.Zhu X, Zhao H, Graveline AR, Buys ES, Schmidt U, Bloch KD, Rosenzweig A, Chao W. MyD88 and NOS2 are essential for toll-like receptor 4- mediated survival effect in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2006;291:H1900–9. doi: 10.1152/ajpheart.00112.2006. [DOI] [PubMed] [Google Scholar]
- 19.Oyama J, Blais C, Jr., Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–9. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 20.Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–81. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 21.Eckle T, Grenz A, Kohler D, Redel A, Falk M, Rolauffs B, Osswald H, Kehl F, Eltzschig HK. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am J Physiol Heart Circ Physiol. 2006;291:H2533–40. doi: 10.1152/ajpheart.00472.2006. [DOI] [PubMed] [Google Scholar]
- 22.Eltzschig HK, Kohler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. 2009;113:224–32. doi: 10.1182/blood-2008-06-165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–3315. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008;111:5571–80. doi: 10.1182/blood-2007-11-126763. [DOI] [PubMed] [Google Scholar]
- 25.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–18. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]