

Abstract
Dihydrofolate reductase (DHFR) has been a well-recognized target for the development of therapeutics for human cancers for several decades. Classical inhibitors of DHFR use an active transport mechanism to gain access to the cell; disabling this mechanism creates a pathway for resistance. In response, recent research focuses on nonclassical lipid-soluble DHFR inhibitors that are designed to passively diffuse through the membrane. Here, a new series of propargyl-linked antifolates are investigated as potential nonclassical human DHFR inhibitors. Several of these compounds exhibit potent enzyme inhibition with 50% inhibition concentration values under 500 nM. Molecular docking investigations show that the compounds maintain conserved hydrogen bonds between the pyrimidine ring and the enzyme as well as form van der Waals interactions with critical residues in the active site. Interestingly, the most potent compound, 2,4-diamino-5-(3-(3,4,5-trimethoxyphenyl)prop-1-ynyl)-6-ethylpyrimidine (compound 35), is 3,500-fold more potent than trimethoprim, a potent inhibitor of bacterial DHFR but weak inhibitor of human DHFR. The two structural differences between compound 35 and trimethoprim show that the propargyl linkage and the substitution at C6 of the pyrimidine ring are critical to the formation of contacts with Thr 56, Ser 59, Ile 60, Leu 22, Phe 31 and Phe 34 and hence, to enhancing potency. The propargyl-linked antifolates are efficient ligands with a high ratio of potency to the number of non-hydrogen atoms and represent a potentially fruitful avenue for future development of antineoplastic agents.
Keywords: Antifolate, dihydrofolate reductase, human DHFR, molecular modeling, propargyl-linked antifolate
Introduction
Inhibitors of dihydrofolate reductase (DHFR), an essential enzyme in the folate biosynthetic pathway, have been pursued for several decades as therapeutics in the treatment of human malignancies. DHFR catalyzes the transfer of a hydride from the cofactor, nicotinamide adenine dinucleotide phosphate (NADPH), to the substrate, dihydrofolate, thus yielding tetrahydrofolate and NADP+. Tetrahydrofolate is an essential cofactor in the production of purines and thymidylate and its deficiency leads to the inhibition of cell growth and proliferation.
DHFR inhibitors fall into two groups: classical and nonclassical. The classical inhibitors, such as methotrexate (Fig. 1), are characterized by a pterin ring, an aromatic ring (p-aminobenzoic acid in the case of methotrexate) and a glutamate tail. Structures of human DHFR bound to several classical inhibitors [1–3] have greatly aided the understanding of the interactions these inhibitors have with active site residues. The pterin ring forms hydrogen bonds with a conserved acidic residue, Glu 30, and the backbone carbonyl oxygen atoms of Ile 7 and Val 115. In addition, the pterin ring also forms hydrophobic contacts with Ala 9, Val 115, Phe 31 and Phe 34. The p-aminobenzoic acid moiety binds in a hydrophobic pocket comprised of Thr 56, Ser 59, Ile 60, Leu 67 and the glutamate tail binds Asn 64, Arg 28 and Arg 32 near the solvent-exposed surface of the enzyme. Owing to these extensive interactions, the classical inhibitors can achieve 50 % inhibition concentrations (IC50) well under 1 μM. For example, methotrexate and PT523 [N-α-(4-amino-4-deoxypteroyl)-N δ-hemiphthaloyl-L-ornithine], both classical inhibitors, have IC50 values of 11.2 and 12.2 nM, respectively [4]. Other experimental human DHFR inhibitors based on the classical scaffold have also achieved potent binding in the low nanomolar range [1, 5]. Owing to the charged glutamate tail, these inhibitors do not passively diffuse across cell membranes and must be actively transported using the reduced folate carrier system. Once inside the cell, they are polyglutamylated. Resistance can arise when the active transport mechanism is disabled.
Figure 1.

Classical (methotrexate) and non-classical (trimetrexate) antifolates
Research to develop nonclassical antifolates that penetrate the membrane by diffusion has attempted to overcome the problems of the classical antifolates. Trimetrexate (Fig. 1), a lipid soluble antifolate, is an effective inhibitor of DHFR and is currently undergoing clinical trials for the treatment of lymphoma [6]. Other compounds such as those based on the piritrexim [7] and diamino-5-methyl-5-deazapteridine [8] scaffolds, are under development.
We have developed a new nonclassical series of DHFR inhibitors based on a propargyl link between the pyrimidine and aryl rings (see Table 1). The straight-forward synthesis of these lipid soluble inhibitors has led to the development of several analogs that are differentially substituted at the C6 position of the pyrimidine ring, the propargylic position and the aryl ring, including biphenyl analogs. During our investigation of these compounds as inhibitors of DHFR from several infectious species [9–14], we measured the in vitro inhibition of human DHFR and found that a number of the propargyl-linked compounds are very effective inhibitors of human DHFR. The best propargyl-linked antifolate shows a 50 % inhibition concentration (IC50) of 57 nM, a value that is within the range of the best known classical inhibitors.
Table 1.
Propargyl-linked analogs inhibit human DHFR in vitro
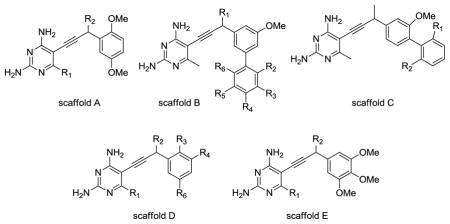 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | Scaffold | R1 | R2 | R3 | R4 | R5 | R6 | Human DHFR IC50 (μM) |
| TMP | - | - | - | - | - | - | - | 198.2±0.003 |
| 1 | A | H | H | - | - | - | - | 3.2±0.053 |
| 2 | A | Me | H | - | - | - | - | 1.3±0.003 |
| 3 | A | Me | OH | - | - | - | - | 1.2±0.20 |
| 4 | A | Me | Me | - | - | - | - | 0.4±0.1 |
| 5 | A | Me | OMe | - | - | - | - | 0.06±0.001 |
| 6 | A | Et | H | - | - | - | - | 1.28±0.015 |
| 7 | A | n-Pr | H | - | - | - | - | 1.18±0.01 |
| 8 | B | H | Me | H | H | H | Me | 1.58±0.009 |
| 9 | B | Me | H | H | H | H | H | 1.7±0.01 |
| 10 | B | Me | H | H | H | H | Me | 1.36±0.05 |
| 11 | B | Me | H | H | Me | H | H | 1.41±0.015 |
| 12 | B | Me | H | H | t-Bu | H | H | 0.21±0.006 |
| 13 | B | Me | H | H | OMe | H | H | 0.18±0.006 |
| 14 | B | Me | H | Me | H | Me | H | 0.75±0.006 |
| 15 | B | Me | Me | H | H | H | Me | 1.25±0.006 |
| 16 | B | Me | i-Pr | H | H | H | i-Pr | 7.2±0.15 |
| 17 | B | OH | H | H | H | H | H | 2.3±0.04 |
| 18 | C | H | H | - | - | - | - | 1.42±0.01 |
| 19 | C | Me | H | - | - | - | - | 2.77±0.06 |
| 20 | C | Me | Me | - | - | - | - | 3.37±0.02 |
| 21 | C | i-Pr | i-Pr | - | - | - | - | 4.2±0.1 |
| 22 | D | Me | Me | H | Br | H | OMe | 3.5 |
| 23 | D | Et | H | OMe | H | H | Ph | 0.3 |
| 24 | D | Et | H | H | H | H | H | 0.36 |
| 25 | E | H | H | - | - | - | - | 1.46±0.02 |
| 26 | E | H | OH | - | - | - | - | 14.3±0.24 |
| 27 | E | H | Me | - | - | - | - | 1.46±0.01 |
| 28 | E | H | OMe | - | - | - | - | 1.16±0.005 |
| 29 | E | Me | H | - | - | - | - | 0.40±0.06 |
| 30 | E | Me | OH | - | - | - | - | 5.71.±0.05 |
| 31 | E | Me | Me | - | - | - | - | 1.38±0.02 |
| 32 | E | Me | OMe | - | - | - | - | 1.22±0.007 |
| 33 | E | Me | Et | - | - | - | - | 0.20±0.01 |
| 34 | E | Me | gem-Me | - | - | - | - | 0.29±0.08 |
| 35 | E | Et | H | - | - | - | - | 0.057±0.002 |
| 36 | E | Et | Me | - | - | - | - | 0.088±0.007 |
In order to further develop this series to inhibit human DHFR, we conducted a molecular docking study to understand the structure-activity relationships. From this study, it is clear that the potent propargyl-linked inhibitors form critical interactions with Leu 22, Thr 56, Ser 59, Ile 60 in addition to the conserved interactions observed with the pyrimidine ring. Interestingly, the most potent propargyl-linked antifolate is only modestly differentially substituted relative to trimethoprim, a potent inhibitor of bacterial DHFR but a weak inhibitor of human DHFR. The stronger interactions with residues Leu 22, Thr 56, Ser 59, Ile 60, relative to the weak interactions observed with trimethoprim, explain this difference. Given that the potent propargyl-linked antifolates are highly efficient ligands [15, 16], they represent a new avenue for the design of human DHFR inhibitors that may be effective antineoplastic agents.
Materials and Methods
Enzyme preparation and assays
The human DHFR protein was expressed and purified as previously described [10]. Enzyme inhibition assays were performed by monitoring changes in absorbance at 340 nm as previously described [14]. Assays were performed in triplicate, from which IC50 values were interpolated.
Docking
Molecular modeling experiments were performed in three parts: in silico preparation of receptors and ligands, docking and validation. The procedure has been rigorously described elsewhere [14]. In brief, a high resolution human DHFR structure extending to 2.10 Å was obtained from the PDB (PDB ID: 1PD8[17]). The receptor structure was prepared for docking by the addition of hydrogens and charges. Since docking to ensembles of receptors has been shown to lead to greater accuracy [14, 18, 19], the receptor was subjected to a molecular dynamics simulation. Residues near the active site (within 3.5 Å) and several other highly flexible residues in a loop region that is contiguous with the active site were included in the simulation. Snapshots were taken along a 30 ps trajectory resulting in an ensemble of 21 structures. Ligand preparation required drawing accurate 3D representations, addition of charges and energy minimization. Sybyl [20] was used for both receptor and ligand preparation.
Docking studies were performed using the Surflex-Dock [21] algorithm as implemented by Sybyl. Sixty-one ligands with empirically determined inhibitory values were docked to each member of the receptor ensemble. Each resultant protein:ligand complex was assessed for adherence to the well established binding mode of all DHFR inhibitors, namely the presence of key hydrogen bonds between the ligand and an acidic residue (Glu 30 for human DHFR). If the correct geometry was found, the complex was accepted. For 90.9 % of the ligands, the conserved binding mode was found to be the top ranked pose. For 4.6 % of ligands, the conserved binding mode was found within poses 2–10, for 3.6 % of ligands the conserved binding mode was found within poses 11–200 and for 0.9 % of ligands, the conserved binding mode was not found after docking 200 poses. The reliability of the docking results were validated by correlating empirical inhibitory concentrations with docking scores using a clustering or neighbor binning technique. Ligands were grouped into six bins according to potency. Each bin contained five to fourteen ligands. If the corresponding docking score placed the ligand in the same or a neighboring bin as the empirical ranking, a positive correlation was identified and the ligand received a score of one. The accuracy of the model is calculated as the percentage of docked ligands receiving a score of one.
Results and Discussion
Stemming from investigations to develop propargyl-linked antifolates effective against several infectious species of DHFR, we have characterized over 70 compounds for their additional capacity to inhibit human DHFR [9–14] (Table 1). Twelve of the compounds exhibit IC50 values less than 1 μM and three (compounds 5, 35 and 36) have IC50 values less than 0.1 μM. All compounds with chiral centers were subjected to assays as racemic mixtures.
In order to understand the structure-activity relationships evident in the series, we took advantage of several high resolution crystal structures available in the PDB and conducted a molecular docking study. This study used previously developed ensemble-based methods to approximate enzyme flexibility and validated docking scores using a binning method [14, 18, 19].
A model of human DHFR (PDB ID: 1PD8 [17]) bound to NADPH and an antifolate compound determined from diffraction data extending to 2.1 Å was chosen as the receptor. Solvent molecules and ligands were removed and the receptor was otherwise prepared as described in Materials and Methods. Since receptor flexibility has been shown to be critical to accurate docking [14, 18, 22–24], a molecular dynamics (MD) simulation was used to introduce this flexibility into the modeling study. Residues within 3.5 Å of the active site and in the loop adjoining the active site were designated as flexible for the course of this molecular dynamics run. Structures collected along the MD trajectory were pooled to create an ensemble for subsequent docking studies. Each member of the ensemble was used in docking and resultant docking scores were averaged across the ensemble as this has been demonstrated to be the most accurate treatment of the composite docking scores [14].
Sixty-one ligands, drawn as single enantiomers, were docked to the ensemble of human DHFR structures and the resulting scores were ranked. The ligand docking mode was validated first by visualization and confirmation of conserved hydrogen bonds between the 2,4-diaminopyrimidine and Glu 30 (Fig. 2a). Additionally, to examine the correlation between docking scores and measured affinity, we used a “binning” method that calculates the percentage of ligands for which the associated docking score falls in the same “bin” as other compounds of similar affinity. This method begins by dividing the ligands into six approximately equal bins based on measured affinities (shown in Supplemental Information). If the docking score for a particular ligand places it within the same or neighboring bin as the measured affinity, it receives a score of 1. If the docking score does not fall within these bins, the compound receives a score of 0. The percentage of correctly ranked compounds is then calculated and reported as the binning score. For the human DHFR docking model comprised of the ensemble of structures, docking scores for 74 % of the compounds fell within the proper bin (compared to 18 % for random placement).
Figure 2.

a) Conserved hydrogen bonds with the enzyme and pyrimidine ring b) the active site of human DHFR bound to compound 35.
These poses of ligands, now validated by a correlation with measured affinity, were examined to illustrate structure-activity relationships. In general, all compounds maintained the conserved hydrogen bonds between the substituted pyrimidine ring and surrounding residues. Nitrogen N1 and the 2-amino group form hydrogen bonds with Glu 30 and Val 8, the 4-amino group forms hydrogen bonds with residues Ile 7, Tyr 121 and Val 115. In addition, the interactions observed during the docking runs shows that the propargyl-linked compounds maintain the same pattern of interactions as observed in crystal structures of human DHFR with other antifolates [1–3, 25]. In addition to the hydrogen bonds formed between the pyrimidine and the active site, there are van der Waals interactions with Phe 31, Ala 9, Ile 7 and Phe 34. The C6 position of the pyrimidine ring forms van der Waals interactions with Phe 31, Phe 34 and Leu 22. The propargyl linker and aryl ring form van der Waals contacts with residues Leu 22, Thr 56, Ser 59, Ile 60, Phe 31 and Phe 34 and NADPH (Figure 2b).
Substitution at the C6 position of the pyrimidine ring is critical for potency. Three compounds with a trimethoxyphenyl ring (scaffold E) and differing substitutions at C6 illustrate this trend. Compounds 25 (C6: hydrogen), 29 (C6: methyl) and 35 (C6: ethyl) have IC50 values of 1.46 μM, 0.4 μM and 0.057 μM, respectively. As the length of the alkyl chain at C6 increases, there are increased van der Waals contacts with a hydrophobic pocket comprised primarily of residues Phe 31 and Phe 34 (Figure 3).
Figure 3.

Docked poses of compounds 25, 29 and 35 bound to the active site of human DHFR. Interactions with residues Phe 31 and Phe 34 are shown.
The most potent compounds, 35 (IC50 = 57 nM), 5 (IC50 = 60 nM) and 36 (IC50 = 88 nM) all have a pyrimidine ring attached via a propargyl linker to a single phenyl ring substituted with methoxy groups either at the 3′, 4′, 5′ positions (compounds 35 and 36) or the 2′,5′ positions (compound 5). The propargyl substitutions are either hydrogen (compound 35) or hydrophobic (methoxy in compound 5 and methyl in compound 36) and interact with the hydrophobic pocket comprised primarily of Leu 22 and Thr 56. Two of the least potent propargyl-linked compounds, 30 and 26 have polar substituents at the propargyl position, verifying the correlation of activity with hydrophobic substituents at this position. Another of the least potent compounds, 16, is a highly substituted biphenyl analog. In general, the biphenyl analogs, represented by scaffolds B and C, are less potent than the monophenyl analogs. Docking results show that as bulky substitution patterns increase on the biphenyl analogs, there is significant steric interference with Pro 61 and Asn 64.
One of the most interesting results from this study is that the value of the propargyl-linked scaffold becomes evident when compared with trimethoprim (Figure 4a). Trimethoprim is an antifolate developed to be effective against several pathogenic bacteria and also shows some efficacy in patients with Pneumocystis pneumonia [26]. Trimethoprim is a highly selective compound that shows no efficacy against human DHFR, most likely owing to a lack of affinity for the human enzyme. The value of the propargyl linker in establishing greater affinity for the enzyme is shown by compound 25, which is very similar to trimethoprim but replaces the methylene linker with a propargyl linker and has an IC50 value 136-fold lower than trimethoprim. Highlighting the value of the hydrophobic C6 substitution, compound 35 has an IC50 value approximately 3500-fold lower than trimethoprim and 26-fold lower than compound 25.
Figure 4.

a) The value of the propargyl linker and substitution at the C6 position is evident in a comparison with trimethoprim b) Surface rendering of the active site of human DHFR bound to TMP (white, from the crystal structure 2W3A), docked compounds 25 (purple) and 35 (tan).
Recently, the crystal structure of human DHFR bound to NADPH and trimethoprim has been reported (PDB 2W3A, [27]). The deposited crystal structure reveals the basis of the low enzyme affinity. The pyrimidine ring of trimethoprim binds in the same orientation as the pyrimidine rings of potent inhibitors, with hydrogen bonds formed between N1 and the 2-amino group with Glu 30 and the 4-amino group and Ile 7 and Val 115. Van der Waals interactions are created between the pyrimidine ring and Ala 9, Ile 7, Val 115 and Phe 34. However, the short methylene linker positions the trimethoxyphenyl ring outside the proper distance for binding interactions with Leu 22, Thr 56, Ser 59 and Ile 60, drastically lowering the potency of this inhibitor. The docking results with human DHFR and the propargyl-linked inhibitors show that the propargyl linker forces the trimethoxyphenyl ring to interact more effectively with Leu 22, Thr 56, Ser 59 and Ile 60 (Figure 4b).
In addition to being potent, the propargyl-linked inhibitors have low molecular weight and are also highly efficient ligands against human DHFR. Ligand efficiency is calculated as the quotient of the overall binding and the count of heavy (non-hydrogen) atoms [15, 16]. Using this calculation, the ligand efficiencies of compounds 35, 5 and 36 are 0.398, 0.413, and 0.373, respectively. The values for ligand efficiencies for the propargyl-linked compounds are much greater than those of the classical inhibitors such as methotrexate, which has a ligand efficiency of 0.33.
Conclusions
Overall, it is apparent that the propargyl-linked antifolates represent a promising scaffold for the development of new inhibitors of human DHFR that may be effective antineoplastic agents. These compounds have notably straightforward synthetic routes for modification, they are nonclassical antifolates that permeate cell membranes and they have high ligand efficiency. Docking reveals the basis of structure-activity relationships and highlights that hydrophobic substitutions at C6 and the propargylic position impart increased potency. Interestingly, the most potent propargyl-linked compound, which is otherwise very closely related to trimethoprim, exhibits 3500-fold greater potency than trimethoprim. The propargyl linker is valuable in extending the trimethoxyphenyl ring closer to critical hydrophobic residues including Leu 22, Thr 56, Ser 59 and Ile 60.
Supplementary Material
Acknowledgments
The authors thank Kathleen Frey for evaluating the inhibition of human DHFR by the propargyl-linked antifolates and the NIH for financial support (GM067542). Oztekin Algul was supported by the Scientific and Technical Research Council of Turkey (TUBITAK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cody V, Galitsky N, Luft J, Pangborn W, Blakley R, Gangjee A. Comparison of ternary crystal complexes of F31 variants of human dihydrofolate reductase with NADPH and a classical antitumor furopyrimidine. Anti-Cancer Drug Des. 1998;13:307–15. [PubMed] [Google Scholar]
- 2.Cody V, Galitsky N, Luft J, Pangborn W, Rosowsky A, Blakley R. Comparison of two independent crystal structures of human dihydrofolate reductase ternary complexes reduced with nicotinamide adenine dinucleotide phosphate and the very tight-binding inhibitor PT523. Biochemistry. 1997;36:13897–903. doi: 10.1021/bi971711l. [DOI] [PubMed] [Google Scholar]
- 3.Lewis W, Cody V, Galitsky N, Luft J, Pangborn W, Chunduru S, Spencer HT, Appleman J, Blakley R. Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of leucine 22. J Biol Chem. 1995;270:5057–64. doi: 10.1074/jbc.270.10.5057. [DOI] [PubMed] [Google Scholar]
- 4.Rosowsky A, Bader H, Wright J, Keyomarsi K, Matherly L. Synthesis and biological activity of N-Hemiphthaloyl-α,ω-diaminoalkanoic acid analogues of aminopterin and 3′,5-dichloroaminopterin. J Med Chem. 1994;37:2167–74. doi: 10.1021/jm00040a008. [DOI] [PubMed] [Google Scholar]
- 5.Gangjee A, Li W, Kisliuk R, Cody V, Pace J, Piraino J, Makin J. Design, synthesis and X-ray crystal structure of classical and nonclassical 2-amino-4-oxo-5-substituted-6-ethylthieno[2,3-d]pyrimidines as dual thymidylate synthase and dihdyrofolate reuctase inhibitors and as potential antitumor agents. J Med Chem. 2009;52:4892–902. doi: 10.1021/jm900490a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarris A, Phan A, Duvic M, Romaguera J, McLaughlin P, Mesina O, King K, Medeiros L, Rassidakis G, Samuels B, Cabanillas F. Trimetrexate in Relapsed T-Cell Lymphoma with Skin Involvement. J Clin Oncol. 2002;20:2876–80. doi: 10.1200/JCO.2002.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Zink M, Lanig H, Troschutz R. Structural variations of piritrexim, a lipophilic inhibitor of human dihydrofolate reductase: synthesis, antitumor activity and molecular modeling investigations. Eur J Med Chem. 2004;39:1079–88. doi: 10.1016/j.ejmech.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Srivastava V, Kumar A, Mishra B, Siddiqi M. Molecular docking studies on DMMP derivatives as human DHFR inhibitors. Bioinformation. 2008;3:180–8. doi: 10.6026/97320630003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beierlein J, Frey K, Bolstad D, Pelphrey P, Joska T, Smith A, Priestley N, Wright D, Anderson A. Synthetic and crystallographic studies of a new inhibitor series targeting Bacillus anthracis dihydrofolate reductase. J Med Chem. 2008;51:7532–40. doi: 10.1021/jm800776a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolstad D, Bolstad E, Frey K, Wright D, Anderson A. A structure-based approach to the development of potent and selective inhibitors of dihydrofolate reductase from Cryptosporidium. J Med Chem. 2008;51:6839–52. doi: 10.1021/jm8009124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frey K, Liu J, Lombardo M, Bolstad D, Wright D, Anderson A. Crystal Structures of Wild-type and Mutant Methicillin-resistant Staphylococcus aureus Dihydrofolate Reductase Reveal an Alternative Conformation of NADPH that may be Linked to Trimethoprim Resistance. J Mol Biol. 2009;387:1298–308. doi: 10.1016/j.jmb.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Bolstad D, Smith A, Priestley N, Wright D, Anderson A. Structure-guided development of efficacious antifungal agents targeting Candida glabrata dihydrofolate reductase. Chem Biol. 2008;15:990–6. doi: 10.1016/j.chembiol.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, Bolstad D, Smith A, Priestley N, Wright D, Anderson A. Probing the active site of Candida glabrata dihydrofolate reductase with high resolution crystal structures and the synthesis of new inhibitors. Chem Biol Drug Des. 2009;73:62–74. doi: 10.1111/j.1747-0285.2008.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulsen J, Liu J, Bolstad D, Smith A, Priestley N, Wright D, Anderson A. In vitro biological activity and structural analysis of 2,4-diamino-5-(2′-arylpropargyl)pyrimidine inhibitors of Candida albicans. Bioorg Med Chem. 2009;17:4866–72. doi: 10.1016/j.bmc.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abad-Zapatero C, Metz J. Ligand efficiency indices as guideposts for drug discovery. Drug Disc Today. 2005;7:464–9. doi: 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- 16.Hopkins A, Groom C, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Disc Today. 2004;9:430–1. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 17.Cody V, Luft J, Pangborn W, Gangjee A. Analysis of three crystal structure determinations of a 5-methyl-6-N-methylanilino pyridopyrimidine antifolate complex with human dihydrofolate reductase. Acta Cryst. 2003;D59:1603–9. doi: 10.1107/s0907444903014963. [DOI] [PubMed] [Google Scholar]
- 18.Bolstad E, Anderson A. In pursuit of virtual lead optimization: the role of the receptor structure and ensembles in accurate docking. Proteins. 2008;73:566–80. doi: 10.1002/prot.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolstad E, Anderson A. In pursuit of virtual lead optimization: Pruning ensembles of receptor structures for increased efficiency and accuracy during docking. Proteins. 2008;75:62–74. doi: 10.1002/prot.22214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.SYBYL 8.1. Tripos Inc; 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: [Google Scholar]
- 21.Jain A. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 22.Hritz J, deRuiter A, Oostenbrink C. Impact of plasticity and flexibility on docking results for cytochrome P450 2D6: a combined approach of molecular dynamics and ligand docking. J Med Chem. 2008;51:7469–77. doi: 10.1021/jm801005m. [DOI] [PubMed] [Google Scholar]
- 23.Huang S, Zou X. Ensemble docking of multiple protein structures: considering protein structural variations in molecular docking. Proteins: Struct, Funct, Bioinf. 2007;66:399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- 24.Knegtel R, Kuntz I, Oshiro C. Molecular docking to ensembles of protein structures. J Mol Biol. 1997;266:424–40. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- 25.Klon A, Heroux A, Ross L, Pathak V, Johnson C, Piper J, Borhani D. Atomic structures of human dihydrofolate reductase complexed with NADPH and two lipophilic antifolates at 1.09 Å and 1.05 Å resolution. J Mol Biol. 2002;320:677–93. doi: 10.1016/s0022-2836(02)00469-2. [DOI] [PubMed] [Google Scholar]
- 26.Nahimana A, Rabodonirina M, Bille J, Francioli P, Hauser P. Mutations of Pneumocystis jirovecii dihydrofolate reductase associated with failure of prophylaxis. Antimicrob Agents Chemother. 2004;48:4301–5. doi: 10.1128/AAC.48.11.4301-4305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leung A, Ross L, Zywno-Van Ginkel S, Reynolds R, Seitz L, Pathak V, Barrow W, White E, Suling W, Piper J, Borhani D. to be published. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.