

Abstract
Genome-wide screening and positional cloning have linked neuropeptide S receptor 1 (NPSR1) with asthma and airway hyperresponsiveness. However, the mechanism by which NPSR1 regulates pulmonary responses remains elusive. Because neuropeptide S and its receptor NPSR1 are expressed in brain regions that regulate respiratory rhythm, and Npsr1-deficient mice have impaired stress and anxiety responses, we aimed to investigate whether neuropeptide S and NPSR1 regulate respiratory function through a central-mediated pathway. After neuropeptide S intracerebroventricular administration, respiratory responses of wildtype and Npsr1-deficient mice were monitored by whole-body or invasive plethysmography with or without serial methacholine inhalation. Airway inflammatory and hyperresponsiveness were assessed in allergen-challenged (ovalbumin or Aspergillus fumigatus) Npsr1-deficient mice. Analysis of breathing patterns by whole-body plethysmography revealed that intracerebroventricular neuropeptide S, as compared with the artificial cerebral spinal fluid control, increased respiratory frequency and decreased tidal volume in an NPSR1-dependent manner but did not affect enhanced pause. Following serial methacholine inhalation, intracerebroventricular neuropeptide S increased respiratory frequency in wildtype mice, but not Npsr1-deficient mice, and had no effect on tidal volume. Intracerebroventricular neuropeptide S significantly reduced airway responsiveness to methacholine as measured by whole-body plethysmography. Npsr1 deletion had no impact on airway inflammation or hyperresponsiveness in ovalbumin- or Aspergillus fumigatus-induced experimental asthma. Our results demonstrate that neuropeptide S and NPSR1 regulate respiratory function through a central nervous system-mediated pathway.
Keywords: Respiration, brain, neuropeptide S, neuropeptide S receptor 1, panting, stress
1. Introduction
Asthma is a complex disease involving the interaction of environmental factors and genetic susceptibility. Through genomic scanning and positional cloning, a series of genes including neuropeptide S receptor 1 (NPSR1, also called GPRA or GPR154) have been identified as asthma susceptibility genes [13, 23]. Single nucleotide polymorphisms (SNPs) and haplotypes of the NPSR1 gene have been associated with asthma or airway hyperresponsiveness (AHR) in Finnish, Canadian, Italian, Chinese, German, European-American, and Hispanic populations [4, 10, 14, 16, 20, 35]. The NPSR1 gene encodes for a G protein-coupled receptor, and an Asn/Ile107 SNP in the first extracellular loop of NPSR1 has been associated with increased asthma susceptibility [16]. However, the functional role of NPSR1 in asthma pathogenesis is still unclear.
Mouse NPSR1 shows high amino acid similarity to human NPSR1 and conserves Ile107. Allen et al. used an Npsr1 gene-targeted mouse, in which exon 4 containing Ile107 was deleted, to assess the role of NPSR1 in asthma development [1]. They found no evidence that NPSR1 contributed to asthma development in an ovalbumin (OVA)-induced experimental asthma model or LPS-mediated airway inflammation model. However, they also did not detect pulmonary expression of NPSR1 in healthy individuals or asthmatic patients or of Npsr1 in OVA-sensitized, saline-challenged or OVA-challenged mice by real-time PCR. These results are in contrast to the initial findings by Laitinen et al., who used an OVA sensitization model (OVA in combination with Stachybotrys chartarum) and found pulmonary upregulation of Npsr1 in mice with allergic lung disease [16]. Although implicated in human and murine models of asthma, the lack of evidence for a functional role of NPSR1 in a known immunological or asthmatic pathway raises questions about its contribution to asthma and the mechanism by which it may operate.
In humans, the Asn/Ile107 SNP in NSPR1 gene is associated with panic disorder in male patients of Japanese ancestry [24]. Interestingly, neuropeptide S (NPS), the endogenous ligand for NPSR1, has been reported as an important modulator of anxiety in rodents [17, 28, 30, 32]. And our earlier study suggests that the NPS/NPSR1 pathway is involved in stress and anxiety response [34]. Clinical observations have provided evidences that there is strong correlation of respiration change with stress response and emotional diseases. For example, patients with panic disorder display enhanced respiratory variability compared to controls [21] and chronic stress exacerbates the symptoms of asthmatic patients [3, 31]. Furthermore, Xu et al. have shown that that Npsr1 and Nps are located in the brain stem, limbic system, and/or cerebral cortex, which establish basic respiratory rhythm [32 33]. In light of these findings, we hypothesized that NPS and NPSR1 might operate through a central nervous system (CNS)-mediated pathway to regulate respiratory function at baseline or in response to a cholinergic stimulus (an inducer of airway responses that are typically exaggerated in asthma).
In the present study we use Npsr1-deficient mice in which exon 2 is deleted to investigate whether NPS alters respiratory function at baseline or in response to methacholine challenge by intracerebroventricular (ICV) administration of NPS and whether this process is NPSR1-dependent. In addition, we confirm whether NPSR1 is directly involved in affecting features of allergen-induced experimental asthma, including AHR using the Npsr1-deficient mice which is different from the mice used in the study from Allen et al. [1]. Experimental asthma is induced by OVA or Aspergillus fumigatus challenge. Compared with conventional OVA model, repeated intranasal challenge of Aspergillus fumigatus is a relatively chronic model [5].
2. Methods
2.1. Mice
A novel strain of Npsr1-deficient mice in which exon 2 was deleted have been described in our previous paper [34]. Mice with the genetic background of 129Sv/J × C57BL/6 were backcrossed 10 generations with BALB/c mice. In all studies, Npsr1-deficient (knock-out (KO)) mice were compared with sex- and age-matched BALB/c WT mice, which were bred on-site. All of the mice were housed as 4 mice with same sex per cage and maintained under pathogen-free conditions and a 14 h light/10 h dark cycle (lights on at 600 h) with temperature (19 ± 1 °C) and humidity (50 ± 10%) controlled. The experiments were performed between 0900-1500 h. All procedures were approved by the Institutional Animal Care and Use Committee of the Cincinnati Children's Hospital Research Foundation. The number of mice per group in each test is given in the figure captions.
2.2. ICV administration of NPS
Cannula implantation and ICV injection were done as previously described [34]. Each mouse was housed individually after cannula implantation. Experiments were initiated following a 7-day recovery period. For ICV injection, 3 μl of artificial cerebrospinal fluid (aCSF at pH 7.4: NaCl 124 mM, KCl 3.0 mM, NaHCO3 26 mM, CaCl2 2.0 mM, MgSO4 1.0 mM, KH2PO4 1.25 mM, D-glucose 10.0 mM) or 1 nmol of NPS (Phoenix Pharmaceuticals, Inc., Burlingame, USA) in 3 μl of aCSF were administered. ICV injection was conducted under light isoflurane anesthesia. The injection was administered over a period of 15 s. To avoid backflow, the injection cannula was left in place for another 60 s before being slowly withdrawn. Mice were returned to their original cage immediately after injection and awoke within 1 minute. For these experiments, 4 mice were processed at the same time.
2.3. Basal respiratory function before and after ICV administration of NPS
Basal respiratory function was measured by whole-body plethysmography (Buxco Electronics, Inc., Sharon, USA) [25] in conscious, unrestrained mice as described in Fig. 1A. Settings were: Maximal expiration time (Te) = 10 sec, minimal inspiration time (Ti) = 0.04 sec, minimal tidal volume (TV) = 0.04 ml, and value balance = 50%. Baseline measurements were made over a 5-min period before NPS ICV injection. Five minutes after NPS ICV injection, respiratory measurements were taken again for a total testing span of 60 min. The average values of respiratory frequency (breath per min, BPM), tidal volume (ml/breath), and enhanced pause (Penh) for each 2-min period were recorded. Minute ventilation (respiratory frequency × tidal volume) (ml/min) was calculated, and the values for the above parameters were plotted for every 10 min.
Figure 1. Role of NPS and NPSR1 in basal respiratory function.
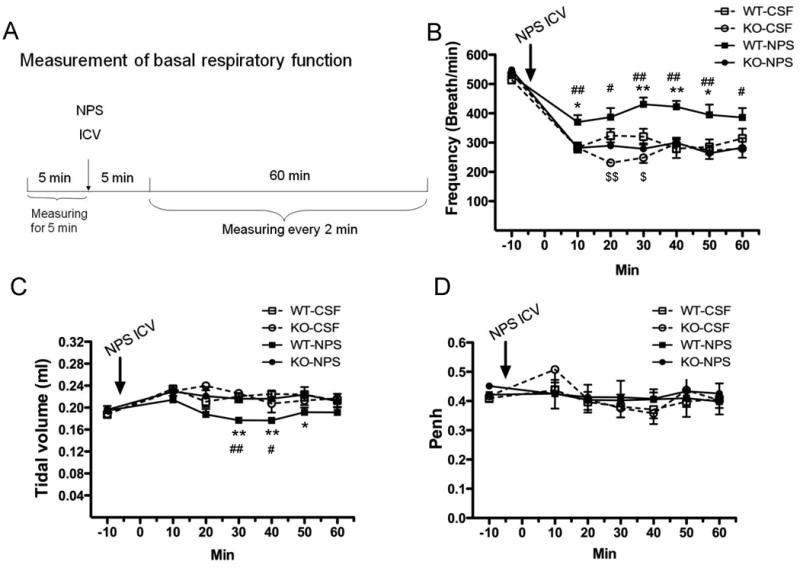
The protocol for measuring basal respiratory function in response to 1 nmol of NPS or aCSF ICV injection by whole-body plethysmography in Npsr1-deficient (KO) and wildtype (WT) mice is outlined in (A). Respiratory frequency (B), tidal volume (C), and enhanced pause (Penh) (D) were recorded every 2 min, and the values were plotted for 10-min intervals (n = 8 - 9 mice for each group). Values are presented as mean ± SEM. * p < 0.05 and ** p < 0.01 indicate the comparison of NPS- and aCSF-treated WT mice; # p <0.05 and ## p < 0.01 indicate the comparison of NPS-treated WT and KO mice; $ p <0.05 and $$ p < 0.01 indicate the comparison of aCSF-treated WT and KO mice.
2.4. Methacholine-challenged respiratory function after ICV administration of NPS
Respiratory response to methacholine challenge was measured by whole-body plethysmography (Buxco Electronics, Inc., Sharon, USA) as described in Fig. 2A. Baseline measurements were made over a 5-min period before NPS ICV injection. The measurement of respiratory response to methacholine challenge started 5 minutes after ICV injection of NPS. Mice were serially exposed to increasing concentrations of aerosolized methacholine (Sigma–Aldrich, St. Louis, USA) in PBS (0.8, 1.6, 3.2, 6.4, 12.8, and 25.6 mg/ml). Each dose of methacholine was delivered for 3 min by inhalation via a DeVilbiss Model 5500D-030 nebulizer (DeVilbiss Healthcare, Somerset, USA), and Penh measurements were made for 5 min, starting 2 min after completion of exposure to the aerosolized methacholine. The average values for the 5-min period of respiratory frequency, tidal volume, minute ventilation, and enhanced pause (Penh) were calculated.
Figure 2. Role of NPS and NPSR1 in methacholine-challenged respiratory function.
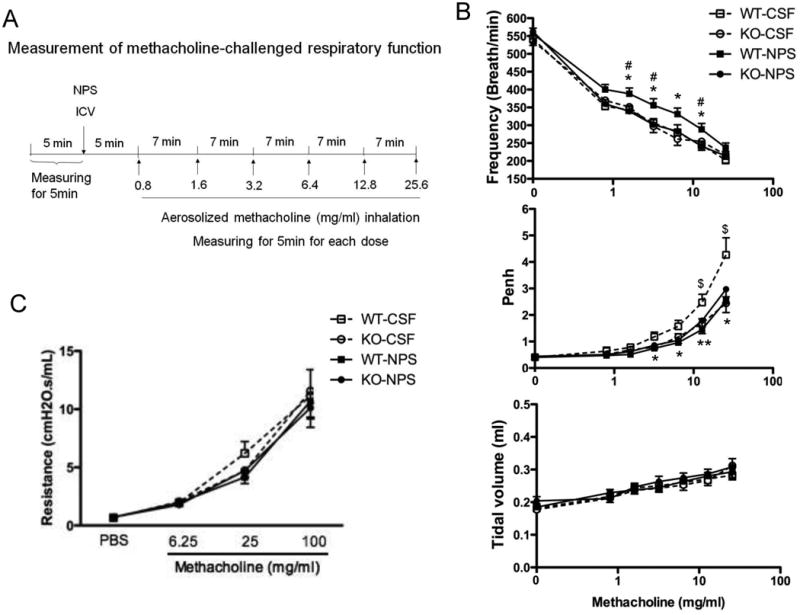
The protocol for using barometric plethysmography to measure methacholine-induced changes in respiratory function after 1 nmol of NPS or aCSF ICV injection in Npsr1-deficient (KO) and wildtype (WT) mice is outlined (A). Respiratory frequency, tidal volume, and enhanced pause (Penh) were assessed in response to methacholine challenge (0.8, 1.6, 3.2, 6.4, 12.8, and 25.6 mg/ml) 5 min after 1 nmol of NPS ICV injection (B). Airway resistance in response to methacholine challenge (6.25, 25, 100 mg/ml) 20 min after 1 nmol of NPS ICV injection was measured using the flexiVent system (n = 11 - 14 mice for each group) (C). Values are presented as mean ± SEM. * p < 0.05 and ** p < 0.01 indicate the comparison of NPS- and aCSF-treated WT mice; # p <0.05 indicates the comparison of NPS-treated WT and KO mice; $ p <0.05 indicates the comparison of aCSF-treated WT and KO mice.
2.5. Methacholine-challenged airway resistance after ICV administration of NPS
Seven days after the whole-body plethysmography, methacholine-challenged airway resistance after NPS ICV injection was measured using the flexiVent system (SCIREQ INC., Montreal, Canada) [22]. Briefly, 20 min after ICV injection of 1 nmol NPS, the mice were weighed and anesthetized with 150 mg/kg ketamine mixed with 10 mg/kg xylazine. A tracheotomy was performed, and a cannula was inserted. Mice were then exposed to aerosolized PBS, followed by increasing concentrations of aerosolized methacholine (Sigma–Aldrich, St. Louis, USA) (6.25, 25, 100 mg/ml). Values for lung resistance were obtained at 5-s intervals for 3 min after each methacholine challenge. The peak responses at each methacholine concentration were used for data analysis.
2.6. Peripheral blood cell counting and flow cytometry
Peripheral blood cells were counted by Hemavet 950 (Drew Scientific Inc., Dallas, USA), and CD3+, B220+, CD4+, CD8+ cells in the peripheral blood were analyzed by flow cytometry.
2.7. Experimental asthma models
Asthma-like lung disease was induced by OVA or Aspergillus fumigatus as previously described [36]. Mice were sacrificed 48 h following the last OVA challenge or 18 h following the last Aspergillus challenge. Bronchoalveolar lavage fluid (BALF), plasma, and left lung tissue were harvested for assessments of airway responsiveness. Right lung was harvested for RNA preparation.
2.8. BALF analysis
The mice were sacrificed by an intraperitoneal injection of pentobarbital (Sodium pentobarbital, 150mg/kg; Vortech Pharmaceuticals, Ltd., Dearborn, USA). Immediately thereafter, BALF was collected, and differential cell counts were determined as previously described [6].
2.9. Characterization of lung morphology
Hematoxylin and eosin (H&E), periodic acid-Schiff (PAS) or Masson's trichrome staining of the lung was performed as previously described [6].
2.10. Measurement of serum total IgE
Total IgE in the serum from OVA- or Aspergillus-challenged mice were determined by ELISA kit (BD Biosciences Pharmingen, San Diego, USA).
2.11. Methacholine-challenged airway resistance after allergen challenge
At 24 h after the last intranasal challenge with either saline or OVA, or at 18h after the last intranasal challenge with either saline or Aspergillus, airway resistance was measured using the flexiVent system (SCIREQ INC., Montreal, Canada) with exposed to aerosolized PBS, followed by aerosolized methacholine (37.5 mg/ml) (Sigma–Aldrich, St. Louis, USA) [22].
2.12. Reverse transcription polymerase chain reaction (RT-PCR)
mRNA expression in the lung and brain of saline- or OVA-challenged Npsr1-deficient and WT mice were examined using RT-PCR. The primers used in the PCR reaction are indicated below: full-length Npsr1 (1300 bp product), sense 5′-TCGTCAGGCAGAACTCTTCA-3′ specific to 5′UTR and antisense 5′-ATCTGCTAGGTGAGGCAGGA-3′ specific to 3′UTR; Npsr1 short fragment (137 bp product), sense 5′-GGCTCATCTCTAAGGCAAAAATCA-3′ specific to exon 8 and antisense 5′-ACGCTCCTTGGTGTCTGGAA-3′ specific to exon 9.
2.13. Statistical analysis
Values were presented as mean ± SEM. The significance of differences between experimental groups was analyzed using paired Student's t-tests. Significance was set at p ≤ 0.05.
3. Results
3.1. Role of NPS and NPSR1 in basal respiratory function
Based on the fact that Nps and Npsr1 are expressed in the brain regions that are responsible for establishing respiratory rhythm [32, 33], we examined whether NPS and/or NPSR1 might have a role in regulating basal respiratory function. Respiratory frequency, tidal volume and Penh were evaluated by whole-body plethysmography in WT and Npsr1-deficient mice before and after ICV administration of NPS or control aCSF (Fig. 1). Before ICV administration, respiratory frequency was similar between genotypes. Mice of both genotypes had a reduction in frequency to approximately 50% of the initial values at 10 min post-ICV aCSF injection under isofluorane anesthesia. The frequency remained dampened during the subsequent 50 min, and there was a greater decrease in frequency in Npsr1-deficient mice than in WT mice at 20 min and 30 min. ICV NPS (1 nmol) significantly increased the dampened frequency in WT, but not in Npsr1-deficient mice, with a significant difference between WT and Npsr1-deficient mice treated with NPS at all time points studied (Fig. 1B). No difference was seen in tidal volume between WT and Npsr1-deficent mice before or after aCSF ICV injection. However, ICV NPS decreased tidal volume in WT, but not in Npsr1-deficient mice, with a significant difference between NPS-treated WT and Npsr1-deficient mice at 30 and 40 min post-injection (Fig. 1C). Penh (Fig. 1D) and minute ventilation (data not shown) were not affected by ICV NPS. Collectively, these data demonstrate that Npsr1 is required for respiratory changes showing increased frequency and decreased tidal volume induced by ICV NPS at baseline.
3.2. Role of NPS and NPSR1 in methacholine-induced changes of respiratory function
Methacholine, a cholinergic stimulus, induces airway constriction that is a typical symptom of asthma. Next, we aimed to define whether ICV administration of NPS attenuates methacholine-induced AHR and whether this process is NPSR1-dependent. After ICV injection of 1 nmol of NPS, the respiratory function was measured by whole-body plethysmography in Npsr1-deficient and WT mice exposed to methacholine (Fig. 2B). Our results show that Npsr1 deficiency prevented the ability of NPS to suppress methacholine-induced decrease in respiratory frequency (upper panel) and obscured the ability of NPS to decrease the Penh response to methacholine by suppressing that response to the same extent in the presence or absence of NPS (middle panel). In contrast, neither NPS treatment nor Npsr1 deficiency affected methacholine-induced increase in tidal volume (lower panel). As measured with a flexiVent apparatus, ICV NPS showed the trend of decreasing airway resistance in response to methacholine, but not significantly. Npsr1 deficiency did not have any effect on airway resistance in response to methacholine challenge (Fig. 2C).
3.3. Role of NPSR1 in allergen-induced experimental asthma
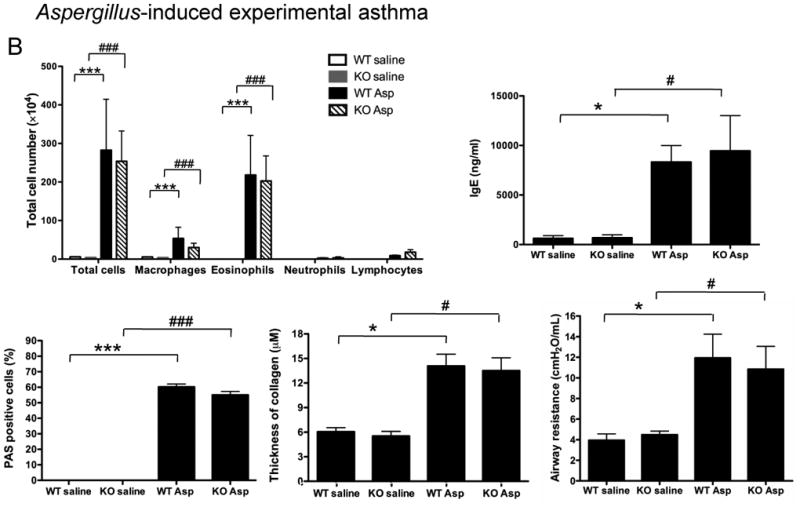
Because a previous study showed that Npsr1 was upregulated in the lung of mice subjected to an OVA/Stachybotrys chartarum mold model of asthma [16], we evaluated the role of NPSR1 in standard allergen-induced model of experimental asthma (OVA or Aspergillus). At baseline, no difference in peripheral blood total or CD3+, B220+, CD4+, CD8+ cell numbers was found between WT and Npsr1-deficient mice (Table 1). In the OVA-induced experimental asthma model (Fig. 3A), there was no significant difference in total cell number or specific cell populations in BALF although all cell populations trended lower in Npsr1-deficient mice between OVA-challenged WT and Npsr1-deficient mice. The Npsr1 deficiency did not alter the IgE level in the serum upon OVA challenge. OVA-induced airway mucus production, examined by PAS staining, and collagen deposition, assessed by trichrome staining, was similar in the Npsr1-deficient and WT mice. Airway resistance in response to 37.5 mg/ml methacholine was not altered in the Npsr1-deficient mice compared with WT mice. Similar results were seen with an Aspergillus fumigatus model of asthma (Fig. 3B): Npsr1 deficiency had no impact on the BALF cell profile, serum IgE, airway mucus, lung collagen, or methacholine-induced airway hyperreactivity. These data demonstrate that Npsr1 does not have a dominant direct role in the development of experimental asthma.
Table 1. Peripheral blood cell number and percent of T cells, B cells, and T cell subsets in the peripheral blood of Npsr1-deficient mice.
Normal blood cell composition was assessed in the Npsr1-deficient (KO) and wildtype (WT) mice (n = 8 for each genotype). WBC, White Blood Cells; NE, Neutrophils; LY, Lymphocytes; MO, Monocytes; EO, Eosinophils; BA, Basophils; RBC, Red blood cells; HGB, Hemoglobin; PLT, Platelets. Flow cytometry was used to analyze T cells, B cells and T cell subtypes in the peripheral blood from WT and KO mice (n = 3 for each genotype) using B220, CD3, CD4, and CD8 markers. These results are representative of three independent experiments. Data represent the mean ± SEM.
| WBC (K/uL) |
NE (K/uL) |
LY (K/uL) |
MO (K/uL) |
EO (K/uL) |
BA (K/uL) |
RBC (M/uL) |
|
|---|---|---|---|---|---|---|---|
| WT | 6.47±0.65 | 1.51±0.16 | 4.59±0.53 | 0.33±0.06 | 0.03±0.01 | 0.01±0.01 | 8.39±0.36 |
| KO | 7.94±0.67 | 1.68±0.20 | 5.86±0.48 | 0.36±0.04 | 0.04±0.02 | 0.01±0.01 | 8.87±0.34 |
| HGB (g/dL) |
PLT (K/uL) |
CD3 (%) |
B220 (%) |
CD4 (%) |
CD8 (%) |
|
|---|---|---|---|---|---|---|
| WT | 13.96±0.49 | 647.38±45.79 | 34±4.73 | 34±3.06 | 27±2.96 | 14±2.40 |
| KO | 14.24±0.53 | 654.13±28.51 | 30±3.18 | 28±3.00 | 26±2.96 | 11±1.53 |
Data expressed as mean ± SEM (n=8 for blood cell counting and n=3 for FACS)
Figure 3. Role of NPSR1 in OVA or Aspergillus -induced experimental asthma.
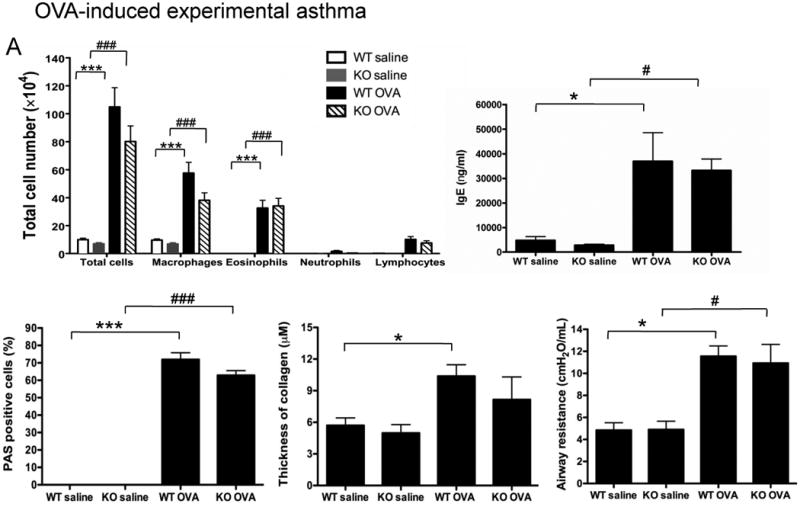
(A) OVA-induced experimental asthma. Forty eight hours after the final challenge, OVA- or saline-challenged Npsr1-deficient (KO) and wildtype (WT) mice were assessed for BALF cell populations (n = 7 - 10 for each group), serum IgE level (n = 5 - 8 for each group), mucus-secreting cells (n = 4 for each group), and collagen deposition in the lung (n = 4 for each group). Airway responsiveness to 37.5 mg/ml methacholine was assessed by flexiVent 24 hours after the final OVA challenge (n = 6 - 8 mice for each group). Data are representative of one of three experiments. Values are presented as mean ± SEM. * p < 0.05 and *** p < 0.001 indicate the comparison of OVA-treated and saline-treated WT mice; # p < 0.05 and ### p < 0.001 indicate the comparison of OVA-treated and saline-treated KO mice.
(B) Aspergillus-induced experimental asthma. Eighteen hours after Npsr1-deficient (KO) and wildtype (WT) mice received a final challenge with Aspergillus or saline, BALF cell populations (n = 7 - 8 for each group), serum IgE level (n = 5 - 6 for each group), mucus-secreting cells (n = 4 for each group), collagen deposition in the lung (n = 4 for each group), and airway responsiveness to 37.5 mg/ml methacholine by flexiVent (n= 8 - 9 mice for each group) were evaluated. Data are representative of one of three experiments. Values are presented as mean ± SEM. * p < 0.05 and *** p < 0.001 indicate the comparison of Aspergillus-treated and saline-treated WT mice; # p < 0.05 and ### p < 0.001 indicate the comparison of Aspergillus-treated and saline-treated KO mice.
3.4. Npsr1 expression in the murine lung and brain
Based on the above results, we examined Npsr1 expression in the lung. Northern blot analysis of several organs from untreated mice and lung from saline- or OVA-challenged mice revealed the presence of detectable Npsr1 mRNA in the brain, whereas no specific hybridization was noted in untreated lung, spleen, heart, kidney or intestine nor in saline- or OVA-challenged lung (data not shown). Subsequently, a more sensitive method, RT-PCR, was used to examine Npsr1 mRNA expression. Full-length Npsr1 (5′ UTR forward primer, 3′ UTR reverse primer) was highly expressed in the brain of untreated WT mice, but not in the lung of saline-challenged or OVA-challenged WT mice (Fig. 4A). Notably, the group who described Npsr1 upregulation in the OVA/Stachybotrys chartarum-challenged lung also showed that the murine RAW 264.7 cell line expressed Npsr1 using PCR primer pair that amplify sequence spanning exon 8 and exon 9 [16, 26]. When we used the same primer pair to examine Npsr1 expression in the lung, a short Npsr1 fragment could be amplified in some, but not in all lung samples from saline- or OVA-challenged WT mice (Fig. 4B). These results suggest that low levels of Npsr1 transcripts may exist in the lung of saline- or OVA-challenged WT mice.
Figure 4. Npsr1 expression in the mouse lung and brain.
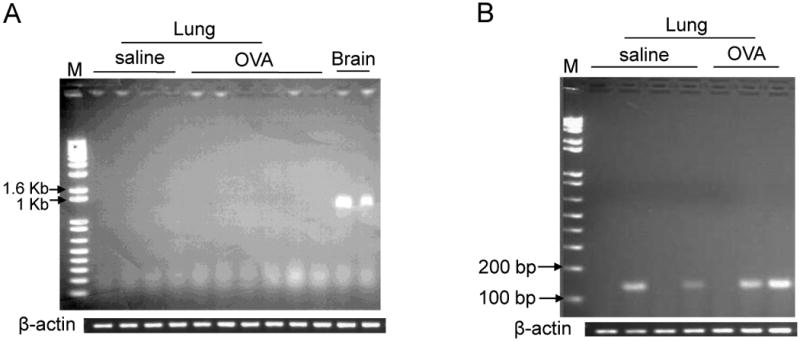
PCR amplification of full-length Npsr1 in the lung of OVA- or saline-challenged WT mice and the brain of untreated WT mice. Full-length Npsr1 was not found in the OVA- or saline-challenged lung (A). PCR amplification of a short Npsr1 fragment in the lung of OVA- or saline-challenged WT mice using primer pair specific for exon 8 and exon 9. A short fragment of Npsr1 was found in some lung samples of OVA- or saline-challenged WT mice (B). The β-actin expression for each set of PCR is shown as control. M indicates standard molecular marker. These results are representative of one of three experiments.
4. Discussion
In humans, the SNPs in the NPSR1 gene have been associated with asthma phenotypes, including AHR. We examined Npsr1-deficient mice using two models of asthma. Consistent with a previous study [1], we found that there were no differences in the lung inflammation and respiration in response to allergen challenge between WT and Npsr1-deficient mice, suggesting that NPSR1 is not directly involved in experimental asthma development. Further, our PCR results showed that Npsr1 has a very low level of expression in the basal or allergen-challenged lung, supporting that the direct role of NPS/NPSR1 in the lung may be limited. Our results are in contrast to the study from Laitinen et al. (16) which suggest that NPSR1 is implicated in human asthma. One of possible explanations is that there may be species differences in biological function of NPSR1 because our study is based in mouse.
Studies from Xu et al. [32, 33] showed that Nps and Npsr1 were mainly expressed in the brain stem, limbic system, and/or cerebral cortex. These regions are involved in regulating respiration, suggesting that NPS and NPSR1 have a role in this process. Unrestrained whole-body plethysmography (Buxco system), a noninvasive method, quantifies pulmonary physiological functions, including respiratory frequency, tidal volume, and Penh, in conscious mice repeatedly (12, 18). However, many researchers have criticized that Penh does not accurately measure airway resistance (2, 19). Although the unrestrained whole-body plethysmography is a useful tool to screen for the presence of lung diseases, it is not suitable to discriminate different mechanisms by which altered patterns of breathing come about in the patients with respiratory diseases,(29). The flexiVent system, an invasive method, allows specific analysis of pulmonary mechanics. In our study, we used these two methods to identify whether ICV NPS had a direct mechanical effect on the airway or changed the respiratory pattern through the CNS. Measured by whole-body plethysmography, mice treated with ICV NPS showed increased respiratory frequency, decreased Penh and no change in tidal volume following methacholine exposure compared with aCSF-treated mice. However, ICV NPS did not change airway mechanics in response to methacholine as measured by flexiVent, further supporting that the ICV NPS-induced decrease in Penh in response to methacholine is attributable to increased minute ventilation by ICV NPS.
NPSR1 has been associated with panic disorders in Japanese males [24]. Besides recurrent spontaneous anxiety attacks, panic disorders are dominated by respiratory symptoms such as hyperventilation, a feeling of being smothered, and shortness of breath, and respiratory dysregulation has been identified as a biological marker for panic disorder [9, 11]. Hypersensitivity of brainstem nuclei regulating respiratory activity has been proposed in patients with panic disorder [7]. Our study demonstrated that ICV NPS could increase respiratory frequency in an NPSR1-dependent manner at baseline or in response to methacholine challenge. In addition, our earlier study showed that ICV NPS induced hyperlocomotion, increased exploration, and plasma corticosterone release in WT mice, not in Npsr1-deficient mice [34]. All these physiological alterations triggered by ICV administration of NPS are part of typical fight-or-flight response, an adaptive response to danger. It is suggested that respiratory change triggered by ICV NPS is a psychophysiological activity in response to stress. As such, we speculate that psychological triggers NPS/NPSR1 mediate changes in respiration and anxiety, possibly modulating asthma symptoms. Further studies are needed to examine the expression of NPS and NPSR1 in the brain in a model of stress. Experimental and clinical evidence has emerged that there is an association of respiration changes with psychological stress and emotional diseases. Experimental exposure to emotional stimuli has been shown to increase respiratory resistance in asthma [27]. Chronic stress can exacerbate the symptoms of asthmatic patients [3, 31]. It will be interesting to define what the role of NPS/NPSR1 is in the exacerbation of asthma by chronic stress in the future as we have shown that NPS/NPSR1 signaling regulates stress and anxiety responses [34].
Our study is limited by testing a dose of ICV NPS (1 nmol) that is indeed a pharmacological dose because our previous paper showed that 1 nmol of ICV NPS had the best effect on anxiety, locomotion and exploration compared with 0.1 and 10 nmol of ICV NPS [34]. In addition, the identified effect of NPS on respiratory frequency was conducted using whole-body plethysmography, and the confinement chamber may have anxiety-promoting effects. However, it is notable that the effect of NPS on respiration is NPSR1-dependent, providing evidence that the triggered pathway is specific to NPS/NPSR1.
It has not escaped our attention that ICV NPS induced increase in respiratory frequency and decrease in tidal volume resembles the respiratory panting behavior observed during hyperthermia [8] and mechanical associated high frequency ventilation that is clinically used to treat lung injury [15]. It will be interesting to determine whether natural panting responses may be NPS/NPSR1 dependent. And our results have implications for potentially understanding the responses to mechanical associated high frequency ventilation.
Taken together, our studies have demonstrated that the direct role of NPSR1 in the development of experimental asthma is limited and the effect of NPS/NPSR1 on respiratory changes is likely mediated via a CNS-mediated pathway, providing a pathway that connects respiratory and stress responses.
Acknowledgments
We would like to thank Drs. Simon Hogan, Nives Zimmermann, Eric Brandt and Carine Blanchard for scientific advice; Drs. Charles Vorhees and Michael Williams for surgery and ICV injection technique assistance; and Dr. Julie Caldwell and Shawna Hottinger for helpful discussions and review of the manuscript. This work was supported by NIH Grant P01 HL076383.
Footnotes
Disclosures
The authors declare no conflict of interest.
Author contributions
H.Z. and M.E.R. designed research; H.Z., C.P., and M.K.M. performed research; H.Z., C.P., M.K.M., F.D.F., and M.E.R. analyzed data; and H.Z., F.D.F., and M.E.R. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allen IC, Pace AJ, Jania LA, Ledford JG, Latour AM, Snouwaert JN, Bernier V, Stocco R, Therien AG, Koller BH. Expression and function of NPSR1/GPRA in the lung before and after induction of asthma-like disease. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1005–17. doi: 10.1152/ajplung.00174.2006. [DOI] [PubMed] [Google Scholar]
- 2.Bates JH, Irvin CG. Measuring lung function in mice: the phenotyping uncertainty principle. J Appl Physiol. 2003;94:1297–1306. doi: 10.1152/japplphysiol.00706.2002. [DOI] [PubMed] [Google Scholar]
- 3.Chen E, Miller GE. Stress and inflammation in exacerbations of asthma. Brain Behav Immun. 2007;21:993–9. doi: 10.1016/j.bbi.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng Y, Hong X, Wang L, Jiang S, Chen C, Wang B, Yang J, Fang Z, Zang T, Xu X, Xu X. G protein-coupled receptor 154 gene polymorphism is associated with airway hyperresponsiveness to methacholine in a Chinese population. J Allergy Clin Immunol. 2006;117:612–7. doi: 10.1016/j.jaci.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 5.Fulkerson PC, Fischetti CA, McBride ML, Hassman LM, Hogan SP, Rothenberg ME. A central regulatory role for eosinophils and the eotaxin/CCR3 axis in chronic experimental allergic airway inflammation. Proc Natl Acad Sci U S A. 2006;103:16418–23. doi: 10.1073/pnas.0607863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulkerson PC, Fischetti CA, Rothenberg ME. Eosinophils and CCR3 regulate interleukin-13 transgene-induced pulmonary remodeling. Am J Pathol. 2006;169:2117–26. doi: 10.2353/ajpath.2006.060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorman JM, Fyer MR, Goetz R, Askanazi J, Liebowitz MR, Fyer AJ, Kinney J, Klein DF. Ventilatory physiology of patients with panic disorder. Arch Gen Psychiatry. 1988;45:31–9. doi: 10.1001/archpsyc.1988.01800250035006. [DOI] [PubMed] [Google Scholar]
- 8.Hales JR, Bligh J. Respiratory responses of the conscious dog to severe heat stress. Experientia. 1969;25:818–9. doi: 10.1007/BF01897895. [DOI] [PubMed] [Google Scholar]
- 9.Hegel MT, Ferguson RJ. Psychophysiological assessment of respiratory function in panic disorder: Evidence for a hyperventilation subtype. Psychosom Med. 1997;59:224–30. doi: 10.1097/00006842-199705000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Hersh CP, Raby BA, Soto-Quirós ME, Murphy AJ, Avila L, Lasky-Su J, Sylvia JS, Klanderman BJ, Lange C, Weiss ST, Celedón JC. Comprehensive testing of positionally cloned asthma genes in two populations. Am J Respir Crit Care Med. 2007;176:849–57. doi: 10.1164/rccm.200704-592OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holt PE, Andrews G. Hyperventilation and anxiety in panic disorder, social phobia, GAD, and normal controls. Behav Res Ther. 1989;27:453–60. doi: 10.1016/0005-7967(89)90016-8. [DOI] [PubMed] [Google Scholar]
- 12.Hoymann HG. Invasive and noninvasive lung function measurements in rodents. J Pharmacol Toxicol Methods. 2007;55:16–26. doi: 10.1016/j.vascn.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Kere J. Mapping and identifying genes for asthma and psoriasis. Philos Trans R Soc Lond B Biol Sci. 2005;360:1551–61. doi: 10.1098/rstb.2005.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kormann MS, Carr D, Klopp N, Illig T, Leupold W, Fritzsch C, Weiland SK, von Mutius E, Kabesch M. G-Protein-coupled receptor polymorphisms are associated with asthma in a large German population. Am J Respir Crit Care Med. 2005;171:1358–62. doi: 10.1164/rccm.200410-1312OC. [DOI] [PubMed] [Google Scholar]
- 15.Krishnan JA, Brower RG. High-frequency ventilation for acute lung injury and ARDS. Chest. 2000;118:795–807. doi: 10.1378/chest.118.3.795. [DOI] [PubMed] [Google Scholar]
- 16.Laitinen T, Polvi A, Rydman P, Vendelin J, Pulkkinen V, Salmikangas P, Mäkelä S, Rehn M, Pirskanen A, Rautanen A, Zucchelli M, Gullstén H, Leino M, Alenius H, Petäys T, Haahtela T, Laitinen A, Laprise C, Hudson TJ, Laitinen LA, Kere J. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304:300–4. doi: 10.1126/science.1090010. [DOI] [PubMed] [Google Scholar]
- 17.Leonard SK, Dwyer JM, Sukoff Rizzo SJ, Platt B, Logue SF, Neal SJ, Malberg JE, Beyer CE, Schechter LE, Rosenzweig-Lipson S, Ring RH. Pharmacology of neuropeptide S in mice: therapeutic relevance to anxiety disorders. Psychopharmacology (Berl) 2008;97:601–11. doi: 10.1007/s00213-008-1080-4. [DOI] [PubMed] [Google Scholar]
- 18.Lomask M. Further exploration of the Penh parameter. Exp Toxicol Pathol. 2006;57:13–20. doi: 10.1016/j.etp.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 19.Lundblad LK, Irvin CG, Adler A, Bates JH. A reevaluation of the validity of unrestrained plethysmography in mice. J Appl Physiol. 2002;93:1198–207. doi: 10.1152/japplphysiol.00080.2002. [DOI] [PubMed] [Google Scholar]
- 20.Malerba G, Lindgren CM, Xumerle L, Kiviluoma P, Trabetti E, Laitinen T, Galavotti R, Pescollderungg L, Boner AL, Kere J, Pignatti PF. Chromosome 7p linkage and GPR154 gene association in Italian families with allergic asthma. Clin Exp Allergy. 2007;37:83–9. doi: 10.1111/j.1365-2222.2006.02615.x. [DOI] [PubMed] [Google Scholar]
- 21.Martinez JM, Kent JM, Coplan JD, Browne ST, Papp LA, Sullivan GM, Kleber M, Perepletchikova F, Fyer AJ, Klein DF, Gorman JM. Respiratory variability in panic disorder. Depress Anxiety. 2001;14:232–7. doi: 10.1002/da.1072. [DOI] [PubMed] [Google Scholar]
- 22.Munitz A, Brandt EB, Mingler M, Finkelman FD, Rothenberg ME. Distinct roles for IL-13 and IL-4 via IL-13 receptor alpha1 and the type II IL-4 receptor in asthma pathogenesis. Proc Natl Acad Sci U S A. 2008;105:7240–5. doi: 10.1073/pnas.0802465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 24.Okamura N, Hashimoto K, Iyo M, Shimizu E, Dempfle A, Friedel S, Reinscheid RK. Gender-specific association of a functional coding polymorphism in the Neuropeptide S receptor gene with panic disorder but not with schizophrenia or attention-deficit/hyperactivity disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1444–8. doi: 10.1016/j.pnpbp.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 25.Perkins C, Wills-Karp M, Finkelman FD. IL-4 induces IL-13-independent allergic airway inflammation. J Allergy Clin Immunol. 2006;118:410–9. doi: 10.1016/j.jaci.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Pulkkinen V, Majuri ML, Wang G, Holopainen P, Obase Y, Vendelin J, Wolff H, Rytilä P, Laitinen LA, Haahtela T, Laitinen T, Alenius H, Kere J, Rehn M. Neuropeptide S and G protein-coupled receptor 154 modulate macrophage immune responses. Hum Mol Genet. 2006;15:1667–79. doi: 10.1093/hmg/ddl090. [DOI] [PubMed] [Google Scholar]
- 27.Ritz T, Steptoe A, DeWilde S, Costa M. Emotions and stress increase respiratory resistance in asthma. Psychosom Med. 2000;62:401–12. doi: 10.1097/00006842-200005000-00014. [DOI] [PubMed] [Google Scholar]
- 28.Rizzi A, Vergura R, Marzola G, Ruzza C, Guerrini R, Salvadori S, Regoli D, Calo G. Neuropeptide S is a stimulatory anxiolytic agent: a behavioural study in mice. Br J Pharmacol. 2008;154:471–9. doi: 10.1038/bjp.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sly PD, Turner DJ, Hantos Z. Measuring lung function in murine models of pulmonary disease. Drug Discovery Today: Disease Models. 2004;1:337–43. [Google Scholar]
- 30.Vitale G, Filaferro M, Ruggieri V, Pennella S, Frigeri C, Rizzi A, Guerrini R, Calò G. Anxiolytic-like effect of neuropeptide S in the rat defensive burying. Peptides. 2008;29:2286–91. doi: 10.1016/j.peptides.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Wright RJ, Cohen RT, Cohen S. The impact of stress on the development and expression of atopy. Current Opinion in Allergy and Clinical Immunology. 2005;5:23–9. doi: 10.1097/00130832-200502000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O. Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004;43:487–97. doi: 10.1016/j.neuron.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Xu YL, Gall CM, Jackson VR, Civelli O, Reinscheid RK. Distribution of neuropeptide S receptor mRNA and neurochemical characteristics of neuropeptide S-expressing neurons in the rat brain. J Comp Neurol. 2007;500:84–102. doi: 10.1002/cne.21159. [DOI] [PubMed] [Google Scholar]
- 34.Zhu H, Mingler MK, McBride ML, Murphy AJ, Valenzuela DM, Yancopoulos GD, Williams MT, Vorhees CV, Rothenberg ME. Abnormal response to stress and impaired NPS-induced hyperlocomotion, anxiolytic effect and corticosterone increase in mice lacking NPSR1. Psychoneuroendocrinology. 2010;35:1119–32. doi: 10.1016/j.psyneuen.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu HY, Wu JM, Cui TP. Study on the association of single nucleotide polymorphisms and haplotypes of GPR154 gene with allergic asthma in Han nationality in Hubei Chinese population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2007;24:48–51. [PubMed] [Google Scholar]
- 36.Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, Muntel EE, Witte DP, Pegg AA, Foster PS, Hamid Q, Rothenberg ME. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–74. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]