

Abstract
The first total synthesis for the (Z)-16-methyl-11-heptadecenoic acid, a novel fatty acid from the sponge Dragmaxia undata, was accomplished in seven steps and in a 44% overall yield. The use of (trimethylsilyl)acetylene was key in the synthesis. Based on a previous developed strategy in our laboratory the best synthetic route towards the title compound was first acetylide coupling of (trimethylsilyl)acetylene to the long-chain protected 10-bromo-1-decanol followed by a second acetylide coupling to the short-chain 1-bromo-4-methylpentane, which resulted in higher yields. Complete spectral data is also presented for the first time for this recently discovered fatty acid and the cis double bond stereochemistry of the natural acid was established. The title compound displayed antiprotozoal activity against Leishmania donovani (IC50 = 165.5 ± 23.4 µM) and inhibited the leishmania DNA topoisomerase IB enzyme (LdTopIB) with an IC50 = 62.3 ± 0.7 µM.
Keywords: Dragmaxia undata, fatty acids, leishmaniasis, (Z)-16-methyl-11-heptadecenoic acid, sponges, synthesis, topoisomerase IB
1. Introduction
Marine organisms continue to yield interesting and unprecedented phospholipid fatty acids with no counterpart in terrestrial organisms. One such class of fatty acids is the iso methyl-branched octadecenoic acids, from which only a handful number of compounds have been reported. To the best of our knowledge, the only known examples of this type of fatty acids are the (Z)-16-methyl-8-heptadecenoic acid, also synthesized for the first time by our group (Carballeira and Pagán, 2001), and the (Z)-16-methyl-6-heptadecenoic acid, which were identified in a Micrococcus bacterium isolated from Lake Pomorie in Bulgaria (Carballeira et al., 2000). Just recently, O. Osorno and coworkers identified the new fatty acid (Z)-16-methyl-11-heptadecenoic acid (1) in the phospholipids (1.1% relative abundance) of the Colombian Caribbean sponge Dragmaxia undata, collected at 21 m depth in Punta de Betin, Santa Marta, Colombia (Rodríguez et al., 2010). This rather unusual fatty acid is the iso methyl-branched octadecenoic acid with the closest double bond to the ω end of the chain, i.e., a (n-6) fatty acid. However, acid 1 was identified in D. undata by means of mass spectrometry on the corresponding methyl ester and pyrrolidide derivative and the identification by GC-MS relied on minute amounts from a complex mixture of 30 fatty acids. Therefore, it was not possible to elucidate if the double bond stereochemistry of the naturally occurring fatty acid was cis or trans.
It is very likely that acid 1 was biosynthesized by a symbiotic bacterium within D. undata, thus implying that having the complete mass spectral data of 1 at hand will be useful to microbiologists seeking to characterize 1 in bacterial samples. The latter statement is backed up by the fact that acid 1 is a two-carbon chain extension of the (Z)-14-methyl-9-pentadecenoic acid, an acid that has been identified in several bacterial sources and most recently in a Streptomyces sp. B6007 originating from mangrove sediment in Papua New Guinea (Stritzke et al., 2004). Of particular interest in this context is also the report by J. H. Jung and collaborators that iso and anteiso fatty acids from a Streptomyces sp. strain KM86-9B displayed topoisomerase I inhibitory activity (Lee et al., 1998). In fact, we have also shown that the acid (Z)-14-methyl-9-pentadecenoic acid inhibits the human placenta DNA topoisomerase I enzyme at concentrations of 500 µM (Carballeira et al., 2007), while the marine fatty acid (Z)-17-methyl-13-octadecenoic acid inhibits the leishmania DNA topoisomerase IB enzyme at concentrations of 50 µM (Carballeira et al., 2009). The latter report indicated that these iso monounsaturated fatty acids do have antiprotozoal potential.
It then becomes of interest to explore both the antileishmanial and the topoisomerase I inhibitory activity of the new acid 1, but it will remain unexplored until enough material becomes available for such studies. However, by applying our previous developed synthetic methodology for unsaturated iso methyl-branched fatty acids (Carballeira et al., 2009) to the synthesis of 1 enough material for such studies could be obtained. Therefore, in order to unequivocally conclude the total characterization of 1, i.e., determine the cis or trans double bond stereochemistry, as well as to study its antileishmanial activity we are reporting herein the first total synthesis for the (Z)-16-methyl-11-heptadecenoic acid (1). This synthesis was accomplished following a synthetic sequence consisting of seven steps and using (trimethylsilyl)acetylene as the key reagent in the synthesis (Scheme 1). We also report that acid 1 displays antiprotozoal activity towards Leishmania donovani promastigotes by a mechanism that probably involves inhibition of the leishmania DNA topoisomerase IB enzyme.
Scheme 1.

Synthesis of (Z)-16-methyl-11-heptadecenoic acid (1).
2. Materials and methods
2.1. Instrumentation
1H NMR (500 MHz) and 13C NMR (125 MHz) were recorded on a Bruker DRX-500 spectrometer. 1H NMR chemical shifts are reported with respect to internal (CH3)4Si, 13C NMR chemical shifts are reported in parts per million relative to CDCl3 (77.0 ppm). Gas chromatography/mass spectrometry (GC/MS) analyses were recorded at 70 eV using either a Hewlett Packard 5972A MS ChemStation or an Agilent 5975C MS ChemStation coupled to an Agilent 7890A GC where both instruments were equipped with a 30 m × 0.25 mm special performance capillary column (HP-5MS) of polymethyl siloxane crosslinked with 5% phenyl methylpolysiloxane. IR spectra were recorded on a Nicolet Magna 750 FT-IR spectrophotometer (Thermo-Nicolet, Madison, WI, USA). High resolution mass spectral data was performed at the Emory University Mass Spectrometry Center on a thermo linear ion trap-Fourier transform mass spectrometer (LTQ-FTMS) using atmospheric pressure chemical ionization (APCI) as the probe.
2.2. 10-Bromo-[(tetrahydropyran-2-yl)oxy]decane (2)
To 2.00 g of 10-bromo-1-decanol in 15 mL of chloroform (CHCl3) was added dropwise 16.9 mmol of 3,4-dihydro-2H-pyran (DHP) and catalytic amounts of p-toluenesulfonic acid (pTSA). The reaction mixture was stirred for 24h at room temperature. The organic layer was washed with water (2 × 20 mL), NaHCO3 (2 × 20 mL), CHCl3 (1 × 20 mL), and dried over MgSO4, filtered, and evaporated in vacuo. The crude product was purified using silica gel column chromatography eluting with hexane/ether (9:1). The pure product 2 (Banaszak et al., 2009) was obtained as a colorless oil 2.35 g (7.32 mmol) for a 100% yield.
2.3. 12-(Trimethylsilyl)-1-[(tetrahydropyran-2-yl)oxy]dodec-11-yn (3)
To a stirred solution of 3.04 mL (22.0 mmol) of (trimethylsilyl)acetylene in 9.5 mL of dry THF and under argon was added 2.36 mL (25.6 mmol) of n-BuLi (2.5 M) in hexane at −78 °C. After 2 min, 3.36 mL of HMPA and 2.35 g (7.32 mmol) of 2 was added to the reaction mixture, maintaining the temperature at −78 °C. After 24h the reaction mixture was quenched with water. The organic product was extracted with brine solution (2 × 20 mL), diethyl ether (2 × 20 mL), dried over MgSO4, filtered, and evaporated in vacuo. The product was purified using silica gel column chromatography eluting with hexane/ether (9:1). The silylated alkyne 3 (Banaszak et al., 2009) was obtained as a colorless oil 2.43 g (7.16 mmol) for a 97% yield.
2.4. 1-[(Tetrahydropyran-2-yl)oxy]dodec-11-yne (4)
To a mixture of 2.35 g (6.93 mmol) of 3 and 12.0 mL of dry THF was added dropwise 6.93 mmol of tetrabutylammonium fluoride (1M) at 0 °C. After 24 h at room temperature the reaction was quenched with HCl (2M), and the organic layer was washed with brine (1 × 20 mL), ether (1 × 20 mL), dried over MgSO4, filtered, and evaporated in vacuo. The crude product was purified using silica gel column chromatography eluting with hexane/ether (9:1). The alkyne 4 was obtained as a colorless oil 1.66 g (6.22 mmol) for a 90% yield. IR (neat) νmax 3312, 2926, 2854, 1466, 1441, 1352, 1323, 1259, 1200, 1136, 1121, 1078, 1026, 988, 904, 870, 814, 721, 627 cm−1; 1H NMR (CDCl3, 500 MHz) δ 4.55 (1H, t, J = 2.7 Hz), 3.84 (1H, m), 3.71 (1H, m), 3.49 (1H, m), 3.36 (1H, m), 2.16 (2H, dt, J = 7.1, 2.6 Hz, H-10), 1.92 (1H, t, J = 2.6 Hz, H-12), 1.81 (2H, m), 1.70 (2H, m), 1.53 (6H, m, -CH2-), 1.27 (12H, m, -CH2-); 13C NMR (CDCl3, 125 MHz) δ 98.80 (d), 84.74 (s, C-11), 68.01 (d, C-12), 67.64 (t, C-1), 62.29 (t), 30.75 (t), 29.71 (t, C-2), 29.48 (t), 29.41 (t), 29.39 (t), 29.04 (t), 28.70 (t), 28.45 (t), 26.19 (t, C-3), 25.48 (t), 19.66 (t, C-10), 18.36 (t); GC-MS (70 eV) m/z (relative intensity) 266 (M+, 1), 237 (1), 225 (1), 211 (1), 195 (1), 165 (1), 149 (1), 135 (1), 123 (1), 109 (4), 107 (2), 101 (27), 97 (2), 95 (11), 93 (4), 91 (2), 86 (6), 85 (100), 84 (10), 81 (19), 79 (9), 67 (21), 57 (9), 56 (17), 55 (28). HRMS (APCI) Calcd for C19H35O2 [M+H]+ 267.2682, found 267.2681.
2.5 16-Methyl-1-[(tetrahydropyran-2-yl)oxy]heptadec-11-yne (5)
To a solution of 1.54 g (5.77 mmol) of 4 and 11.2 mL of THF at 0 °C, 1.87 mL (20.2 mmol) of n-BuLi (2.5 M) in hexane was added while stirring under an argon atmosphere. After 45 min, 2.87 mL of HMPA was added. After additional 15 min, 2.56 mL (17.3 mmol) of 1-bromo-4-methylpentane was added to the reaction mixture and the reaction was left stirring for 24 h at room temperature. After this time the reaction mixture was quenched with water and the organic product was extracted with ether (2 × 25 mL), dried over MgSO4, filtered, and evaporated in vacuo. The crude product was purified using silica gel column chromatography eluting with hexane/ether (9:1). The product 5 was obtained as colorless oil 2.02 g (5.75 mmol) for a 99% yield. IR (neat) νmax 2926, 2854, 1466, 1441, 1383, 1366, 1352, 1259, 1200, 1128, 1121, 1078, 1032, 984, 905, 870, 814, 743 cm−1; 1H NMR (CDCl3, 500 MHz) δ 4.56 (1H, t, J = 2.7 Hz), 3.85 (1H, m), 3.72 (1H, m), 3.49 (1H, m), 3.37 (1H, m), 2.11 (4H, m, H-10, H-13), 1.81 (2H, m), 1.69 (2H,m), 1.51 (7H, m, -CH2-), 1.26 (16H, m, -CH2-), 0.86 (6H, d, J = 6.6 Hz, -CH(CH3)3); 13C NMR (CDCl3, 125 MHz) δ 98.79 (d), 80.22 (s), 80.19 (s), 67.64 (t, C-1), 62.27 (t), 38.18 (t, C-15), 30.75 (t), 29.72 (t, C-2), 29.51 (t), 29.46 (t), 29.44 (t), 29.12 (t), 28.81 (t), 27,61 (d, C-16), 27.04 (t, C-3), 26.20 (t), 25.48 (t), 22.54 (q, C-17, C-18), 19.65 (t), 18.98 (t, C-13), 18.71 (t, C-10); GC-MS (70 eV) m/z (relative intensity) 350 (M+, 1), 294 (1), 279 (1), 277 (1), 265 (1), 221 (1), 207 (1), 195 (1), 165 (1), 151 (1), 149 (2), 138 (1), 137 (2), 135 (3), 123 (6), 121 (5), 111 (3), 109 (27), 107 (6), 101 (24), 96 (12), 95 (31), 93 (9), 86 (6), 85 (100), 84 (20), 83 (18), 82 (20), 81 (34), 79 (15), 69 (41), 68 (12), 67 (32), 57 (13), 56 (14), 55 (46). HRMS (APCI) Calcd for C23H43O2 [M+H]+ 351.3257, found 351.3255.
2.6 16-Methylheptadec-11-yn-1-ol (6)
To a mixture of methanol (20.0 mL) and 1.94 g (6.36 mmol) of 5 was added catalytic amounts of pTSA and the reaction mixture was stirred at 35 °C for 48h. After this time the organic extract was washed with a saturated solution of sodium bicarbonate (2 × 50 mL), ether (2 × 20 mL), dried over MgSO4, filtered, and evaporated in vacuo. The product was purified using silica gel column chromatography eluting with hexane/ether (9:1). The 16-methylheptadec-11-yn-1-ol (6) was obtained as colorless oil 1.41 g (5.31 mmol) for an 83% yield. IR (neat) νmax 3317 (OH, broad), 2926, 2854, 1466, 1385, 1333, 1057, 721, 627 cm−1; 1H NMR (CDCl3, 500 MHz) δ 3.61 (2H, t, J = 6.6, H-1), 2.11 (4H, m, H-10, H-13), 1.54 (2H, m), 1.45 (2H, m), 1.34 (2H, m, -CH2-), 1.26 (16H, m, -CH2-), 0.86 (6H, d, J = 6.6 Hz, -CH(CH3)3); 13C NMR (CDCl3, 125 MHz) δ 80.23 (s), 80.18 (s), 62.95 (t, C-1), 38.17 (t, C-15), 32.74 (t, C-2), 29.70 (t), 29.52 (t), 29.44 (t), 29.38 (t), 29.11 (t), 28.80 (t), 27.70 (d, C-16), 27.03 (t), 25.70 (t), 22.53 (q, C-17, C-18), 19.62 (t, C-13), 18.96 (t, C-10); GC-MS (70 eV) m/z (relative intensity) 266 (M+, 1), 251 (1), 210 (1), 191 (1), 167 (1), 163 (2), 149 (4), 135 (9), 123 (14), 121 (15), 111 (7), 110 (18), 109 (88), 107 (14), 97 (13), 96 (38), 95 (84), 94 (11), 93 (24), 91 (12), 83 (23), 82 (65), 81 (89), 80 (21), 79 (38), 77 (12), 70 (11), 69 (100), 68 (37), 67 (73), 65 (8), 57 (13), 56 (9), 55 (61). HRMS (APCI) Calcd for C18H35O [M+H]+ 267.2682, found 267.2682.
2.7 (Z)-16-Methylheptadec-11-en-1-ol (7)
Into a 50-mL two-necked round-bottomed flask were placed dry hexane, 0.61 g (2.28 mmol) of alkyne 6, quinoline (1.8 mL), and palladium in activated carbon (Lindlar’s catalyst). One of the two necks was capped with a rubber septum and the other was connected via tygon tubing to a 25-mL graduated pipet ending in a 150 mL beaker with distilled water. While stirring at room temperature a 20 mL syringe with needle was used to withdraw air from the system and draw water up into the graduated pipet to the 0.0 mL mark. Hydrogen was then introduced into the system using balloon filled with hydrogen attached to the hose barb-to-luer lock adapter with stopcock and a needle. The reaction mixture consumed 55.9 mL of hydrogen during 1 h. The mixture was filtered and the solvent removed in vacuo. The product was purified under vacuum distillation (Kugelrohr) by removing impurities at 130 °C/3 mmHg. The (Z)-16-methylheptadec-11-en-1-ol (7) was obtained as colorless oil 0.55 g (2.07 mmol) for a 91% yield. IR (neat) νmax 3335, 3005, 2924, 2853, 1464, 1383, 1366, 1119, 1057, 908, 733 cm−1; 1H NMR (CDCl3, 500 MHz) δ 5.52 (2H, m, H-11, H-12), 3.62 (2H, t, J = 6.6 Hz, H-1), 2.00 (4H, m, H-10, H-13), 1.55 (4H, m), 1.31 (16H, m, -CH2-), 1.18 (2H, m, H-15), 0.87 (6H, d, J = 6.6 Hz, -CH(CH3)3); 13C NMR (CDCl3, 125 MHz) δ 129.91 (d), 129.86 (d), 62.30 (t, C-1), 38.64 (t, C-15), 38.78 (t, C-2), 29.74 (t), 29.58 (t), 29.54 (t), 29.51 (t), 29.41 (t), 29.28 (t), 27.87 (d, C-16), 27.54 (t, C-13), 27.46 (t, C-10), 27.18 (t, C-14), 25.72 (t, C-3), 22.60 (q, C-17, C-18); GC-MS (70 eV) m/z (relative intensity) 268 (M+, 1), 250 (4), 223 (1), 208 (1), 195 (1), 151 (3), 149 (2), 137 (7), 135 (5), 125 (5), 123 (18), 121 (6), 111 (17), 109 (37), 98 (15), 97 (37), 96 (54), 95 (70), 83 (55), 82 (82), 81 (65), 80 (10), 79 (15), 70 (29), 69 (100), 68 (41), 67 (63), 65 (4), 57 (48), 56 (69), 55 (96). HRMS (APCI) Calcd for C18H37O [M+H]+ 269.2839, found 269.2838.
2.8 (Z)-16-Methyl-11-heptadecenoic acid (1)
A solution of 3.87 mmol of pyridinium dichromate (PDC) and 2.0 mL of dimethylformamide (DMF) was added under argon to a solution of 0.21 g (0.775 mmol) of alcohol 7 and 5.0 mL of DMF, and the reaction mixture was left stirring at room temperature for 48 h. After this time, the reaction mixture was washed with water (3 × 25 mL), ether (2 × 20 mL), dried over MgSO4, filtered, and the solvent evaporated in vacuo. The crude product was purified using florisil column chromatography eluting with ether. The (Z)-16-methyl-11-heptadecenoic acid (1) was obtained as colorless oil 0.15 g (0.53 mmol) for a 68% yield. IR (neat) νmax 3500-2500, 3005, 2924, 2854, 1709 (C=O), 1464, 1414, 1385, 1366, 1119, 1099, 1024, 721 cm−1; 1H NMR (CDCl3, 500 MHz) δ 5.35 (2H, m, H-11, H-12), 2.34 (2H, t, J = 7.4 Hz, H-2), 2.00 (4H, m, H-10, H-13), 1.64 (2H, m, H- 3), 1.53 (2H, m), 1.31 (12H, m, -CH2-), 1.17 (2H, m, H-15), 0.88 (6H, d, J = 6.6 Hz, - CH(CH3)3); 13C NMR (CDCl3, 125 MHz) δ 179.88 (s, C-1), 129.94 (d), 129.84 (d), 38.64 (t, C-15), 34.05 (t, C-2), 29.73 (t), 29.46 (t), 29.39 (t), 29.24 (t), 29.22 (t), 29.05 (t), 27.88 (d, C-16), 27.55 (t, C-13), 27.43 (t, C-10), 27.18 (t), 24.68 (t, C-3), 22.61 (q, C-17, C-18). HRMS (APCI) Calcd for C18H33O2 [M-H]+ 281.2486, found 281.2488.
Methyl (Z)-16-methyl-11-heptadecenoate
GC-MS (70 eV) m/z (relative intensity) 296 (M+, 5), 265 (15), 264 (25), 249 (3), 241 (11), 222 (11), 221 (5), 209 (10), 191 (8), 185 (2), 165 (4), 153 (4), 152 (5), 151 (6), 141 (6), 139 (7), 137 (9), 125 (13), 124 (10), 123 (15), 121 (6), 111 (24), 109 (18), 98 (27), 97 (46), 96 (34), 95 (31), 87 (40), 85 (10), 84 (34), 83 (53), 82 (21), 81 (31), 79 (11), 74 (54), 70 (22), 69 (100), 68 (19), 67 (36), 59 (22), 57 (32), 56 (50), 55 (98).
2.9 Cell Cultures
L. donovani (MHOM/ET67/L82 strain) promastigotes were propagated in a completely defined medium 199, supplemented with 10% heat inactivated fetal calf serum (FCS) and penicillin/streptomycin cocktail (containing 50 U/mL penicillin, 50 µg/mL streptomycin). The IC50 value was determined with different concentrations of compound 1 dissolved in DMSO (ranging from 3.9 to 1000 µM) added to cultures and cell population assessed by Coulter.
Murine macrophages (RAW 264.7) were grown at 37 °C in RPMI 1640 medium, supplemented with 10% FCS antibiotics (containing 50 U/mL penicillin, 50 µg/mL streptomycin). Toxicity IC50 values were determined after 48 h incubation with different concentrations of the compounds dissolved in DMSO (50 to 2000 µM) using vital alamar-Blue® dye (Invitrogen, Carlsbad, CA, USA), according manufacturer recommendations.
2.10 Purification of recombinant leishmanial TopIB
Expression of a recombinant topoisomerase IB from Leishmania donovani (LdTopIB) in a topoisomerase IB-deficient Saccharomyces cerevisiae strain has been described elsewhere (Villa et al., 2003). Purification of recombinant LdTopIB was done according to Diaz-González and coworkers (Diaz-González et al., 2008). Briefly, LdTopIB overexpressing yeasts were disrupted with one freeze/thaw cycle at −80 °C, with the purpose of weakening the yeast wall; after lysis with 425–600 µm acid-washed glass beads, the extracts were cleared by centrifugation at 15000 × g for 30 min at 4 °C. The protein suspension was loaded onto a phosphocellulose (P-11) column, previously equilibrated as manufacturer indications. LdTopIB was eluted at 4 °C with a discontinuous gradient of KCl (0.2, 0.4, 0.6, 0.8 and 1 M) in TEEG buffer, supplemented with 0.1 mg/mL sodium bisulphite, 0.8 mg/mL NaF and the protease inhibitors cocktail. Active fractions were further loaded onto a phenyl-sepharose column (Sigma-Aldrich, St Louis, US), eluted with a discontinuous inverse gradient of ammonium sulphate (1, 0.8, 0.6, 0.4 and 0.2 M) and then concentrated by Microcon YM-30 (Millipore) before use.
2.11 DNA relaxation assays
DNA topoisomerase IB activity was assayed by the relaxation of negatively supercoiled plasmid DNA. The reaction mixture in a total volume of 20 µl contained 0.2 µg of supercoiled pHOT plasmid, 10 mM Tris-HCl buffer pH 7.5, 5 mM MgCl2, 0.1 mM EDTA, 15 µg/mL bovine serum albumin, 50 mM KCl and various extracts containing altered proteins or wild type enzyme, starting with 1 unit LdTopIB. The reaction mixtures were incubated for 30 min at 37 °C. The enzyme reactions were stopped by the addition of up to 1% SDS - final concentration – and digested by 2 mg/mL proteinase K with 1 h incubation to remove protein bonded to the DNA fragment. The extent of plasmid DNA relaxation was assessed by electrophoresis in a 1% agarose gel in 0.1 M Tris acetate EDTA (TAE) buffer pH 8.0 at 2 V/cm for 14 h. The gels were visualized under UV illumination after being stained with ethidium bromide (0.5 mg/mL) and a posterior electrophoresis in the presence of 0.1 mg/mL ethidium bromide, in order to separate the nicked DNA from the relaxed topoisomers. One unit of LdTopIB is defined as the amount of purified protein able to relax 0.2 µg of pHOT supercoiled DNA per 30 min at 37 °C.
3. Results and discussion
The synthesis of 1 started with commercially available (Aldrich) 10-bromo-1-decanol, which was protected with 3,4-dihydro-2H-pyran (DHP) in the presence of p-toluenesulfonic acid (PTSA) affording the corresponding dihydropyranyl protected alcohol 2 in a 100% isolated yield (Scheme 1). Compound 2 was then submitted to the first acetylide coupling reaction with the versatile reagent (trimethylsilyl)acetylene using n-BuLi in THF-HMPA at −78°C affording the trimethylsilylacetylenic derivative 3 in a 97% yield. Deprotection of the terminal trimethylsilyl group with tetrabutylammonium fluoride (TBAF) afforded the terminal alkyne 4 in a 90% yield. A second acetylide coupling with 1-bromo-4-methylpentane using n-Buli, THF-HMPA at 0 °C resulted in the iso-branched alkyne 5 in a 99% isolated yield (Scheme 1). Deprotection of the dihydropyranyl group in 5 with PTSA in methanol at 35 °C for 48 h afforded the 16-methyl-11-heptadecyn-1-ol (6) in an 83% isolated yield. Catalytic hydrogenation under Lindlar’s conditions of 6 resulted in the (Z)-16-methyl-11-heptadecen-1-ol (7) in a 91% yield with a 100% cis stereochemistry for the double bond. Final oxidation of the alcohol with pyridinium dichromate (PDC) in dimethylformamide (DMF) resulted in the formation of the desired acid 1 in a 68% yield. The overall total yield for the seven steps was 44%, which afforded enough material for its complete spectral characterization.
Acid 1 presented spectral data that confirmed its synthesis and the structure of the natural acid. The iso methyl branching was easily identified by both 1H NMR and 13C NMR spectrometry as well as by infrared spectroscopy (IR). In the 1H NMR spectra both terminal isopropyl methyl groups appeared as a doublet at δ 0.88 ppm, while in the 13C NMR both methyl groups resonated at δ 22.6 ppm. The IR spectrum of 1 was also very characteristic since the terminal isopropyl group was observed as a doublet at 1385 and 1366 cm−1. The presence of the olefinic hydrogens in 1 was easily detected in the 1H NMR spectra by the olefinic signals at δ 5.35 ppm. The 13C NMR of the olefinic region was most interesting, in particular the resonance of the allylic methylene carbons that confirmed the cis double bond stereochemistry. The C-10 allylic carbon resonated at δ 27.4 ppm, while the C-13 allylic carbon was observed at δ 27.5 ppm, confirming the presence of the cis double bond (Gunstone et al., 1977). It is well established that the allylic methylene carbons from cis double bonds in fatty acids resonate in 13C NMR at around 27 ppm, while the allylic methylene carbons from trans double bonds resonate at around 32 ppm (Gunstone et al., 1977). The cis double bond stereochemistry was also further confirmed by IR since a strong absorption at 721 cm−1 was observed for 1. As expected, the carboxylic acid carbonyl was observed in 13C NMR at δ 179.9 ppm and in IR at 1709 cm−1.
The methyl ester of the synthetic acid 1 was also prepared (HCl in MeOH) and its mass spectrum (Fig.1) and GC retention time (ECL value) were compared to the ones reported for the natural methyl ester of 1 (Rodríguez et al., 2010). The mass spectra and GC retention times of both synthetic and natural methyl (Z)-16-methyl-11-heptadecenoates were comparable. For example, in the mass spectrum of the methyl ester of synthetic 1 both the M+-32 and M+-31 fragmentations peaks at m/z 264 (25%) and at m/z 265 (15%) can be easily seen (Fig.1). In addition, a strong M+-55 peak at m/z 241 (11%) as well as the typical McLafferty rearrangement at m/z 74 (54%) were also observed (Rodríguez et al., 2010). The GC retention times (ECL values) for both methyl esters (synthetic and natural 1) were almost identical. For example, the natural methyl ester of 1 was reported to have an ECL value of 17.46, while the synthetic methyl ester of 1 was determined to have an ECL value of 17.47. These results clearly demonstrated the cis stereochemistry of the double bond in the natural fatty acid 1.
Fig. 1.
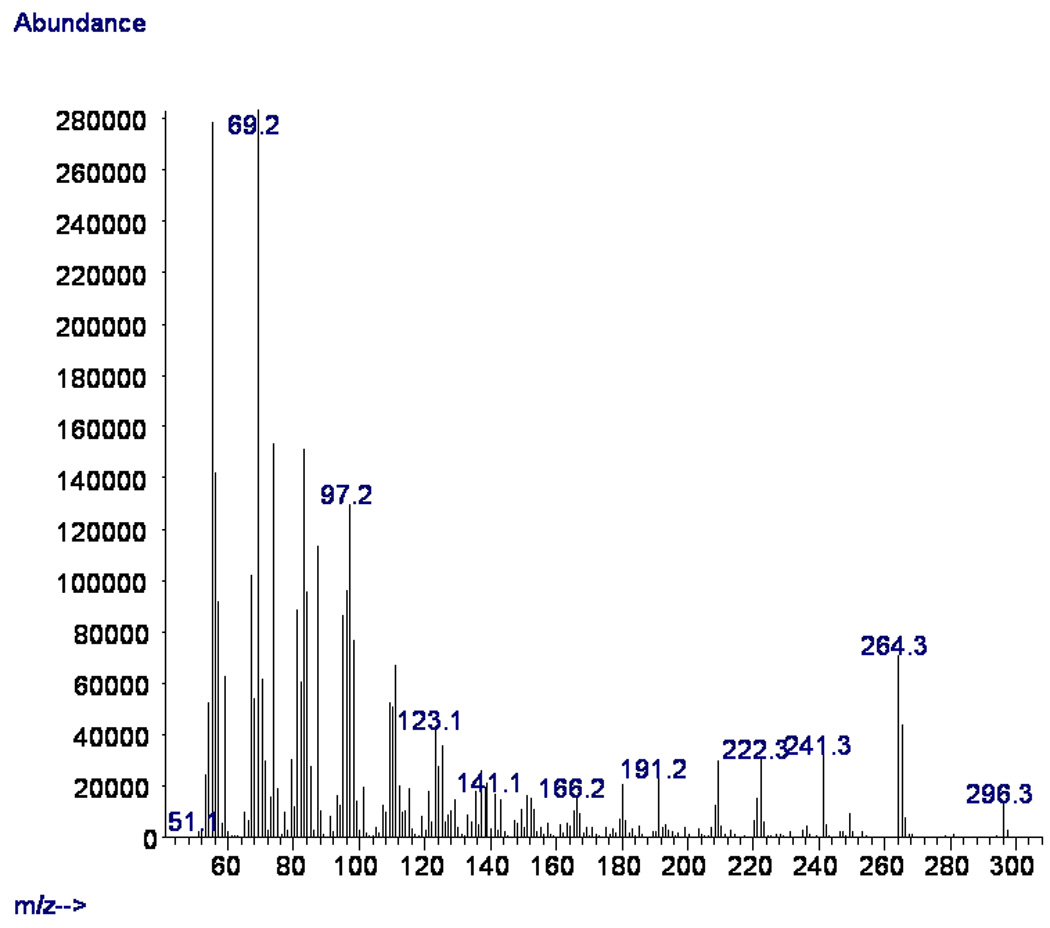
Mass spectrum (70 eV) of methyl (Z)-16-methyl-11-heptadecenoate.
Aimed at exploring the biological activity of acid 1 we studied its antiprotozoal activity towards Leishmania donovani (the causative agent of visceral leishmaniasis) and the inhibition of acid 1 of the leishmania LdTopIB as a possible mechanism of action. We found acid 1 to be cytotoxic to L. donovani at an IC50 of 165.5 ± 23.4 µM, and to murine macrophages at the higher value of IC50 = 604.0 ± 107.9 µM (Fig. 2). Moreover, acid 1 inhibits the LdTopIB with an IC50 = 62.3 ± 0.7 µM. These results tend to indicate that the toxicity of 1 towards L. donovani could be due to the inhibition of its DNA topoisomerase IB. This finding is interesting since it has recently been demonstrated that there are substantial differences between the LdTopIB and the human DNA topoisomerase I (Balaña-Fouce et al., 2006). In this context one might be able to interfere with the protozoan DNA topoisomerase IB without harming the mammalian DNA topoisomerase IB. In fact, we have shown that the acid (Z)-14-methyl-9-pentadecenoic acid, also a monounsaturated iso methyl-branched fatty acid, inhibits the human DNA topoisomerase I but at the higher concentrations of 500 µM (Carballeira et al., 2007). Therefore, the present findings corroborate our previous results that monounsaturated iso methyl-branched fatty acids are more effective towards the LdTopIB than the human DNA topoisomerase I (Carballeira et al., 2009).
Fig. 2.
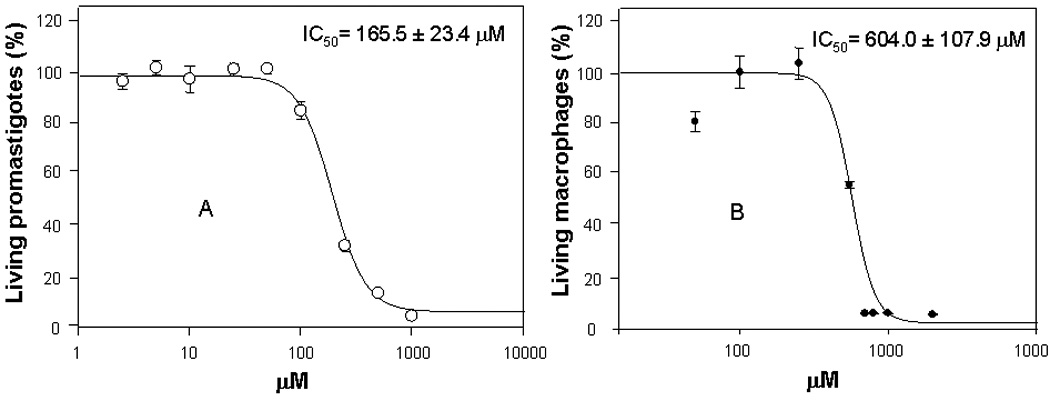
Plots of percent (%) of living L. donovani promastigotes (A) and living percent of murine macrophages (B) versus concentrations (µM) of the fatty acid 1. The calculated therapeutic index (IC50/IC50) for fatty acid 1 is 3.7.
In summary, we can conclude that the first synthesis for the (Z)-16-methyl-11-heptadecenoic acid (1) was accomplished in seven steps and in a 44% overall yield. Certainly, the synthetic strategy of first coupling the (trimethylsilyl) acetylene to the long-chain bromoalcohol (97% yield) followed by a second acetylide coupling to the short-chain iso bromo alkane (99% yield) afforded good overall yields. Complete spectral data is also provided for the first time for acid 1 and the double bond stereochemistry of the natural acid was elucidated. The synthetic route presented furnished enough material to study the antiprotozoal activity of 1 and these preliminary biological studies tend to indicate that fatty acids such as 1 might be good antiprotozoal lead compounds.
Acknowledgements
The project described was supported by Award Number SC1GM084708 from the National Institutes of General Medical Sciences of the NIH. G. Cintrόn thanks the UPR RISE program for an undergraduate fellowship. We thank Dr. Fred Strobel (Emory University) for the high resolution mass spectral data. This research was also partially supported by a grant (Gr238) from Junta de Castilla y León, AGL2009-11935 from MICINN, and the Tropical Diseases Network (RICET) from Ministerio de Salud y Consumo (SPAIN).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balaña-Fouce R, Redondo CM, Pérez-Pertejo Y, Díaz-González R, Reguera RM. Targeting atypical trypanosomatid DNA topoisomerase I. Drug Discov. Today. 2006;11:733–740. doi: 10.1016/j.drudis.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Banaszak E, Xu L-W, Bardeau J-F, Castanet A-S, Mortier J. First syntheses of model long-chain trichloro[ω-(trimethylsilyl)alkynyl]silanes suitable for self-assembled monolayers on silicon surfaces. Tetrahedron. 2009;65:3961–3966. [Google Scholar]
- Carballeira NM, Pagán M, Shalabi F, Nechev JT, Lahtchev K, Ivanova A, Stefanov K. Two novel iso-branched octadecenoic acids from a Micrococcus species. J. Nat. Prod. 2000;63:1573–1575. doi: 10.1021/np000305r. [DOI] [PubMed] [Google Scholar]
- Carballeira NM, Pagán M. Total synthesis of the novel bacterial fatty acid 16-methyl-8(Z)-heptadecenoic acid. Chem. Phys. Lipids. 2001;113:23–27. doi: 10.1016/s0009-3084(01)00137-2. [DOI] [PubMed] [Google Scholar]
- Carballeira NM, Sanabria D, Oyola D. An improved synthesis for the (Z)-14-methyl-9-pentadecenoic acid and its topoisomerase I inhibitory activity. ARKIVOC. 2007;viii:49–57. [PMC free article] [PubMed] [Google Scholar]
- Carballeira NM, Montano N, Balaña-Fouce R, Fernández Prada C. First total synthesis and antiprotozoal activity of (Z)-17-methyl-13-octadecenoic acid, a new marine fatty acid from the sponge Polymastia penicillus. Chem. Phys. Lipids. 2009;161:38–43. doi: 10.1016/j.chemphyslip.2009.06.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-González R, Pérez-Pertejo Y, Pommier Y, Balaña-Fouce R, Reguera RM. Mutational study of the "atalytic tetrad" of DNA topoisomerase IB from the hemoflagellate Leishmania donovani: Role of Asp-353 and Asn-221 in camptothecin resistance. Biochem Pharmacol. 2008;76:608–619. doi: 10.1016/j.bcp.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunstone FD, Pollard MR, Scrimgeour CM, Vedanayagam HS. 13C-Nuclear magnetic resonance studies of olefinic fatty acids and esters. Chem. Phys. Lipids. 1977;18:115–129. doi: 10.1016/0009-3084(77)90031-7. [DOI] [PubMed] [Google Scholar]
- Lee HK, Lee DS, Lim J, Kim JS, Im KS, Jung JH. Topoisomerase I inhibitors from the Streptomyces sp. strain KM86-9B isolated from a marine sponge. Arch. Pharm. Res. 1998;21:729–733. doi: 10.1007/BF02976766. [DOI] [PubMed] [Google Scholar]
- Rodríguez W, Osorno O, Ramos FA, Duque C, Zea S. New fatty acids from Colombian Caribbean Sea sponges. Biochem. Syst. Ecol. 2010;38:774–783. [Google Scholar]
- Stritzke K, Schulz S, Laatsch H, Helmke E, Beil W. Novel caprolactones from a marine Streptomycete. J. Nat. Prod. 2004;67:395–401. doi: 10.1021/np030321z. [DOI] [PubMed] [Google Scholar]
- Taylor J, Parkes JR. The cellular fatty acids of the sulfate-reducing bacteria, Desulfobacter species, Desulfobulbus species and Desulfovibrio desulfuricans. J. Gen. Microbiol. 1983;129:3303–3309. [Google Scholar]
- Villa H, Otero Marcos AR, Reguera RM, Balaña-Fouce R, García-Estrada C, Pérez-Pertejo Y, Tekwani BL, Tyler PJ, Stuart KD, Bjornsti MA, Ordóñez D. A novel active DNA topoisomerase I in Leishmania donovani. J. Biol. Chem. 2003;278:3521–3526. doi: 10.1074/jbc.M203991200. [DOI] [PubMed] [Google Scholar]