

Abstract
Long-chain fatty acids are an important source of energy in muscle and heart where the acyl-CoA dehydrogenases (ACADs) participate in consecutive cycles of β-oxidation to generate acetyl-CoA and reducing equivalents for generating energy. However, the role of long-chain fatty acid oxidation in the brain and other tissues that do not rely on fat for energy is poorly understood. Here we characterize two new ACADs, ACAD10 and ACAD11, both with significant expression in human brain. ACAD11 utilizes substrates with primary carbon chain lengths between 20 and 26, with optimal activity towards C22CoA. The combination of ACAD11 with the newly characterized ACAD9 accommodates the full spectrum of long chain fatty acid substrates presented to mitochondrial β-oxidation in human cerebellum. ACAD10 has significant activity towards the branched-chain substrates R and S, 2 methyl-C15-CoA and is highly expressed in fetal but not adult brain. This pattern of expression is similar to that of LCAD, another ACAD previously shown to be involved in long branched chain fatty acid metabolism. Interestingly, the ACADs in human cerebellum were found to have restricted cellular distribution. ACAD9 was most highly expressed in the granular layer, ACAD11 in the white matter, and MCAD in the molecular layer and axons of specific neurons. This compartmentalization of ACADs in the human central nerve system, suggests that β-oxidation in cerebellum participates in different functions other than generating energy, for example, the synthesis and/or degradation of unique cellular lipids and catabolism of aromatic amino acids, compounds that are vital to neuronal function.
Keywords: Acyl-CoA dehydrogenase, fatty acid oxidation, long chain fat metabolism, CNS specific expression
1.0 Introduction
The acyl-CoA dehydrogenases (ACADs; EC 1.3.99.13) are a group of mitochondrial flavoenzymes that catalyze the α, β-dehydrogenation of acyl-CoA esters using the electron transfer flavoprotein (ETF) as their physiologic electron acceptor. There are at least nine known members of the ACAD gene family active in branched chain amino acid catabolism or fatty acid oxidation. All of the ACADs are nuclear encoded proteins that must first be translated in the cytoplasm then imported into mitochondria and processed into a mature form. Mitochondrial β-oxidation of long chain fatty acyl-CoAs is well recognized as a primary metabolic pathway for maintenance of energy homeostasis and body temperature [1]. However, some of the symptoms in patients with mitochondrial β-oxidation defects (OMIM# 609105) are not readily explained simply by energy deficiency. For example, a high percentage of patients with mitochondrial trifunctional protein deficiency present with neurologic disease, including neuronal demyelination and retinal degeneration, that responds poorly to available therapies designed to restore energy homeostasis [2–5]. Recently we described a new disorder of ACAD9 deficiency (OMIM# 611126) [6]. The presence of chronic abnormalities in the central nervous system in these patients further underscores the importance of non-energy generating, alternative functions of mitochondrial β-oxidation [6]. Three ACADs have been described that are active with long chain acyl-CoA substrates, very long chain acyl-CoA dehydrogenase (VLCAD), acyl-CoA dehydrogenase 9 (ACAD9), and long chain acyl-CoA dehydrogenase (LCAD), but except for the role of VLCAD in energy generation through fatty acid oxidation, the spectra of the physiologic functions of these enzymes remains unknown. ACAD9 has also been implicated in the assembly or stability of complex 1 of the mitochondrial respiratory chain, raising the possibility of a duel roll as a molecular chaperonin for this metabolic enzyme [7]. In this study, we characterize two additional ACADs with long chain acyl-CoA activity, ACAD10 and ACAD11. These new enzymes appear to be compartmentalized mainly in human brain, in contrast to the localization of VLCAD and MCAD to energy generating tissues, suggesting that mitochondrial β-oxidation in human brain is tailored to different fatty acids in different regions of the brain. This in turn implies that β-oxidation of long chain fatty acids in the central nervous system is involved in functions alternative to energy metabolism, such as controlling the composition of fatty acids or catabolizing functional intermediates of aromatic amino acids. Thus the five different long chain ACADs are likely specific to the different functions of mitochondrial β-oxidation in various tissues in humans.
2.0 Material and Methods
2.1 cDNA amplification and subcloning
Total RNA was extracted from frozen post mortem liver using TRIzol reagent (Invitrogen, San Diego, CA) as per the manufacturer’s instructions. Reverse transcription was performed using 1 μg of total RNA and Powerscript™ reverse transcriptase using random hexamer nucleotides as primers. ACAD11 (BC019607) sequences were amplified with the Yieldace PCR kit (Stratagene La Jolla, CA) with primers as follows: 5′CTCGAGACCT GTGGATGCCG CGTTGC3′ and 5′CGACTCGCGG CCGCAAGCTT ATATCTTGGC TGTC. The PCR products were purified using the PCR cleanup kit (Qiagen, Inc., Valencia, CA), subcloned into pcDNA2.1 vector, and sequenced by the Genomics Core Facility at the University of Pittsburgh.
In vitro transcription/translation and mitochondrial import
ACAD 11 proteins were synthesized from 1 μg of plasmid DNA (pcDNA3.1 containing the various cDNAs by coupled in vitro transcription and translation (TNT) in the presence of 35S-labeled methionine (20 μCi, 1250 Ci/mmol) as described [8]. Liver mitochondria were freshly prepared from young male Sprague-Dawley rats by differential centrifugation [8]. The final pellet was resuspended in 500 μl of 5 mM HEPES (pH 7.4), 220 mM D-mannitol, and 70 mM sucrose (HMS buffer) and diluted to a final concentration of 20 mg/ml protein. Thirty-five μl of translation mixture containing 35S-labeled precursor proteins were mixed with 85 μl of mitochondrial suspension and 30 μl of HMS buffer to a total volume of 150 μl and incubated at 30 °C for 75 min. Mitochondria were collected by centrifugation at 9100 xg for 2 min at 4 °C and washed three times in cold HMS buffer. They were then resuspended in HMS buffer and treated with trypsin (0.5 μg/μl) for 0, 10, 20 min. Soybean trypsin inhibitor (3 μg/μl) was added to stop the reaction and mitochondria were re-pelleted as described [9]. The pellets, together with 1 μl of TNT reaction product were solubilized in 30 μl of SDS-PAGE loading buffer, boiled for 10 min, and resolved by SDS-PAGE. Gels were transferred to supported nitrocellulose and exposed to X-ray film overnight. For native PAGE, 4–15% Tris-HCl Criterion gels (Bio-Rad) were used following the manufacture’s instructions.
2.2 Computational Modeling of ACAD11
The ACAD11 model was created with Insight II molecular modeling software, Accelrys Inc. (San Diego, CA), using the atomic coordinates of rat SCAD (1JQI) and human glutaryl-CoA dehydrogenase (GCD) crystal structures (1SIQ) as described previously [10]. An acetyl-CoA and FAD is modeled as the substrate and cofactor respectively.
2.3 Overexpression ACAD10 and 11 in E. coli and partial purification
Full length cDNAs corresponding to the predicted mature mitochondrial protein sequence of ACAD10 and 11 were cloned individually into the E. coli expression vector pET-21a(+) (Novagen, Madison, WI), with E.coli codon biased introduced in the first 100 base pairs and over-expressed as described previously for other ACADs [11, 12]. The lysate was treated with 1% n-Dodecyl β-D-maltoside detergent (Anatrace, Maumee, Ohio, USA) before enzyme activity assay.
2.4 ETF fluorescence reduction assay for ACAD activity
ACAD activities in extracts from cultured skin fibroblasts and frozen tissues obtained 6–10 hours postmortem (Brain and Tissue bank for Developmental Disorders, Baltimore, MD) were measured as described using the sensitive and specific anaerobic ETF fluorescence reduction assay with the indicated substrates at 5 μM final concentration [13]. Mitochondrial matrix and membrane fractions were prepared as previously described and assayed in triplicate using palmitoyl-CoA as substrate [14]. Five normal liver samples were used as controls. Hexanoyl (C6:0)-CoA, octanoyl (C8:0)-CoA, decanoyl (C10:0)-CoA, dodecanoyl (C12:0)-CoA, tetradecanoyl (C14:0)-CoA, palmitoyl (C16:0)-CoA, stearoyl (C18:0)-CoA, and arachidoyl (C20:0)-CoA were purchased from Sigma (St. Louis, MO). Synthesis of racemic 2-methylhexadecanoyl-CoA [15] and 4,8,12-trimethyltridecanoyl-CoA (4,8,12-trimethylC13-CoA)[16] has been described before, S-methyl-hexadecanoyl-CoA (S- methyl C16-CoA), 2S-methylnonadecanoyl-CoA (2S-methyl-C19-CoA) was prepared in a similar way as described for 2S-methyl-pentadecanoyl-CoA (2S-methylC15-CoA) [ref B], using the 1-bromohexadecane as starting point.
2.5 Quantitative Realtime RT-PCR
Reverse transcription was performed with a 1 μg of RNA from a panel of total RNA pooled from 20 human tissues or total RNA from pooled human hippocampus purchased from BD Biosciences Clontech (Palo Alto, CA) and Powerscript™ reverse transcriptase (BD Biosciences Clontech) according to the manufacturer’s instructions. cDNA corresponding to 20 ng of total RNA was used for real-time PCR on an ABI 7900HT Sequence Detection System (PE Applied Biosystems, Foster City, CA) using heat-activated Taq DNA polymerase (BD Biosciences Clontech). Specific primer pairs (sequences available upon request) for VLCAD, ACAD9, LCAD, MCAD, and SCAD were designed to target coding sequences near the catalytic domain of each enzyme. Primers for the housekeeping genes GUSB and 18S rRNA were used as controls. All primers (labeled with Fam fluorescence tag at the 5- end and MGB non-fluorescence quencher at the 3′-end) were synthesized by PE Applied Biosystems (Assay on Demand™). PCR assays were performed in 96 well optical plates following the manufacturer’s recommendations. The amplification efficiency of each primer pair was tested using different dilutions of total RNA, and all were similar (close to 1). For quantitative analysis of the data, the CT (threshold cycle number) values were normalized to those of GUSB or 18S rRNA using the ΔΔCT method [17].
2.6 Western blotting
Mitochondrial membrane and matrix protein fractions from human tissues were separated as described previously and 100 μg of total protein from each tissue lysate were separated on a 12% SDS polyacrylamide gel, transferred to nitrocellulose, and immunostained and visualized with the indicated antibodies as described [12]. Rabbit anti human ACAD11 antibodies were raised by immunization with the synthetic peptide RKGQEVLIKVKHFMK, a peptide unique to human ACAD11 at the predicted surface of its ACAD homologous domain. The resultant antiserum was used at a 1:500 dilution.
2.7 Immunohistochemical and immunofluorescent staining of paraffin-embedded sections of human tissues and human cell lines
Formalin-fixed, paraffin-embedded sections of normal cerebellum and muscle were provided by the Department of Pathology, Children’s Hospital of Pittsburgh. 5 μm sections were cut at regular intervals and mounted on glass slides. Immunohistochemical staining was performed as described previously using ACAD9, ACAD11 and MCAD antibodies at dilutions of 1:200 to 1:300. Staining was performed using the avidin-biotinylated peroxidase complex (ABC) method (Vector Laboratories, Burlingame, CA) and counterstained with hematoxylin. . Human fibroblasts and neuroblastoma SK-N-SH were fixed with 4% formaldehyde on coverslips, permeabilized using either 1% Triton X-100 or digitonin (25 μg/ml), incubated with primary antibody, and visualized with fluorescently-labeled secondary antibodies following manufacture’s recommendation. Mitochondrial ATPase antibody was purchased from Chemicon (Billerica, MA). Secondary antibodies were from Jackson ImmunoResearch (West Grove, PA).
3.0 Results
3.1 Multiple transcripts are made from the ACAD10 and ACAD11 genes
Two genes identified as ACAD10 and ACAD11 have been annotated in the human genome (Fig. 1A and B). They are widely conserved across evolution and share 46% identity. Both genes are located within apparently complicated gene loci containing multiple predicted exons that code for multiple predicted protein domains. Preceding the conserved ACAD domains, there is a predicted aminoglycoside phosphotransferase (APH) domain in both ACAD10 and ACAD11, which also could be a homoserine kinase type II domain. The N-terminus of ACAD10 is longer than ACAD11, and there is a predicted haloacid dehydrogenase-like hydrolase (HDL) domain ahead of the APH/homoserine kinase type II domain. The carboxy end of ACAD11 harbors a peroxisome targeting signal 1 (PTS1). A similar domain structure is found in the non-mammalian orthologues, such as C. elegans [NP_504508], that also contain a PTS1.
Figure 1.
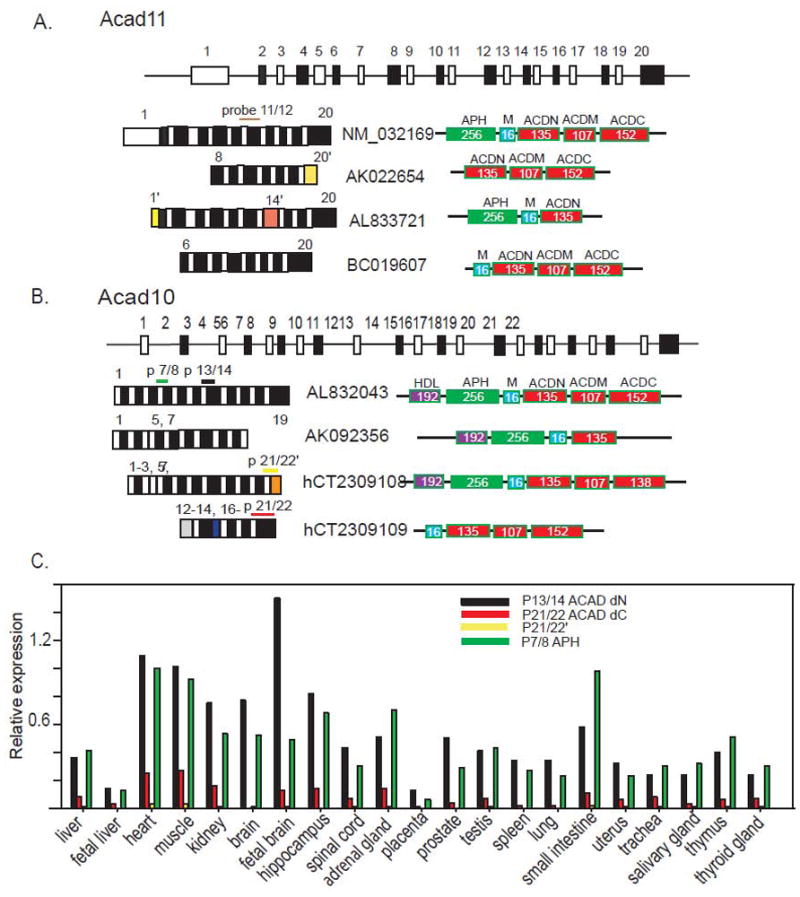
ACAD10 and 11 predicted gene structure, potential transcripts in the EST database, and the level of expression of ACAD10 mRNA in different human tissues. A and B. The human ACAD11 (A) and ACAD10 (B) genes consist of 20 and 22 predicted exons respectively (not drawn to scale), and each has at least four major mRNA species that likely are derived from alternative splicing, according to annotation and evidence from both the public and Celera, Inc. databases. Exons present in only one transcript are marked in yellow or orange. The schematic drawing illustrates different transcripts that can be translated to different proteins with various combinations of conserved protein domains, including an APH domain, mitochondrial targeting signal (M), ACAD N-terminal domain (ACDN), ACAD middle domain (ACDM), and ACAD C-terminal domain (ACDC). There is also a predicted hydrolase superfamily domain (HDL) identified at the N-terminus of some alternative protein forms of ACAD10. These domains are drawn as colored boxes with the number of associated amino acids specified. C. The relative expression of ACAD10 mRNA species vs. the housekeeping gene GUSB in 21 different human tissues. Each colored bar represents the expression of a fragment amplified by a different set of primers as indicated by the legend. The data represents the mean of three qPCR experiments with a robust %CV less than 1.35%.
Database searches of transcribed sequences from these genes identified the presence of multiple transcripts that differed mostly at either the 5′ or 3′ end. To examine the presence of these transcripts in vivo, we used real-time PCR with primer pairs targeted to unique exon junctions in the different transcripts to measure expression level of alternative transcripts of ACAD10 and 11 in 21 different human tissues The probes targeting published unique mRNA product containing alternative exons 20′ and 14′ identified in database entries for ACAD11 (AK022654 and AL833721) did not detect expression in all of the human tissues tested, suggesting that those splicing variants are either rare or do not exist in vivo. Therefore we can not assess further the alternative transcripts in ACAD11. However, multiple transcripts from the ACAD10 gene were detected and the relative abundance of each transcript differed in the various human tissues (Fig. 1C). Primer set 7/8, specific for mRNA AL832043 targets the region corresponding to the predicted APH domain. Primers 13/14 target a region shared by most transcripts in the genetic databases as shown in Fig 1B and translated into the junction between the APH domain and conserved ACAD N-terminal domain. Primers 21/22 amplify a region translated to a conserved C-terminal ACAD domain containing the predicted catalytic base shared by AL832043 and hCT2309109. In contrast, primers 21/22’, unique to AK092356, target an alternatively spliced species that eliminates exon 22, leading to loss of the putative ACAD catalytic domain. In all tissues, expression of the transcripts amplified by primers 7/8 was higher than those by primers 21/22. Thus, a large portion of the transcripts that contain sequences for an APH domain at the N-terminus do not have sequences for the ACAD domain at the C-terminus. Expression of the fragment defined by p21/22 was much higher than p21/22’, showing that the majority of transcripts encoding ACAD domains contain sequences potentially supporting ACAD enzymatic function. However, the expression of AK092356 detected by probe 21/22’ reflects the existence of ACAD10 isoforms without ACAD enzymatic function or mitochondrial targeting signal peptide. Interestingly, in some tissues (e.g., small intestine and thymus) the expression level of transcripts containing fragment, probe 7/8, was higher than those amplified by probe 13/14, suggesting that these tissues may express a previously unrecognized mRNA species that encodes a protein comprised of only an APH domain without ACAD sequences. Information currently available on both genes in the public databases is incomplete, hindering our ability to quantify each of the specific transcripts, particularly for ACAD11. Nevertheless, the expression profile of ACAD10 defined by our probe sets supports the existence of multiple transcripts and proteins as well as possible unique roles in different tissues and cellular compartments. Given that ACAD11 and ACAD10 are highly homologous, it is likely that multiple isoforms also exist for ACAD11.
3.2 Tissue distribution of ACAD10 and ACAD11 transcripts
To begin to understand the in vivo functions of ACAD10 and 11, we first examined the tissue expression pattern of the two genes in 21 different human tissues using real-time RT-PCR. We used probes p13/14 (ACAD10) and p11/12 (ACAD11) to target the sequences that are common to most of the identified transcripts, thus best presenting the total expression of each gene (Fig. 2A and B). ACAD11 had the highest level of expression in adult brain, 1.3 times the expression of the housekeeping gene GUSB (used as the internal control). Thereafter, we normalized expression levels in other tissues to the brain (Fig 2A). ACAD11 additionally had significant levels of expression in adult liver, heart, and kidney. ACAD10 had lower levels of expression than ACAD11 in liver and adult brain, with the highest level in fetal brain (1.5 times the expression of GUSB). Fig 2B shows the expression of ACAD10 in other tissues relative to fetal brain. ACAD10 was expressed more highly in fetal brain than adult, a pattern similar to that seen for LCAD [6] and also was highly expressed in heart and kidney.
Figure 2.
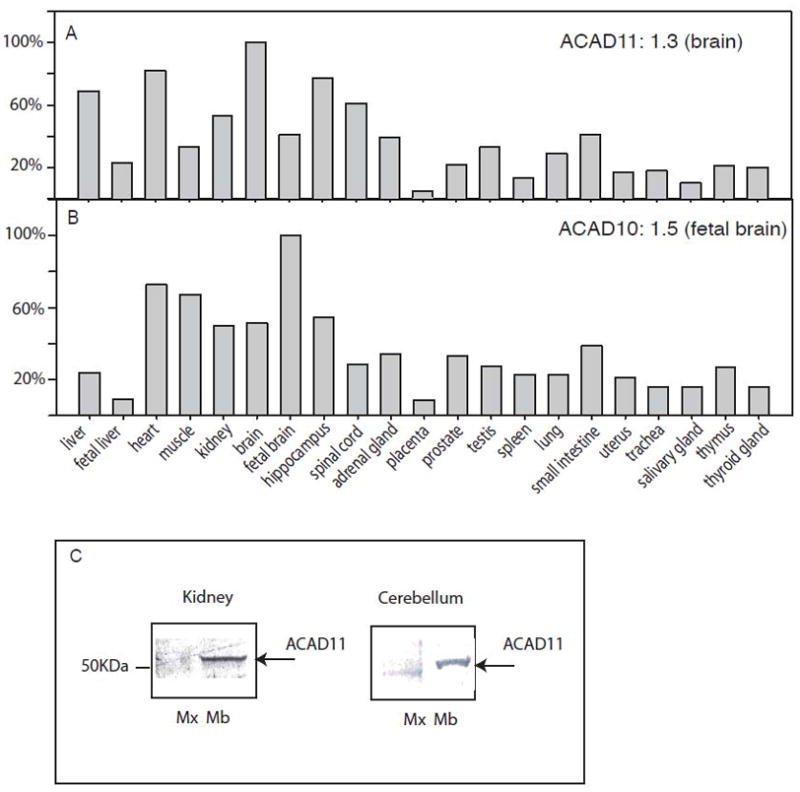
Distribution of ACAD10 and 11 mRNA and protein in human tissues. Levels of ACAD11 (A) and ACAD10 (B) mRNA were measured in 21 human tissues by real-time RT-PCR and normalized to the relative level of expression in the highest expression tissue. All RT-PCR data represent the mean of three experiments with a robust %CV less than 1.35%. Primers P11/12 were used for measuring ACAD11 expression level and primers P13/14 were used for measuring ACAD10 levels. Both probes target the common region among all the known transcripts as described in Fig. 1. The highest expression tissue of ACAD11 was found in brain (ratio to the housekeeping gene GUSB = 1.3), and the highest expression of ACAD10 was in fetal brain (relative expression to GUSB = 1.5); the expression of ACAD10 and 11 in other tissues is shown as % of the fetal brain and brain expression respectively. C. Immunoblotting of mitochondrial subfractions from different tissue lysates with an antibody to an ACAD11 peptide gave the highest signal in human kidney and brain. It appeared mainly in mitochondrial membrane (Mb), rather than matrix fraction (Mx).
3.3 Mitochondrial processing of ACAD11 protein
Among the transcripts of ACAD10 and 11 identified in the public databases, ACAD11 entry BC019607 is similar to entry hCT2309109 originally identified in the proprietary Celera, Inc. cDNA database (Alameda, CA; now also represented in the NCBI public gene database). hCT2309109 is a whole length cDNA and is predicted to translate into an ACAD like protein containing only the conserved ACAD domains. We therefore amplified cDNA sequences corresponding to hCT2309109 sequence from human liver total RNA and cloned them into an in vitro transcription expression vector. In vitro transcription of this plasmid (referred to hereafter as ACAD11 variant 1) followed by translation with rabbit liver lysate produced two stable proteins (Fig. 3A, lane 2). The first one (57 kDa) was of appropriate molecular mass to correspond to a polypeptide starting with the first ATG in the expression insert, while the other protein of ~52 kDa could arise from internal initiation from the third ATG (A142 from the first ATG) of the expression insert. Computer predictions (ATGpr_sim at https://www.jbirc.aist.go.jp/FLJ/ATGpr/atgpr/index.html) indicated that the third ATG could be an equally strong translation start site as the first ATG.
Figure 3.

In vitro transcription and translation coupled with mitochondrial import of ACAD11. A. SDS PAGE separation of in vitro transcription and translation reaction product (TNT) was carried out with a large size PAGE gel made as described [12]. The reaction template was a cDNA fragment (ACAD11 variant 1) corresponding to BC019607/hCT2309109 (Fig 1A) and encoding ACAD11 with a mitochondrial leader sequence before (lane 2) and after (lane 3) in vitro import into mitochondria. (the arrow in lane 3 points to a mitochondrially imported ACAD11), lane 4 is the supernatant from the mitochondrial importing mixture, representing the non-mitochondrial TNT products from the ACAD11 cDNA fragement. B. A criterion XT small bis-tris SDS 4–12% gel was used for electrophoresis: Lane 3–5, The in vitro TNT reaction product of shorter version of the ACDA11 (ACAD11 variant 2 ) (lane 2) and its mitochondrial import reaction mixture were digested with trypsin for 0 min. (lane 3), 10 min. (lane 4), and 20 min. (lane 5). The TNT reaction product of ACAD9 (lane 6) and IVD (lane 8) and their mitochondrial import products after 20 min. of trypsin digestion (lane 7 and 9) were used as controls. Lane 1 shows pre-stained molecular markers of molecular mass 90 and 50 kDa. C. Native polyacrylamide gel electrophoresis of the TNT products of IVD, ACAD9, and ACAD11 before (lane 2, 4, 6, respectively) and after mitochondrial import (lane 3, 5, and 7). Purified IVD protein was used as a control (lane 1) and is shown following immunostaining with IVD antibodies. D. Alignment of the predicted mitochondrial targeting peptide sequence of ACAD10 and 11 from various species. The highly conserved mitochondrial peptide peptidase recognition sequence is highlighted in bold.
Following in vitro transcription/translation of ACAD11 variant 1, the majority of the 52 kDa protein was imported into isolated mitochondria in an in vitro import assay and was processed to a smaller mature protein (Fig. 3B lane 3– 5), while the majority of the 57 kDa protein remained in the supernatant that was free of mitochondria (Fig 3A lane 4). We therefore subcloned the cDNA sequence predicted to correspond to the 52 kDa ACAD11 (designated as ACAD11 variant 2) and performed in vitro transcription and translation followed by mitochondrial import of the translation products. As expected, only the 52 kDa protein was efficiently imported into mitochondria and cleaved to a mature form as shown by its resistance to trypsin digestion after the mitochondrial import reaction (Fig 3B, Lane 3–5). There are a few other smaller trypsin sensitive protein bands identified in the reaction, which likely represent alternative transcript or translation starting sites. A mitochondrial peptide peptidase (MPP) cleavage site in ACAD11 is highly conserved across species (Fig. 3D). Non-denaturing electrophoresis confirmed that, in contrast to the precursor protein (Fig. 3D lane 6), the intra-mitochondrial form of ACAD11 (Figu.3D lane 7) was assembled into a stable multimeric protein as are other ACADs (Fig. 3C, lane 3 and lane 5). This further supports that mature ACAD11 assembled in mitochondrial are likely functional mitochondrial enzyme.
3.4 Prokaryotic expression of ACAD10 and ACAD11
The precursor form of nuclear encoded enzymes that localized to mitochondria, including all of the ACADs, are not usually active when expressed in E. coli cells, but their mature forms can often be produced in this fashion. To facilitate study of the mature form of ACAD11, we cloned the mature coding sequence into a bacterial expression vector with substitution of E. coli codon bias for the first 40 amino acids. Following induction, crude cellular extract was tested for ACAD activity with the ETF (electron transfer flavoprotein) fluorescence assay using a broad range of acyl-CoA substrates. The highest activity was seen with saturated C22-CoA (docosanoyl-CoA) (Fig. 4E) and relative activity of 30%, 63%, 15% and 15% compared to C22-CoA with C20-CoA (eicosanoyl-CoA), C23-CoA (tricosanoyl-CoA), C24-CoA(tetracosanoyl-CoA), and C26-CoA (hexacosanoyl-CoA), respectively at 50 μM substrate concentration. The activity of ACAD11 in crude cell extracts was about 10 mU/mg of cellular protein, similar to the level of expression seen with other ACADs in our system. We also cloned and over expressed the predicted mature mitochondrial coding sequence of ACAD10 in the same fashion. Extracts from cells expressing a predicted mature mitochondrial ACAD10 insert were only active with R and S, 2 methyl-C15-CoA (R and S, 2-methyl-pentadecanoyl-CoA) among a broad series of other substrates including the optimum ones for all of the other ACADs. The measured activity (1.4 mU/mg at 150 μM substrate concentration) is considerably lower than that obtained with other expressed ACADs towards their optimum substrates, suggesting that the dehydrogenation reaction between ACAD 10 and R or S, 2 methyl-C15-CoA is unlikely the optimal function for ACAD 10 in vivo. Unfortunately, we could not purify either of these two proteins further to allow more formal characterization of the enzymes’ kinetic properties.
Figure 4.

ACAD activities in human tissues and of recombinant enzymes measured with a variety of acyl-CoA substrates. A–D. The highly sensitive and specific ETF reduction assay was used to measure ACAD activity in mitochondrial membrane (gray bars; mem) or matrix (black bars; sup) fractions of different human tissues with saturated straight chain acyl-CoA substrates of primary carbon chain length as indicated on the X axis. Tissues tested were cerebellum cortex (A), muscle (B), lung (C), liver (D). E–H with results reported as μM of ETF reduced/min/mg of protein shows the substrate specificity of ACAD9 (the white bar in E), ACAD11 (the gray bar in E), VLCAD (the gray bar in F), LCAD (the black bar in G), SCAD (the white bar in H) and MCAD (the black bar in H). Substrate specificity of each ACAD is present as % of the activity towards their optimal substrate. The pattern of activity with long chain acyl-CoA esters in human cerebellum mitochondrial membrane correlates well with the substrate specificity of ACAD9 and ACAD11; the long chain activity pattern in the mitochondrial membrane of human muscle reflects the substrate specificity of VLCAD; the long chain activity pattern in the mitochondrial matrix of lung reflects LCAD substrate specificity, with no detectable ACAD activity in the membrane fraction of the lung. The major ACAD activity in human liver was in matrix, and its pattern reflected the combination of SCAD and MCAD. All the ACADs used were purified recombinant protein except ACAD11. Substrate specificity for recombinant ACAD11 was measured in crude lysate from E. coli following over expression of ACAD11 as described in the Methods. Purified LCAD was a generous gift from Professor J.J.Kim, Medical College of Wisconsin, Milwaukee, Wisconsin.
3.5 Computational modeling of ACAD10 and ACAD11
ACAD10 and ACAD11 are 20–30% identical to other ACADs (Fig. 5A) and computer modeling predicts that they are structurally similar to previously characterized ACADs (Fig. 5A and B). Interestingly, the catalytic residue, a glutamate in all other ACADs, is not conserved in ACAD 10 and 11. Rather, computer modeling predicts that both ACAD 10 and 11 utilize an aspartate rather than glutamate for this function (Fig. 5C). Mutagenesis studies with other ACAD have confirmed that aspartate can indeed be substituted for the wild type glutamate but that the resultant mutant enzymes have altered substrate specificities [19]. It is likely that the shorter side chain of the aspartate in the catalytic pocket will allow ACAD10 and 11 to utilize substrates with more bulky and/or longer carbon backbones. Of note, the computer modeling revealed two hydrophilic amino acids (Arg 512 and His 509, NP115545 ) in the substrate binding pocket of ACAD11 (Fig. 5C), which are highly conserved in both ACAD 11 and 10 proteins from multiple species.
Figure 5.
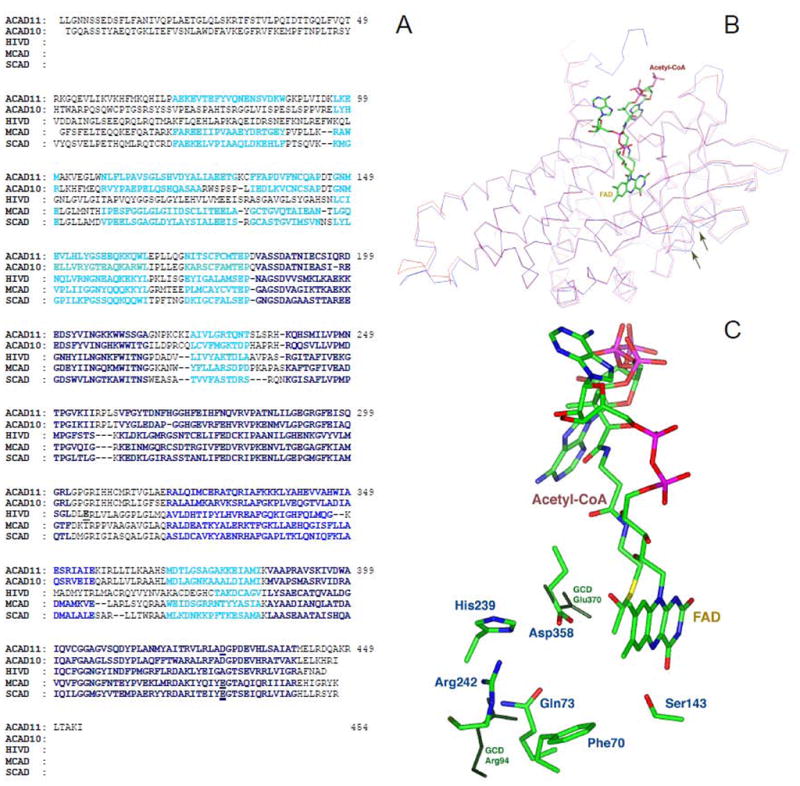
Computer modeling of mature mitochondrial form of ACAD 11 A. Alignment of the predicted mature mitochondrial coding sequences of human ACAD10 (hCT2309109) and 11 (BC019607/hCT2309109) with human IVD (HIVD), MCAD, and SCAD. The sequences predicted by Insight II software to be structurally similar between ACAD 10 and 11, IVD, MCAD and SCAD are highlighted according to known structure of the latter three proteins. The most similar domains are highlighted with black squares, less similar domains are highlighted with blue, and least similar domains are highlighted with cyan. The catalytic bases in known ACADs and the predicted catalytic bases in ACAD 10 and 11 are highlighted with a the red square. B. The ACAD11 predicted model trace (blue) overlays the rat SCAD trace (red) in this representation. An acetyl-CoA and FAD are modeled as the substrate and cofactor, respectively, shown in colors. The areas that are important for FAD binding site and CoA substrate binding pocket show a high degree of structural homology between ACAD11 and rat SCAD molecular models. Areas of weak homology in these models are the middle of β-strand 6 (right arrow) and the loop connecting β-strands 3 and 4 (left arrow), indicating that the bottom the substrate binding pocket for ACAD11 is different from SCAD. C. Comparison of the amino acid residues in the active site of ACAD11 predicted model and GCD crystal structural model. Amino acids in ACAD11 are shown as colored sticks. Dark green highlights the relative positions of Glu370 and Arg94 of GCD in their structural alignment. Arg94 is a unique active residue for GCD and Glu370 is the catalytic residue common to all the ACADs. Asp628 and Arg512 (highlighted red in panel A) in ACAD11 align with Glu370 and Arg94 in GCD, respectively. The key residues for FAD and CoA substrate binding, e.g. Ser413 (shown) and other key residues predicted in substrate binding pocket of ACAD11 (Gln343, His509, Phe340) are also conserved structurally among ACAD11, GCD and other ACADs.
This is unusual since amino acid residues located in the substrate binding pocket of the ACADs are mostly hydrophobic in order to accommodate the hydrophobic fatty acid moiety of the substrates. The exception to this is a highly conserved hydrophilic amino acid, Arg 23, in glutaryl-CoA dehydrogenase (GCD), present in GCD’s substrate binding pocket. This residue has been shown to facilitate catalysis of the CoA ester of the dicarboxylic acid, glutaryl-CoA. Of note in our computer created model, Arg512 in ACAD11 aligns with Arg23 of GCD (Fig. 5C) suggesting a common function. Whether ACAD11 can also act as a decarboxylase as GCD will require further investigation. The predicted structure of the bottom of ACAD11 substrate binding pocket aligns poorly with both GCD and SCAD, indicating it likely uses longer or bulker substrates (Fig. 5B).
3.6 Subcellular location and mitochondrial fractionation of ACAD11 protein isoforms
The presence of multiple ACAD10 and 11 transcripts in various tissues raises the possibility that some might generate protein species that localize to different subcellular compartments. To examine this, a rabbit polyclonal antisera was raised against a synthetic ACAD11 peptide corresponding to a portion of the sequence of the N-terminal domain of the mature mitochondrial ACAD11. The peptide sequence is common to potential ACAD11 protein isoforms translated from all identified ACAD11 transcripts, but not homologous to other ACADs including ACAD10. Western blot analysis with this antiserum identified an ACAD11 protein species in mitochondrial fractions of both human cerebellum and kidney corresponding in size to the mature mitochondrial form seen in our in vitro import studies. This form primarily localized to the membrane fraction of mitochondria isolated from both human cerebellum and kidney (Fig. 2C), but not in the mitochondrial matrix fraction. Multiple ACAD11 positive bands were seen in other tissues, such as liver (date not shown). The specificity of our antibody toward the ACAD11 peptide was validated by peptide competition analysis, however, purified ACAD11 isoforms were not available to further characterize antibody interactions. Immunofluorescence staining of ACAD11 in human neuroblastoma and skin fibroblasts revealed a marked difference in subcellular location of ACAD11 proteins (Fig. 6A–J). ACAD11 protein in neuroblastoma cells co-localized with mitochondrial ATPase (Fig. 6A–C). In contrast, the majority of the ACAD11 signal in fibroblasts did not co-localize with mitochondrial ATPase (Fig. 6D–F). Interestingly, in both human skin fibroblasts and human neuroblastoma cells, additional ACAD11 specific staining was observed in punctate cytoplasmic vesicles. These were most evident with digitonin treatment (which preferentially permeabilizes the plasma membrane but not organellar membranes). Finally, ACAD11 antibodies strongly stained nuclei regardless of cell permeabilization conditions (Fig. 6H–J), a pattern also seen for ACAD9. The striking difference in subcellular location of ACAD11 among various cell and tissue types emphasizes that expression of the mitochondrial form of ACAD11 as the product of alternative splicing is tissue specific, in agreement with our RNA expression studies.
Figure 6.

Immunofluorescent staining of ACAD11 in human neuroblastoma and skin fibroblast. Human neuroblastoma (SK-N-SH) cells (A) and skin fibroblasts (D) were permeabilized with Triton X-100 and immunostained (green fluorescence) with antibodies to an ACAD11 peptide. (B and E) Fluorescent staining of the same human cells against mitochondrial ATPase antibody is shown in red. C is the merged image of A and B, and F is the merged image of D and E. The nucleus is indicated by the white arrow. Human skin fibroblasts were also treated with digitonin which selectively permeabilizes the plasma membrane but not internal membranes, and then stained with ACAD11 peptide antibody (H), mitochondrial ATPase antibody (I). J is the merged image of H and I. DAPI was used for nuclear staining in panels H–J. In SK-N-SH cells, there was significant co-localization of ACAD11 with mitochondria. In fibroblasts, ACAD11 was primarily found associated with membranes of intracellular vesicles or organelles, and was oriented facing the cytoplasm.
3.7 Mitochondrial fatty acid acyl-CoA oxidation in human cerebellum
In light of the unusual tissue distribution of ACAD11 and the growing menu of ACADs active with long chain substrates, we next examined the full spectrum of ACAD activity in fractionated extracts from various tissues using the highly specific ETF fluorescent reduction assay (Fig. 4). Most of the activity measured with long chain substrates in cerebellum (Fig. 4A) and muscle (Fig. 4B) was detected in the mitochondrial membrane fraction, however, the chain length optima were different. The muscle mitochondrial membrane fraction was most active with substrates with carbon backbones of 12–18. In contrast, cerebellum had a sharp peak of activity with C16- and C18-CoA and significant C22CoA activity. Of note, mitochondrial membranes from human cerebellum showed no detectable activity with C10- or C12-CoA as substrate. The spectrum of membrane bound activity in human muscle corresponds to that of purified human VLCAD [6] (Fig. 4F), while the spectrum of activity in human cerebellum is consistent with a combination of ACAD9 and ACAD11 (Fig. 4E). In contrast to VLCAD and ACAD9, LCAD is a mitochondrial matrix protein and is the only long chain ACAD with significant expression in human lung [6]. ACAD activity in human lung (Fig 6C) was detected only in the mitochondrial supernatant fraction with a substrate pattern matching that of LCAD (Fig 6G). ACAD activity with short and medium chain substrates predominated in human liver mitochondrial matrix (Fig 6D), matching the activity profiles of MCAD and SCAD (Fig 6H). The C22/C20 activity ratio in a liver mitochondrial membrane fraction was much greater than that seen for purified VLCAD or ACAD9 enzymes (1.0 vs. 0.6, respectively), suggesting a likely contribution from ACAD11 (C22/C20 >3) in human liver as well. Our ACAD11 peptide derived antibody did not inhibit native ACAD11 activity, precluding additional immunoinactivation experiments.
3.8 ACAD distribution in human cerebellum is compartmentalized
To better characterize the distribution of ACADs in human cerebellum, immunohistochemical staining was performed using antisera against MCAD, ACAD9, and ACAD11 (Fig 7. A–L). We have previously shown that VLCAD immunostaining in human cerebellum reacts mostly surrounding the vascular regions (He et al, 2007). ACAD11 antisera uniquely stained the white matter of cerebellum (Fig. 7A & C) but only minimally stained Purkinje neurons or granular layer (Fig. 7B), and this was especially strong in oligodendric cells as determined by cell morphology (Fig. 7C). ACAD9 uniquely strongly stained the granular layer (Fig. 7E & F), particularly the dendrites of granular neurons (Fig. 7F), but not the white matter (Fig. 7G). Both ACAD11 and ACAD9 antisera showed a high level of staining in the dentate nucleus in the deep white matter of cerebellum (Fig. 7D and H), while MCAD specific staining was much weaker (Fig. 7L). MCAD uniquely stained the molecular layer (Fig. 7I), certain axons in the white matter (Fig. 7K), and some astrocytes scattered in different layers of cerebellum (Fig. 7J–L). Both ACAD9 and MCAD also strongly stained Purkinje neurons (Fig. 7F and J). Overall the specific and compartmentalized pattern of distribution of the three major ACADs in human cerebellum suggests that each cell type is utilizing mitochondrial β-oxidation for distinct functions.
Figure 7.

Immunofluorescent and immunohistochemical staining of human cerebellum with anti-ACAD11 peptide, anti-ACAD9, and anti-MCAD antibodies. Immunofluorescent staining of human cerebellar cortex with ACAD11 (A), ACAD9 (E) and MCAD (I) antibodies (red). Immunohistochemical staining of cerebellar cortex (B, F, J), cerebellar white matter (C, G, K), and dentate nucleus (D, H, L) with anti-ACAD11 peptide antisera (B–D), anti-ACAD9 antisera (F–H), and anti-MCAD antisera (J–L) (brown). Sections were counterstained with a blue nuclear dye.
In contrast to cerebellum, MCAD and VLCAD both stained muscle fibers in a similar punctate pattern indicative of mitochondrial localization (Fig. 8A and B). ACAD9 and ACAD 11 stained muscle fibers less strongly, however, both were present in the perivascular and vascular regions (Fig 8C and D) indicating a different function in muscle for these enzymes than for VLCAD.
Figure 8.

Immunohistochemical staining of human muscle with anti-MCAD, VLCAD, ACAD9 and ACAD11 antisera. The ACAD specific staining is brown and the sections were counterstained with a blue nuclear dye. Panels are as follows: MCAD (A), VLCAD (B), ACAD9 (C), and ACAD11 (D).
4.0 Discussion
A majority of the genes in humans are now recognized to generate multiple functional transcripts through alternative splicing and it is especially common for genes with primary expression in the central nervous system [20]. In this study, we characterize expression of two new genes with homology to the mitochondrial ACAD family (ACAD10 and 11). Both genes exhibit a complicated pattern of alternative splicing from complicated loci and generate at least one protein species that can be imported into mitochondria. As demonstrated by real-time PCR, the majority of the transcripts generated from the ACAD10 and 11 genes translate to proteins with a predicted APH domain, at the N-terminus and an N-terminal ACAD domain and a C-terminal ACAD catalytic domain at the C-terminus (Fig. 1). These predicted proteins do not have mitochondrial targeting sequences at their N-termini and therefore are likely to be localized to alternative cellular locations. Consistent with this observation, a global proteomic survey of subcellular organelles of six major organs in mouse localized ~70% of predicted ACAD10 protein to the membrane (microsomal) fraction and 30% to mitochondria [21]. A typical glycosylation site (NTO) is present between the APH and ACAD N-terminal domains in ACAD11 protein, suggesting that this large ACAD11 isoform may target to microsomes or the cytoplasmic membrane. But this predicted protein also contains a canonical peroxisomal targeting signal at its C-terminus that could override other localization signals. In support of this, one published proteomic study has identified ACAD11 variants with an APH domain in peroxisomes [22]. In the current study, although a small portion of ACAD11 in skin fibroblasts may be localized to peroxisomes (data not shown), the majority of the staining appeared in a punctate pattern, adjacent to the cytoplasmic membrane of the cell (Fig. 6). This finding suggests that some ACAD11 isomers may be localized to membrane associated vesicles, a location it shares in part with ACAD10. In contrast, we found that the majority of ACAD11 localized to mitochondria in human neuroblastoma cells (Fig. 6), highlighting the importance of considering tissue of origin when examining the physiological role of the products of these genes. Recent global analysis of alternately spliced genes has indicated that groups of tissue-specific alternative splicing events often function in a coordinated manner in a specific pathway or interaction network [20]. Of note, we have observed a similar tissue pattern of alternative splicing for the ACAD9, 10 and 11 genes in human cerebellum, fibroblasts, and liver [6]. Moreover, mitochondrial forms of ACAD9, and 11 all were found to be present in certain neuronal cell populations in human CNS (Figs. 7 and 8), while VLCAD was not. Therefore, we speculate that these three long chain ACADs and their isomers of unknown physiologic function act in a coordinated pathway in the CNS not related to energy metabolism, but related to their specificity towards certain groups of functional fatty acid CoA esters. Recently, Zolman et al. indirectly showed that the Arabadopsis homologue of ACAD10 is involved in the β-oxidation of indole-3-butyric acid to indole-3-acetic acid, a metabolite of tryptophan metabolism [23]. Both compounds function as plant hormones known as auxins. The metabolism of auxins is localized to peroxisomes in plants but is independent of the more general fatty acid β-oxidation pathway [23]. In humans, weak auxins such as phenylbutyrate and phenylacetate are currently in the spotlight for their roles as drugs, especially in treatment of neoplastic disorders and may provide a novel target for ACAD10 function [24–26]. It seems likely that in mammals, the function performed by Arabadopsis ACAD10 has been transferred to mitochondria and may be involved in metabolism of tryptophan derivatives such as serotonin, an important neurotransmitter. Little is known of the role of β-oxidation in aromatic amino acid catabolism, although it is well known that MCAD is active with substrates with aromatic rings such as phenylpropionyl-CoA, a tyrosine metabolite from gut bacteria.
The identification of multiple isoforms of ACAD10 and 11 led us to further investigate the enzymatic activity of the mitochondrial forms of these enzymes (Fig. 4). Using in vitro mitochondrial import experiments and homology searches, we constructed prokaryotic expression inserts for the predicted mature mitochondrial forms of both enzymes. Expressed mitochondrial ACAD11 utilized very long chain acyl-CoA substrates with optimal activity with C22CoA as substrate while ACAD10 had low activity with 2S-methyl C15-CoA and 2-methyl C16-CoA (both the R and S forms). Minimal, but detectable, activity was seen with ACAD10 using very long and branched chain substrates such as 2-methyl C19-CoA or long chain substrates with branch at other positions such as 3-methyl iso C15-CoA. Thus, we postulate ACAD10 will have maximal activity with substrates of shorter chain length than ACAD11 with specific branched structures. Unfortunately, such substrates were not available for further study of this enzyme. Very long chain fatty acids are not found in significant quantities in the human diet, however, they are abundant in neuronal tissues and are important components of myelin [27]. This is consistent with the tissue expression pattern of the mitochondrial form of ACAD11, which is highly expressed in adult brain, especially in the oligodendrocytes. Expression of ACAD10 in fetal brain may reflect a role in metabolism of the fatty acids unique to brain development [27].
Most interestingly, our results indicate that different ACADs predominate in mitochondrial β-oxidation of long chain fatty acids activity in human brain than are used in most other tissues. Immunostaining of human adult cerebellum identified the presence of MCAD, ACAD9 and ACAD11 as major ACADs in this tissue (Fig 7.A–L). Thus, these three ACADs are likely responsible for the three major activity peaks seen at C18, C22 and C8 in cerebellar extracts (Fig. 4A). Interestingly, each of these enzymes has a distinct cellular distribution pattern in cerebellum rather than all residing in the same cells types as is typical of VLCAD and MCAD in other tissues such as human muscle (Fig. 7A–L and Fig 8. A–D). We hypothesize that the unique cerebellar cellular distribution pattern reflects physiological function. For example, ACAD9 predominates in Purkinje neurons and granular neurons of the grey matter (Fig. 7E and F), while ACAD11 is mainly present in the oligodendrocyte-like cells of the white matter (Fig. 7A and C). Recent studies have revealed that the fatty acid composition of glycosphingolipid sulfatide, a component of myelin and some non-myelin forming cells in the brain, differs between white and grey matter. Neurons contain mostly long chain fatty acids with an emphasis on stearic acid (C18), corresponding to the optimal substrate of ACAD9. In contrast, sulfatides in myelin, normally synthesized by oligodendrocytes in white matter, have a majority of very long chain fatty acids (C22-26). We hypothesize that C22-CoA is metabolized in mitochondria by ACAD11, filling a recognized gap in long chain fatty acid catabolism by peroxisomal acyl-CoA oxidases, which have substrate optima for C24 and 26 substrates [28] Moreover, we suggest that multiple ACAD9 and ACAD11 protein isoforms are involved in controlling the fatty acid composition of cellular lipids through lipid recycling and synthesis and thus play a major role in determining the functions of these lipids. It is interesting that even in human muscle, both ACAD9 and 11 were found mostly in the perivascular regions, differentiating them from VLCAD and MCAD (Fig. 8A–D). Therefore we suspect that their roles even in tissues outside the CNS are other than energy production. Of note, the majority of long chain fatty acids in humans are actually recycled by β-oxidation rather than being consumed as fuel [29].
In the CNS, the cellular location of MCAD is strikingly different from ACAD9 and 11, being uniquely present in the axons of certain neurons (Fig. 7K) and the molecular layer of the cerebellum (which contain mostly dendrites of purkinje neurons) (Fig. 7I). Numerous proteins and processes critical for CNS plasticity require ATP including cytoskeletal components, ion-motive ATPase, and a myriad of protein phosphorylation reactions. In turn, it is mitochondria that produce the bulk of ATP in neurons, and concentration gradients of ATP are greatest immediately adjacent to the mitochondria [30]. Therefore, it is likely that the high expression of MCAD in neuronal axons and dendrites reflects a primary role for β-oxidation in ATP production in these cells. This of course is the major function of β-oxidation in tissues such as muscle and heart, but it is not well established for brain. Since medium chain fatty acids can readily pass the blood-brain barrier, they serve as an important potential fuel for ATP production in maximizing neuroplasticity, which could be important in neurodegenerative disorders.
ACAD9 has recently been postulated to have an alternative structural role as a respiratory chain complex 1 stability or assembly factor [7]. We have also demonstrated the direct, physical interaction of multifunctional fatty acid oxidation complex and the respiratory chain supercomplexes [31]. Thus the possible existence of non-enzymatic functions for both the mitochondrial and non-mitochondrial forms of ACAD10 and 11 must be considered in future studies.
5.0 Conclusions
In summary, we have identified two new mitochondrial ACADs, one of several protein isoforms produced by the ACAD10 and 11 genes. Our studies indicate that in combination with the recently characterized ACAD9, these enzymes are likely involved in serving novel physiologic functions in the central nervous system such as controlling fatty acid composition of cellular lipids and metabolizing aromatic amino acids intermediates. These findings provide an impetus for further studies on the role of β-oxidation in the metabolism of these novel groups of substrates with neuronal and immune functions.
Acknowledgments
We thank Professor J. J. Kim for providing purified human LCAD. We thank Dr. Ronald Jaffe for providing the control human tissues. This work was supported in part by National Institutes of Health (NIH) Public Health Service grant R01 DK54936 and DK78755 (to J.V.), and by a grant Geconcerteerde Onderzoeksacties (GOA2004/08) from the Flemish Government (to P.P.V.V)
Abbreviations
- ACAD9
acyl-CoA dehydrogenase 9
- APH
aminoglycoside phosphotransferase
- C22-CoA
docosanoyl-CoA
- C20-CoA
eicosanoyl-CoA
- C23-CoA
tricosanoyl-CoA
- C24-CoA
tetracosanoyl-CoA
- C26-CoA
hexacosanoyl-CoA
- C6-CoA
hexanoyl-CoA
- C8CoA
octanoyl-CoA
- C10CoA
decanoyl-CoA
- C12CoA
dodecanoyl-CoA
- C14CoA
tetradecanoyl-CoA
- C16CoA
palmitoyl-CoA
- R and S
2methylC15-CoA, R or S, 2-methyl-pentadecanoyl-CoA
- VLCAD
very-long-chain acyl-CoA dehydrogenase
- ACAD
acyl-CoA dehydrogenase
- LCAD
long-chain acyl-CoA dehydrogenase
- MCAD
medium-chain acyl-CoA dehydrogenase
- SCAD
short-chain acyl-CoA dehydrogenase
- ETF
electron transfer flavoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vockley J, Whiteman DA. Defects of mitochondrial beta-oxidation: a growing group of disorders. Neuromuscul Disord. 2002;12:235–246. doi: 10.1016/s0960-8966(01)00308-x. [DOI] [PubMed] [Google Scholar]
- 2.Bertini E, Dionisivici C, Garavaglia B, Burlina AB, Sabatelli M, Rimoldi M, Bartuli A, Sabetta G, Didonato S. Peripheral sensory-motor polyneuropathy, pigmentary retinopathy, and fatal cardiomyopathy in long-chain 3-hydroxy-acyl-CoA dehydrogenase deficiency. Eur J Pediatr. 1992;151:121–126. doi: 10.1007/BF01958956. [DOI] [PubMed] [Google Scholar]
- 3.Tein I, Donner EJ, Hale DE, Murphy EG. Clinical and neurophysiologic response of myopathy and neuropathy in long-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency to oral prednisone. Pediatr Neurol. 1995;12:68–76. doi: 10.1016/0887-8994(94)00109-f. [DOI] [PubMed] [Google Scholar]
- 4.Lundy CT, Shield JP, Kvittingen EA, Vinorum OJ, Trimble ER, Morris AA. Acute respiratory distress syndrome in long-chain 3-hydroxyacyl-CoA dehydrogenase and mitochondrial trifunctional protein deficiencies. J Inherit Metab Dis. 2003;26:537–541. doi: 10.1023/a:1025995813914. [DOI] [PubMed] [Google Scholar]
- 5.Das AM, Illsinger S, Lucke T, Hartmann H, Ruiter JP, Steuerwald U, Waterham HR, Duran M, Wanders RJ. Isolated mitochondrial long-chain ketoacyl-CoA thiolase deficiency resulting from mutations in the HADHB gene. Clin Chem. 2006;52:530–534. doi: 10.1373/clinchem.2005.062000. [DOI] [PubMed] [Google Scholar]
- 6.He M, Rutledge SL, Kelly DR, Palmer CA, Murdoch G, Majumder N, Nicholls RD, Pei Z, Watkins PA, Vockley J. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am J Hum Genet. 2007;81:87–103. doi: 10.1086/519219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nouws J, Nijtmans L, Houten SM, van den Brand M, Huynen M, Venselaar H, Hoefs S, Gloerich J, Kronick J, Hutchin T, Willems P, Rodenburg R, Wanders R, van den Heuvel L, Smeitink J, Vogel RO. Acyl-CoA Dehydrogenase 9 Is Required for the Biogenesis of Oxidative Phosphorylation Complex I. Cell Metab. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Vockley J, Nagao M, Parimoo B, Tanaka K. The variant human isovaleryl-CoA dehydrogenase gene responsible for type-II isovaleric acidemia determines an RNA splicing error, leading to the deletion of the entire 2nd coding exon and the production of a truncated precursor protein that interacts poorly with mitochondrial import receptors. J Biol Chem. 1992;267:2494–2501. [PubMed] [Google Scholar]
- 9.Pedersen CB, Bross P, Winter VS, Corydon TJ, Bolund L, Bartlett K, Vockley J, Gregersen N. Misfolding, degradation, and aggregation of variant proteins. The molecular pathogenesis of short chain acyl-CoA dehydrogenase (SCAD) deficiency. Journal of Biological Chemistry. 2003;278:47449–47458. doi: 10.1074/jbc.M309514200. [DOI] [PubMed] [Google Scholar]
- 10.He M, Burghardt TP, Vockley J. A novel approach to the characterization of substrate specificity in short/branched chain Acyl-CoA dehydrogenase. J Biol Chem. 2003;278:37974–37986. doi: 10.1074/jbc.M306882200. [DOI] [PubMed] [Google Scholar]
- 11.Aoyama T, Souri M, Ushikubo S, Kamijo T, Yamaguchi S, Kelley RI, Rhead WJ, Uetake K, Tanaka K, Hashimoto T. Purification of human very-long-chain acyl-coenzyme A dehydrogenase and characterization of its deficiency in seven patients. J Clin Invest. 1995;95:2465–2473. doi: 10.1172/JCI117947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ensenauer R, He M, Willard JM, Goetzman ES, Corydon TJ, Vandahl BB, Mohsen AW, Isaya G, Vockley J. Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids. J Biol Chem. 2005;280:32309–32316. doi: 10.1074/jbc.M504460200. [DOI] [PubMed] [Google Scholar]
- 13.Vockley J, Parimoo B, Tanaka K. Identification of the molecular defects responsible for the various genotypes of isovaleric acidemia. Prog Clin Biol Res. 1992;375:533–540. [PubMed] [Google Scholar]
- 14.Battaile KP, McBurney M, VanVeldhoven PP, Vockley J. Human long chain, very long chain and medium chain acyl-Coa dehydrogenases are specific for the S-enantiomer of 2-methylpentadecanoyl-CoA. Biochimica et Biophysica Acta - Lipids & Lipid Metabolism. 1998;1390:333–338. doi: 10.1016/s0005-2760(97)00185-9. [DOI] [PubMed] [Google Scholar]
- 15.Van Veldhoven PP, Van hove G, Vanhoutte F, Dacremont G, Parmentier G, Eyssen HJ, Mannaerts GP. Identification and Purification of a Peroxisomal Branched Chain Fatty Acyl-CoA Oxidase. J Biol Chem. 1991;266:24676–24683. [PubMed] [Google Scholar]
- 16.Van Veldhoven PP, Croes K, Asselberghs S, Herdewijn P, Mannaerts GP. Peroxisomal Beta-Oxidation Of 2-Methyl-Branched Acyl-Coa Esters - Stereospecific Recognition Of the 2s-Methyl Compounds By Trihydroxycoprostanoyl-Coa Oxidase and Pristanoyl-Coa Oxidase. FEBS Lett. 1996;388:80–84. doi: 10.1016/0014-5793(96)00508-x. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Ye X, Ji C, Zhou C, Zeng L, Gu S, Ying K, Xie Y, Mao Y. Cloning and characterization of a human cDNA ACAD10 mapped to chromosome 12q24.1. Mol Biol Rep. 2004;31:191–195. doi: 10.1023/b:mole.0000043622.57408.6b. [DOI] [PubMed] [Google Scholar]
- 19.Binzak B, Willard J, Vockley J. Identification of the Catalytic Residue Of Human Short/Branched Chain Acyl-CoA Dehydrogenase By In Vitro Mutagenesis. Biochim Biophys Acta. 1998;1382:137–142. doi: 10.1016/s0167-4838(97)00161-1. [DOI] [PubMed] [Google Scholar]
- 20.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 21.Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, Rossant J, Hughes TR, Frey B, Emili A. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 22.Kikuchi M, Hatano N, Yokota S, Shimozawa N, Imanaka T, Taniguchi H. Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J Biol Chem. 2004;279:421–428. doi: 10.1074/jbc.M305623200. [DOI] [PubMed] [Google Scholar]
- 23.Zolman BK, Nyberg M, Bartel B. IBR3, a novel peroxisomal acyl-CoA dehydrogenase-like protein required for indole-3-butyric acid response. Plant Mol Biol. 2007;64:59–72. doi: 10.1007/s11103-007-9134-2. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Li J, Sidell N. Modulation by phenylacetate of early estrogen-mediated events in MCF-7 breast cancer cells. Cancer Chemother Pharmacol. 2007;59:217–225. doi: 10.1007/s00280-006-0260-3. [DOI] [PubMed] [Google Scholar]
- 25.Greco O, Dachs GU, Tozer GM, Kanthou C. Mechanisms of cytotoxicity induced by horseradish peroxidase/indole-3-acetic acid gene therapy. J Cell Biochem. 2002;87:221–232. doi: 10.1002/jcb.10292. [DOI] [PubMed] [Google Scholar]
- 26.Maslak P, Chanel S, Camacho LH, Soignet S, Pandolfi PP, Guernah I, Warrell R, Nimer S. Pilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndrome. Leukemia. 2006;20:212–217. doi: 10.1038/sj.leu.2404050. [DOI] [PubMed] [Google Scholar]
- 27.Svennerholm L, Stallberg-Stenhagen S. Changes in the fatty acid composition of cerebrosides and sulfatides of human nervous tissue with age. J Lipid Res. 1968;9:215–225. [PubMed] [Google Scholar]
- 28.Wanders RJ, Vreken P, Ferdinandusse S, Jansen GA, Waterham HR, van Roermund CW, Van Grunsven EG. Peroxisomal fatty acid alpha- and beta-oxidation in humans: enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem Soc Trans. 2001;29:250–267. doi: 10.1042/0300-5127:0290250. [DOI] [PubMed] [Google Scholar]
- 29.Cunnane SC, Ryan MA, Nadeau CR, Bazinet RP, Musa-Veloso K, McCloy U. Why is carbon from some polyunsaturates extensively recycled into lipid synthesis? Lipids. 2003;38:477–484. doi: 10.1007/s11745-003-1087-8. [DOI] [PubMed] [Google Scholar]
- 30.Mattson MP. Mitochondrial Regulation of Neuronal Plasticity. Neurochem Res. 2006 doi: 10.1007/s11064-006-9170-3. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Mohsen AW, Mihalik SJ, Goetzman ES, Vockley J. Evidence for physical association of mitochondrial Fatty Acid oxidation and oxidative phosphorylation complexes. J Biol Chem. 2010;285:29834–29841. doi: 10.1074/jbc.M110.139493. [DOI] [PMC free article] [PubMed] [Google Scholar]