

Abstract
Sco is a red copper protein that plays an essential yet poorly understood role in the metalation of the CuA center of cytochrome oxidase, and is stable in both the Cu(I) and Cu(II) forms. To determine which oxidation state is important for function, we constructed His135 to Met or selenomethionine (SeM) variants that were designed to stabilize the Cu(I) over the Cu(II) state. H135M was unable to complement a scoΔ strain of Bacillus subtilis, indicating that the His to Met substitution abrogated cytochrome oxidase maturation. The Cu(I) binding affinities of H135M and H135SeM were comparable to that of the WT and 100-fold tighter than that of the H135A variant. The coordination chemistry of the H135M and H135SeM variants was studied by UV/vis, EPR, and XAS spectroscopy in both the Cu(I) and the Cu(II) forms. Both oxidation states bound copper via the S atoms of C45, C49 and M135. In particular, EXAFS data collected at both the Cu and the Se edges of the H135SeM derivative provided unambiguous evidence for selenomethionine coordination. Whereas the coordination chemistry and copper binding affinity of the Cu(I) state closely resembled that of the WT protein, the Cu(II) state was unstable, undergoing autoreduction to Cu(I). H135M also reacted faster with H2O2 than WT Sco. These data, when coupled with the complete elimination of function in the H135M variant, imply that the Cu(I) state cannot be the sole determinant of function; the Cu(II) state must be involved in function at some stage of the reaction cycle.
Keywords: Copper, Sco, Cytochrome oxidase, Selenomethionine, X-ray absorption
Introduction
Cells tightly regulate the trafficking of metal ions inside the cell via metalloregulatory proteins which sequester the metals and deliver them safely to the appropriate protein receptors [1, 2]. One such protein, Sco1, localized in the mitochondria of eukaryotes, is involved in the assembly of cytochrome c oxidase (Cox), the terminal enzyme of the respiratory chain, and also in the regulation of cellular copper levels along the mitochondrial pathway together with its paralog Sco2 [3–6]. Sco1 is a copper binding protein [7, 8] that receives Cu(I) from Cox17 [9] and is proposed to insert the copper ion into the Cox2 CuA site, perhaps via the intermediacy of a transient protein–protein complex [10–12]. Recently, this paradigm for Sco function has been challenged, and other functions such as the reduction of metal-binding cysteine residues in Cox2 [13, 14] or the redox buffering of metal ions in the immature oxidase have been proposed [15].
Sco is a red copper protein with the Cu ion coordinated by two cysteines, a histidine, and a fourth as-yet undetermined endogenous protein ligand [7, 14, 16–19]. The coordination environment is able to stabilize the copper ion in either the Cu(II) state or the Cu(I) state, which differ with respect to their coordinated ligands. In Bacillus subtilis Sco (BSco), EXAFS and EPR data from our laboratory suggested that Cu(II) is coordinated by C45 and C49 in a cis configuration with H135 and the additional O/N protein ligand completing the coordination sphere [15]. Cu(I) is strongly coordinated to C45 and C49, but forms a weaker substoichiometric interaction with H135, leading to the description of the site as an equilibrium between His-on and His-off states [19, 20]. Mutation of the His or Cys ligands leads to loss of function, as documented previously for yeast [7], human [8], B. subtilis [12] and Agrobacterium tumefaciens [21], but this is not due to loss of copper binding [15, 20]. Loss of function appears to be related to the destabilization of the Cu(II) rather than the Cu(I) forms, due at least in part to a unique conformation of the H135 coordinating residue [20]. Substitution of H135 with ala-nine led to a variant that was three orders of magnitude more sensitive to hydrogen peroxide reduction in the Cu(II) form, and could not be rescued by the exogenous coordination of imidazole. Cu(I)-H135A, on the other hand, was spectroscopically indistinguishable from the Cu(I) state of the WT protein.
Central to the question of Sco function is the issue of which oxidation state(s) of the copper ion are essential to function. The destabilization of the Cu(II) but not the Cu(I) state in the nonfunctional H135A variant suggests an important role for the Cu(II) state [20]. On the other hand, in yeast, Sco1 has been shown to be metalated in the Cu(I) form from Cox17 [9], emphasizing the accepted paradigm that copper chaperones transfer their copper in the Cu(I) state. To further explore the dependence of Sco function on copper oxidation state, we created the His135 to Met variant, which was expected to stabilize the Cu(I) form of BSco. Methionine ligands are known to play an important role in stabilizing Cu(I) sites in copper chaperones, particularly in more oxidizing environments [22–27]. Therefore, the substitution of Met for His would be expected to enhance or leave unchanged the function of a Cu(I)-transferring chaperone, but would abrogate a Cu(II)-dependent function. In the present paper we describe the results of a structural and functional study of the H135M variant of BSco. Like its H135A counterpart, the variant is unable to complement a scoΔ strain of B. subtilis with respect to the assembly of the CuA-containing caa3 terminal oxidase. However, using XAS of S-methionine (S-Met)- and selenomethionine (SeM)-substituted forms, we demonstrate the binding of the Met ligand in the Cu(I) state with a Cu(I) binding affinity that is close to that of the WT Cu(I) protein. The Cu(II) state also binds the Met ligand but is unstable with respect to autoredox chemistry. These studies provide evidence that a stable Cu(I) state is insufficient to impart functionality, and clearly point to a role for the Cu(II) state of BSco in function.
Experimental procedures
Construction and functional analysis of BSco and its H135M mutant
The BSco mutant H135M was constructed and its functional analysis was carried out as described previously [20]. The cell cultures of WT and H135M B. subtilis strains were grown in LB medium at 37 °C to a final OD600 of between 0.6 and 0.8. For Western blot analysis, proteins were separated on a gradient 8–16% SDS-PAGE gel following the method of Schagger and von Jagow [28]. After electrophoresis was performed, the proteins were transferred to a polyvinylidene difluoride membrane (Bio-Rad) in a wet blot using 25 mM tris–glycine buffer in 20% (v/v) methanol. Anti-BSco rabbit serum obtained by immunizing rabbits with a purified BSco (Josman LLC, CA, USA) was used to recognize the WT and H135M variant. Antibodies were visualized using the secondary antibody, goat anti-rabbit immunoglobulin G-alkaline phosphatase (AB Applied Biosystems) and a detection developer (Bio-Rad) containing a 1:1 mixture of nitroblue tetrazolium and 5-bromo-4-chloro-3-indolylphosphate.
Cloning and purification of BSco and its His135 to Met mutants
The H135M single mutant and the triple mutant M52I/M56I/H135M (I-H135M) were obtained via site-directed mutagenesis, and their mutation was confirmed by DNA sequence analysis as previously described [15, 20]. Mutant DNA was inserted into the pTXB3 vector (New England Biolabs), from which the recombinant proteins were expressed as self-cleaving inteins fused to a chitin binding domain for affinity purification. For the S-Met-containing variants, cell cultures were grown in 1 L LB-glucose medium containing 100 μg/mL of ampicillin at 37 °C to a final OD600 of between 0.6 and 0.8, as previously described [15, 20]. Cell cultures of SeM variants were grown in the Met minus strain B834(DE3) according to a protocol reported earlier [29]. The cells were initially grown as small cultures (10 mL) in LB media with ampicillin overnight at 37 °C. One hundred microliters of this were used to inoculate 10 mL of enhanced minimal M9 medium supplemented with 2 mM MgSO4, 0.1 mM CaCl2, 0.2% glucose, 0.05% thiamine, and 2 mL of a 50× stock of L-amino acids minus methionine, which also contained 50 μg/mL ampicillin and 20 μg/mL normal methionine. This culture was incubated overnight at 37 °C. After the cells had begun to grow in this medium, 10 mL of this culture were then transferred to 1 L of M9 enhanced minimal medium containing 20 μg/mL Se-Met plus ampicillin, and growth was continued to a final OD600 of between 0.6 and 0.8. The cultures were induced with 1 mM isopropylthio-β-D-galactoside for apoprotein production. The apoproteins were purified by affinity chromatography on chitin beads followed by intein cleavage by mercaptoethanesulfonic acid, as described previously [19].
The purified proteins were analyzed for protein concentration by Bradford assay, and their purity was checked by SDS-PAGE on an Amersham Biosciences PHAST system (20% homogeneous gel). H135M and H135SeM were further analyzed by electrospray mass spectrometry and were consistent with the expected masses with the N-terminal Met residue retained (Electronic supplementary material (ESM) Table S1). When necessary, proteins were concentrated using Amicon Centricon centrifugal concentrators of 5 kDa CO. The protein concentrations were determined either by Bradford assay or from the absorption at 280 nm using an extinction coefficient of 19,180 M−1 cm−1 [18]. Buffers used were reagent grade, and water was purified to a resistivity of 17–18 MΩ with a Barnstead Nanopure deionizing system.
Reconstitution with Cu(I) and Cu(II)
Apoproteins were reduced anaerobically with 2 mM dithiothreitol (DTT). Excess DTT was removed by exhaustive dialysis in an anaerobic chamber. To obtain Cu(I)-loaded proteins, the methionine and selenomethionine variants were reconstituted with tetrakis(acetonitrile)copper(I) hexafluorophosphate, as described earlier [19, 30]. For Cu(II) reconstitution, proteins were loaded by in situ addition of 0.9 M equiv. of CuSO4(aq) to the reduced apoprotein. The Cu(I) reconstituted proteins were concentrated to a final volume of ~1 mL and routinely analyzed for copper content by inductively coupled plasma optical emission spectrometry (ICP-OES) on a Perkin Elmer Optima 2000 spectrometer.
Determination of Cu(I) association constant (KA)
Cu(I) binding constants of BSco proteins were measured at 23 °C by competitive titration against bicinchoninic acid (BCA), following the protocol of Rosenzweig and coworkers [31]. Aliquots containing ~0.05–0.1 M equiv. of reduced apoproteins (1–3 μL, 1.0 mM in 50 mM Na–P buffer, pH 7.2) were titrated into 1.0 mL of ~10 μM CuI(BCA)2 solution that had been prepared by mixing 10 μM of tetrakis(acetonitrile)copper(I) hexafluorophosphate and 50–500 μM BCA. The additions were carried out under strictly anaerobic conditions, and the reaction mixture was incubated for 15 min after each addition to attain equilibrium. The reaction progress was monitored by UV/vis spectroscopy by following the decrease in the absorption band of CuI(BCA)2 at 562 nm. The reactions were also measured in the reverse direction by titrating the competitor BCA with the Cu(I)-loaded proteins. In this case, the increase in the band at 562 nm was monitored. Individual absorption spectra collected after each addition of protein were corrected for dilution factors, and data sets collected at different starting concentrations of BCA (50–500 μM) were analyzed using the program DYNAFIT [32], which calculates binding constants based on a model that includes the concentration and absorbance of all possible species in solution. β2 for the formation of the Cu(I)(BCA)2 complex was taken as 4.6 × 1014 M−1.
UV/vis and EPR spectroscopic characterization
UV/vis spectra were measured under anaerobic conditions in septum-sealed 1 cm path-length cuvettes using a Cary 50 spectrophotometer at 23 °C. For UV/vis measurements, the reduced apoproteins were reconstituted in situ in 50 mM sodium phosphate buffer, pH 7.2, with one molar equivalent CuSO4(aq), if found to be unstable. Quantitative X-band (9.4 GHz) EPR spectra were recorded on a Bruker Elexsys E500 spectrometer equipped with a Bruker ER049X SuperX microwave bridge and an E27H lock-in detector. Temperature control was provided by a continuous nitrogen flow cryostat system in which the temperature was monitored with a Bruker W1100321 thermocouple probe. Frozen Cu(II) protein samples at concentrations of between 100 and 500 μM in 50 mM sodium phosphate buffer at pH 7.2 were measured in 4 mm silica tubes. Spectra were collected at 90–110 K under nonsaturating power conditions, and the total spin due to Cu(II) was integrated. The integrated spin was then calibrated against a standard curve from the integrated Cu(II)–EDTA spins of known concentration (100–1,000 μM) to give the number of copper atoms per protein. EPR spectra were simulated using the program SIMPIP, developed at the University of Illinois and described in detail elsewhere [33].
X-ray absorption spectroscopy
Cu K edge (8,980 eV) and Se K edge (12,658 eV) extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES) data for copper-substituted BSco mutants were collected at the Stanford Synchrotron Radiation Lightsource at 3 GeV with currents of between 100 and 80 mA. Data were collected on beam line 9-3 using a liquid nitrogen cooled Si[220] monochromator and a Rh-coated mirror upstream of the monochromator with a 13 keV energy cutoff to reject harmonics. A second Rh mirror downstream of the monochromator was used to focus the beam. Data were collected in fluorescence mode using a high-count-rate Canberra 30-element Ge array detector with maximum count rates of below 120 kHz. A Z-1 metal oxide (Ni, As) filter and Soller slit assembly were placed in front of the detector to reduce the elastic scatter peak. Six to nine scans of a sample containing only sample buffer (50 mM Naphos, pH 7.2) were collected at each absorption edge, averaged, and subtracted from the averaged data for the protein samples to remove Z-1 Kβ fluorescence and produce a flat pre-edge baseline. The samples (80 μL) were measured as aqueous glasses in 20% ethylene glycol at 15 K. Energy calibration was achieved by reference to the first inflection point of a copper foil (8,980.3 eV) for Cu K edges and a selenium foil (12,658.0 eV) for Se K-edges, placed between the second and third ionization chamber. Data reduction and background subtraction were performed with the program modules of EXAFSPAK [34]. Data from each detector channel were inspected for glitches or drop-outs before inclusion in the final average. Spectral simulation was carried out with EXCURVE 9.2 [35–38], as previously described [39, 40]. This allowed for the inclusion of multiple scattering pathways between the metal center and the atoms of imidazole rings of the histidine residues. The experimental threshold energy, E0, was chosen as 8,985 eV. The structural parameters refined during the fitting process included coordination numbers (N), bond distances (R), and the Debye–Waller factor (2σ2), which results from both thermal motion and the static disorder of the absorber–scatter pair. The nonstructural parameter ΔE0, equivalent to a small correction to the threshold energy, was also allowed to vary, but was restricted to a common value for every component in a given fit.
Results
Loss of function in the H135A variant
In previous work we demonstrated that the H135A variant, while still able to bind copper stoichiometrically, was nonfunctional in the assembly of the B. subtilis cytochrome caa3 oxidase [20]. To assess whether methionine substitution (H135M) restores the Sco activity in B. subtilis, we used the assay of colony-staining with the cytochrome caa3-specific substrate tetramethylphenylenediamine (TMPD). In this assay, the presence of fully assembled functional caa3 oxidase causes the TMPD electron donor to oxidize and change color from the colorless reduced form to the deep-blue oxidized monocationic form [12]. The sco knockout strain ORB6556 (sco::erm) was TMPD-oxidation negative (as expected), giving rise to colorless colonies, whereas the ORB6625 strain where the WT sco gene together with its promoter was integrated at the amyE locus of the sco knockout produced blue colonies, indicating that TMPD-oxidation ability was restored. In contrast, the mutant sco (H135M, ORB6893) gene was unable to complement the sco knockout mutation, indicating that the H135M mutation results in the loss of cytochrome caa3 activity. The results are shown in Fig. 1a, where sco knockout (colorless), functional (WT complimented, blue) and nonfunctional H135M (colorless) colonies are compared.
Fig. 1.
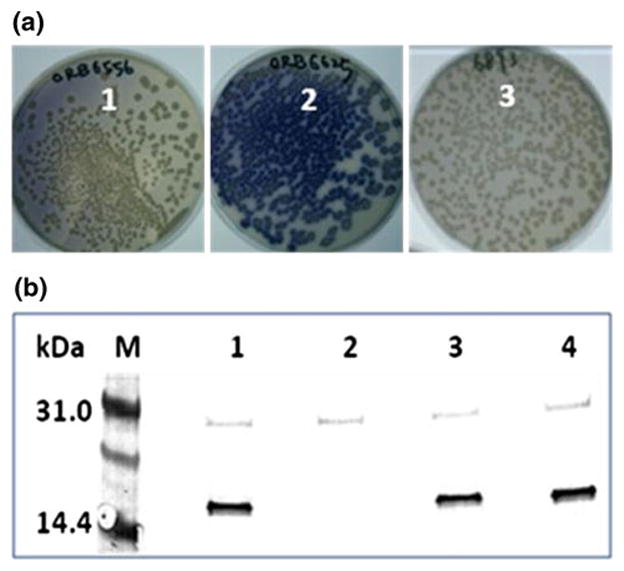
Colony-staining assay with the cytochrome caa3-specific substrate TMPD. a The B. subtilis strains: ORB6556, Sco::erm (Sco knockout mutant) (1); ORB6625, Sco::erm amyE::Sco (Sco knockout complemented with WT Sco gene) (2); ORB6893, Sco::erm amyE:: Sco[H135M] (Sco knockout complemented with H135M Sco gene) (3). b Western blot analysis of Sco expression in B. subtilis. Lane 1 JH642 (WT), lane 2 ORB6556 (sco knockout), lane 3 ORB6625 (sco knockout complemented with WT Sco gene), and lane 4 ORB6893 (Sco::erm amyE:: Sco[H135M])
In order to confirm that the H135M mutation abrogates the cytochrome caa3 activity by affecting copper insertion into the oxidase, but not by affecting the stability of the Sco protein, we next examined the intracellular levels of Sco by Western blot analysis (Fig. 1b). The results showed that the level of Sco in extracts of the Sco knockouts complemented with WT and H135M genes were similar and comparable to the level produced in the parental strain (JH642). This result indicated that the H135M mutation affects the function of Sco that is essential for the cytochrome caa3 activity, rather than affecting levels of protein synthesis.
Cu(I) binding affinity of WT BSco and its H135A and H135M variants
For various biochemical studies, the soluble domain of BSco was expressed in Escherichia coli and purified as reported earlier [19]. The H135M single mutants (H135M, H135SeM) and the triple mutants in which M52 and M56 were mutated to Ile (I-H135M and I-H135SeM) were constructed and purified by similar methods. Association constants for Cu(I) binding were determined by competitive titration with the Cu(I) chelating ligand BCA as a competitive ligand [23, 31]. Reactions were carried out in both the forward and reverse directions by titrating apo BSco proteins into a solution containing the fully formed CuI(BCA)2 complex, or in reverse by titrating BCA into the fully loaded Cu(I) BSco protein complexes. Representative UV/vis spectral traces for the H135M variant are shown in the forward and reverse directions in Fig. 2a and b, and the fits to the experimental data are shown in Fig. 2c and d, respectively. Average values of the binding constants are listed in Table 1, with values derived for different concentrations of [Cu(I)] and/or BCA for both the forward and reverse titrations listed in Table S2 of the ESM. The WT protein was found to bind Cu(I) with an affinity of 1.1 × 1013 M−1. Removal of the conserved and essential histidine residue His135 led to weaker binding by approximately two orders of magnitude (8.0 × 1010 M−1), whereas the Cu(I) binding constants of the methionine and the corresponding selenomethionine derivatives are essentially the same (within experimental error) as that of the WT protein.
Fig. 2.
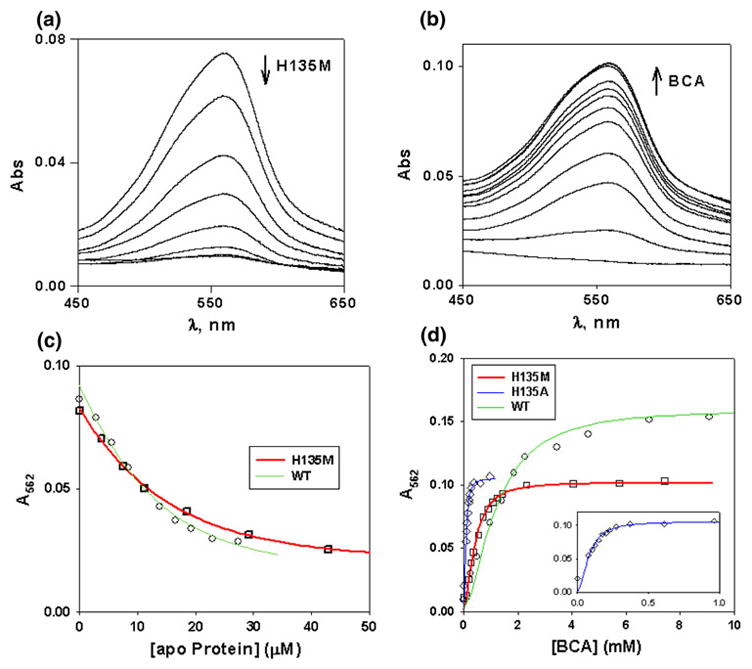
Determination of the association constant for Cu(I) binding to H135M BSco. a Competitive titration of the [CuIBCA)2] complex with apo H135M (forward titration). Aliquots containing 0.05–0.1 M equiv. of reduced apoproteins (1–3 μL of 1 mM protein in 50 mM Na–P buffer, pH 7.2) were titrated into 1.0 mL of a 10 μM CuI(BCA)2 solution prepared by mixing 10 μM [Cu(CH3CN)4](PF6) and differing amounts (50–500 μM) of BCA. b Titration of Cu(I)-loaded H135M with increasing concentrations of the BCA ligand (reverse titration). In general, reverse titrations were carried out in a 1 mL volume containing 10 μM [Cu(CH3CN)4](PF6), 25 μM Sco variant, with the addition of 1 μL aliquots of a stock 290 mM BCA solution. c Simulation of the data for the forward titrations of WT (green) and H135M (red) variants for concentrations of 10 μM Cu(I) + 500 μM BCA, with DYNAFIT used to calculate the association constant. The data were simulated assuming that K1 = K2 = 2.145 × 107 M−1 (β2 = 4.6 × 1014 M−2) for the Cu(I)–BCA complex. d Simulation of the reverse titration data for WT (green), H135A (blue), and H135M (red) using DYNAFIT. The inset shows an expanded view of the H135A variant which binds Cu(I) more weakly and is thus out-competed by BCA at lower BCA concentrations
Table 1.
Cu(I) association constants of the WT and H135 mutants of BSco proteins
| Sample | KA |
|---|---|
| Wild type | 1.1 ± 0.8 × 1013 |
| H135A | 8.0 ± 2.2 × 1010 |
| H135M | 6.0 ± 3.5 × 1012 |
| H135SeM | 2.1 ± 0.2 × 1012 |
| I-H135M | 3.1 ± 1.2 × 1012 |
| I-H135SeM | 2.8 ± 0.3 × 1012 |
Binding constants are the average of a number of titrations in both the forward and reverse directions, as listed in Table S1 of the ESM
EXAFS of Cu(I) complexes of the H135 variants
The structures of the Cu(I) complexes of the H135M and SeM variants were investigated by X-ray absorption spectroscopy. Binding affinity data (vide supra) demonstrated that substituting selenomethionine for methionine has a negligible effect on the binding affinity, thus validating its use as an XAS probe of structure. The selenium EXAFS of the H135SeM derivative can therefore act as a powerful indicator of coordination of the S/Se scatterer to the copper center. Figure 3 shows Cu K-EXAFS and absorption edge data for the Cu(I) forms of H135M and its SeM derivative, while Fig. 4 (top panel) shows Se K-EXAFS data for Cu(I)-H135SeM. Previous crystallographic and spectroscopic data have described the Cu(I) center in the WT BSco protein as being coordinated by two S donors from C45 and C49, and (in many but not all forms) the N atom from H135 [7, 14, 16–19]. In our earlier EXAFS study of BSco, the outer shell scattering from the His ligand was absent from the Cu(I) EXAFS, and the data were best simulated by two Cys residues and 60% partial occupancy of a third low-Z (O/N) scatterer, suggesting that H135 exists in His-on and His-off conformations, or that the third coordination position was occupied by solvent [19]. In support of this premise, the EXAFS of the Cu(I)-H135A variant was almost identical to that of the Cu(I)-bound WT protein [20]. These studies imply that in the H135M variant, the Cu(I) center is expected to be coordinated by three S ligands, two from cysteines and one from Met135, although the latter could possibly be substoichiometric. Therefore, we simulated the data using an all S-scatterer model which refined to a best fit of three S scatterers at 2.22 Å and a Debye–Waller (DW, 2σ2) of 0.014 Å2. When both shell occupancy and DW factor were allowed to float independently, a slight improvement in the fit (7%) was obtained with 2.7 S scatterers and a DW factor of 0.013 Å2. Given the correlation between coordination number and DW factor, these fits are probably equivalent, but they are also consistent with substoichiometric coordination of the Met residue, similar to what was found for the WT protein.
Fig. 3.

Cu K-EXAFS of Cu(I)-H135M and Cu(I)-H135Sem. Phase-corrected Fourier transforms and EXAFS (top insets) for Cu(I) complexes of H135M (top panel, 1.16 mM) and H135SeM (bottom panel, 0.2 mM). Experimental data are shown in black and simulated data in red. Parameters used in the fits are listed in Table 2. Bottom insets in each panel show the XANES region of the spectrum with the 8,983 eV peak characteristic of 3-coordination
Fig. 4.

Se K-EXAFS of H135SeM. Phase-corrected Fourier transforms and EXAFS (top insets) for Cu(I) (top panel, 0.6 mM Se) and Cu(II) (bottom panel, 1.8 mM Se) complexes of H135SeM. Experimental data are shown in black and simulated data in red. Parameters used in the fits are listed in Table 2. Bottom insets in each panel show the XANES region of the spectrum
Coordination of Met residues to Cu(I) can also be probed using SeM substitution, where the increased scattering amplitude of the heavier Se atom and the longer Cu–Se bond provide characteristic spectroscopic markers for the presence or absence of a Cu–SeM interaction. In addition, data at the Se edge yield an independent observation of the Se–Cu interaction. Analysis of the Cu(I)-H135SeM at the Cu edge required two shells of scatterers, 2 Cu–S interactions at 2.24 Å, and 0.6 Cu–Se at 2.38 Å. Fits using higher Se shell occupancies invariably led to higher values of the fitting parameter F. Therefore, these data also support substoichiometric coordination of the SeM ligand with a shell occupancy identical to that found for the third ligand in the WT protein.
Analysis of data at the Se edge was complicated by the presence of three noncoordinating methionine residues, M52, M56 and the N-terminal Met residue, which (from the mass spectral data) was not cleaved during protein expression. Since SeM substitution is global, the Se EXAFS will have contributions from all sources of Se in the protein. Consequently, the Se edge data is less sensitive to the presence of Cu coordination, as only one in four Se atoms see scattering from Cu. The Se edge data for Cu(I)-H135SeM are shown in Fig. 4 (top panel). The Fourier transform shows an intense first shell peak from the two covalent Se–C bonds at ~2.0 Å, and an outer shell feature due to Se–Cu at ~2.4 Å. The data simulate to 2 Se–C at 1.96 Å and 0.3 Se–Cu at 2.39 Å, which, when renormalized to take account of the four Met residues in the protein (multiplying by 4), gives 1.2 Se–Cu per protein. This value is larger than that found from the Cu edge data, but is likely to be less accurate due to the renormalization procedure. Notwithstanding, the data are unambiguous in the conclusion that Met135 coordinates to Cu(I), albeit with some uncertainty in the coordination number. Parameters used in the fits are listed in Table 2. We also examined the EXAFS of the I-H135SeM variant where two of the three noncoordinating Met residues were mutated to Ile. Surprisingly, the data did not indicate any increase in Se–Cu shell occupancy, suggesting that this mutation is nonconservative and may affect the binding affinity of the M135 residue. This conclusion was supported by measurements of the Cu(I) binding constants for the I-H135M and I-H135SeM variants, which were five- to eightfold lower than the single M135M/SeM variants (Table 1).
Table 2.
Fits obtained to the Cu and Se EXAFS of reconstituted Cu(I) and Cu(II) proteins of H135 mutants by curve-fitting using the program EXCURV 9.2
| Sample/fit | Fa | Cu–S |
Cu–Se |
−E0 | ||||
|---|---|---|---|---|---|---|---|---|
| Nob | R (Å)c | DW (Å2) | Nob | R (Å)c | DW (Å2) | |||
| Copper EXAFS | ||||||||
| Cu(I)Met | 0.282 | 2.7 | 2.22 | 0.013 | 1.92 | |||
| Cu(I)SeM | 0.472 | 2 | 2.24 | 0.009 | 0.6 | 2.38 | 0.010 | 4.20 |
| Cu(II)Metd | 0.321 | 3 | 2.22 | 0.012 | 1.89 | |||
| Cu(II)SeMd | 0.391 | 2 | 2.21 | 0.009 | 1.0 | 2.36 | 0.011 | 2.00 |
| Sample/fit | Fa | Se–C |
Se–Cud |
−E0 | ||||
| Nob | R (Å)c | DW (Å2) | Nob | R (Å)c | DW (Å2) | |||
| Selenium EXAFS | ||||||||
| Cu(I)SeM | 0.718 | 2 | 1.96 | 0.005 | 1.2 | 2.39 | 0.009 | 5.96 |
| Cu(II)SeM | 1.031 | 2 | 1.95 | 0.005 | 1.0 | 2.36 | 0.011 | 5.04 |
F is a least-squares fitting parameter defined as
Coordination numbers are generally considered accurate to ±25%
In any one fit, the statistical error in bond lengths is ±0.005 Å. However, when errors due to imperfect background subtraction, phase-shift calculations, and noise in the data are compounded, the actual error is probably closer to ±0.02 Å
Shell occupancy values have been renormalized by multiplying by 4 (the total number of methionines, both coordinating and noncoordinating, in the protein) so as to represent the number of Cu scatterers interacting with the coordinated Se atom
Properties of the Cu(II) H135M and H135SeM derivatives
The H135 mutants H135M and H135SeM can be reconstituted with Cu(II) by the in situ addition of 0.9 M equiv. of CuSO4(aq). They all form Cu(II) complexes, and their UV/vis spectral features are tabulated in Table 3. As found previously for the H135A, C45A, and C49A variants, the Cu(II) complexes are unstable and undergo an autoredox process leading to bleaching of the UV/vis thiolate → Cu(II) charge transfer spectra in a single exponential process. The autoreduction of Cu(II)-H135M monitored by UV/vis spectroscopy is shown in Fig. 5a, while that of the H135SeM derivative is shown in Fig. S1 of the ESM. Methionine and selenomethionine derivatives autoreduce at the same rate (Table 3).
Table 3.
Optical absorption features and autoreduction rates (k, s−1) of the Cu(II) species of the H135 mutants
| S. no | Cu(II) proteins | UV/vis: λmax, nm (ε, M−1 cm−1)a | k (s−1) |
|---|---|---|---|
| 1 | H135M | 400 (3,145), 495 (1,140), 565 (800) | 0.4 × 10−3 |
| 2 | H135-SeM | 415 (2,050), 495 (1,126), 565 (635) | 0.3 × 10−3 |
| 3 | I-H135M | 400 (2,605), 495 (1,200), 565 (885) | 2.4 × 10−3 |
| 4 | I-H135SeM | 415 (1,185), 495 (715), 565 (395) | 2.5 × 10−3 |
The extinction coefficients are limited to 10% error due to the autoreducing nature of the Cu(II) complexes
Fig. 5.

Top UV/visible spectra of H135M (black) and H135SeM (red). Bottom autoreduction of the H135M variant (40 μM), showing bleaching of the spectrum associated with conversion to the Cu(I) form. The inset shows the time dependence of the process fitted to a single exponential decay with rate constant 0.4 × 10−3 s−1
EPR of Cu(II) derivatives
Figure 6 shows the X-band EPR spectrum of natural-abundance Cu reconstituted H135M. The spectrum integrates to 52% Cu(II), with the remaining copper in the EPR-detectable Cu(I) form due to autoreduction. The spectrum resembles that obtained for the H135A variant in the respect that no N hyperfine splitting is observed in the perpendicular region, but is more complex, exhibiting splitting of the parallel copper hyperfine peaks due to the presence of at least two components. The spectrum could be simulated using two species with very similar EPR parameters, which are listed in Table 4 along with those obtained earlier for WT and the H135A variant. Comparison of the g and A values indicate lower values of gz in both components relative to that found for the WT protein, suggesting an increase in the total covalency of the H135M variant. In azurin, it has been shown that the 2.9 Å Cu–S(thioether) interaction between Cu(II) and M121 makes almost no contribution to the overall covalency of the Cu(II) center (which arises almost exclusively from the Cu(II)–C112 bond) [41]. An increase in covalency in the H135M variant of Sco therefore suggests either a shorter, stronger Cu–S(thioether) bond with greater 3d-S(thioether) overlap, an increase in Cu–S(Cys) covalency, or both. The identities of the two components in the H135M EPR spectrum are unclear, but they may arise from the coordination of methionine in two or more conformations [25]. The X-band EPR spectrum of the H135SeM derivative is similar to that of the S-Met form and is shown (without simulation) in Fig. S2 of the ESM.
Fig. 6.

Experimental (black) and simulated (red) X-band EPR spectrum of Cu(II)-H135M (0.17 mM Cu(II)). The parameters used in the simulation are listed in Table 4. Microwave frequency 9.40 GHz, modulation frequency 100 kHz, modulation amplitude 4 G, microwave power 0.8 mW, temperature 110 K. The inset shows an expanded view of the first two low-field hyperfine lines
Table 4.
Simulated CW X-EPR data for the Cu(II) species of the H135M variant in comparison with the WT and H135A Cu(II) species
| WT | H135A | H135M | ||
|---|---|---|---|---|
| gx | 2.0342(3) | 2.0324(2) | 2.0203(5) | 2.0300(5) |
| gy | 2.0288(3) | 2.0355 (2) | 2.0306(5) | 2.0260(5) |
| gz | 2.1503(3) | 2.1646(2) | 2.1142(5) | 2.1273(5) |
| Ax (Cu) | −135(2) | −145(2) | −130(3) | −166(5) |
| Ay (Cu) | −115(2) | −113(2) | −144(5) | −94(3) |
| Az (Cu) | −572(2) | −569(2) | −510(5) | −524(5) |
| Ax (14N) | 30 | |||
| Ay (14N) | 41 | |||
| Az (14N) | 31 | |||
| Ax (H) | 21 | 16 | 46 | 13 |
| Ay (H) | 16 | 14 | 19 | 16 |
| Az (H) | 18 | 11 | 56 | 26 |
| wx | 5.3 | 7.2 | 27 | 15 |
| wy | 6.9 | 5.5 | 15 | 9 |
| wz | 5.9 | 8.9 | 51 | 49 |
Hyperfine principal values in MHz (for units of 10−4 cm−1, divide by 3). Linewidths are in Gauss
EXAFS of Cu(II) H135M derivatives
To investigate the coordination of M135 or SeM135 to Cu(II), we carried out XAS studies at both the Cu and the Se edges. Experimental and simulated spectra for the Cu and Se K edges are shown in Fig. 7a and b, with metrical parameters listed in Table 2. Good fits to the Cu data of the H135S(Met) variant were obtained with three Cu–S interactions at 2.22 Å, consistent with the coordination of two Cys residues (C45, C49) and one Met residue (M135). The Cu(II) center is expected to be at least four-coordinate, but inclusion of additional O/N ligands did not improve the fit. This is similar to the case of nitrosocyanin, where the equatorial water ligand is not observed in the EXAFS [42]. However, given the sensitivity of Cu(II)-H135M to auto-redox chemistry (vide supra), we cannot exclude photoreduction in the X-ray beam as one possibility leading to a more “Cu(I)-like” EXAFS spectrum. To explore the binding of the Met ligand further, we measured the EXAFS data of the H135SeM variant. The Cu edge data could be simulated with two Cu–S(Cys) and one Cu–Se(SeM) interactions at 2.21 and 2.36 Å, respectively. The Se edge data contain contributions from all four Met residues in the protein, and since only one of these is expected to coordinate, the extraction of the Se–Cu coordination number and bond length is less reliable. When the Se–Cu Debye–Waller factor was fixed at the value obtained from the Cu edge data, a good fit was obtained with 0.25 ± 0.1 Se–Cu at 2.36 Å. Renormalizing this number to take account of the four Met residues in the protein (multiplying by 4) gives a Se–Cu coordination number of 1. Therefore, both Cu and Se edge data support the binding of one S-Met at 2.22, or one SeM ligand at 2.36 Å.
Fig. 7.

Cu K-EXAFS of Cu(II)-H135M and Cu(II)-H135Sem. Phase-corrected Fourier transforms and EXAFS (top insers) for Cu(II) complexes of H135M (top panel, 0.7 mM Cu2+) and H135SeM (bottom panel, 0.6 mM Cu2+). Experimental data are shown in black and simulated data in red. Parameters used in the fits are listed in Table 2
Peroxide reactivity of H135M
Hydrogen peroxide reacts with the Cu(II) centers in BSco, reducing the copper to Cu(I) and generating dioxygen [20]. This reaction is slow in WT BSco, but occurs at a rate that is three orders of magnitude faster in the H135A variant. Whereas the WT protein appears to react in a simple bimolecular fashion (first order in Cu(II)–BSco and in H2O2), H135A shows saturation kinetics suggestive of pre-equilibrium binding of peroxide to the Cu(II) center. Reaction of the H135M variant with hydrogen peroxide (Fig. 8) showed similar kinetic behavior to the WT protein, but with an increased rate. The reaction was first order in both the concentration of variant and hydrogen peroxide over the range 0–5 mM, with no indication of saturation behavior. This suggests a simple bimolecular mechanism in which peroxide reacts with the H135M variant in an outer sphere process, and has been attributed in the WT protein to the absence of an open equatorial coordinating position at the Cu(II) center. This is consistent with the spectroscopic data, and suggests that in the H135M variant all equatorial coordination positions are likewise occupied by protein-derived ligands, including the methionine at position 135. However, the redox potential of the variant is more positive, resulting in an increase in the rate of peroxide oxidation from 4.3 to 11.6 M−1 s−1.
Fig. 8.

Kinetics of the reaction of the WT and H135M variant BSco proteins with hydrogen peroxide. The figure shows the re-plotting of the pseudo-first-order rate constants for the reduction of the Cu(II) forms of the WT and the H135M variant (measured at 357 and 400 nm, respectively, 90 μM Cu(II)-loaded protein) as a function of H2O2 concentration. Black circles WT, red circles H135M
Discussion
The generally accepted paradigm for copper chaperone function is that these proteins sequester and transport the metal in the Cu(I) form. This paradigm has been demonstrated for ATOX1 [30, 43, 44] and its bacterial homologs [45], CCS [46, 47], and the periplasmic copper transporter CusF [24, 25, 48], where X-ray absorption spectroscopy in conjunction with crystallography and NMR has been seminal in establishing the binding and coordination geometry of the Cu(I) center. The metalation of the dinuclear CuA center of cytochrome oxidase has likewise been proposed to occur via transfer of Cu from the putative chaperone Sco1 [3, 6, 7, 10, 12, 49], generating the di-Cu(I) form, a premise that is supported by the finding that Sco1 is itself metalated in its Cu(I) form by Cox17 [9]. However, recent studies using NMR failed to demonstrate the transfer of Cu(I) from Sco to CuA, and instead suggested that Sco functioned as a thiol-disulfide oxidoreductase that converts the bis-cysteinyl moiety of the CuA center to the reduced bis-thiolate in preparation for the insertion of copper [13]. Alternatively, since the CuA center is an electron-delocalized mixed-valence Cu(II)–Cu(I) species, it is possible that in vivo metalation requires copper to be inserted in both oxidation states [20]. These considerations prompted us to examine which oxidation state of Sco was most important for function via the construction of copper-site mutants that were designed to favor the Cu(I) over the Cu(II) state. If the Sco function was merely that of a metallochaperone, then we would predict little effect on function since CuA would continue to be metalated as the di-Cu(I) form. This state of CuA is easily obtained from the mixed-valence form by dithionite reduction, and has been fully characterized by previous EXAFS studies from our laboratory [29, 50]. On the other hand, if the function was redox, the destabilization of the Cu(II) form was predicted to lead to loss of function. The approach involved the construction of variants at residue 135, a histidine residue in the WT protein which imparts special stability to the Cu center [20]. In previous work we studied the properties of the H135A variant, which was found to have a similar structure and stability to the WT protein in the Cu(I) form (two coordinated Cys residues and weaker substoichiometric coordination of a third ligand) but was significantly destabilized in the Cu(II) form with respect to reduction to Cu(I) through either autoredox or hydrogen peroxide [20]. In the present study we have extended these studies to the H135M and H135Sem variants, reasoning that replacement of His by Met or SeM should lead to similar or somewhat strengthened Cu(I) coordination, which in turn should have a minimal effect on activity if only the Cu(I) state was important for function. Using a functional assay originally developed by Hill and coworkers [12], we showed that neither H135A or H135M produced functional caa3 oxidase. This assay measures the oxidation of the dye TMPD by the CuA center of intact caa3 oxidase in B. subtilis, and therefore reports directly on the functional metalation of the CuA center. ScoΔ cell lines could be complemented with the WT Sco gene to give blue colonies, while cells complemented with either variant were colorless.
While these results suggested that factors in addition to the stability of the Cu(I) state are important for Sco function, it was necessary to investigate the relative binding affinity of Cu(I) for WT and variant proteins, and to establish that Met135 coordinated to Cu(I) in a similar fashion to the native His. Measurement of Cu(I) affinity for the WT, H135A and H135M forms showed that replacement of the native His by a noncoordinating Ala residue resulted in a decrease in association constant of two orders of magnitude, from 1.1 ± 0.8 × 1013 to 8.0 ± 2.2 × 1010 M−1, implying that the His residue is clearly increasing the Cu(I) binding affinity. Unlike H135A, the H135M variant was found to have the same affinity within experimental error as the WT protein, with an association constant of 6.0 ± 3.5 × 1012 M−1. This binding constant is based on the β2 value of 4.6 × 1014 for the Cu(I)–(BCA)2 complex reported by Rosenzweig and coworkers [31], rather than the higher value reported by Wedd and coworkers [23, 51], and as such may underestimate the absolute value of the Cu(I)–Sco association constant. Notwithstanding, it is the relative values of KA that are relevant to the present discussion of the differences in affinity between the WT, H135A and H135M variants. The near-coincidence of KA for the WT and M135M forms suggests that the Met residue replaces the His as the third ligand with similar affinity.
Methionine coordination has been shown to be a powerful method of stabilizing Cu(I), particularly in oxidizing environments such as vesicular compartments or the periplasmic spaces of Gram-negative bacteria [52, 53]. Generally, these sites are built from Met and His residues, and span ligand sets from His2Met in the cuproenzymes DBM [54], PHM [40, 55] and TBM [56], His–(Met)2 in CusF [24, 25], His–(Met)3 in CopC [57] and pcoC [22], to the (Met)3 and (Met)4 sites of CusB [27] and CopK [58], respectively. For three-coordinate sites, the EXAFS-derived Cu–S(met) distance is close to 2.31 Å, significantly longer than that generally observed for Cu–S(Cys) coordination, which is usually in the 2.22–2.25 Å range [46, 59]. Two coordinate sites containing Met coordination have yet to be reported, perhaps because of the poor donor capacity of the thioether ligand, whereas two-coordinate Cu–S(thiolate)2 with short (2.12–2.15 Å Cu–S bonds) are common in metallochaperones such as ATOX1 [30], N-terminal domains of P-type ATPases [60], and transcription factors such as CueR [61]. Recently, a mixed two-coordinate Cu(I)–(Cys)(His) center was reported in the C49A variant of BSco [15].
Against this background, spectroscopic measurements were used to determine structural details of the Met135 coordination. EXAFS studies on the Cu(I) form established the presence of a Cu–Met interaction in both the S-Met and the Se-Met forms. The occupation number for the single Met ligand was close to stoichiometric. The Cu–Se bond length of 2.38 Å was ~0.03 Å shorter than that determined previously for the SeM derivative of the periplasmic chaperone CusF [48]. The Cu–S(met) bond length was more difficult to determine, as it is contained within the overall Cu–S shell, which is dominated by the shorter 2.22 Å Cu–S(Cys) distance. The EXAFS data were fully consistent with a stable Cu(I)–(Cys)2Met coordination, with bond lengths typical of a stable three-coordinate Cu(I) environment.
The Cu(II) forms of the M135M and M135SeM variants are readily formed by the stoichiometric addition of Cu(II) ions, but are less stable than the Cu(I) congeners. The Cu(II)-reconstituted proteins undergo autoredox to the Cu(I) form, and react faster with hydrogen peroxide than the WT Cu(II) protein. The peroxide reactivity is similar to the WT in that it appears to be bimolecular, suggesting that the peroxide may react by an outer sphere mechanism. This contrasts with the behavior observed for the H135A variant, which showed saturation kinetics typical of the binding of peroxide to a site on the protein, presumably the Cu(II) center [20]. These data can be rationalized by the formation of a four-coordinate Cu(II) complex in the H135M and H135SeM derivatives with two Cys, one Met, and one endogenous protein residue as ligands. EXAFS data at both the Cu and the Se edges supports the assignment of the Cys and Met ligands but provides little evidence for a fourth ligand. The 0.02 Å decrease in Cu–Se bond length between Cu(II) and Cu(I) states is consistent with the higher effective charge on the Cu(II) center. The UV/vis data for the H135M and H135SeM variants show Cu–S(pσ) CT bands at 400 and 415 nm, respectively, which are redshifted by 43 and 58 nm relative to the WT protein. This is consistent with the observed increase in ease of reduction due to the easier transfer of charge from S to Cu(II). EPR data also support this conclusion, since both of the species have gz values below that of the WT (Table 4). These trends are suggestive of an increased covalency of the site as the result of the substitution of His by the more polarizable Met and (to a greater extent) SeM ligands, which would clearly favor the Cu(I) over the Cu(II) form.
Our results show that we have succeeded in creating a BSco variant that has a destabilized Cu(II) center but also a Cu(I) center that closely resembles the electronic, geometric and thermodynamic properties of the native protein. The complete elimination of function in this variant then implies that the properties of the Cu(I) state cannot be the sole determinant of function in Sco proteins; the Cu(II) state must be involved in function at some stage of the reaction cycle. These findings therefore rule out simple metallochaperone function in which copper is inserted into CuA in the Cu(I) form. Possible roles for Cu(II) in Sco function include direct transfer to CuA to generate the mixed-valence form, or an as-yet undiscovered redox role [20]. On the other hand, a role involving only thiol-disulfide oxidoreduction [13] seems unlikely unless it was coupled to copper redox, as proposed for Cox17 metalation [62]. Further studies are underway to probe these possibilities.
Supplementary Material
Acknowledgments
We gratefully acknowledge the use of facilities at the Stanford Synchrotron Radiation Lightsource, which is supported by the National Institutes of Health Biomedical Research and Technology Program Division of Research Resources, and by the US Department of Energy Office of Biological and Environmental Research. The work was supported by a grant from the National Institutes of Health (R01 GM54803) to NJB.
Abbreviations
- BCA
Bicinchoninic acid
- BSco
Bacillus subtilis Sco
- Cox
Cytochrome oxidase
- DW
Debye–Waller factor
- EXAFS
Extended X-ray absorption fine structure
- FT
Fourier transform
- I-H135M
Triple mutant M52I/M56I/H135M
- I-H135SeM
Triple mutant M52I/M56I/H135SeM
- SeM
Selenomethionine
- TMPD
Tetramethylphenylenediamine
- XAS
X-ray absorption spectroscopy
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00775-010-0725-z) contains supplementary material, which is available to authorized users.
Contributor Information
Gnana S. Siluvai, Division of Environmental and Biomolecular Systems, Oregon Health and Sciences University, 20000 NW Walker Road, Beaverton, OR 97006, USA
Michiko Nakano, Division of Environmental and Biomolecular Systems, Oregon Health and Sciences University, 20000 NW Walker Road, Beaverton, OR 97006, USA.
Mary Mayfield, Division of Environmental and Biomolecular Systems, Oregon Health and Sciences University, 20000 NW Walker Road, Beaverton, OR 97006, USA.
Ninian J. Blackburn, Email: ninian@comcast.net, Division of Environmental and Biomolecular Systems, Oregon Health and Sciences University, 20000 NW Walker Road, Beaverton, OR 97006, USA
References
- 1.Kim BE, Nevitt T, Thiele DJ. Nat Chem Biol. 2008;4:176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 2.Boal AK, Rosenzweig AC. Chem Rev. 2009;109:4760–4779. doi: 10.1021/cr900104z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glerum DM, Shtanko A, Tzagoloff A. J Biol Chem. 1996;271:20531–20535. doi: 10.1074/jbc.271.34.20531. [DOI] [PubMed] [Google Scholar]
- 4.Cobine PA, Pierrel F, Winge DR. Biochim Biophys Acta. 2006;1763:759–772. doi: 10.1016/j.bbamcr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Carr HS, Winge DR. Acc Chem Res. 2003;36:309–316. doi: 10.1021/ar0200807. [DOI] [PubMed] [Google Scholar]
- 6.Leary SC, Kaufman BA, Pellecchia G, Guercin GH, Mattman A, Jaksch M, Shoubridge EA. Hum Mol Genet. 2004;13:1839–1848. doi: 10.1093/hmg/ddh197. [DOI] [PubMed] [Google Scholar]
- 7.Nittis T, George GN, Winge DR. J Biol Chem. 2001;276:42520–42526. doi: 10.1074/jbc.M107077200. [DOI] [PubMed] [Google Scholar]
- 8.Horng YC, Leary SC, Cobine PA, Young FB, George GN, Shoubridge EA, Winge DR. J Biol Chem. 2005;280:34113–34122. doi: 10.1074/jbc.M506801200. [DOI] [PubMed] [Google Scholar]
- 9.Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. J Biol Chem. 2004;279:35334–35340. doi: 10.1074/jbc.M404747200. [DOI] [PubMed] [Google Scholar]
- 10.Lode A, Kuschel M, Paret C, Rodel G. FEBS Lett. 2000;485:19–24. doi: 10.1016/s0014-5793(00)02176-1. [DOI] [PubMed] [Google Scholar]
- 11.Rigby K, Cobine PA, Khalimonchuk O, Winge DR. J Biol Chem. 2008;283:15015–15022. doi: 10.1074/jbc.M710072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mattatall NR, Jazairi J, Hill BC. J Biol Chem. 2000;275:28802–28809. doi: 10.1074/jbc.M002741200. [DOI] [PubMed] [Google Scholar]
- 13.Abriata LA, Banci L, Bertini I, Ciofi-Baffoni S, Gkazonis P, Spyroulias GA, Vila AJ, Wang S. Nat Chem Biol. 2008;4:599–601. doi: 10.1038/nchembio.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banci L, Bertini I, Calderone V, Ciofi-Baffoni S, Mangani S, Martinelli M, Palumaa P, Wang S. Proc Natl Acad Sci USA. 2006;103:8595–8600. doi: 10.1073/pnas.0601375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siluvai GS, Mayfield M, Nilges MJ, DeBeer George S, Blackburn NJ. J Am Chem Soc. 2010;132:5215–5226. doi: 10.1021/ja910759v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balatri E, Banci L, Bertini I, Cantini F, Ciofi-Baffoni S. Structure. 2003;11:1431–1443. doi: 10.1016/j.str.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Williams JC, Sue C, Banting GS, Yang H, Glerum DM, Hendrickson WA, Schon EA. J Biol Chem. 2005;280:15202–15211. doi: 10.1074/jbc.M410705200. [DOI] [PubMed] [Google Scholar]
- 18.Ye Q, Imriskova-Sosova I, Hill BC, Jia Z. Biochemistry. 2005;44:2934–2942. doi: 10.1021/bi0480537. [DOI] [PubMed] [Google Scholar]
- 19.Andruzzi L, Nakano M, Nilges MJ, Blackburn NJ. J Am Chem Soc. 2005;127:16548–16558. doi: 10.1021/ja0529539. [DOI] [PubMed] [Google Scholar]
- 20.Siluvai GS, Nakano M, Mayfield M, Nilges MJ, Blackburn NJ. Biochemistry. 2009;48:12133–12144. doi: 10.1021/bi901480g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saenkham P, Vattanaviboon P, Mongkolsuk S. FEMS Microbiol Lett. 2009;293:122–129. doi: 10.1111/j.1574-6968.2009.01516.x. [DOI] [PubMed] [Google Scholar]
- 22.Peariso K, Huffman DL, Penner-Hahn JE, O’Halloran TV. J Am Chem Soc. 2003;125:342–343. doi: 10.1021/ja028935y. [DOI] [PubMed] [Google Scholar]
- 23.Djoko KY, Xiao Z, Huffman DL, Wedd AG. Inorg Chem. 2007;46:4560–4568. doi: 10.1021/ic070107o. [DOI] [PubMed] [Google Scholar]
- 24.Xue Y, Davis AV, Balakrishnan G, Stasser JP, Staehlin BM, Focia P, Spiro TG, Penner-Hahn JE, O’Halloran TV. Nat Chem Biol. 2008;4:107–109. doi: 10.1038/nchembio.2007.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loftin IR, Franke S, Blackburn NJ, McEvoy MM. Protein Sci. 2007;16:2287–2293. doi: 10.1110/ps.073021307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loftin IR, Blackburn NJ, McEvoy MM. J Biol Inorg Chem. 2009;14:905–912. doi: 10.1007/s00775-009-0503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM. J Biol Chem. 2007;282:35695–35702. doi: 10.1074/jbc.M703937200. [DOI] [PubMed] [Google Scholar]
- 28.Schagger H, von Jagow G. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 29.Blackburn NJ, Ralle M, Gomez E, Hill MG, Patsuszyn A, Sanders D, Fee JA. Biochemistry. 1999;38:7075–7084. doi: 10.1021/bi982500z. [DOI] [PubMed] [Google Scholar]
- 30.Ralle M, Lutsenko S, Blackburn NJ. J Biol Chem. 2003;278:23163–23170. doi: 10.1074/jbc.M303474200. [DOI] [PubMed] [Google Scholar]
- 31.Yatsunyk LA, Rosenzweig AC. J Biol Chem. 2007;282:8622–8631. doi: 10.1074/jbc.M609533200. [DOI] [PubMed] [Google Scholar]
- 32.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 33.Nilges MJ, Matteson K, Belford RL. In: ESR spectroscopy in membrane biophysics. Hemminga MA, Berliner LJ, editors. Springer; New York: 2006. [Google Scholar]
- 34.George GN. Exafspak. Stanford Synchrotron Radiation Laboratory; Stanford: 1990. [Google Scholar]
- 35.Binsted N, Gurman SJ, Campbell JW. Excurve, 9.2. Daresbury Laboratory; Warrington: 1998. [Google Scholar]
- 36.Gurman SJ, Binsted N, Ross I. J Phys C. 1984;17:143–151. [Google Scholar]
- 37.Gurman SJ, Binsted N, Ross I. J Phys C. 1986;19:1845–1861. [Google Scholar]
- 38.Binsted N, Hasnain SS. J Synchrotron Radiat. 1996;3:185–196. doi: 10.1107/S0909049596005651. [DOI] [PubMed] [Google Scholar]
- 39.Eisses JF, Stasser JP, Ralle M, Kaplan J, Blackburn NJ. Biochemistry. 2000;39:7337–7342. doi: 10.1021/bi000690j. [DOI] [PubMed] [Google Scholar]
- 40.Blackburn NJ, Rhames FC, Ralle M, Jaron S. J Biol Inorg Chem. 2000;5:341–353. doi: 10.1007/pl00010663. [DOI] [PubMed] [Google Scholar]
- 41.Sarangi R, Gorelsky SI, Basumallick L, Hwang HJ, Pratt RC, Stack TD, Lu Y, Hodgson KO, Hedman B, Solomon EI. J Am Chem Soc. 2008;130:3866–3877. doi: 10.1021/ja076495a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basumallick L, Sarangi R, DeBeer George S, Elmore B, Hooper AB, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2005;127:3531–3544. doi: 10.1021/ja044412+. [DOI] [PubMed] [Google Scholar]
- 43.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 44.Walker JM, Tsivkovskii R, Lutsenko S. J Biol Chem. 2002;277:27953–27959. doi: 10.1074/jbc.M203845200. [DOI] [PubMed] [Google Scholar]
- 45.Singleton C, Le Brun NE. Biometals. 2007;20:275–289. doi: 10.1007/s10534-006-9068-1. [DOI] [PubMed] [Google Scholar]
- 46.Stasser J, Siluvai GS, Barry AN, Blackburn NJ. Biochemistry. 2007;46:11845–11856. doi: 10.1021/bi700566h. [DOI] [PubMed] [Google Scholar]
- 47.Stasser JP, Eisses JF, Barry AN, Kaplan JH, Blackburn NJ. Biochemistry. 2005;44:3143–3152. doi: 10.1021/bi0478392. [DOI] [PubMed] [Google Scholar]
- 48.Bagai I, Rensing C, Blackburn NJ, McEvoy MM. Biochemistry. 2008;47:11408–11414. doi: 10.1021/bi801638m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bengtsson J, von Wachenfeldt C, Winstedt L, Nygaard P, Hederstedt L. Microbiology. 2004;150:415–425. doi: 10.1099/mic.0.26691-0. [DOI] [PubMed] [Google Scholar]
- 50.Blackburn NJ, de Vries S, Barr ME, Houser RP, Tolman WB, Sanders D, Fee JA. J Am Chem Soc. 1997;119:6135–6143. [Google Scholar]
- 51.Djoko KY, Chong LX, Wedd AG, Xiao Z. J Am Chem Soc. 2010;132:2005–2015. doi: 10.1021/ja9091903. [DOI] [PubMed] [Google Scholar]
- 52.Kim EH, Rensing C, McEvoy MM. Nat Prod Rep. 2010;27:711–719. doi: 10.1039/b906681k. [DOI] [PubMed] [Google Scholar]
- 53.Davis AV, O’Halloran TV. Nat Chem Biol. 2008;4:148–151. doi: 10.1038/nchembio0308-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blackburn NJ, Hasnain SS, Pettingill TM, Strange RW. J Biol Chem. 1991;266:23120–23127. [PubMed] [Google Scholar]
- 55.Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 56.Hess CR, Klinman JP, Blackburn NJ. J Biol Inorg Chem. 2010 doi: 10.1007/s00775-010-0677-3. http://www.springerlink.com/content/31u6186j43q443g0/ [DOI] [PMC free article] [PubMed]
- 57.Arnesano F, Banci L, Bertini I, Mangani S, Thompsett AR. Proc Natl Acad Sci USA. 2003;100:3814–3819. doi: 10.1073/pnas.0636904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarret G, Favier A, Coves J, Hazemann JL, Mergeay M, Bersch B. J Am Chem Soc. 2010;132:3770–3777. doi: 10.1021/ja9083896. [DOI] [PubMed] [Google Scholar]
- 59.Pickering IJ, George GN, Dameron CT, Kurz B, Winge DR, Dance IG. J Am Chem Soc. 1993;115:9498–9505. [Google Scholar]
- 60.Ralle M, Cooper MJ, Lutsenko S, Blackburn NJ. J Am Chem Soc. 1998;120:13525–13526. [Google Scholar]
- 61.Chen K, Yuldasheva S, Penner-Hahn JE, O’Halloran TV. J Am Chem Soc. 2003;125:12088–12089. doi: 10.1021/ja036070y. [DOI] [PubMed] [Google Scholar]
- 62.Arnesano F, Balatri E, Banci L, Bertini I, Winge DR. Structure. 2005;13:713–722. doi: 10.1016/j.str.2005.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.