

The molecular millennium has bestowed researchers with the essential tools to identify the underlying genetic substrates for thousands of genetic disorders, most of which are rare and follow Mendelian inheritance patterns. The genetic basis of potentially lethal and heritable cardiomyopathies and cardiac channelopathies including hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), left ventricular non-compaction (LVNC), arrhythmogenic right ventricular cardiomyopathy (ARVC), long QT syndrome (LQTS), short QT syndrome (SQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), Brugada syndrome (BrS), and familial atrial fibrillation (FAF) have been identified and are now better understood.
Marked genetic and clinical heterogeneity are hallmark features of these disorders with multiple genes and allelic variants conferring their underlying pathogenic mechanisms. To date, thousands of gene mutations at the single nucleotide level have been discovered for this group of divergent cardiovascular disorders of the heart. Most mutations represent pathogenic disease-causing mutations only discovered in disease cohorts while others are common or rare genetic polymorphisms identified in disease and in health that may or may not provide the precise pathogenic substrate. Genetic testing for several of these heritable cardiomyopathies and channelopathies has made its transition from discovery through translation and are now commercially available clinical genetic tests.
The purpose of this review is to provide the reader with a foundational understanding of genetic testing in clinical practice. Here, we will present some general principles of genetic testing, the need for careful interpretation of genetic testing results, the importance of genetic counseling, and some points on the ethical, legal, and societal implications of genetic testing, and conclude by reviewing the state of clinical genetic testing for four of the principle cardiomyopathies/channelopathies – HCM, LQTS, CPVT, and BrS.
GENERAL PRINCIPLES OF GENETIC TESTING
Mutation Types in Human Genetic Disease
Genes contain an encrypted genetic message for the assembly of polypeptides or proteins that serve the biological function of the cell. Polypeptides or proteins are polymers of linear repeating units called amino acids. The amino acid-encoding sequences within genes are called exons and between the exons are intervening DNA sequences called introns which are not a part of the genetic code and are removed during splicing. The assembly of a polypeptide is directed by a triplet genetic code or codon (three consecutive bases), of which there are 64 codons that encode for either 20 distinct amino acids or the termination (stop codon) of protein assembly. Each codon is decoded sequentially to give a specific sequence of amino acids that are covalently linked through peptide bonds and ultimately comprise a protein.
The DNA of the human genome is highly stable from generation to generation, but not immutable. Instead, it is vulnerable to an array of different types of germline (heritably transmitted) and somatic mutations. In general, mutations can be classified into three categories: genome mutations, chromosome mutations, and gene mutations1. Genome mutations involve the abnormal segregation of chromosomes during cell division (for example, Trisomy 21 or “Down syndrome”). Chromosome mutations involve the structural breakage and rearrangement of chromosomes during cell division, or major deletions or insertions of portions of a particular chromosome (for example, chromosome 22q11.2 microdeletion syndrome). Finally, gene mutations involve nucleotide alterations that disrupt the normal function of a single gene product. This review will focus mainly on genetic testing for gene mutations. Such single gene mutations are classified into three primary categories: single nucleotide substitutions, deletions, and insertions.
Single Nucleotide Substitutions
Single nucleotide substitutions represent the most common type of pathogenic mutation for the various genetic heart diseases to be reviewed here accounting for approximately two-thirds of the pathogenic mutations. If a single nucleotide substitution occurs in the coding region (exon), the result may be either a synonymous (silent) mutation whereby the new codon still encodes the same amino acid (Figure 1) or a non-synonymous mutation whereby the altered codon encodes for a different amino acid or terminates further protein assembly (i.e. introduces a premature stop codon). The term “missense” mutation is used to specify a single nucleotide substitution that disrupts the open reading frame and replaces the normal (wildtype, WT) amino acid with a different one (Figure 1). Importantly, a missense mutation may or may not result in a functionally perturbed protein that leads to a disease phenotype. The functional consequence of a missense mutation may depend on the differences in biochemical properties between the amino acids that are being substituted and/or the location in the protein at which the exchange occurs. A “nonsense” mutation refers to a non-synonymous, single nucleotide substitution that mutates a codon that encodes one of 20 amino acids to one of the three stop codons (Figure 1). A nonsense mutation results in a truncated (shortened) gene product at the location of the new stop codon. The functional effects could range from no appreciable difference to functional lethality (a non-functioning protein) depending again on where in the protein a nonsense mutation occurs.
Figure 1. Nucleotide Substitutions.

Compared to the depicted normal DNA, amino acid (single letter abbreviation) sequence and resulting peptide sequence are examples of nucleotide substitutions and deletion mutations. The amino acids of the peptide sequence are color coded to represent their unique biophysical properties where yellow represents nonpolar hydrophobic amino acids, green are polar hydrophilic, pink are negatively charged acidic residues, and blue represents positively charged basic amino acid residues. A nucleotide change resulting in a new codon that encodes for: A) the same amino acid as the normal sequence is a silent mutation, B) a different amino acid is a missense mutation, and C) for a termination codon is a nonsense mutation. Illustrated in D) is a deletion of a single nucleotide (G) that results in a shift of the open reading frame of the transcript thus representing a frameshift mutation. Note how the sequence of amino acids has been altered from this point forward. Although not illustrated here, frameshift mutations as a result of a deletion or insertion of nucleotides often lead to a premature stop codon and thus a truncated protein. Illustrated in E), the deletion of three nucleotides (GAC) produced an in-frame deletion of a single amino acid (aspartic acid, Asp) in the protein. The remaining amino acid sequence is unaltered. Three nucleotide insertions (not shown) can have a similar affect whereby an amino acid is inserted into the protein product. Figure adapted from Tester and Ackerman82
Intronic (non-coding) base substitutions may also result in an altered gene product. The normal process by which intronic sequences are excised to give a mature protein encoding transcript is reliant on specific nucleotide sequences located at the intron/exon (acceptor site) and exon/intron (donor site) boundaries. Base substitutions within these highly conserved sequences can result in abnormal splicing. In some cases, entire exons can be skipped (deleted) or entire introns may be included in the mature transcript, most often resulting in a “frame-shift”.
Insertions and Deletions
Gene mutations may also involve insertions and deletions of nucleotides that can be as small as a single nucleotide insertion/deletion or as large as several hundreds to thousands of nucleotides in length. Most of these insertions and deletions occurring in the exon alter the “reading frame” of translation at the point of the insertion/deletion and produce a new sequence of amino acids in the finished product, a so-called “frame-shift” mutation (Figure 1). Many frame-shift mutations often result in a different product length from the normal gene product by creating a new stop codon, which produces either a shorter or longer gene product depending on the location of the new stop codon. When a multiple of three nucleotides is either inserted or deleted, in-frame insertions and deletions result in single or multiple amino acids being removed or added without affecting the remainder of the protein (Figure 1).
Mutation Nomenclature
The standard nomenclature for numbering nucleotides and codons within a gene begins with the A of the initiation or “start” codon (ATG) representing nucleotide 1 and ATG as codon 12. Generally, only consecutive nucleotides constituting the coding region of the gene are numbered. For an extensive review of mutation nomenclature, see www.hgvs.org/mutnomen.html or reference #2. Universally accepted nomenclature exists for describing mutations/genetic variants at both the DNA and protein levels.
The DNA level description of a nucleotide alteration begins with the affected nucleotide number(s) followed by the original nucleotide, the type of change (substitutions are designated as “>”, deletions as “del”, insertions as “ins”), and the new nucleotide. For example, a single nucleotide substitution of an A (alanine) to a G (guanine) at nucleotide position 100 would be written as 100 A>G or 100 a>g. A deletion of A at nucleotide 100 would be written as 100delA, and a single nucleotide insertion of a C between nucleotide 100 and 101 would be written as 100_101insC. Intronic nucleotides are numbered relative to either the first or last nucleotide in the exon preceding or following the intron. When a nucleotide change occurs in the intron preceding the exon (i.e. the acceptor splice site), the nucleotide position is considered a negative position relative to the first nucleotide of the exon. When the nucleotide change occurs in the intron that follows an exon (i.e. donor splice site), the position would be a positive position relative to the last nucleotide of the exon. For example, the KCNQ1 exon 2, nucleotide substitution: 477 +5 G>A, results from a G to A substitution in the intron, 5 nucleotides following exon 2, where nucleotide 477 is exon 2’s final nucleotide3.
The protein level description of a nucleotide substitution begins with the original amino acid (using either the amino acid’s single or three letter abbreviation while an “X” or “ter” denotes a termination or “stop” codon) in which the affected codon encodes for, followed by the codon number, then the amino acid for which the mutated codon (resulting from the nucleotide exchange) encodes. For example, a nucleotide alteration that results in the exchange of a methionine (M or met) to a leucine (L or leu) at codon 56 (a missense mutation) would be written as M56L or MET56LEU. A nucleotide substitution that results in Tyrosine (Y or tyr) being mutated to a stop codon (X or ter) at codon 57 (a nonsense mutation) is indicated as Y57X or TYR57TER. A nucleotide substitution where the new codon at position 58 does not specify a new amino acid (silent or synonymous mutation) would be written as Y58Y. A nucleotide substitution altering normal splicing would be written as M159sp where at position 159, methionine (M) is the last normal amino acid prior to the splicing error.
Nucleotide insertions and deletions may result in either in-frame or frameshift alterations in the transcript. An in-frame deletion (del) of the methionine at position 56 would be written as M56del, and a deletion of both M56 and Y57 would be written as M56_Y57del. An in-frame insertion (ins) of a serine (S) between methionine at position 56 and tryosine at 57 would be written as M56_Y57insS. Frame-shift (fs) mutations as a result of either a deletion or insertion of nucleotides are generally written as the amino acid that is affected followed by “fsX” and then the codon number preceding the new termination or stop codon (X). For example, M56fsX62 or M56fs + 5X represents a frameshift mutation where methionine (M) at 56 is the first amino acid that is “frame-shifted” followed by 5 additional frame-shifted amino acids before the protein prematurely truncates with a new stop codon at position 62.
Genotyping Jargon
Inherited variation in the genome is the basis of human and medical genetics. Reciprocal forms of genetic information at a specific locus (location) along the genome are called alleles. An allele can refer to a segment of DNA or even a single nucleotide. The normal version of genetic information is often considered the “wild-type” or “normal” allele. The vast majority of the human genome represents a single version of genetic information. The DNA from one person is mostly made up of the same exact nucleotide sequence as another person. However, there are many small sections of sequence or even single nucleotides that differ from one individual to another. These normal variations at distinct loci in the DNA sequence are called polymorphisms1.
Some polymorphisms are very common and others represent rare genetic variants. In medical genetics, a disease-causing mutation refers to a DNA sequence variation that represents an abnormal allele and is not found in the normal healthy population but exists only in the disease population and produces a functionally abnormal product. However, this definition presumes a depth of understanding of a particular “mutations” presence or absence among “normals” and the production of a biologically perturbed protein. In general, a single nucleotide substitution that occurs with a measurable frequency (i.e. > 0.5% allelic frequency) among a particular ethnic population(s) is designated as a single nucleotide polymorphism (SNP) while those single nucleotide polymorphisms that occur less frequently than this threshold are termed mutations.
Unfortunately, this terminological distinction has probably brought about more confusion than clarity as SNPs do not necessarily have to be benign and a mutation may not be pathogenic. In fact, SNPs can represent functional, biological, and clinically relevant disease/treatment modifying biomarkers. In addition, a “mutation” (i.e. a rare, < 0.5% frequency variant) can be “just there, just rare, just because.” As will be detailed later, distinguishing pathogenic mutations from simply rare ones is critical to genetic test interpretation and presently represents genetic testing’s “Achilles heel”.
A person is said to be homozygous when they have a pair of identical alleles, one paternal (from father) and one maternal (from mother). When the alleles are different, then that person is said to be heterozygous for that particular allele. The term genotype refers to a person’s genetic or DNA sequence composition at a particular loci, or at a combined body of loci and the term phenotype refers to a person’s observed clinical expression of disease in terms of a morphological, biochemical, or molecular trait4. Penetrance is the likelihood that a pathogenic mutation will have a discernible expression among a population of mutation-positive subjects. For example, a gene mutation with an ascribed penetrance of 75% means that 75% of patients that possess that particular gene mutation will exhibit a measurable, diagnostic feature (phenotype) indicative of its presence such as echocardiographic evidence of left ventricular hypertrophy in HCM or electrocardiographic evidence of QT prolongation in LQTS. The vast majority of heritable cardiomyopathies and channelopathies are associated with disease-susceptibility genes characterized by incomplete penetrance. Expressivity refers to the level of phenotypic expression, and when the phenotypic manifestations of individuals who have the same genotype are diverse, then there is variable expressivity. Reduced penetrance and variable expressivity create a significant challenge for the appropriate diagnosis, pedigree interpretation, and risk stratification for genetic disorders.
Genetic variants may be inherited (familial) or represent spontaneously derived (sporadic) de novo variants occurring for the first time in an individual. Variants/mutations are said to be germline if present in the gametes (sperm or ovum) or somatic if in cells other then gametes. Germline mutations may be transmitted to offspring while somatic mutations can not. Mosaicism refers to a condition were different cells in the same individual have a different genetic makeup. For example, an individual may have a mutation occur within his or her gametes, but either not in other specific cell types at all or in limited numbers of other cells of the body (i.e. in his sperm or her ovum (egg) but not in his or her peripheral blood lymphocytes). Germline or gonadal mosaicism is beginning to emerge as a recognized, albeit rare, pattern of transmission in some disorders, including LQTS5.
Genetic disorders are characterized by their patterns of transmission within families. There are four basic modes: autosomal dominant, autosomal recessive, X-linked dominant, and X-linked recessive1. These modes of inheritance are based mostly on two factors: 1) what type of chromosome (autosome or X-chromosome) the gene is located on and, 2) whether the disease phenotype is expressed only when both chromosomes have the abnormal allele (recessive) or if the phenotype can be expressed even when only one chromosome hosts the mutant allele (dominant). The majority of heritable cardiomyopathies and channelopathies are familial, rather than sporadic, and autosomal dominant rather than autosomal recessive.
Genetic Testing Methods
Biological Material Used
Typically, 5 to 15 cc of whole blood obtained from venipuncture placed in EDTA-containing tubes (“purple top”) is requested as the genomic DNA source for either research or clinical based genetic testing4. For some genetic tests, a 50 μl blood spot on a Guthrie DNA filter card maybe sufficient source for genomic DNA6. While not providing a sufficient amount of DNA for comprehensive genetic testing, DNA isolated from a buccal (mouth cheek) swab may be adequate for mutation-specific confirmatory testing of family relatives. Umbilical cord blood may be acquired at the time of birth for newborn screening. DNA derived from sperm or ovum (egg) may be helpful in assessing for gonadal mosaicism. For autopsy negative sudden unexplained death (SUD) cases, genetic testing can be completed on DNA isolated from EDTA-blood or from a piece of frozen ventricle myocardium tissue or tissue from any other organ (liver, spleen, thymus) with a high nucleus to cytoplasm ratio7. DNA from tissue in formalin or from paraffin embedded tissue however remains an unreliable source and may yield a false positive genetic test result8. Both research based and clinical genetic testing typically require a signed and dated informed consent to accompany the samples to be tested. Importantly, some clinical laboratories are currently accepting only certain sources of DNA for specific tests, usually venipuncture or buccal swabs.
General Techniques used in Genetic Testing at the Single Gene Level
In genetic testing, the elucidation of gene mutations usually involves the polymerase chain reaction (PCR) technique. PCR is used to amplify many copies of a specific region of DNA sequence within the gene of interest, defined by the location of uniquely designed PCR primers. Typically, 20–25 base pair (bp) forward and reverse single-stranded DNA oligonucleotide primers are designed to be complementary to reciprocal intronic DNA sequences flanking the exon of interest in order to produce PCR products or “amplicons” (200 to 400 bp in length) containing the desired DNA sequence to be analyzed. It is important that primers be designed properly in this mutation detection process as false negatives secondary to poor primer design and possible allelic drop-out can occur9. A well optimized PCR reaction will yield millions of copies of only the specific sequence of interest10.
Among research laboratories, PCR amplification is often followed by an intermediate mutation detection platform, such as single stranded conformational polymorphism, denaturing gradient gel electrophoresis, or denaturing high performance liquid chromatography. The sensitivity of detection with these methods range from around 80 – 95% and are highly dependent on proper optimization of conditions. Failure to optimize correctly will result in mutation detection failures. Following identification of a possible abnormality, direct DNA sequencing must be used to determine the precise underlying DNA alteration(s). Review and comparison of the resulting sequence chromatograms and the published “wild-type” DNA and amino acid sequence for the gene/protein of interest will allow for the determination of whether the underlying DNA change is protein altering and a potentially pathogenic or a non-pathogenic normal variant.
In contrast, for most commercially available genetic tests, the intermediate mutation detection platform is bypassed for direct DNA sequencing of all samples examined. Though this direct approach to mutational analysis is presently more expensive, its sensitivity, specificity, and speed for mutation detection are superior to the typical research-based platforms. Recent advances in DNA sequencing methodologies and technical instrumentation have rapidly evolved DNA sequencing capacity and genetic information output. For example, through the use of “next-generation” sequencing, massively parallel sequencing, and oligonucleotide hybridization-chip based technologies, the molecular interrogation of an individual’s complete library of specific disease associated protein-coding sequences in a single or a few reactions with remarkable cost-effectiveness11, 12 is now available in a limited number of laboratories. At the current pace, it is conceivable that specific gene panel genetic testing for specific heritable diseases will become extinct by 2020, being replaced by comprehensive human genome testing.
Together these techniques provide excellent precision and accuracy to detect i) single nucleotide substitutions that produce missense, nonsense, and splice site mutations and ii) small insertion/deletions. However, large whole gene, multiple exon, or single exon deletions or duplications elude detection by this approach. Another technique, multiple ligation probe analysis (MLPA), however, enables identification of such large gene rearrangements13–15. Besides MLPA, other technologies for copy number variant (CNV) detection exist.
Genetic Testing in a Research Versus Clinical Setting
Research studies are those in which patient samples are collected based on study design and inclusion or exclusion criteria. Research-based genetic tests are performed principally for discovery purposes and the advancement of science with direct benefit to the “research subject” as an ancillary feature whereas commercial, clinical, fee-based genetic testing is a patient-centric test ordered by a referring physician based on his/her clinical index of suspicion and clinical objective to establish/refute a considered diagnosis, obtain further risk stratifying information, and guide clinical decision making. The cost of research testing is generally covered by an investigator’s research program. Research laboratories, in accordance with their institutional review board (IRB), may be granted permission to inform a study participant of their genetic test result. Importantly, research-derived test results are provided initially to the research subject rather than to his/her health care provider. When applicable, research laboratories typically quote research subjects 6–24 months as the time frame to “expect” learning of a positive result from examination of established disease-susceptibility genes (if the known genes are even being examined) and of course, an indefinite and indeterminate time frame when research testing is strictly focused on novel discovery. In some cases, several years have elapsed, after submission of a blood sample, before the research participants have been the direct beneficiary of the testing results. In some research settings, patients may never be informed of either a positive or negative genetic test result. Genetic testing in the research environment often rely on assays developed in-house rather than on commercially available kits approved by the Food and Drug Administration (FDA)16. In the research setting, genetic tests are often available before analytical and clinical validity are established. Furthermore, quality control mechanisms and “good laboratory practices” are not under the same strict guidelines and regulations of a clinical diagnostic test.
In contrast, a clinical genetic test is a fee based test performed with the intent to inform both the health care provider and patient of the test result with a definite diagnostic, prognostic, and/or therapeutic goal in mind. The charge of the clinical genetic test is generally based on the complexity of the test and is generally proportional to the amount of genetic material being “scanned”. In the United States, a clinical laboratory must be Clinical Laboratory Improvement Amendments (CLIA) approved and regulated by the Centers for Medicaid and Medicare Services (CMS)17. In order to be CLIA-approved, the testing laboratory must meet quality control and proficiency testing standards in the accordance with “good laboratory practices”. On-site inspections are conducted to assure proper personnel qualifications, clinical testing procedures, and quality control measures and documentation.
Typically, the clinically available genetic test is a high throughput, automated, direct DNA sequencing based assay performed with 2–4-fold redundancy to maximize diagnostic accuracy. Unlike the research laboratory environment, the clinical laboratory is usually a highly ordered environment with personnel in specifically designated roles to ensure maximum efficiency and quality control measures of the genetic test. Rather than reporting results to the research participant after a long period of time as with research testing efforts, results (both positive and negative) from the clinical genetic test are reported to the ordering physician in a written report within approximately 4 to 8 weeks for index case testing and 2 to 4 weeks for mutation specific confirmatory testing for family members.
Genetic testing for several cardiomyopathies and cardiac channelopathies has made the quantum leap from research-based discovery and translation to the highly regulated clinical laboratory environment. Genetic testing for heritable cardiomyopathies and channelopathies has been recognized by leading cardiovascular societies throughout the world as clinically relevant tests18. Further, in the United States, many third party payers have begun to acknowledge their clinical utility and have implemented favorable reimbursement policies. For example, in April 2008, the Blue Cross Blue Shield Technology Evaluation Center concluded that LQTS genetic testing was no longer “investigational”19.
Clinically Available Genetic Testing for Cardiovascular Diseases
Once confined to the research laboratory, genetic testing for HCM, DCM, ARVC, LQTS, CPVT, and BrS-associated mutations has matured into clinically available diagnostic tests for physicians evaluating and treating patients with these diseases. An international index of clinically available genetic tests and testing centers is inventoried at GeneTests, a continually updated, publically funded, on-line medical genetics information resource (www.genetests.org) for physicians and other health care providers20. Table 1 lists genetic testing centers currently offering commercially available clinical genetic testing for the major cardiomyopathies and cardiac channelopathies. In the United States, GeneTests lists PGxHealth (www.pgxhealth.com), GeneDx (www.genedx.com), and Correlagen Diagnostics (www.correlagen.com) as three CLIA-approved clinical laboratories that offer fee-based clinical genetic testing for both cardiomyopathies (HCM, DCM, and ARVC) and cardiac channelopathies (LQTS, CPVT, and BrS). Presently, only PGxHealth is a recognized Medicare and Medicaid (40 of 50 states) provider. In addition, Partners Healthcare Centers for Personalized Genetic Medicine (www.hpcgg.org) at Harvard Medical School offers genetic testing for HCM, DCM, and ARVC.
Table 1.
Clinical Genetic Testing Centers
| HCM | LQTS | CPVT | BrS | |
|---|---|---|---|---|
| NORTH AMERICA | ||||
|
Correlagen Diagnostics, Inc. Waltham, MA www.correlagen.com |
Y | Y | Y | Y |
|
GeneDx Gaithersburg, MD www.genedx.com |
Y | Y | Y | Y |
|
Masonic Medical Research Laboratory, Molecular Genetics Program Utica, NY www.mmrl.edu |
- | - | - | Y |
|
Partners Healthcare, Harvard Medical School, Laboratory for Molecular Medicine Boston, MA www.hpcgg.org |
Y | - | - | - |
|
PGxHealth New Haven, CT www.pgxhealth.com |
Y | Y | Y | Y |
|
PreventionGenetics, Molecular Diagnostics and BioBanking Marshfield, WI www.preventiongenetics.com |
Y | - | Y | - |
| EUROPE | ||||
|
Academic Medical Centre, University of Amsterdam, DNA Diagnositcs Laboratory Amsterdam, Netherlands www.cardiogenetica.nl |
Y | Y | Y | Y |
|
Centogene GmbH, Institute of Molecular Diagnostics Rostock, Germany www.centogene.com |
Y | - | - | - |
|
Centre Hospitalier Universitaire de Grenoble, Biochimie et Genetique Moléculaire Grenoble, France |
- | - | Y | - |
|
CGC Genetics Porto, Portugal www.cgcgenetics.com |
Y | - | - | - |
|
Diagenom GmbH, Medical Genetics Laboratory Rostock, Germany www.digenom.de |
Y | - | - | - |
|
IRCCS Fondazione Salvatore Maugeri, Molecular Cardiology Laboratories Pavia, Italy |
- | Y | Y | Y |
|
Institute of Human Genetics Goettingen, Germany |
Y | - | - | - |
|
Rikshospitalet, Molecular Genetics Laboratory Oslo, Norway |
Y | Y | Y | Y |
|
Sistemas Genomics SL Medical Genetics Unit Paterna, Comunidad Valenciana, Spain www.sistemasgenomics.com |
- | Y | - | Y |
|
St Mary’s Hospital Manchester, Regional Molecular Genetics Service Manchester, United Kingdom www.mangen.co.uk |
- | - | Y | - |
|
University Hospital of Umea, Department of Clinical Genetics Umea, Sweden |
Y | Y | Y | Y |
| NEW ZEALAND | ||||
|
LabPLUS, Auckland City Hospital, Molecular Genetics Laboratory (Diagnostics Genetics) Auckland, New Zealand |
- | Y | - | Y |
Centers represent those listed on GeneTests.org as of May 201020 offering fee-based clinical genetic testing. The genes tested and methods used for each disorder will vary between centers. For more details and contact information for each center, see www.GeneTests.org.
These commercial laboratories utilize a variety of technologies including oligonucleotide hybridization-based chip methodology, traditional dideoxy sequencing direct DNA sequencing, or high-throughput “next-generation” or “massively parallel” sequencing. The analytical sensitivity of these test are typically 95 to 100% for the detection of nucleotide substitutions and small insertion/deletion mutations. Some laboratories also currently offer MLPA analysis for the detection of large gene rearrangements that would escape detection when using standard DNA sequencing methodologies. While most laboratories provide genetic testing for the most common susceptibility genes for each of the disease, some laboratories offer genetic testing for even the rarest forms of each disorder (Tables 2 and 3). While some genetic test vendors comprehensively analyze the full protein-encoding region of each gene, others analyze only targeted regions of genes where mutations have been shown to reside.
Table 2.
Molecular Basis of Hypertrophic Cardiomyopathy
| Gene | Locus | Protein | Frequency |
|---|---|---|---|
| Myofilament (Sarcomeric) Hypertrophic Cardiomyopathy (HCM) | |||
| Giant Filament | |||
| TTN | 2q31 | Titin | Rare |
| Thick Filament | |||
| MYH7* | 14q11.2-q12 | β-Myosin heavy chain | 25–35% |
| MYH6* | 14q11.2-q12 | α-Myosin heavy chain | Rare |
| MYL2* | 12q23-q24.3 | Regulatory myosin light chain | Rare |
| MYL3* | 3p21.2-p21.3 | Essential myosin light chain | Rare |
| Intermediate Filament | |||
| MYBPC3* | 11p11.2 | Cardiac myosin-binding protein C | 25–35% |
| Thin Filament | |||
| TNNT2* | 1q32 | Cardiac troponin T | 3–5% |
| TNNI3* | 19p13.4 | Cardiac troponin I | 1–5% |
| TPM1* | 15q22.1 | α-Tropomyosin | 1–5% |
| ACTC* | 15q14 | α-Cardiac actin | Rare |
| TNNC1* | 3p21.1 | Cardiac troponin C | Rare |
| Z-Disc HCM | |||
| ACTN2 | 1q42-q43 | α-Actinin 2 | Rare |
| CSRP3* | 11p15.1 | Muscle LIM protein | Rare |
| LBD3 | 10q22.2-q23.3 | LIM binding domain 3 | Rare |
| MYOZ2 | 4q26-q27 | Myozenin 2 | Rare |
| TCAP | 17q12-q21.1 | Telethonin | Rare |
| VCL | 10q22.1-q23 | Vinculin/metavinculin | Rare |
| Calcium-Handling HCM | |||
| CALR3 | 19p13.11 | Calreticulin 3 | Rare |
| CASQ2 | 1p13.3-p11 | Calsequestrin | Rare |
| JPH2 | 20q13.12 | Junctophilin 2 | Rare |
| PLN | 6q22.1 | Phospholamban | Rare |
| Metabolic Cardiac Hypertrophy (HCM Mimickers) | |||
| FXN | 9q13 | Frataxin | Rare |
| GLA* | Xq22 | α-Galactosidase A | Rare |
| LAMP2* | Xq24 | Lysosome-associated membrane protein 2 | Rare |
| PRKAG2* | 7q35-q36.36 | AMP-activated protein kinase | Rare |
| RAF1 | 3p25.2 | RAF serine/threonine kinase | Rare |
Genes available as a commercially available genetic test
Rare is defined as < 1% anticipated contribution to the syndrome.
Note that the rare disease-susceptibility genes are indicated in alphabetical order.
Table 3.
Molecular Basis of Cardiac Channelopathies
| Gene | Locus | Protein | Frequency |
|---|---|---|---|
| Long QT Syndrome (LQTS) | |||
| KCNQ1 (LQT1)* | 11p15.5 | Kv7.1 | 30–35% |
| KCNH2 (LQT2)* | 7q35-36 | Kv11.1 | 25–30% |
| SCN5A (LQT3)* | 3p21-p24 | NaV1.5 | 5–10% |
| AKAP9* (LQT11) | 7q21-q22 | Yotiao | Rare |
| ANKB* (LQT4) | 4q25-q27 | Ankyrin B | Rare |
| CACNA1C* (LQT8) | 12p13.3 | L-type calcium channel | Rare |
| CAV3* (LQT9) | 3p25 | Caveolin 3 | Rare |
| KCNE1* (LQT5) | 21q22.1 | MinK | Rare |
| KCNE2* (LQT6) | 21q22.1 | MiRP1 | Rare |
| KCNJ2* (LQT7 and ATS1) | 17q23 | Kir2.1 | Rare |
| KCNJ5 (LQT13) | 11q23.3 | Kir3.4 | Rare |
| SCN4B* (LQT10) | 11q23.3 | Sodium channel β 4 subunit | Rare |
| SNTA1* (LQT12) | 20q11.2 | Syntrophin α 1 | Rare |
| Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) | |||
| RYR2 (CPVT1)* | 1q42.1-q43 | Ryanodine receptor 2 | 50–60% |
| KCNJ2 (CPVT3)* | 17q23 | Kir2.1 | 10% |
| CASQ2* (CPVT2) | 1p13.3-p11 | Calsequestrin 2 | 1–2% |
| Brugada Syndrome (BrS) | |||
| SCN5A (BrS1)* | 3p21-p24 | NaV1.5 | 20–30% |
| CACNA1C* (BrS3) | 2p13.3 | L-type calcium channel | Rare |
| CACNB2* (BrS4) | 10p12 | L-type calcium channel β 2 subunit | Rare |
| GPD1L* (BrS2) | 3p22.3 | Glycerol-3-phosphate dehydrogenase 1-like | Rare |
| KCNE3* (BrS6) | 11q13.4 | MiRP2 | Rare |
| KCNJ8 (BrS8) | 12p12.1 | Kir6.1 | Rare |
| SCN1B* (BrS5) | 19q13.1 | Sodium channel β 1 subunit | Rare |
| SCN3B (BrS7) | 11q24.1 | Sodium channel β 3 subunit | Rare |
Genes available as a commercially available genetic test.
Rare is defined as < 1% anticipated contribution to the syndrome.
Note that the rare disease-susceptibility genes are indicated in alphabetical order.
Although the disease genotypic subtype is provided in parentheses for these rare disease susceptibility genes, we recommend annotating only the common genotypes with such a numerical designation and refer to the rare ones with their gene descriptor such as AKAP9-LQTS rather than LQT11 and GPD1L-BrS rather than BrS2.
Once a disease-causing mutation is identified within a proband, mutation-specific genetic testing is offered for family members. Where published, the index test ranges in price from about $3000 – $5500 (USA) depending on the test and mutation-specific confirmatory testing costs $250 to $900, depending on the genetic testing vendor. As previously indicated, the insurance reimbursement landscape is rapidly changing with favorable coverage policies being adopted akin to the same “evolutionary” process that other genetic tests like cystic fibrosis and breast cancer genetic testing have undergone previously.
Importantly, as discussed in the subsequent sections for each particular disorder, the anticipated yield from genetic testing is disease-specific ranging from a low of 25–30% for BrS genetic testing to a 75% yield for LQTS genetic testing. As such, patients with a robust, unequivocal clinical phenotype for their respective disorder who have a “negative” genetic test should at least be informed of on-going research efforts and directed towards research centers specializing in the study of their particularly disorder. Continued genetic analysis on a research basis of well phenotyped patient cohorts will allow for new gene discovery and continued enhancement, expansion, and refinement of genetic testing in clinical practice.
Potential Benefits and Indications for Genetic Testing
Genetic testing provides clear diagnostic, prognostic, and therapeutic implications for some disorders21–23. Genetic testing may 1) offer diagnostic value for symptomatic individuals by elucidating the exact molecular basis for the disorder, 2) establish a definitive molecular diagnosis or disease prediction when the clinical evaluation for the disorder is inconclusive, 3) confirm or exclude the presence of a disease-causing mutation in pre-symptomatic family members/relatives with a family history of a genetic disorder, and 4) help personalize treatment recommendations and management of a patient’s specific disorder by characterization of the precise genotype24, 25. Genetic testing may also provide “carrier status” for those concerned about recessively inherited disorders like cystic fibrosis.
As an aside, the term carrier should not be used for the majority of the genetic heart diseases to be discussed in the following section. A carrier generally refers to someone who solely carries a disease-susceptibility mutation but he/she has no risk of expressing a phenotype associated with that particular pathogenic mutation. For the majority of the cardiomyopathies/channelopathies, a single mutated allele (autosomal dominant) is sufficient for disease expression in contrast to cystic fibrosis. As such, a mutation-positive subject is not simply a “carrier” but potentially has risk for disease expression. For clarity, the term “carrier” should be reserved for autosomal recessive conditions and should not be used to describe patients with diseases like HCM and LQTS.
As will be discussed in more detail in future sections of this review, specific indications for and utility of genetic testing vary depending on the disorder. For example, while a recommendation for universal neonatal genetic testing for LQTS would be utterly irresponsible as would pre-sports participation HCM genetic testing, clinical suspicion and first degree relative confirmatory testing are clear indications for genetic testing in these various disorders (Figure 2). Most importantly, rather than proceeding with shotgun-based genetic testing, clinical evaluation should be used to phenotypically guide genetic testing and provide specific indicators for which genetic test(s) to order. For illustrative purposes, Figure 3 provides an example of a work flow decision tree for possible indicators for genetic testing when evaluating a patient with exercise-induced syncope. Further, the degree to which genetic testing contributes to the triad of diagnostic, prognostic, and therapeutic utility is also disease specific with LQTS genetic testing contributing to the entire triad while genetic testing for other diseases may be of diagnostic value only for the index case and his/her relatives (Figure 4).
Figure 2. Indications for Genetic Testing.

Provided is a table of possible indications for genetic testing for hypertrophic cardiomyopathy (HCM), long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BrS). LVH = left ventricular hypertrophy, TdP = torsade de pointes, SUD = sudden unexplained death. The + symbol represents a positive indication for genetic testing. The − symbol represents an indicator that does not warrant for genetic testing for the specific disorder. The +/− symbol represents an indicator that may or may not warrant genetic testing.
Figure 3. Possible Indicators for Genetic Testing in Exercise-Induced Syncope.

Depicted for illustrative purposes is a work flow decision tree for indicating which genetic test would be appropriate to order based on the clinical evaluation of an individual manifesting with exercise-induced syncope. The blue boxes represent specific clinical evaluation tests. The yellow boxes represent specific genetic testing panels. The + symbol represents a positive evaluation and the − symbol represents a negative evaluation for the respective clinical test. The key point is that genetic testing for these disorders should be phenotype-guided, NOT universal.
Figure 4. Utility of Genetic Testing.

Shown is the current diagnostic, prognostic, and therapeutic utility of genetic testing for hypertrophic cardiomyopathy (HCM), long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BrS). The + symbol indicates the test has utility, the − symbol indicates no current measurable utility, and +/− indicates the test may have some utility.
INTERPRETATION OF GENETIC TEST RESULTS – THE “VARIANT OF UNCERTAIN/UNCLEAR SIGNIFICANCE (VUS)” ISSUE
Because of the potential enormity and severe consequences surrounding the misdiagnosis and mismanagement of patients with these potentially lethal cardiac disorders, the clinical evaluation and management of a patient and family suspected of having genetic heart disease should be conducted under the supervision of a pediatric or adult cardiologist with specific expertise in heritable channelopathies/cardiomyopathies4. Due to the issues associated with incomplete penetrance and variable expressivity, the genetic test result must be interpreted cautiously and incorporated into the overall diagnostic evaluation for these disorders. Even when a genetic variant has been published previously as a putative pathogenic mutation, assignment of a specific genetic variant as a true pathogenic disease-causing mutation still requires vigilant scrutiny. To be sure, genetic tests are fundamentally probabilistic tests rather than absolutely deterministic ones. To illustrate further, in contrast to rare, pathogenic LQTS-associated channel mutations present in less than 1 in 2500 persons (0.04%) and in 75% of clinically strong LQTS cases, comprehensive genetic testing of KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) for over 1300 ostensibly healthy volunteers has demonstrated that approximately 4% of Caucasians and up to 8% non-Caucasians host rare (< 0.5% allelic frequency) non-synonymous (amino acid altering) genetic variants in these cardiac channel genes26. Some of the variants observed in this healthy population may represent sub-clinical disease modifiers while the vast majority must represent benign background “genetic noise”.
Although exposed for the LQTS genes, this observation of background non-pathogenic missense variants is not confined to this disorder, but likely extends throughout all of the heritable cardiomyopathies and channelopathies detailed here and may well extend to virtually every gene in the human genome. However, while there are currently over 1500 clinically available genetic tests for various disorders, only for a handful of diseases is the “signal-to-noise” ratio regarding genetic variation known as it is for the major LQTS-susceptibility genes. For LQTS genetic testing, this effort has enabled a case/control mutational analysis of the properties and localization of case-associated mutations compared to the compendium of presumably innocuous variants26. Here, algorithms based on mutation location may assist in distinguishing pathogenic mutations from an otherwise rare VUS and perhaps allow for the assignment of an estimated probability of pathogenicity of each novel mutation identified within a specific gene26. This probabilistic rather than binary nature of genetic testing is depicted in Figure 5 showing that while rare nonsense, insertion/deletions, and splice site mutations are high probability LQTS-susceptibility mutations, the probability of pathogenicity for missense mutations is strongly predicated on location. For example, missense mutations localizing to the transmembrane spanning/pore domains of the LQT1- and LQT2-associated potassium channels are high probability disease mutations whereas a similarly rare missense mutation that localizes to the domain I–II linker of the LQT3-associated sodium channel is absolutely uncertain (i.e. a VUS). Without co-segregation or functional data, such a mutation has a point estimate for probability of pathogenicity of < 50% which necessitates extreme caution when interpreting the genetic test.
Figure 5. Probabilistic Nature of LQTS Genetic Testing.
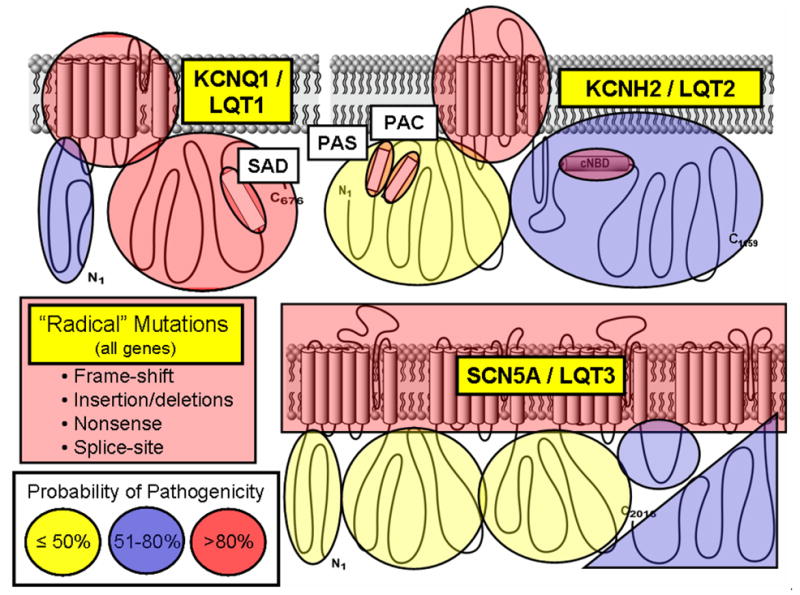
Depicted are the three major ion channels causative for LQTS with areas of probability of pathogenicity shown for mutations localizing to these respective areas. While “radical” mutations have a greater then 90% probability of being a true pathogenic mutation, the level of probability for missense mutations vary depending on their location for each channel protein. Missense mutations residing in red shaded areas have a high probability (>80%) of being pathogenic, those in blue are possibly (51–80%) pathogenic, and those in yellow shaded areas truly represent variants of uncertain significance (VUS, ≤50% probability) clinically26. Figure adapted from Tester and Ackerman83.
GENETIC COUNSELING
If the ordering cardiologist, heart rhythm specialist, or cardiomyopathy/channelopathy sub-specialist lacks genetic expertise with regard to the particular disorder being considered, it is essential to have a masters-trained board certified genetic counselor, preferably with specialized training in cardiovascular genetics, as part of the team to be involved in the communication process with the patient concerning the implications of genetic testing and genetic test results. A genetic counselor may be instrumental in 1) collecting a family history comprising at least three–four generations, 2) providing information as to the clinical presentation of the disorder, mode of inheritance, and implications in family planning, 3) explaining the benefits, limitations, risk, availability, costs, and potential outcomes of genetic testing, and 4) discussing the possible psychosocial impact of these potentially lethal disorders with the patient and their family27, 28.
ETHICAL, LEGAL, AND SOCIETAL IMPLICATIONS
While benefits such as diagnostic certainty and greater awareness of prophylactic treatment and risk stratification may be achieved, genetic testing may also contribute to an increase in risk for depression, anxiety, guilt, stigmatization, discrimination, family conflict, and unnecessary or inappropriate use of risk-reducing strategies29. Therefore, it is imperative that patients are well informed on genetic testing implications and must not be coerced into providing a sample for genetic analysis. Full disclosure must be given as to the research or clinical intent of the genetic test, the results of the analysis, and who will have access to the results4.
Genetic information must be considered private and personal information with the potential for mishandling30, 31. Confidential information disclosure to third parties, including insurance companies or employers can have genetic discriminatory consequences to the patient. However, in May 2009, the Genetic Information Nondiscrimination Act (GINA) was signed into federal law barring employers and health insurers from denying employment or insurance to a healthy individual based on genetic test results32. Despite this welcomed advance, the law failed to extend discriminatory protection over either life insurance or disability insurance.
Genetic testing should be considered both a family and individual experience29. While genetic testing is performed on an individual’s genetic material, both the individual’s decision to undergo genetic testing and that individual’s test results may have substantial implications for other family members, especially in sudden cardiac death related disorders. However, under current practice anchored in the principle of autonomy, only the individual being tested or the legal guardian, if a minor, has to be informed of their genetic test results. The decision or responsibility to inform unsuspecting relatives of the potential for genetic predisposition for sudden cardiac death, resides solely on that informed patient4.
GENETIC TESTING FOR SPECIFIC HERITABLE CARDIOMYOPATHIES AND CHANNELOPATHIES
Cardiomyopathies and Channelopathies-Clinical Descriptions, Genetics, and Indications/Benefits of Genetic Testing
Although commercially available genetic testing exists for HCM, DCM, ARVC, LVNC, LQTS, SQTS, CPVT, and BrS, this review will focus on the four genetic cardiomyopathies/channelopathies (HCM, LQTS, CPVT, and BrS) where the clinical utility is arguably the greatest at the present time. DCM and ARVC genetic testing is evolving rapidly and though it is commercially available, genetic testing for DCM and ARVC is presently handicapped by low yields, unclear clinical impact apart from first degree relative confirmatory mutation testing, and comparatively high “background noise rates” that make DCM and ARVC genetic test interpretations extremely challenging.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized as asymmetrical left ventricular hypertrophy in the absence of a clinically identifiable cause. HCM affects 1 in 500 people and more importantly is the most common cause of premature sudden cardiac death in the young, especially the young athlete. Clinically, HCM is viewed principally as disease of incomplete penetrance and variable expressivity, as the clinical course varies extremely, ranging from an asymptomatic lifelong course to chest pain, exertion-related dyspnea, syncope, progressive exercise intolerance, or sudden death as the sentinel, event occurring at any age.28, 33–35 Morphologically, HCM may present with negligible to extreme hypertrophy, minimal to extensive fibrosis and myocyte disarray, and absent to severe left ventricular outflow tract obstruction. Distinct septal contours/morphologies such as reverse curve-, sigmoidal-, and apical variant-HCM have been identified.33 HCM is mostly inherited in an autosomal dominant fashion.
The most common genetic subtype of HCM is sarcomeric- or myofilament-HCM, secondary to mutations in genes encoding for proteins of the thick and thin myofilaments of the cardiac sarcomere (Table 2). The yield from HCM gene testing that involves the 8 or 9 established HCM-susceptibility genes for sarcomeric-HCM ranges from 35 – 65% among several different, international research cohorts of unrelated patients who met the clinically accepted definition of HCM21, 28, 36, 37. In addition, echocardiography may provide echo-guided genetic testing to the clinician by providing an estimate of the apriori probability of a positive genetic test based upon morphologic subtyping.38 Among the two most common morphological subtypes of HCM, reverse septal curvature or reverse curve-HCM and HCM with a sigmoidal shaped ventricular septum or sigmoidal-HCM, patients with reverse curve-HCM have a 80% chance of a positive genetic test whereas only a minority of patients with sigmoidal-HCM will test positive.
MYBPC3 and MYH7 are, by far, the two most common HCM-associated genes with an estimated prevalence of 25 – 35% for each gene and account for the majority of positive research based genetic tests. Additionally, genes encoding components of the cardiac Z-disc, calcium (Ca2+)-handling, and regulatory proteins have been associated with HCM pathogenesis. Mutations in several genes encoding proteins responsible for cellular metabolic processes have been linked to unexplained left ventricular hypertrophy which can mimic the HCM-phenotype. To date, hundreds of mutations have been identified in at least 27 putative HCM-susceptibility genes (Table 2).
In terms of the diagnostic, prognostic, and therapeutic implications of genetic testing, HCM genetic testing contributes primarily to the diagnostic part of the triad and secondarily to prognosis and therapeutic impact (Figure 4). While a negative genetic test can not “rule out” HCM, a positive genetic test may play a important role in distinguishing HCM from exercise adaptive hypertrophy (so called athlete’s heart) or from HCM phenocopies such as Anderson-Fabry’s, glycogen (PRKAG2) and lysosomal (LAMP2) storage, mitochondrial, Noonan and LEOPARD syndromes, for which there are definitive and alternative therapies distinct from HCM treatments35. Additionally, genetic testing of the proband may provide the diagnostic gold standard for his/her relatives. A positive genetic test would enable systematic scrutiny of an HCM-affected proband’s relatives to identify those relatives who are mutation positive irrespective of their currently manifest clinical phenotype, allowing for early detection and appropriate targeting of relatives for ongoing surveillance while dismissing those mutation negative/phenotype negative family members and thereby the need for regular cardiac evaluations and echocardiograms. Further, “chemoprevention” may be possible in the future whereby strategies to prevent or delay the onset of hypertrophy among those patients who are genotype positive but hypertrophy negative. Drawing from promising animal data, a randomized study called “DELITE” that involves prophylactic diltiazem pharmacotherapy is underway for humans with mutation positive/LVH negative-HCM39.
Since the seminal observation in 1992, there has been much debate as to whether the genetic test result provides an independent risk factor for either degree of hypertrophy or susceptibility for sudden death. Watkins and colleagues noted that within families hosting particular MYH7 mutations, some, like MYH7-R453C, were associated with a 50% mortality at 40 years while others seemed to confer immortality40. This observation set into motion the notion of “malignant” mutations and “benign” mutations. However, among unrelated patients, each discrete HCM-causative mutation is rare and the previous genotype-phenotype associations may not hold firm. In fact, among the first 400 unrelated patients with HCM that we tested, only 3 possessed one of the mutations coined as “malignant” and 7 patients had one of the “benign” mutations for which 4 already underwent a surgical myectomy, 3 had a positive family history of premature, sudden death, and 1 patient received a heart transplant at the age of 16 despite her “benign” form of HCM41, 42. Presently, great restraint must be exercised when tempted to make a prophylactic implantable cardioverter defibrillator decision based upon the genetic test result. Instead, the current clinical evidence supports a prognostic “mutation class effect” whereby unrelated patients with a positive genetic test for sarcomeric-HCM exhibit a more severe phenotype in terms of degree of hypertrophy, age at diagnosis, and likelihood to progress to end stage disease then do clinically diagnosed HCM patients with a negative genetic test43, 44. However, it is not clear how this observation translates to the bedside in terms of therapeutic approach for the patient with sarcomeric/myofilament-HCM (i.e. a positive genetic test) compared to the patient with negative genetic test-HCM.
Long QT Syndrome
With prevalence as high as 1 in 2500 persons45, congenital long QT syndrome (LQTS), comprises a distinct group of cardiac channelopathies characterized by delayed cardiac repolarization manifesting as QT prolongation on a resting 12-lead surface electrocardiogram (ECG) in the setting of a structurally normal heart. Patients have an increased risk for syncope, seizures, and SCD, usually precipitated by triggers such as physical exertion (swimming), emotion, auditory stimuli (alarm clocks, door bells, etc.), or during the postpartum period22, 23. While typically the heart’s normal sinus rhythm spontaneously returns, resulting in “just” an episode of “torsadogenic” syncope, 5% of untreated and unsuspecting LQTS individuals succumb to a fatal arrhythmia as their first event. As much as 20% of autopsy negative SUD46 and 10% of sudden infant death syndrome (SIDS) may be attributed to LQTS47. Like all the cardiac channelopathies discussed in this review, incomplete penetrance and variable expressivity are hallmark features of LQTS.
LQTS is a genetically heterogeneous disorder most often inherited in an autosomal dominant manner (previously known as Romano-Ward syndrome). Rarely, LQTS presents as the recessive trait first described by Drs. Jervell and Lange-Nielsen and is characterized by a severe cardiac phenotype and sensorineural hearing loss. In addition, 5–10% of LQTS results from a spontaneous/sporadic germline mutation rather than familial inheritance. Hundreds of mutations have now been identified in 13 LQTS-susceptibility genes (Table 3) with approximately 75% of clinically strong LQTS due to mutations in three genes: KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) that critically orchestrate the cardiac action potential of the ventricular myocytes22, 23, 48. The remainder of genotype-positive LQTS stems from mutations in genes that encode either other cardiac channels, channel interacting proteins, or structural membrane scaffolding proteins that modulate channel function. These minor LQTS-susceptibility genes associated with LQT4-13 contribute < 5% of LQTS.
The majority of LQTS-causing mutations are coding region single nucleotide substitutions or small insertion/deletions 48–50. However, recently a few large gene rearrangements involving hundreds to thousands of nucleotides resulting in single or multiple whole exon deletions/duplications have been described13, 51. Importantly to note, 20 – 25% of LQTS remains genetically elusive. Thus, continued genetic testing on a research basis of clinically strong yet genotype negative (negative for LQT1-13) patients will be critical for novel gene discovery.
Phenotype-genotype associations may facilitate phenotype-directed genetic testing for LQTS. For example, swimming and exertion-induced cardiac events strongly indicate mutations in KCNQ1, whereas auditory triggers and events occurring during the postpartum period should prompt suspicion for LQT2, and an event during sleep is associated with LQT3 (Figure 6)22, 23, 52–56. In contrast, exercise-induced syncope in the setting of a normal QTc (< 460 ms) should move suspicion away from “concealed LQT1” and instead point towards a phenotypic mimicker of concealed LQT1, namely Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)57. Characteristic gene-suggestive ECG patterns have been described previously22, 23. LQT1 is associated with a broad-based T wave, LQT2 with a low amplitude notched or biphasic T wave, and LQT3 with a long isoelectric segment followed by a narrow-based T wave. These gene-suggestive ECG patterns may be helpful in guiding genetic testing, however exceptions to these relatively gene-specific T-wave patterns exist and due caution must be exercised with making a pre-genetic test prediction of the particular LQTS subtype. For example, the most common clinical mimicker of the LQT3-looking ECG is seen among patients with LQT1. The clinical management in the “mind set of LQT3” for a patient who is actually LQT1 could prompt an unnecessarily aggressive prophylactic internal cardioverter defibrillator (ICD) recommendation.
Figure 6. Genotype-Phenotype Correlations in Long QT syndrome.

Seventy-five percent of clinically strong LQTS is due to mutations in three genes (35% KCNQ1, 30% KCNH2, and 10% in SCN5A) encoding for ion channels that are critically responsible for the orchestration of the cardiac action potential. Genotype-phenotype correlations have been observed, including swimming/exertion and LQT1, auditory triggers/postpartum period and LQT2, and sleep/rest and LQT3. The bar graphs represent genotype-phenotype data from reference 59. Also illustrated is the relative gene-specific effectiveness in β blocker therapy where β blockers are extremely protective in LQT1 patients, moderately protective in LQT2, and may not provide sufficient protection for those with LQT3. The late sodium current blockers like mexiletine, ranolazine, and propranolol may be protective in LQT3.
As such, genetic testing not only has significant diagnostic implications, but also has prognostic and therapeutic implications in LQTS (Figure 4). For example, the underlying genetic basis heavily influences the response to standard LQTS pharmacotherapy (β blockers), where β blockers are extremely protective in LQT1 patients, moderately protective in LQT2, and may not provide sufficient protection for those with LQT3 (Figure 6)22, 23, 58. However, targeting the pathologic, LQT3-associated late sodium current with agents such as mexiletine, flecainide, ranolazine, or propranolol may represent a gene-specific therapeutic option for LQT3 (Figure 6) 22, 23, 59, 60.
In addition, intra-genotype risk stratification has been realized for LQT1 and LQT2 based upon mutation type, mutation location, and cellular function61–66. For example, patients with LQT1 transmembrane localizing missense mutations have a two-fold greater risk of a LQT1-triggered cardiac event than LQT1 patients with C-terminal localizing mutations. Trumping location, LQT1 patients with mutations resulting in a greater degree of ion channel loss-of-function at the cellular in vitro level (i.e. dominant negative) have a two-fold greater clinical risk than those mutations that damage the biology of the channel less severely (haploinsufficiency). Patients with LQT2 secondary to pore region mutations have a longer QTc, a more severe clinical manifestation of the disorder, and have significantly more arrhythmia-related cardiac events occurring at a younger age than those LQT2 patients with non-pore mutations61. Additionally, LQT2 patients with mutations involving the transmembrane pore region had the greatest risk for cardiac events, those with frame-shift/nonsense mutations in any region had an intermediate risk, and those with missense mutations in the C-terminus had the lowest risk for cardiac events66. In 2010, LQTS genotyping fully satisfies the triad in terms of diagnostic (move towards/move away from diagnosis in index case, gold standard rule in/rule out in family members), prognostic (both between and within genotypes), and therapeutic impact (LQT1 patients clinically approached with a fundamentally distinct treatment plan than LQT3 patients for example).
From a clinical test standpoint, any patient with a suspected clinical LQTS diagnosis should be offered clinical genetic testing even when the QTc is unequivocally prolonged (i.e. ≥ 500 ms) and the clinical diagnosis is not in doubt. As indicated earlier, such genotyping additionally risk stratifies and guides treatment decisions. Further, genotyping of such an index case has a 75% likelihood of yielding the index case’s possible/probable LQTS-associated mutation which if positive, genotyping of appropriate relatives in concentric circles of first degree relatedness can definitively rule in or rule out the genetic diagnosis regardless of its phenotypic expression in terms of symptoms or QT interval. To be sure, when genetic LQTS has been established in a family, a screening ECG is insufficient to assess the presence/absence of the familial LQTS genetic trait. Additionally, LQTS clinical genetic testing may also be warranted for patients with unexplained, exertional syncope or drug-induced QT prolongation/torsade de pointes who do not meet full LQTS diagnostic criteria and are stuck with a rendered diagnosis of “possible” or “borderline” LQTS. Here, genetic testing could help upgrade the diagnosis from clinically equivocal to genetically probable.
Catecholaminergic Polymorphic Ventricular Tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is another heritable arrhythmia syndrome that classically manifests with exercise-induced syncope or sudden death57, 67, 68. CPVT is associated with a normal resting ECG, with possible bradycardia and U waves, and is clinically suspected based on history and documentation of significant ventricular ectopy, including bidirectional VT, during either treadmill or catecholamine stress testing. Like LQTS, CPVT is generally associated with a structurally normal heart. However, CPVT is more lethal with a positive family history of juvenile (< 40 years) SCD for more than 30% of CPVT individuals and up to 60% in families hosting mutations in RYR267. In addition, 15% of autopsy negative SUD may be attributed to CPVT7, 46. Like LQT1, swimming is a potentially lethal arrhythmia precipitating trigger in CPVT. In fact, both LQT1 and CPVT have been shown to underlie several cases of unexplained drowning or near-drowning involving previously healthy young swimmers.
CPVT stems from genetic mutations in genes encoding components of the macromolecular intracellular Ca2+ release channel complex within the sarcoplasmic reticulum (SR) of the cardiocyte (Figure 7). Mutations in the RYR2-encoded cardiac ryanodine receptor 2/calcium release channel represents the most common genetic subtype of CPVT (CPVT1) accounting for approximately 50 to 60% of CPVT cases (Table 3)57, 67. The 105 exon containing RYR2 gene encodes for one of the largest proteins in the human proteome and the largest ion channel, comprising 4,967 amino acids. While there does not appear to be any specific mutation “hot-spots”, there are 3 regional “hot-spots” or “domains” where unique mutations reside (Figure 7)57. This observation has lent itself towards the possibility of targeted genetic testing of RYR2 rather than a comprehensive 105 exon analysis (Figure 7). However, currently there is no consensus or clear definition of the most appropriate, targeted RYR2 scan. Presently, two-thirds of detectable RYR2 mutations localize to 16 exons while all of the currently published mutations are confined to 45 of RYR2’s 105 translated exons57. While greater than 90% of RYR2 mutations discovered to date represent missense mutations; as much as 5% of unrelated CPVT patients have large RYR2 gene rearrangements consistent with large, whole exon deletions that would not be detected using standard direct DNA sequencing technology, similar to what has been observed in LQTS57. A rare autosomal recessive subtype of CPVT is mediated by mutations in CASQ2-encoded calsequestrin 2 (CPVT2)69.
Figure 7. Catecholaminergic Polymorphic Ventricular Tachycardia: a Disorder of Intracellular Calcium Handling.

A) Perturbations in key components of the calcium-induced calcium release (CICR) mechanism responsible for cardiac excitation-contraction coupling are the pathogenic basis for CPVT. At the center of this mechanism is the RYR2-encoded cardiac ryanodine receptor/calcium release channel located in sarcoplasmic reticulum membrane. Mutations in RyR2 are clustered and distributed in three “hot-spot” regions of this 4967 amino acid (AA) protein; Domain I or N-terminal Domain (AA 57-1141), Domain II or the Central Domain (AA 1638-2579), and Domain III or Channel Region (AA 3563-4967). B) Because of this clustering of mutations, a tiered strategy for genetic testing of the 105 exon RYR2 gene has been proposed61. C) Illustrated is the percent yield of RYR2 mutation detection in cases of strong CPVT, possible CPVT, and LQT1-3 gene-negative “LQTS” referral cases manifesting with exercise-induced cardiac events61.
Strikingly, nearly a third of “possible/atypical” LQTS (QTc < 480ms) cases with exertion-induced syncope have also been identified as RYR2 mutation positive (Figure 7)57, accordingly a clinical presentation of exercise-induced syncope and a QTc < 460 ms should always prompt first consideration of CPVT rather than so-called “concealed” or “normal QT interval” LQT1. In fact, it has been reported that nearly 30% of patients with CPVT have been misdiagnosed as “LQTS with normal QT intervals” or “concealed LQTS”, indicating the critical importance of properly distinguishing between CPVT and LQTS at the clinical level, as risk assessments and treatment strategies of these unique disorders may vary. Similarly, some patients diagnosed with CPVT based on the presence of bi-directional VT on exercise, have been identified to possess KCNJ2 mutations which are associated with the rarely lethal Andersen-Tawil syndrome (ATS1, LQT7)70. The misdiagnosis of ATS as the potentially lethal disorder CPVT, may lead to a more aggressive prophylactic therapy (i.e. ICD implantation) than necessary. Genetic testing may provide a clear differential diagnosis between atypical LQT1 and CPVT and CPVT and ATS1.
Specific indications of clinical genetic testing for CPVT include all patients with a suspected clinical diagnosis of CPVT and those patients with exercise-induced syncope/cardiac arrest or near-drowning in the setting of a QTc less than 460 ms along with all immediate family members of genetically proven CPVT index case.
Brugada Syndrome
Brugada syndrome (BrS) is another heritable arrhythmia syndrome characterized by an ECG pattern of coved type ST-segment elevation in the right precordial leads (V1 through V3) and an increased risk for sudden death resulting from episodes of polymorphic ventricular tachyarrhythmias22, 71. BrS is considered generally a disorder involving young male adults with arrhythmogenic manifestation first occurring at an average age of 40 years with sudden death typically occurring during sleep. However, like the previously mentioned channelopathies, the penetrance and expressivity are highly variable, ranging from entirely asymptomatic individuals to SCD during the first year of life. In addition idiopathic ventricular fibrillation (IVF) in many cases may be clinically, genetically, and mechanistically most closely linked with BrS72 and progressive cardiac conduction defect (PCCD) is often an overlapping phenotype with BrS73.
While gain-of-function mutations in SCN5A cause approximately 5–10% of LQTS (LQT3), approximately 20 – 25% of BrS results from loss-of-function mutations in the SCN5A-encoded cardiac sodium channel (NaV1.5, BrS1; Table 3 and Figure 8). In 2009, an international compendium of SCN5A mutations derived from over 2000 patients referred for BrS genetic testing yielded nearly 300 distinct mutations in 438 of 2111 (21%) unrelated patients and the mutation detection yield ranged from 11% to 28% across the 9 testing centers74. In 2009, Meregalli et. al. illustrated that SCN5A mutation type can anticipate the severity of PCCD and BrS75. Their data suggest that those mutations with a more deleterious loss of sodium current produce a more severe phenotype of syncope and conduction defect, providing the first evidence for intra-genotype risk stratification associated with SCN5A loss-of-function disease. Akin to LQTS, knowing the SCN5A mutation status may assist in the risk stratification of patients with BrS.
Figure 8. Sodium Channelopathies.

Depicted is the linear topology of the 2016-amino acid containing cardiac sodium channel isoform with mutation location and their associated disorders. Circles denote missense mutations and squares represent “radical” mutations including, frame-shift insertion/deletion mutations, in-frame insertions/deletions, nonsense, and splice-site mutations. Figure adapted from Tester and Ackerman83.
In addition to mutations involving the Nav1.5 sodium channel, mutations in genes (GPD1L76, SCN1B77, and SCN3B78) that modulate the sodium channel function have been associated with BrS. Recently, mutations involving the L-type calcium channel α (CACNA1C) and β (CACNB2B) subunits have been implicated in nearly 10% of BrS cases79 while mutations in the putative β subunit of the transient outward potassium channel (Ito, KCNE380) and in the KCNJ8-encoded ATP-sensitive potassium channel81 have been reported as a rare cause. Still, nearly two-thirds of BrS remains genetically elusive.
Patients suspected of having BrS, IVF, or PCCD could undergo clinical genetic testing as long as it is recognized that the yield from currently available BrS genetic tests is approximately 25 – 35%. Further, except for the recent association between SCN5A mutation type and clinical severity, the primary role of BrS genetic testing is diagnostic confirmation of root cause for an index case followed by confirmatory genetic testing of immediate family members to distinguish those needing ongoing clinical surveillance and preventative measures (www.brugadadrugs.org) from those who can be dismissed as clinically, electrocardiographically, and genetically unaffected (Figure 4).
CONCLUSIONS
Genomic advances have propelled cardiologists into the era of personalized genomic medicine. As novel genes are discovered, the compendium of available genetic tests will surely increase. For the four specific heritable cardiomyopathies/channelopathies detailed in this review, the bench-to-bedside continuum of research from scientific discovery to translation to clinical implementation and even post implementation test interpretation has been completed or is nearing completion. Now with genetic testing being added to our armamentarium of diagnostic/prognostic tests, it will behoove the community of cardiologists to not only be conversant in the language of genomic medicine but to also be wise users and even wiser interpreters of genetic testing so that wise decisions can be rendered for those patients/families being evaluated with respect to the presence or absence of one of these potentially lethal yet highly treatable genetic cardiomyopathies/channelopathies.
Footnotes
Conflicts of Interest: Dr. Ackerman is a consultant for PGxHealth with respect to the FAMILION™ genetic test for cardiac ion channel mutations. Intellectual property derived from MJA’s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals).
References
- 1.Nussbaum RLMR, Willard HF. Thompson & Thompson Genetics in Medicine. 6. Philadelphia, Pa: WB Saunders Co; 2001. pp. 51–94. [Google Scholar]
- 2.den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Human Genetics. 2001;109(1):121–124. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- 3.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2(5):507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 4.Tester DJ, Ackerman MJ. Genetic Testing. In: Gussak I, Antzelevitch C, editors. Electrical diseases of the heart: genetics, mechanisms, treatment, prevention. London: Springer; 2008. pp. 444–458. [Google Scholar]
- 5.Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, Ahearn ME, Baumbach L, Kurlansky P, Wolff G, Bishopric NH. Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome.[see comment] Circulation. 2004;109(24):3029–3034. doi: 10.1161/01.CIR.0000130666.81539.9E. [DOI] [PubMed] [Google Scholar]
- 6.Gladding PA, Evans CA, Crawford J, Chung SK, Vaughan A, Webster D, Neas K, Love DR, Rees MI, Shelling AN, Skinner JR. Posthumous diagnosis of long QT syndrome from neonatal screening cards. Heart Rhythm. 2010;7(4):481–486. doi: 10.1016/j.hrthm.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 7.Tester DJ, Ackerman MJ. The role of molecular autopsy in unexplained sudden cardiac death. Current Opinion in Cardiology. 2006;21(3):166–172. doi: 10.1097/01.hco.0000221576.33501.83. [DOI] [PubMed] [Google Scholar]
- 8.Carturan E, Tester DJ, Brost BC, Basso C, Thiene G, Ackerman MJ. Postmortem genetic testing for conventional autopsy-negative sudden unexplained death: an evaluation of different DNA extraction protocols and the feasibility of mutational analysis from archival paraffin-embedded heart tissue. American Journal of Clinical Pathology. 2008;129(3):391–397. doi: 10.1309/VLA7TT9EQ05FFVN4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tester DJ, Cronk LB, Carr JL, Schulz V, Salisbury BA, Judson RS, Ackerman MJ. Allelic dropout in long QT syndrome genetic testing: a possible mechanism underlying false-negative results.[see comment] Heart Rhythm. 2006;3(7):815–821. doi: 10.1016/j.hrthm.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 10.Tester DJ, Will ML, Ackerman MJ. Mutation detection in congenital long QT syndrome: cardiac channel gene screen using PCR, dHPLC, and direct DNA sequencing. Methods in Molecular Medicine. 2006;128:181–207. doi: 10.1385/1-59745-159-2:181. [DOI] [PubMed] [Google Scholar]
- 11.Mardis ER. Next-generation DNA sequencing methods. Annual Review of Genomics & Human Genetics. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 12.Porreca GJ, Zhang K, Li JB, Xie B, Austin D, Vassallo SL, LeProust EM, Peck BJ, Emig CJ, Dahl F, Gao Y, Church GM, Shendure J. Multiplex amplification of large sets of human exons.[see comment] Nature Methods. 2007;4(11):931–936. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- 13.Eddy C-A, MacCormick JM, Chung S-K, Crawford JR, Love DR, Rees MI, Skinner JR, Shelling AN. Identification of large gene deletions and duplications in KCNQ1 and KCNH2 in patients with long QT syndrome.[see comment] Heart Rhythm. 2008;5(9):1275–1281. doi: 10.1016/j.hrthm.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 14.Koopmann TT, Alders M, Jongbloed RJ, Guerrero S, Mannens MMAM, Wilde AAM, Bezzina CR. Long QT syndrome caused by a large duplication in the KCNH2 (HERG) gene undetectable by current polymerase chain reaction-based exon-scanning methodologies.[see comment] Heart Rhythm. 2006;3(1):52–55. doi: 10.1016/j.hrthm.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Tester DJ, Ackerman MJ. Novel gene and mutation discovery in congenital long QT syndrome: let’s keep looking where the street lamp standeth.[comment] Heart Rhythm. 2008;5(9):1282–1284. doi: 10.1016/j.hrthm.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Constantin CM, Faucett A, Lubin IM. A primer on genetic testing. Journal of Midwifery & Women’s Health. 2005;50(3):197–204. doi: 10.1016/j.jmwh.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Ehrmeyer SS, Laessig RH. Regulatory compliance for point-of-care testing: 2009 United States perspective. Clinics in Laboratory Medicine. 2009;29(3):463–478. doi: 10.1016/j.cll.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Maron BJ, Moller JH, Seidman CE, Vincent GM, Dietz HC, Moss AJ, Towbin JA, Sondheimer HM, Pyeritz RE, McGee G, Epstein AE. Impact of laboratory molecular diagnosis on contemporary diagnostic criteria for genetically transmitted cardiovascular diseases: hypertrophic cardiomyopathy, long-QT syndrome, and marfan syndrome: A statement for healthcare professionals from the councils on clinical cardiology, cardiovascular disease in the young, and basic science, american heart association. Circulation. 1998;98(14):1460–1471. [PubMed] [Google Scholar]
- 19.Association BCBS. TEC report. Genetic Testing for Long QT Syndrome. Assement Program. 2008 February;22(9) Available at URL address: http://www.bcbs.com/blueresorces/tec/vols/22/22_09.html.
- 20.GeneTests: Medical Genetics Information Resource (database online) Copyright, University of Washington; Seattle: 1993–2009. [Accessed [June 2010]]. Available at http://www.genetests.org. [Google Scholar]
- 21.Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. Journal of the American College of Cardiology. 2009;54(3):201–211. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu W. Clinical impact of genetic studies in lethal inherited cardiac arrhythmias. Circulation Journal. 2008;72(12):1926–1936. doi: 10.1253/circj.cj-08-0947. [DOI] [PubMed] [Google Scholar]
- 23.Ruan Y, Liu N, Napolitano C, Priori SG. Therapeutic strategies for long-QT syndrome: does the molecular substrate matter? Circulation: Arrhythmia and Electrophysiology. 2008;1(4):290–297. doi: 10.1161/CIRCEP.108.795617. [DOI] [PubMed] [Google Scholar]
- 24.Tester DJ, Ackerman MJ. Genetic testing for cardiac channelopathies: ten questions regarding clinical considerations for heart rhythm allied professionals. Heart Rhythm. 2005;2(6):675–677. doi: 10.1016/j.hrthm.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 25.Priori SG, Napolitano C. Role of genetic analyses in cardiology: part I: mendelian diseases: cardiac channelopathies. Circulation. 2006;113(8):1130–1135. doi: 10.1161/CIRCULATIONAHA.105.563205. [DOI] [PubMed] [Google Scholar]
- 26.Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AAM, Ackerman MJ. Genetic testing for long-QT syndrome distinguishing pathogenic mutations from benign variants. Circulation. 2009;120(18):1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charron P. Clinical genetics in cardiology. Heart. 2006;92(8):1172–1176. doi: 10.1136/hrt.2005.071308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hershberger RE, Cowan J, Morales A, Siegfried JD. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation: Heart Failure. 2009;2(3):253–261. doi: 10.1161/CIRCHEARTFAILURE.108.817346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Riper M. Genetic testing and the family. Journal of Midwifery & Women’s Health. 2005;50(3):227–233. doi: 10.1016/j.jmwh.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Thomas SM. Society and ethics - the genetics of disease. Current Opinion in Genetics & Development. 2004;14(3):287–291. doi: 10.1016/j.gde.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 31.Lea DH, Williams J, Donahue MP. Ethical issues in genetic testing. Journal of Midwifery & Women’s Health. 2005;50(3):234–240. doi: 10.1016/j.jmwh.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 32.Abiola S. Recent developments in health law. The Genetic Information Nondiscrimination Act of 2008: “First major Civil Rights bill of the century” bars misuse of genetic test results. Journal of Law, Medicine & Ethics. 2008;36(4):856–860. doi: 10.1111/j.1748-720X.2008.00344.x. [DOI] [PubMed] [Google Scholar]
- 33.Bos JM, Ommen SR, Ackerman MJ. Genetics of hypertrophic cardiomopathy: one, two, or more diseases? Current Opinion in Cardiology. 2007;22(3):193–199. doi: 10.1097/HCO.0b013e3280e1cc7f. [DOI] [PubMed] [Google Scholar]
- 34.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MRG, Towbin JA Heart Failure Society of A. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. Journal of Cardiac Failure. 2009;15(2):83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 35.Robin NH, Tabereaux PB, Benza R, Korf BR. Genetic testing in cardiovascular disease. Journal of the American College of Cardiology. 2007;50(8):727–737. doi: 10.1016/j.jacc.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 36.Marian AJ, Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2001;33:655–670. doi: 10.1006/jmcc.2001.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Driest SLOS, Tajik AJ, Gersh BJ, Ackerman MJ. Sarcomeric Genotyping in Hypertrophic Cardiomyopathy. Mayo Clinic Proceedings. 2005;80(4):463–469. doi: 10.1016/S0025-6196(11)63196-0. [DOI] [PubMed] [Google Scholar]
- 38.Binder J, Ommen SR, Gersh BJ, Van Driest SL, Tajik AJ, Nishimura RA, Ackerman MJ. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clinic Proceedings. 2006;81(4):459–467. doi: 10.4065/81.4.459. [DOI] [PubMed] [Google Scholar]
- 39.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, Seidman CE, Seidman JG. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. Journal of Clinical Investigation. 2002;109(8):1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. New England Journal of Medicine. 1992;326(17):1108–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 41.Ackerman MJ, Van Driest SV, Ommen SR, Will ML, Nishimura RA, Tajik AJ, Gersh BJ. Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin T gene in hypertrophic cardiomyopathy: a comprehensive outpatient perspective. Journal of the American College of Cardiology. 2002;39(12):2042–2048. doi: 10.1016/s0735-1097(02)01900-9. [DOI] [PubMed] [Google Scholar]
- 42.Van Driest SV, Ackerman MJ, Ommen SR, Shakur R, Will ML, Nishimura RA, Tajik AJ, Gersh BJ. Prevalence and severity of “benign” mutations in the beta myosin heavy chain, cardiac troponin-T, and alpha tropomyosin genes in hypertrophic cardiomyopathy. Circulation. 2002;106:3085–3090. doi: 10.1161/01.cir.0000042675.59901.14. [DOI] [PubMed] [Google Scholar]
- 43.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Yield of Genetic Testing in Hypertrophic Cardiomyopathy. Mayo Clinic Proceedings. 2005;80:739–744. doi: 10.1016/S0025-6196(11)61527-9. [DOI] [PubMed] [Google Scholar]
- 44.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, Ommen SR, Theis JL, Vaubel RA, Re F, Armentano C, Poggesi C, Torricelli F, Cecchi F. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clinic Proceedings. 2008;83(6):630–638. doi: 10.4065/83.6.630. [DOI] [PubMed] [Google Scholar]
- 45.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, Rimini A, Rosati E, Salice P, Spazzolini C. Prevalence of the Congenital Long-QT Syndrome. Circulation. 2009;120(18):1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young.[see comment] Journal of the American College of Cardiology. 2007;49(2):240–246. doi: 10.1016/j.jacc.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 47.Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Wang DW, Rhodes TE, George AL, Jr, Schwartz PJ. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115(3):361–367. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- 48.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 49.Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, Bottelli G, Cerrone M, Leonardi S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice.[see comment] JAMA. 2005;294(23):2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 50.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AAM, Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION (R) long QT syndrome genetic test. Heart Rhythm. 2009;6(9):1297–1303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koopmann TT, Alders M, Jongbloed RJ, Guerrero S, Mannens M, Wilde AAM, Bezzina CR. Long QT syndrome caused by a large duplication in the KCNH2 (HERG) gene undetectable by current polymerase chain reaction-based exon-scanning methodologies. Heart Rhythm. 2006;3(1):52–55. doi: 10.1016/j.hrthm.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 52.Ackerman MJ, Tester DJ, Porter CJ. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clinic Proceedings. 1999;74(11):1088–1094. doi: 10.4065/74.11.1088. [DOI] [PubMed] [Google Scholar]
- 53.Wilde AA, Jongbloed RJ, Doevendans PA, Duren DR, Hauer RN, van Langen IM, van Tintelen JP, Smeets HJ, Meyer H, Geelen JL. Auditory stimuli as a trigger for arrhythmic events differentiate HERG- related (LQTS2) patients from KVLQT1-related patients (LQTS1) Journal of American College of Cardiology. 1999;33(2):327–332. doi: 10.1016/s0735-1097(98)00578-6. [DOI] [PubMed] [Google Scholar]
- 54.Moss AJ, Robinson JL, Gessman L, Gillespie R, Zareba W, Schwartz PJ, Vincent GM, Benhorin J, Heilbron EL, Towbin JA, Priori SG, Napolitano C, Zhang L, Medina A, Andrews ML, Timothy K. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. American Journal of Cardiology. 1999;84(8):876–879. doi: 10.1016/s0002-9149(99)00458-0. [DOI] [PubMed] [Google Scholar]
- 55.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 56.Khositseth A, Tester DJ, Will ML, Bell CM, Ackerman MJ. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 2004;1:60–64. doi: 10.1016/j.hrthm.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 57.Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, Mannens MMAM, Wilde AAM, Ackerman MJ. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. Journal of the American College of Cardiology. 2009;54(22):2065–2074. doi: 10.1016/j.jacc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khositseth A, Ackerman MJ. Clinical evaluation, risk stratification, and management of congenital long QT syndrome. In: IGe, et al., editors. Contemporary Cardiology: Cardiac Repolarization: Bridging Basic and Clinical Science. Totowa: Humana Press, Inc; 2003. [Google Scholar]
- 59.Moss AJ, Windle JR, Hall WJ, Zareba W, Robinson JL, McNitt S, Severski P, Rosero S, Daubert JP, Qi M, Cieciorka M, Manalan AS. Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (DeltaKPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Annals of Noninvasive Electrocardiology. 2005;10(4 Suppl):59–66. doi: 10.1111/j.1542-474X.2005.00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome.[see comment] Journal of Cardiovascular Electrophysiology. 2008;19(12):1289–1293. doi: 10.1111/j.1540-8167.2008.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, Towbin JA, Keating MT, Priori SG, Schwartz PJ, Vincent GM, Robinson JL, Andrews ML, Feng C, Hall WJ, Medina A, Zhang L, Wang Z. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105(7):794–799. doi: 10.1161/hc0702.105124. [DOI] [PubMed] [Google Scholar]
- 62.Jons C, Moss AJ, Lopes CM, McNitt S, Zareba W, Goldenberg I, Qi M, Wilde AAM, Shimizu W, Kanters JK, Towbin JA, Ackerman MJ, Robinson JL. Mutations in conserved amino acids in the KCNQ1 channel and risk of cardiac events in type-1 long-QT syndrome. Journal of Cardiovascular Electrophysiology. 2009;20(8):859–865. doi: 10.1111/j.1540-8167.2009.01455.x. [DOI] [PubMed] [Google Scholar]
- 63.Shimizu W, Horie M, Ohno S, Takenaka K, Yamaguchi M, Shimizu M, Washizuka T, Aizawa Y, Nakamura K, Ohe T, Aiba T, Miyamoto Y, Yoshimasa Y, Towbin JA, Priori SG, Kamakura S. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in the LQT1 form of congenital long QT syndrome: multicenter study in Japan. Journal of the American College of Cardiology. 2004;44(1):117–125. doi: 10.1016/j.jacc.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 64.Moss AJ, Shimizu W, Wilde AAM, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, McNitt S. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115(19):2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagaoka I, Shimizu W, Itoh H, Yamamoto S, Sakaguchi T, Oka Y, Tsuji K, Ashihara T, Ito M, Yoshida H, Ohno S, Makiyama T, Miyamoto Y, Noda T, Kamakura S, Akao M, Horie M. Mutation site dependent variability of cardiac events in Japanese LQT2 form of congenital long-QT syndrome. Circulation Journal. 2008;72(5):694–699. doi: 10.1253/circj.72.694. [DOI] [PubMed] [Google Scholar]
- 66.Shimizu W, Moss A, Wilde A, Towbin J, Ackerman M, Tester D, January C, Zareba W, Robinson J, Qi M, Vincent G, Kaufman E, Hofman N, Noda T, Kamakura S, Miyamoto Y, Shah S, Amin V, Goldenberg I, Andrews M, McNitt S. Genotype-Phenotype Aspects of Type-2 Long-QT Syndrome. Journal of American College of Cardiology. 2009;54(22):2052–2062. doi: 10.1016/j.jacc.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia.[see comment] Circulation. 2002;106(1):69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 68.Tester DJ, Kopplin LJ, Will ML, Ackerman MJ. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm. 2005;2(10):1099–1105. doi: 10.1016/j.hrthm.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 69.Eldar M, Pras E, Lahat H. A Missense Mutation in the CASQ2 Gene Is Associated with Autosomal- Recessive Catecholamine-Induced Polymorphic Ventricular Tachycardia. Trends Cardiovascular Medicine. 2003;13:148–151. doi: 10.1016/s1050-1738(03)00025-2. [DOI] [PubMed] [Google Scholar]
- 70.Tester DJ, Arya P, Will M, Haglund CM, Farley AL, Makielski JC, Ackerman MJ. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm. 2006;3(7):800–805. doi: 10.1016/j.hrthm.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 71.Chen PS, Priori SG. The Brugada syndrome. Journal of the American College of Cardiology. 2008;51(12):1176–1180. doi: 10.1016/j.jacc.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 72.Sarkozy A, Brugada P. Sudden cardiac death: what is inside our genes? Canadian Journal of Cardiology. 2005;21(12):1099–1110. [PubMed] [Google Scholar]
- 73.Probst V, Denjoy I, Meregalli PG, Amirault JC, Sacher F, Mansourati J, Babuty D, Villain E, Victor J, Schott JJ, Lupoglazoff JM, Mabo P, Veltmann C, Jesel L, Chevalier P, Clur SA, Haissaguerre M, Wolpert C, Le Marec H, Wilde AA. Clinical aspects and prognosis of Brugada syndrome in children.[see comment] Circulation. 2007;115(15):2042–2048. doi: 10.1161/CIRCULATIONAHA.106.664219. [DOI] [PubMed] [Google Scholar]
- 74.Kapplinger J, Tester D, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, Kamakura S, Kyndt F, Koopmann T, Miyamoto Y, Pfeiffer R, Pollevick G, Probst V, Zumhagen S, Vatta M, Towbin J, Shimizu W, Schulze-Bahr E, Antzelevitch C, Salisbury B, Guicheney P, Wilde A, Brugada R, Schott J, Ackerman M. An International Compendium of Mutations in the SCN5A-Encoded Cardiac Sodium Channel in Patients Referred for Brugada Syndrome Genetic Testing. Heart Rhythm. 2009;7(1):33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meregalli PG, Tan HL, Probst V, Koopmann TT, Tanck MW, Bhuiyan ZA, Sacher F, Kyndt F, Schott J-J, Albuisson J, Mabo P, Bezzina CR, Le Marec H, Wilde AAM. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm. 2009;6(3):341–348. doi: 10.1016/j.hrthm.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 76.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC., Jr Mutation in Glycerol-3-Phosphate Dehydrogenase 1 Like Gene (GPD1-L) Decreases Cardiac Na+ Current and Causes Inherited Arrhythmias. Circulation. 2007;116(20):2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Watanabe H, Koopmann TT, Le SS, Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F, Escande D, Wiesfeld AC, Pfeufer A, Kaab S, Wichmann HE, Hasdemir C, Aizawa Y, Wilde AA, Roden DM, Bezzina CR. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. Journal of Clinical Investigation. 2008;118:2260–2268. doi: 10.1172/JCI33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, Pfeiffer R, Koopmann TT, Cordeiro JM, Guerchicoff A, Pollevick GD, Antzelevitch C. A mutation in the beta3 subunit of the cardiac sodium channel associated with brugada ECG phenotype. Circulation Cardiovascular Genetics. 2009;2(3):270–278. doi: 10.1161/CIRCGENETICS.108.829192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Delpon E, Cordeiro JM, Nunez L, Bloch Thomsen PE, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Burashnikov E, Christiansen M, Antzelevitch C. Functional Effects of KCNE3 Mutation and Its Role in the Development of Brugada Syndrome 10.1161/CIRCEP.107.748103. Circulation Arrhythmia Electrophysiology. 2008;1(3):209–218. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Medeiros-Domingo A, Tan BH, Crotti L, Tester DJ, Eckhardt L, Cuoretti A, Kroboth SL, Song C, Zhou Q, Kopp D, Schwartz PJ, Makielski JC, Ackerman MJ. Gain-of-function mutation, S422L, in the KCNJ8-encoded cardiac K ATP channel Kir6.1 as a pathogenic substrate for J wave syndromes. Heart Rhythm. doi: 10.1016/j.hrthm.2010.06.016. Epub ahead of print – June 14, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tester DJ, Ackerman MJ. Electrophysiological Disorders of the Heart. Elselvier; 2010. Genomics and Prinicples of Clinical Genetics. [Google Scholar]
- 83.Tester DJ, Ackerman MJ. Genetics of Cardiac Arrhythmias. In: Bonow RO, Libby P, Mann DL, Zipes DP, Braunwald E, editors. Braunwald’s Heart Diseases: A Textbook of Cardiovascular Medicine. 9. Elsevier; 2010. pp. 0000–0000. [Google Scholar]