

Abstract
Oxygen therapy is a promising treatment strategy for ischemic stroke. One potential safety concern with oxygen therapy, however, is the possibility of increased generation of reactive oxygen species (ROS), which could exacerbate ischemic brain injury. Our previous study indicated that normobaric hyperoxia (NBO, 95% O2 with 5% CO2) treatment during ischemia salvaged ischemic brain tissue and significantly reduced ROS generation in transient experimental stroke. In this follow-up study, we tested the hypothesis that suppression of NADPH oxidase is an important mechanism for NBO-induced reduction of ROS generation in focal cerebral ischemia. Male Sprague-Dawley rats were given NBO (95% O2) or normoxia (21% O2) during 90-min filament occlusion of the middle cerebral artery, followed by 22.5-hour reperfusion. NBO treatment increased the tissue oxygen partial pressure (pO2) level in the ischemic penumbra close to the pre-ischemic value, as measured by electronic paramagnetic resonance (EPR), and led to a 30.2% reduction in magnetic resonance imaging (MRI) apparent diffusion coefficients (ADC) lesion volume. Real time PCR and western blot analyses showed that the mRNA and protein expression of NADPH oxidase catalytic subunit gp91phox were upregulated in the ischemic brain, which was significantly inhibited by NBO. As a consequence of gp91phox inhibition, NBO treatment reduced NADPH oxidase activity in the ischemic brain. Our results suggest that NBO treatment given during ischemia reduces ROS generation via inhibiting NADPH oxidase, which may serve as an important mechanism underlying NBO’s neuroprotection in acute ischemic stroke.
Keywords: NADPH oxidase, neuroprotection, oxygen, free radical, stroke
1. Introduction
Tissue hypoxia plays a critical role in the primary and secondary events leading to neuronal cell death after cerebral ischemia (Zauner et al. 2002). Therefore, improving brain tissue oxygenation is a logical and important treatment strategy for ischemic stroke. In the past several years, we and others have demonstrated that normobaric hyperoxia (NBO) treatment effectively reduces both infarct volume and neurological deficits in rodents with ischemic stroke (Singhal et al. 2002a; Singhal et al. 2002b; Liu et al. 2004; Kim et al. 2005; Liu et al. 2006; Henninger et al. 2007; Shin et al. 2007). In human studies, NBO treatment was associated with improvement in clinical deficits and survival in selected patients with acute ischemic stroke (Singhal et al. 2005; Chiu et al. 2006). Because NBO is readily available, inexpensive and can be initiated within minutes after stroke symptom onset, the observed neuroprotective effects of NBO may have important clinical implications. However, based on observations associated with reperfusion injury, there have been some concerns that increased oxygen delivery, particularly during the phase of reperfusion after stroke, could increase the generation of oxygen free radicals, which may lead to lipid peroxidation, disruption of cell membranes, and increases in blood-brain barrier damage and brain hemorrhage. Interestingly, several recent studies demonstrated that NBO treatment salvaged ischemic brain tissue without increasing the risk of oxygen free radical injury (Singhal et al. 2002b; Kim et al. 2005; Liu et al. 2006), and in fact, the generation of free radicals was reduced (Liu et al. 2006). However, how NBO affects the generation of oxygen free radical in the ischemic brain remains largely unknown.
NADPH oxidase is a principal enzyme for the production of superoxide (Cai et al. 2003). It is expressed in neural and nonneural cell types of the central nervous system, including neurons, glia, cerebral blood vessels, monocytes, and neutrophils (Anthony et al. 1997; Bianca et al. 1999; Serrano et al. 2003; Abramov et al. 2005). NADPH oxidase is a multiunit enzyme that is composed of membrane bound subunits (p22phox, gp91phox), cytoplasmic subunits (p40phox, p47phox, and p67phox), and the small G protein Rac-1 (Babior 2004). Expression of gp91phox, which is the catalytic subunit of NADPH oxidase, has been shown to be upregulated in ischemic stroke (Hirabayashi et al. 2000). Conversely, gp91phox mutation or inhibition of NADPH oxidase resulted in reduced cerebral infarction in the ischemic brain (Walder et al. 1997; Ostrowski et al. 2006; Wang et al. 2006). These findings implicate an important role of NADPH oxidase in neuronal damage in ischemic stroke. Using electron paramagnetic resonance (EPR) oximetry, we have recently shown that NBO treatment given during ischemia raised the penumbral interstitial pO2 levels close to the pre-ischemic value in a rat model of transient focal cerebral ischemia (Liu et al. 2006). Furthermore, NBO treatment also significantly decreased reactive oxygen species generation in the ischemic brain. In the present study, we tested the hypothesis that NBO treatment protects against cerebral infarction via inhibition of NADPH oxidase using the same rat model of focal cerebral ischemia as described in our previous study (Liu et al. 2006).
2. RESULTS
2.1 NBO treatment increases penumbral tissue pO2 during ischemia
Focal ischemia leads to the reduction of blood flow, and consequently, the tissue oxygenation of the affected areas. Using the EPR oximetry method that is well established in our lab to measure brain tissue pO2 (Liu et al. 2004), we found that occlusion of the middle cerebral artery led to the reduction of cerebral tissue pO2 from 40 mmHg to 18 mmHg in the ischemic penumbra, while NBO treatment raised the pO2 back to the pre-ischemic level, i.e., at 38 mmHg (Figure 1B). These results further confirm our earlier report (Liu et al. 2006), and demonstrate that NBO can elevate cerebral oxygenation in the ischemic brain tissue.
Figure 1.
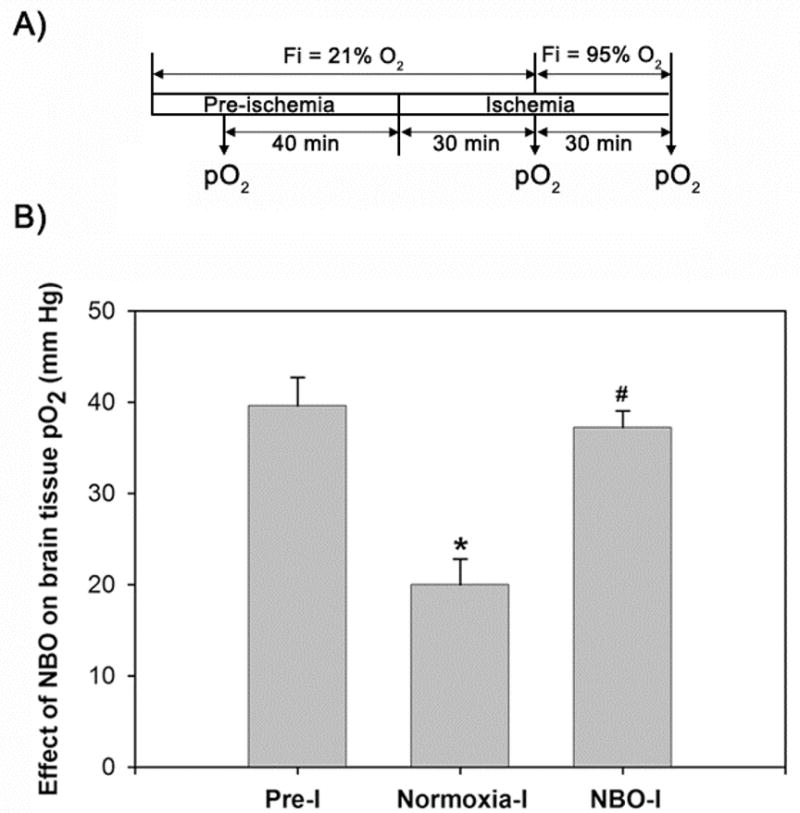
NBO during ischemia raises the cerebral tissue oxygenation pO2 in the ischemic penumbra to the pre-ischemic level. A) The sketch shows that the penumbral pO2 values were measured in vivo by EPR oximetry in the anesthetized rats 40 min before ischemia (FiO2: 21% O2, Pre-I), 30 min after the start of ischemia (FiO2: 21% O2, Normoxia-I), and then 30 min after the start of NBO treatment (FiO2: 95% O2, NBO-I). FiO2: inspired oxygen fraction. B) Bar graph shows the changes of the pO2 levels in the ischemic penumbra. Data were expressed as mean ± SEM, n = 5. *p < 0.05 versus Pre-I, #p < 0.05 versus Normoxia-I.
2.2 NBO treatment reduces MRI ADC lesion volume in the ischemic brain
At 24 hrs after the onset of middle cerebral artery occlusion (MCAO), MRI ADCs were calculated to quantify tissue lesion volume. Typical T2 weighed (T2W) images and ADC maps of the brains from normoxic and NBO-treated rats are shown in Figure 2A. Average ADC lesion volume was significantly lower in the NBO-treated rats (235.7 ± 14.8 mm3, n = 6) compared to the normoxic rats (374.1 ± 36.6 mm3, n = 6) (P < 0.05), representing a reduction of 37.0% (Figure 2B). These results are consistent with our previous findings that NBO treatment during 90-min MCAO significantly decreased the infarction in the ischemic brain, which was measured by 2,3,5-triphenyltetrazolium chloride (TTC) staining (Liu et al. 2006).
Figure 2.
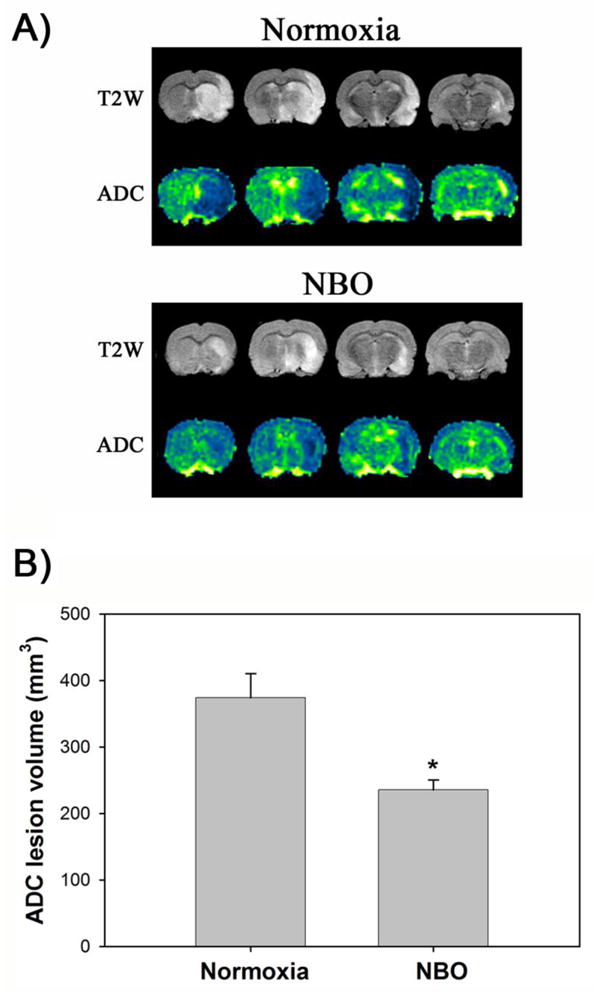
NBO treatment significantly reduced infarction volume in the ischemic brain after 90-min MCAO with 22.5 hrs of reperfusion. NBO (95% O2) was delivered during 90-min ischemia. A) Representative T2W images and ADC color maps showing tissue infarction in the ischemic hemispheres (right) of the normoxic and NBO-treated rats. B) Total infarction volume was quantified as described in the text. Data were expressed as mean ± SEM, n = 7 for the normoxic group, and n = 6 for the NBO group. *P < 0.05 versus normoxic group.
2.3 NBO treatment inhibits the upregulation of gp91phox mRNA
Gp91phox, the catalytic unit of NADPH oxidase, is upregulated and critically implicated in neuronal death during cerebral ischemia (Walder et al. 1997; Cheng et al. 2006), therefore, we examined whether NBO treatment could inhibit gp91phox expression in the ischemic brain. Real time RT-PCR showed that ischemia and reperfusion significantly increased gp91phox mRNA expression in the ischemic hemispheric tissue as compared to the non-ischemic hemispheric tissue (3.50 ± 0.56 versus 0.96 ± 0.04, n = 8, P < 0.05). NBO treatment significantly inhibited gp91phox mRNA upregulation in the ischemic brain tissue (2.1 ± 0.8 versus 3.5 ± 0.6, P < 0.05, n = 8), but did not change its expression in the non-ischemic hemispheric tissue (Figure 3).
Figure 3.

The effect of NBO treatment on gp91phox mRNA expression in the ischemic hemisphere after 90-min MCAO with 22.5 hrs of reperfusion. NBO (95% O2) was delivered during 90-min ischemia. Gp91phox mRNA level was analyzed by real time RT-PCR on extracted total RNA. Threshold cycles (Ct values) were normalized to rpl 32 and the comparative mRNA levels were determined by the ΔΔCt method. Data were expressed as mean ± SEM, n =8. *P < 0.05 versus non-ischemic hemisphere, #P<0.05 versus normoxic group.
2.4 NBO treatment inhibits the overexpression of gp91phox protein
We next examined whether downregulation of gp91phox mRNA by NBO could result in a decreased gp91phox protein expression in the ischemic brain. Western blot analysis showed that gp91 phox protein was expressed at a low level in the non-ischemic hemispheric tissue, but its level was remarkably elevated in the ischemic hemispheric tissue in the normoxic rats. Similar to its effect on gp91phox mRNA, NBO treatment inhibited gp91phox protein increase in the ischemic hemispheric tissue, but did not change its level in the non-ischemic hemisphere (Fig 4A). Gp91phox protein band intensity was quantified and expressed as hemispheric gp91phox ratio. The quantitative results showed that NBO significantly inhibited gp91phox protein increase in the ischemic hemisphere as compared to the normoxic rats (hemispheric gp91phox ratio: 4.31± 0.68 versus 8.04 ± 0.98, P < 0.05, n = 8) (Fig 4B).
Figure 4.

NBO treatment significantly reduced the increase of gp91phox protein in ischemic brain tissue after 90-min MCAO with 22.5 hrs of reperfusion. NBO (95% O2) was delivered during 90-min ischemia. Gp91phox protein level was analyzed by western blot. The membrane was stripped and re-blotted with β-actin antibody. A) Representative western blots of gp91phox and β-actin. B) Hemispheric ratio of band intensity of gp91phox protein in the ischemic hemisphere versus non-ischemic hemisphere after normalization to β-actin. *P < 0.05 versus normoxic group, n = 8.
2.5 NBO treatment impedes NADPH oxidase activity
Our results above showed that NBO inhibited the increase of gp91phox mRNA and protein expression in the ischemic hemisphere (Figure 3 and 4), we next examined whether gp91phox inhibition by NBO could result in a reduction in NADPH oxidase activity. Lucigenin-enhanced chemiluminescence was used to measure NADPH oxidase activity. Since no obvious difference of gp91phox expression was observed (Figure 4) in the non-ischemic hemispheres between the normoxic or NBO-treated rats, we only compared NADPH oxidase activity in their ischemic hemispheres. In consistent with the lower level of gp91phox observed in the NBO-treated rats, its NADPH oxidase activity was also significantly lower than that of the normoxic rats (5054.4 ± 533.6 versus 7798.3 ± 948.7 RLU/min/mg, P < 0.05, n = 6) (Figure 5).
Figure 5.

NBO treatment significantly reduced the activity of NADPH oxidase in ischemic brain tissue after 90-min MCAO with 22.5 hrs of reperfusion. NBO (95% O2) was delivered during 90-min ischemia. Data are expressed as mean ± SEM, n = 6, *P < 0.05 versus the normoxic group.
3. Discussion
Tissue hypoxia is a key event leading to cell death after cerebral ischemia (Zauner et al. 2002). Therefore, improving brain tissue oxygenation could be a logical and important stroke treatment strategy. A growing body of evidence has shown that normobaric hyperoxia (NBO) treatment is neuroprotective in experimental cerebral ischemia across a wide range of species including mice, rats, rabbits and baboons (Branston et al. 1976; Kaminogo 1989; Singhal et al. 2002a; Singhal et al. 2002b; Liu et al. 2004; Kim et al. 2005; Liu et al. 2006; Henninger et al. 2007; Shin et al. 2007). However, a potential safety concern with oxygen therapy is the possibility of increased generation of reactive oxygen species (ROS), which, in the setting of ischemic stroke, could exacerbate brain edema, post-ischemic hemorrhage and neuronal death (Singhal 2007). Additionally, oxygen’s effect on ROS generation in ischemic stroke is still not conclusive because controversial results have been reported about ROS production in stroke animals exposed to hyperbaric hyperoxia (HBO) (Mink and Dutka 1995; Elayan et al. 2000; Sunami et al. 2000; Oter et al. 2005) or NBO (Mickel et al. 1987; Singhal et al. 2002b; Kim et al. 2005). There are several factors potentially contributing to these inconsistent results, such as severity and duration of cerebral ischemia, timing and duration of hyperoxia treatment, oxygen pressure and concentration, and animal species. In our recent study, we showed that NBO (95% O2 with 5% CO2) treatment during ischemia increased brain tissue pO2 level, but significantly reduced ROS generation in a rat model of middle cerebral artery occlusion (MCAO) (Liu et al. 2006). As a follow-up study, we herein further investigated the mechanism underlying the inhibitory action of NBO on ROS generation using the same rat model of ischemic stroke. Our results demonstrate that NBO significantly improves tissue oxygenation in the peri-infarct area and reduces infarction size. Cerebral ischemia and reperfusion induced gp91phox mRNA and protein expression in the ischemic brain, which was significantly inhibited by NBO treatment. As a consequence of gp91phox inhibition, NBO treatment reduced NADPH oxidase activity in the ischemic brain. These results suggest that inhibition of NADPH oxidase may be an important mechanism for NBO-induced reduction of ROS generation in focal cerebral ischemia.
ROS may be excessively generated when tissue oxygen is too high (hyperoxic status) or too low (hypoxic status) (Kulkarni et al. 2007). Consistent with our previous results (Liu et al. 2006), we found that cerebral ischemia decreased peri-infarct tissue pO2 level to approximately 40% of the pre-ischemic level at 30 min after MCAO. NBO treatment during ischemia significantly reversed this ischemia-induced decrease of tissue pO2 level, maintaining tissue pO2 level close to the pre-ischemic value (Fig 1). In another study using a mouse stroke model of distal MCAO, NBO (100% O2) treatment partially recovered brain tissue oxygenation as manifested by the increase of oxyhaemoglobin in the ischemic core and peri-infarct area (Shin et al. 2007). Under these experimental conditions, NBO treatment significantly improved the hypoxic status of the ischemic brain, but did not cause an overshoot of tissue pO2. Therefore, it is conceivable that NBO treatment would decrease, rather than increase, ROS generation in the ischemic brain.
We further investigated the mechanism of the inhibitory action of NBO on ROS generation in cerebral ischemia. Accumulating experimental evidence indicates that NADPH oxidase is an important contributor to the oxidative damage in ischemic stroke. Several studies have demonstrated that gp91phox, the catalytic subunit of this enzyme, is upregulated in ischemic stroke, and inhibition of NADPH oxidase limits infarction size and blood brain barrier disruption in different animal stroke models (Walder et al. 1997; Hirabayashi et al. 2000; Cheng et al. 2006; Ostrowski et al. 2006; Wang et al. 2006; Kahles et al. 2007; Liu et al. 2008; Chen et al. 2009; Kim et al. 2009). In addition, NADPH oxidase has been suggested as a possible oxygen sensor in cells (Schumacker 2003). Therefore, brain tissue pO2 alternation due to ischemia and NBO treatment could change the expression and activity of NADPH oxidase. We tested this hypothesis by analyzing gp91phox expression and NADPH oxidase activity in the present study. Our results indicate that NBO treatment significantly inhibited ischemia-induced upregulation of gp91phox mRNA and protein expression in the ischemic brain. As a consequence of gp91phox inhibition, NADPH oxidase activity was also inhibited by NBO. Given the important role of NADPH oxidase in ROS generation in the ischemic brain (Suh et al. 2008), inhibiting NADPH oxidase may represent an important mechanism underlying NBO’s protection against oxidative damage in the ischemic brain, reflected as reduced 8-OHdG formation in NBO-treated rats in our earlier study (Liu et al. 2006). However, further research is necessary to precisely determine the importance of NADPH oxidase in mediating 8-OHdG formation, as well as oxidative stress mediated signaling pathways, in transient cerebral ischemia.
In conclusion, the present study suggests that NBO therapy with 95% O2 given during ischemia reduces ROS generation in the ischemia brain via inhibiting NADPH oxidase, which may serve as an important mechanism mediating the neuroprotective effect of NBO in acute ischemic stroke.
4. Experimental Procedure
4.1 Rat model of focal cerebral ischemia and reperfusion
All experimental protocols are approved by the Laboratory Animal Care and Use Committee of the University of New Mexico. Sprague–Dawley rats (Charles River Laboratory) weighing 290 to 320 g were anesthetized with isoflurane (4% for surgical induction, 1.75% for maintenance) in N2:O2 (70%:30%) during surgical procedures. Body temperature was monitored continuously with a rectal probe and maintained at 37.5°C ± 0.5°C using a heating pad. Middle cerebral artery occlusion (MCAO) was established by proximal occlusion of the right middle cerebral artery with a 4-0 silicone-coated nylon monofilament, as we previously described (Liu et al. 2008). After 90-min occlusion, reperfusion was accomplished by careful withdrawal of the monofilament and the rats were returned to their cages for 22.5 h before sacrificing. The normoxic and NBO rats were ventilated (3 liter/min) with medical air (21% O2) or a gas mixture of 95% O2 + 5% CO2, respectively, during the 90-min ischemic period.
4.2 Electron paramagnetic resonance (EPR) oximetry measurement of tissue pO2
Interstitial partial pressure of oxygen (pO2) in the ischemic penumbra was measured using Bruker EleXsys E540 EPR spectrometer equipped with an L-band bridge (Bruker Instruments, Billerica, MA, USA) exactly as described in our previous studies (Liu et al. 2004). In brief, lithium phthalocyanine crystal (approximate diameter 0.2 mm) was placed at 0.5 mm posterior, 2 mm lateral to bregma, and 5.5 mm down from skull 72 h before induction of cerebral ischemia. Interstitial pO2 was measured with the EPR spectrometer using the following acquisition parameters: microwave power 10 mW, microwave frequency 1.07 GHz, magnetic field center 380 G, scan range 2.0 G, and modulation amplitude at less than one-third of the EPR line-width. The lithium phthalocyanine material was a gift from Dr Harold Swartz (NIH In Vivo EPR Center, Dartmouth College, NH, USA). Three tissue pO2 measurements were made for each rat at the same location before and during ischemia as indicated in Figure 1A.
4.3 Magnetic resonance imaging measurements for infarction volume
Thirty minutes before the end of reperfusion, rats were subjected to MRI analysis on a 4.7T Biospec dedicated research MR scanner (Bruker-Biospin, Billerica), equipped with 500 mT/m (rise time 80–120 μs) gradient set (for performing small animal imaging) and a small bore linear RF coil (ID 72 mm). Infarction volume was estimated by calculating the apparent diffusion coefficient (ADC) of water on ADC maps obtained with MRI, as previously described (Sood et al. 2009). In brief, a tripilot imaging sequence was used for reproducible positioning of the animal in the magnet at each MRI session. T2 weighted (T2W) and diffusion weighted imaging (DWI) were acquired with the following parameters: T2W TR/TE 4s/65ms, FOV 3.2cm × 3.2 cm, matrix 256 ×128, number of slices 20, slice thickness 2 mm, number of averages 22; DWI- TR/TE 2s/50ms, number of averages 15, d = 5ms, D = 20ms, b=0 and 927 s/mm2. The DWI images were slices matched to the T2 weighted images. At the end of the MRI experiments, the raw MRI data was transferred to an offline workstation. ADC maps were reconstructed from DWI using commercial software MRVision (MRVision, Winchester, MA). Lesion size was measured using various ADC thresholds and was defined as area with ADC values <80% of the mean contralateral hemisphere values on the ADC map. The analysis was performed by a single operator and the procedure was repeated twice. Lesion size was calculated as number of pixels × pixels size, where the number of pixels correspond to pixels with values less than the ADC threshold value. Infarction volume was calculated as the sum of lesion size on each prescribed slices.
4.4 Postischemic tissue processing
Rats were sacrificed by decapitation at the end of reperfusion. Brains were quickly removed and chilled in ice-cold PBS for 5 minutes. A 8-mm thick brain region 3 mm away from the tip of the frontal lobe were then cut into four 2-mm thick coronary slices, which contained the main infarction area according to our earlier studies with 2% 2,3,5-triphenyltetrazolium chloride (TTC) staining (Liu et al. 2004). The 2-mm thick brain slices were carefully cleaned of meninges, and then a longitudinal cut was made 2 mm away from the midline between two hemispheres to exclude tissue primarily supplied by the anterior cerebral artery. Non-ischemic and ischemic hemispheric tissue were then collected from each brain slice, and used for analyzing the protein and mRNA expression of gp91phox and the enzymatic activity of NADPH oxidase.
4.5 SYBER® Green real time RT-PCR analysis for gp91phox mRNA
Brain tissue collected as described above was homogenized in Trizol reagents (Invitrogen, Carlsbad, CA). Total RNA was isolated and purified using the DNA-Free kit (Ambion, Austin, TX) according to manufacturer’s protocols. 1 μg RNA was reverse-transcribed with random primers in a 20 μl final reaction volume using TaqMan® Reverse Transcription Kits (Applied Biosystems, Foster City, CA). 1.0 μl RT products were amplified with the 7900HT Fast Real-time PCR system (PerkinElmer Life Sciences, Boston, MA) in a 10 μl final reaction volume in 361-well plate using SYBR® Green PCR Master Mix (Applied Biosystems). The thermal cycling condition comprised an initial incubation at 95 °C 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, and annealing and extension at 60 °C for 1 min. Oligonucleotide primers (Integrated DNA Technologies, Inc., Coralville, IA) for gp91phox (Accession number: AF298656) and ribosomal protein 32 (rpl 32) (Accession number: NM_013226) were as follows: gp91phox, 5′-CAGCCTGCCTGAATTTCAACT-3′ (sense), 5′-GGAGAGGAGATTCCGACACACT-3′ (antisense); rpl 32, 5′-AGACCTGAATGTGAAGGAAG-3′ (sense), 5′-CCTTGGGATTGGTGACTCTGA -3′ (antisense). No template control or melting curve analysis was used to eliminate non-specific reaction. The fluorescence threshold value (Ct value) was calculated using the 7900HT Fast Real-time PCR system software. Triplicate determinations were averaged for each data point. The relative value of mRNA expression was calculated by the comparative ΔΔCt method described in the 7900HT manual. In brief, mean Ct values were normalized to the internal control rpl 32 and the difference was defined as ΔCt. The difference between the mean ΔCt values of compared samples was calculated and defined as ΔΔCt. The comparative mRNA expression level was expressed as 2−ΔΔCt.
4.6 Western blot analysis for gp91Phox protein expression
Non-ischemic and ischemic hemispheric brain tissue was homogenized in RIPA buffer containing protease inhibitor cocktail (Sigma). After centrifugation at 14,000 g for 15 min at 4 °C, the supernatant was stored at −80 °C until use. Protein concentration was determined by using a BCA protein assay kit (Pierce, Rockford, IL). Samples (75 μg of total protein) were boiled for 5 min and then electrophoresed in 10% SDS-PAGE acrylamide gels, transferred onto nitrocellulose membranes (Bio-Rad), and incubated for 1 hr in TBS-T (Tris-buffered saline and 0.1% Tween 20) containing 5% nonfat milk at room temperature. Membranes were then incubated overnight with monoclonal anti-gp91phox antibody (1:1000 dilution; BD Transduction Laboratories, Forest Grove, OR), washed in TBS-T, incubated for 1 hr at room temperature with HRP-conjugated anti-mouse antibody (1:1000; Santa Cruz Biotech). The protein was detected using the SuperSignal West Pico chemiluminescent kit (Pierce, Rockford, IL) according to the manufacturer’s instructions, and the bands were visualized and quantified on a Kodak Image Station 4000 Digital Imaging System. To control sample loading and protein transfer, the membranes were stripped and rehybridized for β-actin (1:4000, Sigma, St. Louis, MO). Gp91phox protein expression was standardized to an equivalent β-actin protein amount and expressed as a hemispheric ratio of band intensity (ischemic hemisphere versus non-ischemic hemisphere).
4.7 Measurement of NADPH oxidase activity
NADPH oxidase activity was determined based on superoxide-induced lucigenin photoemission as described previously (Ostrowski et al. 2006). Briefly, ischemic hemispheric brain tissue was homogenized in ice-cold modified Krebs HEPES buffer of the following composition (in mmol/L): (NaCl 119, HEPES 20, KCl 4.6, MgSO4 1.0, CaCl2 1.2, Na2HPO4 0.15, KH2PO4 0.4, NaHCO3 5, and glucose 5.5 (pH 7.4) with phenylmethylsulphonyl fluoride and a protease inhibitor cocktail (Sigma). To assay NADPH oxidase enzymatic activity in each sample, 20 μL homogenate was transferred to a luminometer tube, which contained 80 μL Krebs buffer supplemented with 6.25 μmol/L of lucigenin (Sigma). The reaction was initiated by the addition of 1 μL NADPH solution (Sigma) to a final concentration of 0.1 mmol/L. Chemiluminescence counts were recorded every 30 seconds continuously for 5 min using a Turner Designs TD20/20 luminometer. The respective background counts were subtracted and chemiluminescence was expressed in relative light units (RLU) per min per mg protein.
4.8 Statistical analysis
The Data are presented as means ± SEM. Statistical analysis was performed using ANOVA and Student’s t-test. A value of P < 0.05 was considered statistically significant.
Acknowledgments
The work was supported in part by grants from National Institutes of Health (P20 RR15636 and R01AG031725), American Heart Association (0555669Z and 0765461Z), and PhD Programs Foundation of the Ministry of Education of China (Grant NO.20070533060).
List of abbreviations
- NBO
Normobaric hyperoxia
- ROS
Reactive oxygen species
- MCAO
middle cerebral artery occlusion
- TTC
2,3,5-triphenyltetrazolium chloride
- pO2
tissue oxygen partial pressure
- EPR
electronic paramagnetic resonance
- MRI
magnetic resonance imaging
- T2W
T2 weighted
- ADC
apparent diffusion coefficient
- DWI
diffusion weighted imaging
References
- Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR. Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci. 2005;25:9176–9184. doi: 10.1523/JNEUROSCI.1632-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony DC, Ferguson B, Matyzak MK, Miller KM, Esiri MM, Perry VH. Differential matrix metalloproteinase expression in cases of multiple sclerosis and stroke. Neuropathol Appl Neurobiol. 1997;23:406–415. [PubMed] [Google Scholar]
- Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J Biol Chem. 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- Branston NM, Symon L, Crockard HA. Recovery of the cortical evoked response following temporary middle cerebral artery occlusion in baboons: relation to local blood flow and PO2. Stroke. 1976;7:151–157. doi: 10.1161/01.str.7.2.151. [DOI] [PubMed] [Google Scholar]
- Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernandez JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- Chiu EH, Liu CS, Tan TY, Chang KC. Venturi mask adjuvant oxygen therapy in severe acute ischemic stroke. Arch Neurol. 2006;63:741–744. doi: 10.1001/archneur.63.5.741. [DOI] [PubMed] [Google Scholar]
- Elayan IM, Axley MJ, Prasad PV, Ahlers ST, Auker CR. Effect of hyperbaric oxygen treatment on nitric oxide and oxygen free radicals in rat brain. J Neurophysiol. 2000;83:2022–2029. doi: 10.1152/jn.2000.83.4.2022. [DOI] [PubMed] [Google Scholar]
- Henninger N, Bouley J, Nelligan JM, Sicard KM, Fisher M. Normobaric hyperoxia delays perfusion/diffusion mismatch evolution, reduces infarct volume, and differentially affects neuronal cell death pathways after suture middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 2007;27:1632–1642. doi: 10.1038/sj.jcbfm.9600463. [DOI] [PubMed] [Google Scholar]
- Hirabayashi H, Takizawa S, Fukuyama N, Nakazawa H, Shinohara Y. Nitrotyrosine generation via inducible nitric oxide synthase in vascular wall in focal ischemia-reperfusion. Brain Res. 2000;852:319–325. doi: 10.1016/s0006-8993(99)02117-4. [DOI] [PubMed] [Google Scholar]
- Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- Kaminogo M. The effects of mild hyperoxia and/or hypertension on oxygen availability and oxidative metabolism in acute focal ischaemia. Neurol Res. 1989;11:145–149. doi: 10.1080/01616412.1989.11739880. [DOI] [PubMed] [Google Scholar]
- Kim GS, Jung JE, Niizuma K, Chan PH. CK2 is a novel negative regulator of NADPH oxidase and a neuroprotectant in mice after cerebral ischemia. J Neurosci. 2009;29:14779–14789. doi: 10.1523/JNEUROSCI.4161-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Singhal AB, Lo EH. Normobaric hyperoxia extends the reperfusion window in focal cerebral ischemia. Ann Neurol. 2005;57:571–575. doi: 10.1002/ana.20430. [DOI] [PubMed] [Google Scholar]
- Kulkarni AC, Kuppusamy P, Parinandi N. Oxygen, the lead actor in the pathophysiologic drama: enactment of the trinity of normoxia, hypoxia, and hyperoxia in disease and therapy. Antioxid Redox Signal. 2007;9:1717–1730. doi: 10.1089/ars.2007.1724. [DOI] [PubMed] [Google Scholar]
- Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ. Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:343–349. doi: 10.1097/01.WCB.0000110047.43905.01. [DOI] [PubMed] [Google Scholar]
- Liu S, Liu W, Ding W, Miyake M, Rosenberg GA, Liu KJ. Electron paramagnetic resonance-guided normobaric hyperoxia treatment protects the brain by maintaining penumbral oxygenation in a rat model of transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:1274–1284. doi: 10.1038/sj.jcbfm.9600277. [DOI] [PubMed] [Google Scholar]
- Liu W, Sood R, Chen Q, Sakoglu U, Hendren J, Cetin O, Miyake M, Liu KJ. Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem. 2008;107:1196–1205. doi: 10.1111/j.1471-4159.2008.05664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickel HS, Vaishnav YN, Kempski O, von Lubitz D, Weiss JF, Feuerstein G. Breathing 100% oxygen after global brain ischemia in Mongolian Gerbils results in increased lipid peroxidation and increased mortality. Stroke. 1987;18:426–430. doi: 10.1161/01.str.18.2.426. [DOI] [PubMed] [Google Scholar]
- Mink RB, Dutka AJ. Hyperbaric oxygen after global cerebral ischemia in rabbits does not promote brain lipid peroxidation. Crit Care Med. 1995;23:1398–1404. doi: 10.1097/00003246-199508000-00014. [DOI] [PubMed] [Google Scholar]
- Ostrowski RP, Tang J, Zhang JH. Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke. 2006;37:1314–1318. doi: 10.1161/01.STR.0000217310.88450.c3. [DOI] [PubMed] [Google Scholar]
- Oter S, Korkmaz A, Topal T, Ozcan O, Sadir S, Ozler M, Ogur R, Bilgic H. Correlation between hyperbaric oxygen exposure pressures and oxidative parameters in rat lung, brain, and erythrocytes. Clin Biochem. 2005;38:706–711. doi: 10.1016/j.clinbiochem.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Schumacker PT. Current paradigms in cellular oxygen sensing. Adv Exp Med Biol. 2003;543:57–71. doi: 10.1007/978-1-4419-8997-0_5. [DOI] [PubMed] [Google Scholar]
- Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003;988:193–198. doi: 10.1016/s0006-8993(03)03364-x. [DOI] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Lo EH, Moskowitz MA, Ayata C. Normobaric hyperoxia improves cerebral blood flow and oxygenation, and inhibits peri-infarct depolarizations in experimental focal ischaemia. Brain. 2007;130:1631–1642. doi: 10.1093/brain/awm071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal AB. A review of oxygen therapy in ischemic stroke. Neurol Res. 2007;29:173–183. doi: 10.1179/016164107X181815. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Dijkhuizen RM, Rosen BR, Lo EH. Normobaric hyperoxia reduces MRI diffusion abnormalities and infarct size in experimental stroke. Neurology. 2002a;58:945–952. doi: 10.1212/wnl.58.6.945. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Wang X, Sumii T, Mori T, Lo EH. Effects of normobaric hyperoxia in a rat model of focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2002b;22:861–868. doi: 10.1097/00004647-200207000-00011. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Benner T, Roccatagliata L, Koroshetz WJ, Schaefer PW, Lo EH, Buonanno FS, Gonzalez RG, Sorensen AG. A pilot study of normobaric oxygen therapy in acute ischemic stroke. Stroke. 2005;36:797–802. doi: 10.1161/01.STR.0000158914.66827.2e. [DOI] [PubMed] [Google Scholar]
- Sood R, Yang Y, Taheri S, Candelario-Jalil E, Estrada EY, Walker EJ, Thompson J, Rosenberg GA. Increased apparent diffusion coefficients on MRI linked with matrix metalloproteinases and edema in white matter after bilateral carotid artery occlusion in rats. J Cereb Blood Flow Metab. 2009;29:308–316. doi: 10.1038/jcbfm.2008.121. [DOI] [PubMed] [Google Scholar]
- Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol. 2008;64:654–663. doi: 10.1002/ana.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunami K, Takeda Y, Hashimoto M, Hirakawa M. Hyperbaric oxygen reduces infarct volume in rats by increasing oxygen supply to the ischemic periphery. Crit Care Med. 2000;28:2831–2836. doi: 10.1097/00003246-200008000-00025. [DOI] [PubMed] [Google Scholar]
- Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–189. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Zauner A, Daugherty WP, Bullock MR, Warner DS. Brain oxygenation and energy metabolism: part I-biological function and pathophysiology. Neurosurgery. 2002;51:289–301. discussion 302. [PubMed] [Google Scholar]