

Abstract
Bacterial ADP-ribosyltransferases (ADPRTs) are toxins that play a significant role in pathogenicity by inactivating host proteins through covalent addition of ADP-ribose. In this study we used ADP-ribosylated Kemptide as a standard to examine the effectiveness of three common tandem mass spectrometry fragmentation methods for assignment of amino acid sequence and site of modification. Fragmentation mechanisms investigated include low-energy collision induced dissociation (CID), infrared multiphoton dissociation (IRMPD), and electron capture dissociation (ECD); all were performed on a hybrid linear ion trap Fourier transform ion cyclotron resonance mass spectrometer. We show that ECD, but neither CID nor IRMPD, of ADP-ribosylated-Kemptide produces tandem mass spectra that are interpretable with regard to amino acid sequence assignment and site of modification. Examination of CID and IRMPD tandem mass spectra of ADP-ribosylated Kemptide revealed that fragmentation was primarily focused to the ADP-ribose region, generating several potential diagnostic ions for use in discovery of ADP-ribosylated proteins. Due to the lower relative sensitivity of ECD during data-dependent acquisition to CID, we suggest a two-fold strategy where CID and IRMPD are first used to detect ADP-ribosylated peptides, followed by sequence assignment and location of modification by ECD analysis.
Introduction
ADPRTs catalyze the transfer of ADP-ribose from β-NAD+ to target proteins (Scheme I), commonly at arginine residues[1]. Members of the ADPRT family modify a diverse set of target proteins and share little sequence homology. The mammalian ADP-ribosyltransferase ART-1 modifies defensin-1 which ultimately blocks the antimicrobial and cytotoxic effects of the defensin HNP-1[2]. Many ADPRTs found in bacteria have been implicated in pathogenicity through the inactivation of host proteins (e.g. Vibrio cholerae, Bordetella pertussis and Streptococcus pyogenes[1]). S. pyogenes produces the ADPRT toxin SpyA, which modifies vimentin, actin and tropomyosin, all proteins involved in cytoskeletal structure[3]. ADP-ribosylation of these proteins prevents polymerization of the cytoskeleton leading to collapse of intermediate filaments. Other ADPRT toxins are known to target proteins involved in signal transduction and regulatory functions[1]. Although there are numerous molecular assays demonstrating that ADP-ribosylation is an important step in pathogenicity, to date the exact site of ADP-ribosylation and number of modification targets remains unknown for many ADPRTs. A high-throughput mass spectrometry assay designed for the identification of target proteins as well as sites of ADP-ribosylation would provide insight into mechanisms of infection.
Scheme I.
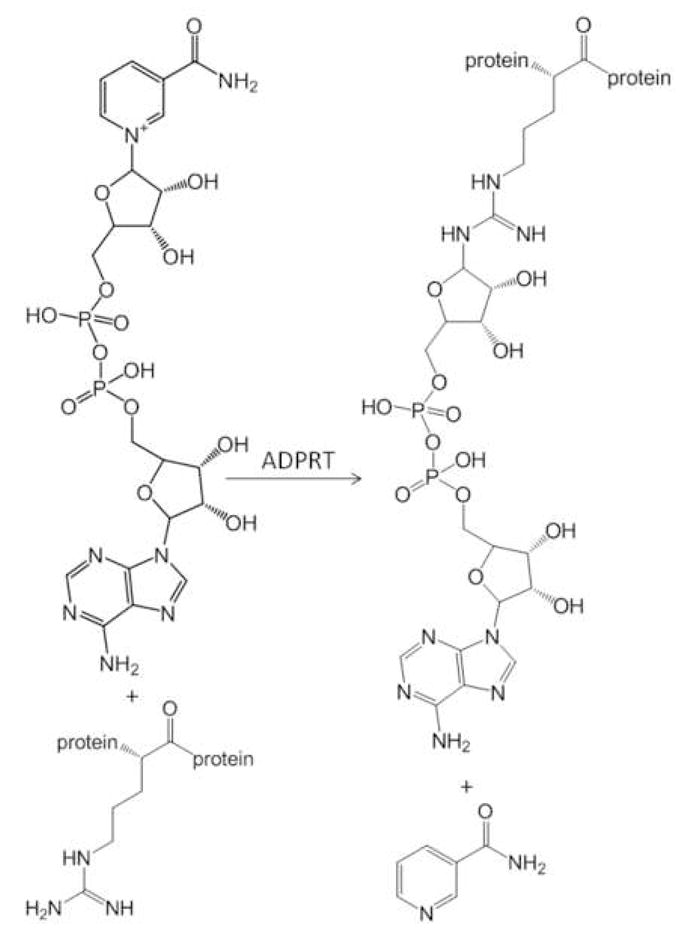
ADPRTs catalyze the transfer of ADP-ribose to acceptor proteins from NAD+.
Often LC-MS/MS methods are used to identify sites and protein targets of post-translational modifications (PTMs) in complex biological samples[4, 5]. Collision induced dissociation (CID) is typically employed as the main fragmentation method during LC-MS/MS analyses because the instrumental duty cycle is high and acquired peptide tandem mass spectra may be easily sequenced. This primarily results from the predictable peptide fragmentation pathways[6], allowing for the identification of the parent proteins[7]. However, the presence of a PTM on a peptide can redirect CID fragmentation patterns such that sequence may not be assigned. Previously, Margarit et al. (2006)[8] demonstrated that CID of an ADP-ribosylated peptide did not result in typical’ peptide fragmentation, interfering with facile sequence interpretation. Additionally, CID analysis of SpyA ADP-ribosylated Vimentin[3] verified similar fragmentation trends, preventing modified peptide sequence assignment (SM Hengel unpublished data). The inability to directly assign peptide sequences of ADP-ribosylated peptides using CID led us to undertake a detailed mass spectrometry fragmentation pattern analysis of a standard, ADP-ribosylated-Kemptide (LRRASLG). Three common fragmentation methods were investigated: 1) CID, 2) electron capture dissociation (ECD) and 3) infrared multiphoton dissociation (IRMPD).
The goals of this study were to determine the most efficient technique for producing reliable and predictable fragment-ion spectra for the identification of ADP-ribosylated peptides and to determine if diagnostic ions exist that might be used in future studies to identify new ADPRT targets. ADP-ribosylated Kemptide was chosen as a standard for examining which of the three methods, CID, ECD and IRMPD, were best suited to 1) assign amino acid sequence and 2) produce ions diagnostic of ADP-ribosylation. Here we discuss the results of this comparison and how these methods may be used alone and in combination to identify ADP-ribosylated proteins in complex mixtures.
Experimental
ADP-Ribosylation of Peptide Standard
Reagents used for peptide modification were obtained from Sigma (St. Louis, MO). A 5 mM solution of Kemptide (Leu-Arg-Arg-Ala-Ser-Leu-Gly, MW 771.4715) was modified as previously described[9], in the presence of 10 mM ADP-ribose, 20 mM DTE and 100 mM phosphate buffer at 33° C for four hours. After incubation, the extent of ADP-ribosylation was determined by comparing peak height and area of extracted ion chromatograms for modified and unmodified Kemptide precursor masses following separation on a C18 column described below.
Electrospray Ionization LTQ-FT-based Mass Spectrometry
Peptides were analyzed using electrospray ionization in a linear ion trap Fourier transform ion cyclotron resonance mass spectrometer (LTQ-FT ICR) (Thermo Electron Corp., San Jose, CA). HPLC columns were packed in-house (0.75 μm i.d. × 11cm; 100 Ǻ Magic C18AQ: Michrom Bioresources, Auburn, CA), and separations were performed with an inline Agilent 1100 binary HPLC pump using a linear gradient of 5 to 35% ACN over one hour. ESI voltage was applied with a liquid junction prior to the analytical column with a gold wire and a micro-tee junction[10]. All ions were measured in positive ion mode, both precursor and fragment masses were detected in the FT mass analyzer. Source parameters were set as follows: ESI voltage, 2.5 kV; capillary temperature, 265° C; capillary voltage, 13 V; tube lens, 35 V. For all fragmentation methods the resolution was set to 5.0e4 and 2.0e4 for MS and MS/MS respectively. Automatic Gain Control (AGC) was used to maintain constant ion populations at 5e5 and 2e5 for MS and MS/MS respectively.
Fragmentation Methods
Reported tandem mass spectra for both modified and unmodified Kemptide are from doubly charged precursor masses 657.27 and 386.74 m/z respectively, tandem mass spectra were acquired in the FT-ICR cell during LC-MS/MS analysis. Fragment ion spectra acquired with CID were produced using 35% collision energy and a 2.0 Da isolation window. ECD tandem mass spectra were acquired using 5 eV fragmentation energy for 70 ms with a delay of 0.3 msec and a 3.0 Da isolation window. IRMPD was performed by irradiating precursor ions with photons produced by a CO2 laser [Synrad firestar series V20, model FSV20SFB; 75 Watts (10.2–10.8 μm)] with a pulse duration of 50 msec a power of 60% and an isolation window of 5.0 Da.
Limit of Detection
To determine the limit of detection of ADP-ribosylated Kemptide in a complex mixture, a BSA peptide standard was used to dilute the analyte (Michrom Bioresource). BSA was maintained at a concentration of 5 pmol/μL and serial dilutions of the analyte were initiated with a 1:1 ratio of BSA to ADP-ribosylated Kemptide. ADP-ribosylated Kemptide concentrations ranged from 2.5 pmol/μL to 250 amol/μL in 5-fold serial dilution increments (i.e. 1:1; 1:10; 1:50; 1:100…). Samples were analyzed using data-dependent LC-MS/MS conditions described above, selecting the five most intense peaks observed in the MS spectrum for MS/MS, acquiring both CID and IRMPD data for each dilution.
Results and Discussion
Collision Induced Dissociation (CID)
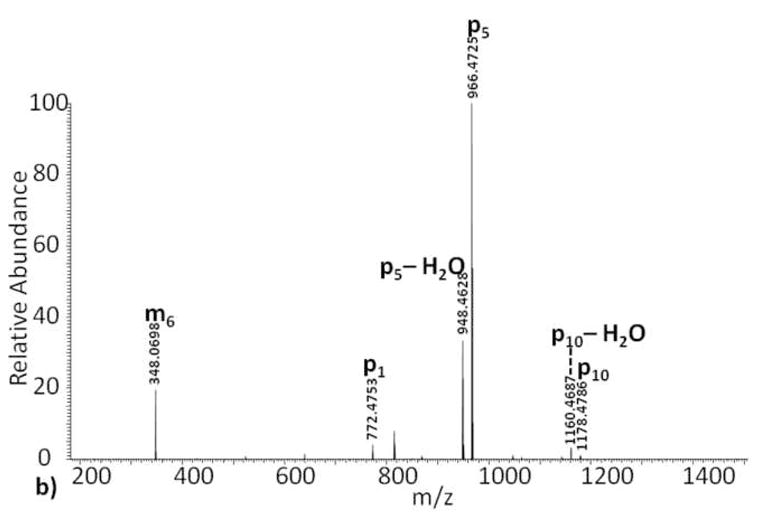
The process of conducting CID combined with automated, data-dependent ion selection provides a fast duty cycle that is well suited for LC-based analysis of mixtures and is therefore commonly used in proteomics[7]. However, the CID generated tandem mass spectrum of ADP-ribosylated Kemptide contains none of the observed b or y ions in the unmodified Kemptide CID tandem mass spectrum (Figure 1a) making sequence and site of ADP-ribosylation assignment impossible (Figure 1b). Close examination of the APD-ribosylated Kemptide CID tandem mass spectrum revealed several ions not accountable by Kemptide peptide backbone fragmentation (Figure 1b). Based on accurate mass measurements, the known structure of ADP-ribose, and Kemptide amino acid sequence, most of these ions appear to be formed through fragmentation of the ADP-ribose region.
Figure 1.

Collision induced dissociation tandem mass spectra of (a) Kemptide, and (b) ADP-ribosylated Kemptide.
ADP-ribose contains ten single bonds, excluding cross ring fragmentation, which can yield fragment ions. In order to keep track of fragment ions occurring on the ADP-ribose backbone, we suggest the nomenclature shown in Scheme II. Fragment ions with charge retained on the ADP-ribose group, but with loss of the peptide, are labeled m1 … m10 for modification. Fragment ions that contain part of the ADP-ribose region with the peptide backbone intact are labeled p1 … p10. We do not show any cross-ring fragmentation sites labeled in Scheme II because none were observed in our studies. However, we realize that such fragments might be formed by some future fragmentation method or combination of current methods. For these potential fragment ions we propose the use of a variation on the carbohydrate fragmentation nomenclature proposed by Domon and Costello[11]. In place of the subscript number for single bond fragments, rings are assigned a Roman numeral (i.e. I for the ribose closest to the peptide, then II, etc.), and cross ring fragments are noted using numbers indicating which cross ring bonds are broken. Similar to the single bond fragments along ADP-ribose, m and p will be used to designate on which side the charge is observed. For example, a fragmentation event at the ribose ring, adjacent to the guanidine functional group, between bonds zero and two would be designated as 0,2pI or 0,2mI depending on which side the charge is observed. The ADP-ribosylated Kemptide CID tandem mass spectrum contains an observed ion at m/z 348, labeled m6 (Scheme II), which results from fragmentation of the APP-ribose pyrophosphate bond. This fragmentation generates two types of ions explained with structures where charge is retained on both sides of the cleavage site. The measured mass difference for the ion labeled m6 and that of the putative structural assignment is calculated to be 1.7 ppm, strongly suggesting the assignment is correct. We also note that previously reported results of CID ADP-ribosylated peptides[8] yielded similar fragment ions localized between ribose and the phosphate group proximal to the peptide backbone, again suggesting that the ADP-ribose modification “directs” CID fragmentation process of ADP-ribosylated peptides.
Scheme II.
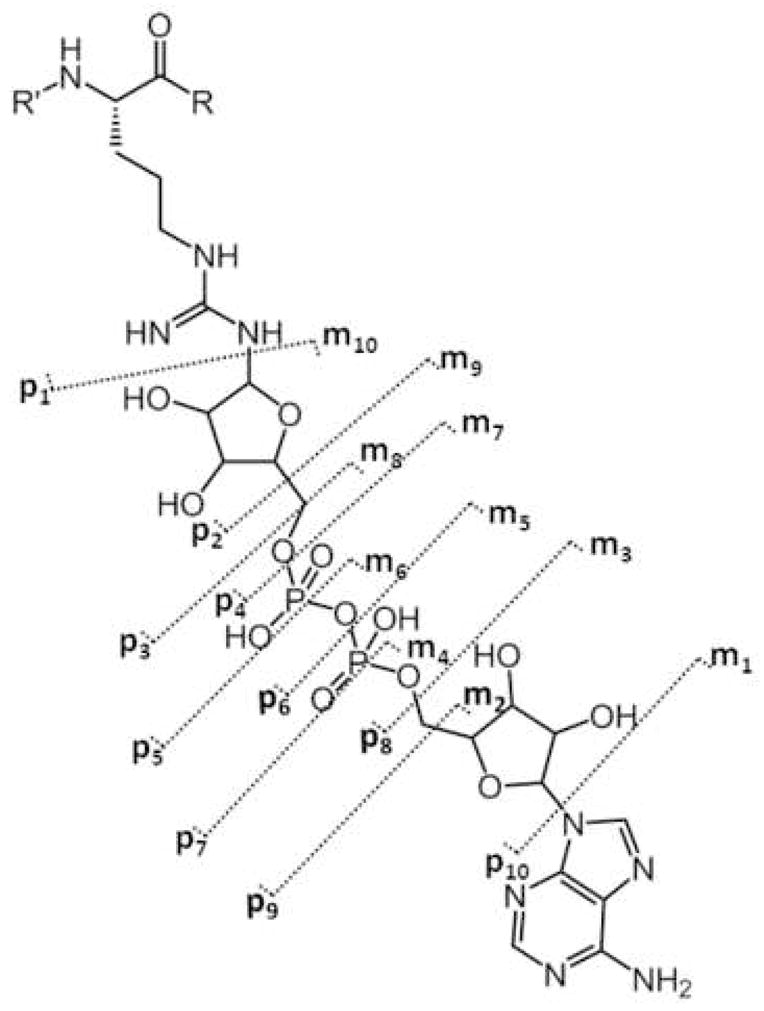
ADP-ribose fragmentation scheme: fragment ions containing the intact peptide backbone plus a portion of the modification are labeled p1 through p10. Fragment ions independent of the peptide sequence, only containing the ADP-ribose modification, are labeled m1 through m10.
Further examination into the origin of other prominent ions in Figure 1b, i.e. the base peak, suggests that such ions are complimentary fragments to the ion at 348 m/z and an additional fragment near the adenine portion of the ADP-ribose group (fragments labeled p5, p10; Scheme II). Unlike the ion at 348 m/z, which appears to be charged fragments of ADP-ribose without peptide attached, ions p5 and p10 contain a mass equal to that of Kemptide plus a residual mass from ADP-ribose fragmentation. Charge states of this type of fragment ion are equivalent to the initial charge state minus the loss of the protonated ADP-ribose fragment. Because it is not a neutral loss, often seen in CID with other post translational modifications like phosphorylation[5], the charge state of the observed fragment ions may vary. Therefore, we predict that CID spectral complexity of ADP-modified peptides will increase directly with amino acid number and charge state of the precursor peptide.
While the CID tandem mass spectrum of ADP-ribosylated Kemptide contains ions that may be diagnostic of ADP-ribose, the spectrum does not contain any information on peptide sequence as seen in the associated CID tandem mass spectrum of Kemptide (Figure 1a). In an attempt to understand what single tandem mass spectrometry method, or combination of methods, provides full interpretation of peptide sequence, site of modification, and generates potential diagnostic ions, we investigated the fragmentation of Kemptide and ADP-ribosylated Kemptide by ECD and IRMPD.
Electron Capture Dissociation (ECD)
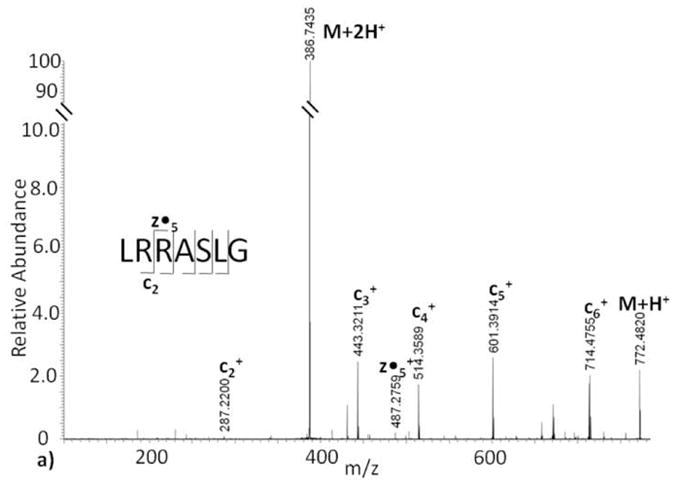
In comparison to the CID tandem mass spectrum (Figure 1a), a more complete ion series is observed with ECD fragmentation on the unmodified Kemptide standard (Figure 2a). During ECD one charge on the peptide is neutralized due to the capture of an electron. As a result, peptides which are initially doubly charged retain a charge on only one of the fragment ion products, thus both complete c or z ion series may not be detected (Figure 2a). Therefore, larger peptides with higher charge states should have a greater probability to produce both c and z ions upon ECD fragmentation, increasing the opportunity for assigning post-translational modification sites.
Figure 2.
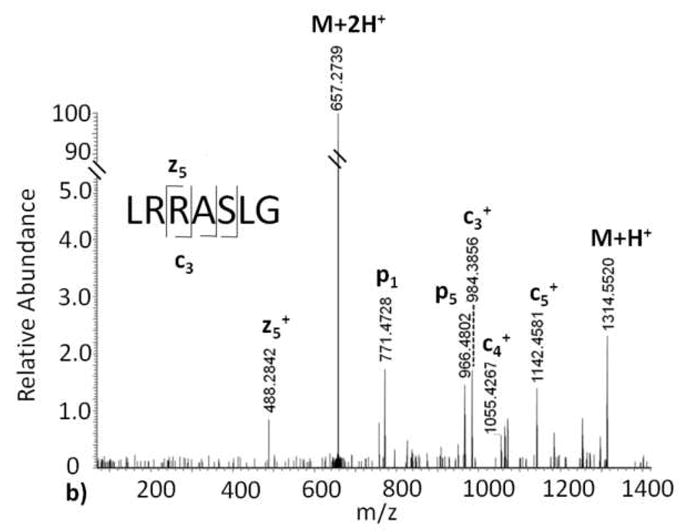
Electron capture dissociation tandem mass spectra of (a) Kemptide, and (b) ADP-ribosylated Kemptide.
A distinguishing feature of ECD is the retention of peptide side chain modifications [12–14]. Predictably, we see in our ECD data of ADP-ribosylated Kemptide (Figure 2b) that ADP-ribose is retained on the arginine side chain. Additionally, the ECD tandem mass spectrum of ADP-ribosylated Kemptide contains three of six predicted c ions and one of six z peptide fragment ions within 10 ppm mass accuracy of the calculated theoretical values. As a result, peptide sequence and the site of ADP-ribosylation can be elucidated from the ECD tandem mass spectrum. The Kemptide amino acid sequence contains two arginine residues, and we were able to localize the site of modification to the second amino acid, i.e. LR*RASLG where R* = arginine ribosylated on the guanidinium amino group, in the sequence using ECD. In contrast to the CID tandem mass spectrum, only two observed ions of lower intensity correlate to fragmentation of the ADP-ribose group in the ECD tandem mass spectrum (fragment p1 and p5, Figure 2b). This indicates that, unlike CID of ADP-ribosylated Kemptide, a minimal amount of the ECD energy is directed to ADP ribose and the majority of energy is directed to fragmentation of the peptide N-Cα amide backbone in a predictable manner. Thus, ECD is a reliable fragmentation method that can provide information for peptide sequencing and site-assignment of ADP-ribosylated Kemptide.
Infrared Multiphoton Dissociation (IRMPD)
As expected, the IRMPD fragment ion spectrum of the unmodified Kemptide peptide standard contains b and y ions, as well as a diagnostic immonium ion (Figure 3a). Despite the fact that both CID and IRMPD generate fragment ions through an increase in vibrational energy, there is a noticeable difference between the unmodified Kemptide CID and IRMPD tandem mass spectra. e One likely source of this variation is 10 the time of flight affect observed for CID fragment ions generated in the LTQ and subsequently transmitted to the ICR cell, as opposed to IRMPD fragment ions that are both generated and measured in the ICR-cell[15]. Compared to the unmodified Kemptide ECD tandem mass spectrum, the observed IRMPD ion series is less complete, and peptide sequencing of this short peptide would be challenging. Implementation of IRMPD fragmentation on ADP-ribosylated Kemptide produced a tandem mass spectrum with only one fragment ion, y5, corresponding to peptide backbone fragmentation (Figure 3b). Upon further analysis, the IRMPD tandem mass spectrum base peak, 136 m/z, is not a result of peptide backbone fragmentation, but instead is the compliment of fragment ion p10 (Scheme II) found in the CID spectrum. This ion, m1, appears to result from the loss of adenine from ADP-ribose, is independent of peptide sequence, and confirmed again using less than 1 ppm mass error.
Figure 3.
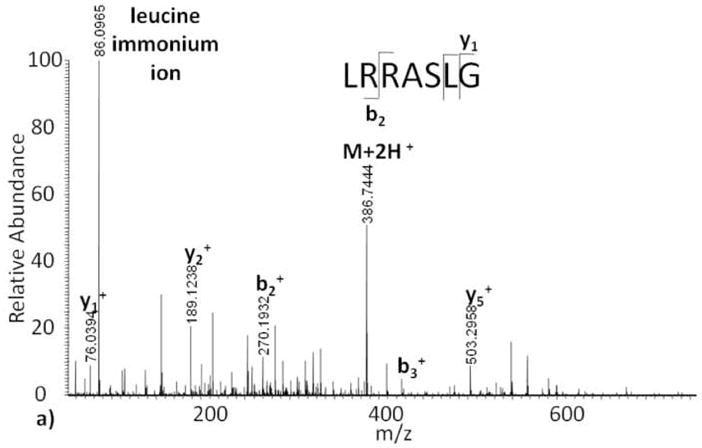
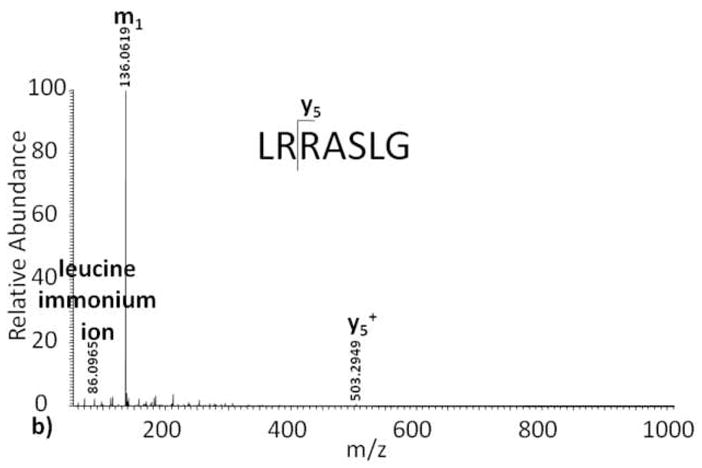
Infrared multiphoton dissociation tandem mass spectra of (a) Kemptide, and (b) ADP-ribosylated Kemptide.
Distinct from the ADP-ribosylated Kemptide ECD spectrum and the unmodified Kemptide IRMPD spectrum, the precursor ion is no longer observed when using IRMPD to fragment ADP-ribosylated Kemptide. This may be due to the ease in which the ADP-ribose group fragments, as seen in the CID tandem mass spectrum, and an increase of photon absorbtivity[16], making the fragmentation more efficient for an ADP-ribose group when compared to a peptide backbone.
Potential Diagnostic Ions of ADP-ribosylated Peptides
Both IRMPD and CID fragmentation techniques generated ions independent of the Kemptide amino acid sequence, i.e. 136 m/z and 348 m/z respectively, yet specific to ADP-ribose. One of these ions, 348 m/z, was previously observed using CID to study an ADP-ribosylated actin peptide[8]. In addition, CID tandem mass spectrometry generated fragment ions that are indicative of an ADP-ribose modification and dependent on peptide sequence. Ions such as these are potentially diagnostic of ADP-ribose modifications on a peptide and may be useful in discovery based proteomic screens that seek to identify targets of ADPRTs. However, because neither CID nor IRMPD produced fragment ions that lead to peptide sequence assignment, precursor ions identified containing ADP-ribose in an initial CID or IRMPD screening-analysis would need to be further subjected to ECD fragmentation. There are two primary benefits of using CID over ECD or IRMPD in the initial discovery of ADP-ribosylated peptide precursor ion masses: 1) ions that are dependent on and independent of peptide precursor ion mass to charge ratios are produced providing multiple fragment ions indicative of ADP-ribosylated peptides, thus reducing the false discovery rate and 2) duty cycle is high, when acquired in an LTQ, relative to IRMPD and ECD providing better analytical sampling for detection of ADP-ribosylated peptides. However, in this study all fragment ions were measured in the FT-ICR cell to generate high confidence structure assignments. When analyzing samples with a large dynamic range where sensitivity is crucial to detection, IRMPD should provide two advantages: 1) the IRMPD-produced diagnostic ion at 136 m/z is the base peak of the tandem mass spectrum and 2) ions are measured at high mass accuracy in the ICR cell. Both CID and IRMPD have unique benefits when searching for diagnostic ions in tandem mass spectra. Provided sample availability and sufficient instrument access, we suggest performing both CID and IRMPD LC-MS/MS experiments measuring fragment masses in the ICR cell generating high mass accuracy data. If CID fragments were generated and measured in the LTQ while IRMPD fragmentation occurs in the FT-ICR, then both data could be acquired in a single run with minimal reduction in duty cycle. However, compared to measuring fragment ions in the ICR cell, the loss of mass accuracy for CID fragment ions acquired in the LTQ would be substantial. Given that no neutral loss trigger is possible with the observed type of fragment ions, a discovery-based screen designed to detect ADP-ribosylated peptides would need to be conducted in two LC-MS/MS experiments. First, a typical data-dependent experiment would be performed using CID, and or IRMPD, and second an ECD experiment using an inclusion list based on the first experiment would be completed. When MS-driver software advances to allow on-the-fly switching from CID/IRMPD to ECD with this type of diagnostic ion, then the discovery and identification of ADP-ribosylated peptides could be conducted in a single LC-MS/MS experiment.
Testing Diagnostic Ions
Spiking ADP-ribosylated Kemptide into a complex mixture simulated an experiment designed to discover new targets of ADPRTs. These analyses also allowed us to test the proposed diagnostic ions and establish a limit of detection. Three proposed ions diagnostic of ADP-ribosylated peptides were investigated as marker ions for the purpose of rapid identification of unknown ADPRTs targets. These ions were selected based on intensity and observed frequency from MS analyses of ADP-ribosylated Kemptide. Two of the ions, m6 and p5 (Scheme II), are observed in CID tandem mass spectra, and the third, m1, is observed when using IRMPD fragmentation.
Varying amounts of ADP-ribosylated Kemptide were spiked into a BSA protein digest (see Limit of Detection) for two reasons: 1) to determine a limit of detection 2) to assess whether m1, m6 and p5 fragment ions could be used to accurately and reproducibly detect the tandem mass spectra of ADP-ribosylated Kemptide in a mixture of non-ribosylated peptides from BSA. Data was acquired via a data-dependent ion selection scheme that selected the top five ions per precursor ion scan for tandem mass spectrometry; i.e. CID or IRMPD. A limit of detection was determined for both precursor ions and all three potential diagnostic fragment ions. In all cases, the limit of detection was determined to be femtomoles of ADP-ribosylated Kemptide in the presence of picomoles of the protein digest at a signal to noise ratio of 2:1.
Tandem mass spectra from the spiking experiments were sorted using PERL scripts that identified dta files containing each of the three specified marker ions within 10 ppm mass accuracy. Precursor ion masses of the dta files containing any one of these three diagnostic ions were then compared to the known ADP-ribosylated Kemptide mass. All observed precursor masses from the three PERL script lists that were outside a mass tolerance of 10 ppm were considered false positives. Using this measure, initial false positive rates of each ion independent of the others were high: m6 95% (CID), p5 66% (CID), and m1 92% (IRMPD). This is likely a reflection of the commonality of these ions in many tandem mass spectra. However, when precursor masses identified from dta files selected by the three PERL scripts were used in combination rather than independently, the resulting overall false positive rate was reduced to 0%. This same type of calculation changed the false negative rate from 0% to 8%. This suggests that when trying to blindly screen tandem mass spectral data sets for dta files containing ions diagnostic of ADP-ribose that one should include all three diagnostic ions to maintain a low false positive rate. Using fragment ions diagnostic of ADP-ribose from both CID and IRMPD data, we were able to successfully identify in an automated manner (subsequently confirmed manually) dta files of ADP-ribosylated Kemptide spiked into a digest of BSA. The same approach should be useful for discovery work in more complex whole cell lysates, however this may result in a higher false positive rate.
Conclusions
Examination of three different fragmentation techniques for the purpose of understanding ADP-ribosylated Kemptide fragmentation patterns yielded tandem mass spectra with both unique and shared fragment ions. Although CID and IRMPD produced ions diagnostic of ADP-ribose useful for determining whether a parent mass contains ADP-ribose, the tandem mass spectra lack significant peptide backbone fragmentation preventing identification of peptide sequence and modification site. Because ECD has been recognized as a technique that provides peptide backbone fragmentation while allowing for retention of post-translational modifications[13, 14], we investigated its utility for sequencing ADP- ribosylated peptides. Our data clearly demonstrated that ECD is the best fragmentation method for determining peptide sequence and the site of modification of ADP-ribosylated peptides. Electron transfer dissociation[17], ETD, a similar fragmentation method to ECD may also provide sequence information for an ADP-ribosylated peptide, but has not been examined in this study. Disadvantages of ECD, however, include a longer duty cycle and low fragmentation efficiency relative to CID. Because of these drawbacks, we recommend first using CID and IRMPD fragmentation techniques to discover ADP-ribosylated peptides with diagnostic ions, followed by targeted ECD analyses to decipher the location of the modification and the sequence of the peptide. This two step assay will allow for a more thorough analysis when attempting to identify ADP-ribosylated targets compared to only using shotgun proteomic analysis with ECD alone.
Acknowledgments
The authors wish to thank the NIH Pharmaceutical Sciences Training Grant (T32 GM07750) and NCRR (1S10RR017262-01) for funding and support. Additional thanks go to Dr. Alex Sherl for PERL script assistance, and Dr. Priska Van Haller at the University of Washington South Lake Union Mass Spec Facility. This work was supported in part by the University of Washington’s Proteomics Resource (UWPR95794).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Corda D, Di Girolamo M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003;22(9):1953–8. doi: 10.1093/emboj/cdg209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paone G, Wada A, Stevens LA, Matin A, Hirayama T, Levine RL, Moss J. ADP ribosylation of human neutrophil peptide-1 regulates its biological properties. Proc Natl Acad Sci U S A. 2002;99(12):8231–5. doi: 10.1073/pnas.122238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coye LH, Collins CM. Identification of SpyA, a novel ADP-ribosyltransferase of Streptococcus pyogenes. Mol Microbiol. 2004;54(1):89–98. doi: 10.1111/j.1365-2958.2004.04262.x. [DOI] [PubMed] [Google Scholar]
- 4.Huddleston MJ, Annan RS, Bean MF, Carr SA. Selective Detection of Phosphopeptides in Complex-Mixtures by Electrospray Liquid-Chromatography Mass-Spectrometry. Journal of the American Society for Mass Spectrometry. 1993;4(9):710–717. doi: 10.1016/1044-0305(93)80049-5. [DOI] [PubMed] [Google Scholar]
- 5.Hunter AP, Games DE. Chromatographic and Mass-Spectrometric Methods for the Identification of Phosphorylation Sites in Phosphoproteins. Rapid Communications in Mass Spectrometry. 1994;8(7):559–570. doi: 10.1002/rcm.1290080713. [DOI] [PubMed] [Google Scholar]
- 6.Dookeran NN, Yalcin T, Harrison AG. Fragmentation reactions of protonated alpha-amino acids. Journal of Mass Spectrometry. 1996;31(5):500–508. [Google Scholar]
- 7.Spahr CS, Davis MT, McGinley MD, Robinson JH, Bures EJ, Beierle J, Mort J, Courchesne PL, Chen K, Wahl RC, Yu W, Luethy R, Patterson SD. Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometryI. Profiling an unfractionated tryptic digest. Proteomics. 2001;1(1):93–107. doi: 10.1002/1615-9861(200101)1:1<93::AID-PROT93>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 8.Margarit SM, Davidson W, Frego L, Stebbins CE. A steric antagonism of actin polymerization by a salmonella virulence protein. Structure. 2006;14(8):1219–29. doi: 10.1016/j.str.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 9.Kharadia SV, Graves DJ. Relationship of phosphorylation and ADP-ribosylation using a synthetic peptide as a model substrate. J Biol Chem. 1987;262(36):17379–83. [PubMed] [Google Scholar]
- 10.Yi EC, Lee H, Aebersold R, Goodlett DR. A microcapillary trap cartridge-microcapillary high-performance liquid chromatography electrospray ionization emitter device capable of peptide tandem mass spectrometry at the attomole level on an ion trap mass spectrometer with automated routine operation. Rapid Commun Mass Spectrom. 2003;17(18):2093–8. doi: 10.1002/rcm.1150. [DOI] [PubMed] [Google Scholar]
- 11.Domon B, Costello CE. A Systematic Nomenclature for Carbohydrate Fragmentations in FAB-MS/MS Spectra of Glycoconjugates. Glycoconjugate Journal. 1988;5:13. [Google Scholar]
- 12.Breuker K, Oh H, Lin C, Carpenter BK, McLafferty FW. Nonergodic and conformational control of the electron capture dissociation of protein cations. Proc Natl Acad Sci U S A. 2004;101(39):14011–6. doi: 10.1073/pnas.0406095101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelleher NL, Zubarev RA, Bush K, Furie B, Furie BC, McLafferty FW, Walsh CT. Localization of labile posttranslational modifications by electron capture dissociation: the case of gamma-carboxyglutamic acid. Anal Chem. 1999;71(19):4250–3. doi: 10.1021/ac990684x. [DOI] [PubMed] [Google Scholar]
- 14.Kjeldsen F, Haselmann KF, Budnik BA, Sorensen ES, Zubarev RA. Complete characterization of posttranslational modification sites in the bovine milk protein PP3 by tandem mass spectrometry with electron capture dissociation as the last stage. Anal Chem. 2003;75(10):2355–61. doi: 10.1021/ac026295b. [DOI] [PubMed] [Google Scholar]
- 15.Scherl A, Shaffer SA, Taylor GK, Hernandez P, Appel RD, Binz PA, Goodlett DR. On the benefits of acquiring peptide fragment ions at high measured mass accuracy. J Am Soc Mass Spectrom. 2008;19(6):891–901. doi: 10.1016/j.jasms.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowe MC, Brodbelt JS. Infrared multiphoton dissociation (IRMPD) and collisionally activated dissociation of peptides in a quadrupole ion trap with selective IRMPD of phosphopeptides. J Am Soc Mass Spectrom. 2004;15(11):1581–92. doi: 10.1016/j.jasms.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 17.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101(26):9528–33. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]