

Abstract
Iron-sulfur clusters are an important class of protein-bound prosthetic center that find wide utility in nature. Roles include electron transfer, enzyme catalysis, protein structure stabilization, and regulation of gene expression as transcriptional and translational sensors. In eukaryotes their biosynthesis requires a complex molecular machinery that is located within the mitochondrion, while bacteria exhibit up to three independent cluster assembly pathways. All of these paths share common themes. This review summarizes some key structural and functional properties of three central proteins dedicated to the Fe-S cluster assembly process: namely, the sulfide donor (cysteine desulfurase); iron donor (frataxin), and the iron-sulfur cluster scaffold protein (IscU/ISU).
Keywords: Iron-sulfur cluster, assembly, biosynthesis, ISU/IscU, frataxin, NFS/IscS
1. Introduction
Iron-sulfur clusters are one of the most ancient, ubiquitous and versatile classes of metal cofactor in nature. They are exemplified by three common forms ([2Fe-2S], [3Fe-4S] and [4Fe-4S], Figure 1), although more complex cluster arrays are known, such as the [8Fe-7S] center in nitrogenase [1, 2]. In these clusters the iron ions are typically bridged by inorganic sulfide and ligated to the protein by cysteine residues, although non-cysteinyl ligands, such as histidine, arginine, lysine, serine and water [3-6] are occasionally used. Eukaryotic iron-sulfur clusters are localized in the mitochondrion, cytosol, endoplasmic reticulum and nucleus. The variability of oxidation state between +2 and +3 valency allows for a complex biological redox chemistry, and potentials ranging from -500 mV to 300 mV [7]. The functional chemistry includes both redox and non-redox roles in pathways as diverse as mitochondrial respiration, enzymology, gene regulation and DNA/RNA metabolism.
Figure 1.

Prototypical structures of the most common iron-sulfur centers. [2Fe-2S], [3Fe-4S] and [4Fe-4S] clusters with cysteines typically providing the additional ligand contacts to iron.
Three distinct pathways have been identified for bacterial Fe-S cluster biosynthesis [2, 8-11]. The ISC system provides for general Fe-S cluster assembly [12, 13], the NIF system is dedicated to cluster maturation for nitrogenase [14] and the SUF system [15] is suggested to serve as an independent pathway that is activated in times of oxidative stress. The components of the ISC machinery include a cysteine desulfurase IscS (the sulfide donor), frataxin (the iron donor), IscU and IscA (scaffold proteins), ferredoxin (redox chemistry), and chaperones. The NIF and SUF systems possess many homologous proteins and domains for the IscS, IscU and IscA families. The iron donor protein(s) for the NIF and SUF pathways remains uncertain, although an iron donor role for IscA-type proteins has been suggested [16-18]. Certain classes of bacteria contain both the ISC and SUF operons, with SUF associated with harsh cellular environments, such as oxidative stress or limited iron availability, while the ISC operon appears most relevant for cells functioning under normal conditions [19]. The fundamental cluster biosynthesis mechanisms in all three pathways appear very similar and two of these three systems, ISC and SUF, have adapted to eukaryotic use. Mitochondria possess a set of gene products that are homologous to those associated with ISC, while homologues to SUF components are found in plastids [20, 21]. The mitochondrial machinery appears to be essential for cellular cluster biosynthesis in eukaryotes, irrespective of the final destination of the cluster (cytosol, nucleus etc.). Additional protein components that constitute part of a cytosolic iron-sulfur cluster assembly machinery (CIA) have been identified, and understanding the CIA and its linkage to mitochondrial cluster export is an emerging area of investigation [7, 13, 20, 22]. Key members of the CIA include a Cfd1-Nbp35 scaffold complex [23-25] that bind clusters with the assistance of other CIA component proteins, Nar1 and Cia1 [26, 27]. The former two are ATPases that bind [4Fe-4S] centers in a C-terminal Cys-rich domain in each protein, and an N-terminal site in Nbp35. The most plausible explanation for this linkage is for the CIA to be a family of proteins engaged in the transport and transfer of clusters to cytosolic (or nuclear etc.) targets following export from the mitochondrion. Although the molecular details of how clusters are exported from the mitochondria remains uncertain [7], and in particular the chemical formulation of the transported species, nevertheless several important components have been successfully identified. Atm1p/ABCB7 has been identified as a key element of the export apparatus and is located on the inner mitochondrial membrane [22, 28-30]. The exported compound is believed to involve thiol functional groups [30-32]. Genetic studies suggest another component to be Erv1, which is located in the intermembrane space and catalyzes the formation of disulfide bonds [33].
Iron-sulfur cluster biosynthesis can be viewed in terms of three major steps (Figure 2) [11]; namely cluster assembly on the scaffold protein ISU/IscU, mitochondrial export (where necessary) [28, 30, 34], and transport and delivery to a target protein. This review will focus on the first step of cluster assembly on the scaffold protein, including NFS/IscS mediated sulfide delivery following desulfurization of cysteine amino acid [35], [36, 37], and frataxin-promoted iron delivery to IscU/ISU [38].
Figure 2.

Schematic illustration of the key steps and proteins involved in iron-sulfur cluster biosynthesis. In the first step, IscU/ISU receives iron and sulfide from the iron donor protein frataxin and sulfur donor protein IscS, respectively, and assembles the [2Fe-2S] iron-sulfur cluster. Subsequently the assembled cluster is transferred to an apo target protein.
2. Scaffold Protein-IscU/ISU
The IscU family is one of the most conserved and ubiquitous classes of protein in nature, and homologues have been identified in essentially all cellular forms. While a [4Fe-4S]-bound cluster has been characterized in the specific case of A. vinelandii IscU, the general holo state and most likely biologically relevant form is clearly the [2Fe-2S] bound cluster. Three conserved cysteines, together with a non-cysteinyl ligand serve to bind the cluster. This section summarizes the structural and conformational chemistry of the IscU class of protein, characterization of the non-cysteinyl ligand, and molecular pathways for cluster formation.
2.1 Conformational flexibility of IscU/ISU
Knowledge of the structure of the scaffold protein is clearly important for the understanding of their function in iron-sulfur cluster biosynthesis. However, it is also increasingly evident that this class of protein displays unusual conformational dynamics that is both important for a complete understanding of their functional chemistry, and has complicated the process of structure determination. Until recently only a very limited number of representative structures had been elucidated – a reflection of the conformational flexibility and dynamic behavior associated with this class of protein.
The first structure of a representative IscU was determined by NMR in the case of H. influenzae IscU [39]. These studies were performed under reducing conditions in the presence of bound zinc in the cluster binding site. While the bound zinc has no clear physiological function, it did appear to stabilize a specific conformer of the protein to facilitate structure determination. In the zinc-bound form Hi IscU is monomeric, having a compact core structure of an α-β sandwich with three antiparallel β-sheets packed between four α helixes. Upon removal of zinc from the binding site, Hi IscU demonstrated substantial loss of structure and greater structural flexibility [39]. The NMR solution structures for the zinc-bound forms of Mus musculus(Mm) IscU (PDB entry 1WFZ), Bacillus subtilis (Bs) IscU (PDB entry 1XJS) and the crystal structure of Thermus thermophilus(Tt) IscU (PDB entry 2QQ4) all show the same core structure with variable N-terminal conformation (Figure. 3). The crystal structure of Aquifex aeolicus(Aa) IscU reveals a unique asymmetric trimer with only one subunit carrying a [2Fe-2S] center [40]. The three subunits in the Aa IscU trimer exhibit similar core structures, with observed differences between the N-terminus and the L2, L4 and L7 loops. These differences reflect the coordination of a cluster in one of the subunits, and the relative interactions between them, consistent with the structural flexibility of IscU.
Figure 3.

IscU core structures. Crystallographic and NMR solution studies show very similar core structures for Hi IscU, Mm IscU, Bs IscU, Tt IscU and Aa IscU.
In earlier efforts to structurally characterize the Thermotoga maritima (Tm) IscU, various secondary structure elements were clearly identified by CD and NMR [36, 41], however, the paucity of long-range NOE's made it difficult to define the overall fold of the protein. Both NMR and various biochemical studies led to the conclusion that Tm IscU is a conformationally flexible protein that samples multiple conformations with unusually slow dynamics [41, 42]. The experimental evidence was consistent with a model wherein the protein could exist in multiple discrete conformations in the absence of an external stabilizing factor, such as binding to a metal cofactor or protein partner that would stabilize a specific conformer appropriate for that interaction.
A number of related studies also support the conformational flexibility of this family of proteins across the range of bacterial to eukaryotic organisms. X-ray structural characterization of the complex of the IscU-derived peptide ELPPVKIHC [43], a chaperone recognition motif, and the substrate binding domain of HscA [44, 45] indicates a significant conformational change of the peptide upon binding to HscA. NMR studies of the interaction between E. coli IscU and E. coli HscB have also demonstrated IscU to exist in solution in two major conformations that exchange slowly, and where binding of HscB stabilizes the ordered state of IscU [46]. Crystallographic studies of the E. Coli IscS/IscU complex reveals major conformational changes of the N-terminal residues (Glu5-Glu12), following complex formation, which are largely disordered in all available structures, but become ordered and folded into an α-helix following complex formation and provide the crucial contacts with IscS [47].
The structural flexibility of IscU-type proteins appears to be necessary for their physiological role of assembling a [2Fe-2S] cluster building block via interactions with several key partner proteins (including frataxin, NFS/IscS, chaperones, holo Fd's [48], as a minimal set). The holo form of the assembly protein must then recognize a wide range of apo target proteins, to which the cluster is transferred intact [48], including ferredoxin [48, 49], and aconitase [50]. The complexity of the cluster transfer chemistry, with residues on the target protein required to effect ligand substitution reactions of the IscU-bound cluster, coupled with the range of partner proteins and distinct functional chemistries that each performs, all presumably underlie the need for conformational flexibility of the IscU protein. In certain cases specific chaperones may also be required to promote selectivity or facilitate individual cluster transfer steps. For example, human Ind1 is required for effective cluster assembly in complex I [51, 52]. It is also expected that binding of IscU to a specific target protein would induce the stabilization of a specific conformation, as suggested by IscU-IscS docking studies [53] described in a later section.
2.2 Iron and sulfide transfer
Assembly of the iron-sulfur cluster requires the scaffold protein to receive iron and sulfide from appropriate donor proteins. Uptake of free iron and sulfide is precluded by the cellular toxicity of these isolated species, and so NFS/IscS and frataxin serve as sulfide and iron donors, respectively. While earlier reports had suggested direct sulfur delivery from the IscS-type sulfur donor to form persulfide bonds with cluster binding Cys residues of IscS and subsequent delivery of iron from the iron donor protein [35, 54], later studies demonstrated the persulfide adduct of IscS to be incapable of conversion to the cluster-bound form following addition of iron [55]. Both fluorescence quenching and isothermal titration calorimetry demonstrate Tm IscU to bind ferrous ion [36], which is subsequently converted to cluster following sulfide delivery, either by direct addition or via IscS. Either method results in the rapid formation of holo IscU, although the latter is clearly more physiologically relevant. Additional mechanistic details for sulfide and iron delivery will be described in later sections that discuss NFS/IscS and frataxin, respectively.
2.3 Non-cysteinyl ligand
Both site-directed mutagenesis and more recent crystallographic studies suggest the IscU-bound cluster to be ligated by three well conserved cysteine residues and one non-cysteinyl ligand [56] (Figure. 4). In the NMR structure of zinc bound Hi IscU, zinc ion is ligated by these three conserved cysteine residues (Cys37, Cys63 and Cys107) and a fourth ligand which appears to be His105 [39]. Although zinc is presumably bound at the cluster binding site, His105 is not necessarily the ligand used to coordinate a bound [2Fe-2S] cluster, since other potential coordinating ligands are also available in the pocket, including Asp39 and Ser65. Indeed the NMR structure of Bs IscU (PDB entry 1XJS), suggests the fourth ligand to bound zinc to be the neighboring aspartate residue instead of histidine (Figure. 5). However, point substitutions of this conserved residue with alanine result in a stabilized cluster [48, 57], and so this aspartate is unlikely to serve as the fourth non-cysteinyl ligand to the cluster. This aspartate has been implicated in stabilization of bound iron prior to sulfide incorporation to form cluster [48, 50, 57] and has also been implicated in the conformational isomerism noted for the native apo structure [46]. In the crystal structure of the trimeric Aa IscU, where one of the subunits has a bound [2Fe-2S] cluster, the cluster is coordinated by three conserved cysteine residues and one histidine [40] (Figure. 5). Substitution of this semi-conserved histidine in human IscU does not cause IscU to lose the ability to coordinate cluster [58], but does result in a decrease in iron binding affinity to IscU and a slower cluster reconstitution rate. Studies suggest that this histidine facilitates interaction with the iron donor protein frataxin, and also mediates iron delivery to the cluster binding site [58] (Figure. 6). However, the amino acid sequences of IscU's from gram-positive bacteria and archaebacteria have lysine at this position, rather than histidine, and it has been suggested that the lysine promotes contact with the iron donor frataxin, while the previously mentioned conserved aspartate mediates iron delivery [58]. In other words, the roles accomplished by His alone in gram-negative bacteria and eukaryotes are divided between two residues in the case of gram-positive and archaebacteria.
Figure 4.

Sequence alignment for IscU-type proteins. Three conserved cysteine residues that are directly coordinated to the cluster are highlighted in yellow. The semi-conserved histidine and corresponding lysine residues are marked as green or purple, respectively. Other possible non-cysteinyl ligands include Asp39 and Ser65 (numbering as in H. influenzae) are marked in blue.
Figure 5.

Cluster coordination. Solution and crystal structures of zinc-bound (A and B) or cluster-bound (C) forms of IscU proteins provide evidence that the cluster is ligated by three conserved cysteine residues and a fourth non-cysteinyl ligand. (A) The structure of the zinc-bound Hi IscU shows that the fourth ligand is His106. (B) In the structure of zinc-bound Bs IscU, the fourth ligand is Asp43. (C) Structure of one subunit of the trimeric Aa IscU, which has a bound [2Fe-2S] cluster with His106 (Aa numbering) as the fourth ligand.
Figure 6.

Iron transfer from frataxin to ISU. In human ISU, residue His106 appears to play a role in promoting interaction between ISU and the iron donor protein frataxin (schematically illustrated), as well as mediating iron delivery to the cluster binding site [58].
2.4 Role as a Carrier Protein
While ISU/IscU proteins are viewed as [2Fe-2S] assembly scaffolds, alternative scaffold proteins have been identified, including the ISA, IscA and NFU type proteins [59-62]. The distinct roles of cluster assembly versus cluster transport or carriers are at this time often intertwined. While both the ISU and ISA classes of proteins have been demonstrated to serve as assembly proteins, they have also been shown to give up the cluster to a target ferredoxin [48, 62], potentially serving as carriers. As discussed later, members of the NFU class of protein and glutaredoxin families have also been implicated with cluster assembly and carrier roles. This remains an emerging field of study, and whether the roles of assembly or carrier proteins are truly distinct, and which class each protein might fall into remains a question to be resolved.
3 Iron Donor Protein – Frataxin
Frataxin is a conserved protein found in prokaryotes and eukaryotes. A deficiency of functional frataxin is related to Friedreich's ataxia, a degenerative disease characterized by neurodegeneration and cardiomyopathy [63-66]. Although the varied roles for cellular frataxin remain a matter of debate, there is substantial evidence in support of a major role in cellular iron homeostasis, and as a mediator of iron delivery to the iron-sulfur cluster scaffold protein IscU/ISU, as well as ferrochelatase in heme biosynthesis. Deletion of the frataxin gene in yeast results in the accumulation of mitochondrial iron and mitochondrial dysfunction, resulting in hypersensitization to oxidative stress [63]. There is also a loss of aconitase activity and a deficiency of mitochondrial iron-sulfur cluster proteins [65, 67, 68], while iron-dependent proteins that do not require clusters are not affected [69, 70]. These results suggest that frataxin plays a direct and central role in iron-sulfur cluster biosynthesis. In vitro studies also demonstrate iron binding by yeast, human and bacterial frataxins [71, 72]. Co-immunoprecipitation experiments suggest yeast frataxin Yfh1p to bind to the ISU1/NFS1 complex and play a crucial role for iron-sulfur cluster assembly in ISU1 [73]. Iron appears to promote binding of frataxin to ISU in the sub-micromolar range, while apo frataxin binds very weakly, suggesting a role for iron as a bridging agent between each protein [38]. Yeast frataxin and E. coli frataxin were also characterized as iron donor proteins [74, 75], while kinetic studies have quantified the rate of frataxin-mediated delivery of iron to human ISU to form a bound [2Fe-2S] cluster [38]. Frataxin, as an iron donor, has also been implicated in modulating aconitase activity [76] and heme biosynthesis [77, 78]. While deficient in substantial sequence homology across species, the general role for frataxin in the homeostatic control of cellular iron chemistry is supported by an apparent structural homology of frataxin, irrespective of sequence homology (Section 3.3).
3.1 Truncation of human frataxin
Following mitochondrial import in human cells, the targeting sequence (residues 1-55) is removed in a two-step process by mitochondrial processing peptidases located in the mitochondrial matrix [79]. Recent studies have demonstrated human frataxin to be truncated beyond the expected residue site following removal of the mitochondrial targeting sequence through the action of two mitochondrial processing peptidases (MPP's). Such cleavage has been suggested to occur by natural degradation during purification [80] or by an unknown protease [79]. Recent cellular studies by Testi and coworkers suggest that this truncated form of human frataxin is a principal active form within the cell [81], and that it also is formed by cleavage of a longer precursor protein in vivo. In fact the truncated form is similar in overall size to that of yeast and other bacterial frataxins or structural homologues of frataxin (Figures 7 and 8). It has been suggested that this truncation is the result of a catalytic self-cleavage reaction rather than a result of protease activity. Moreover, the self-cleavage activity appears to be stimulated by iron ion [82].
Figure 7.

Sequence alignment for frataxin homologs. Conserved acidic residues on the α1-β1 surface that are proposed to be responsible for iron binding are marked as blue.
Figure 8.
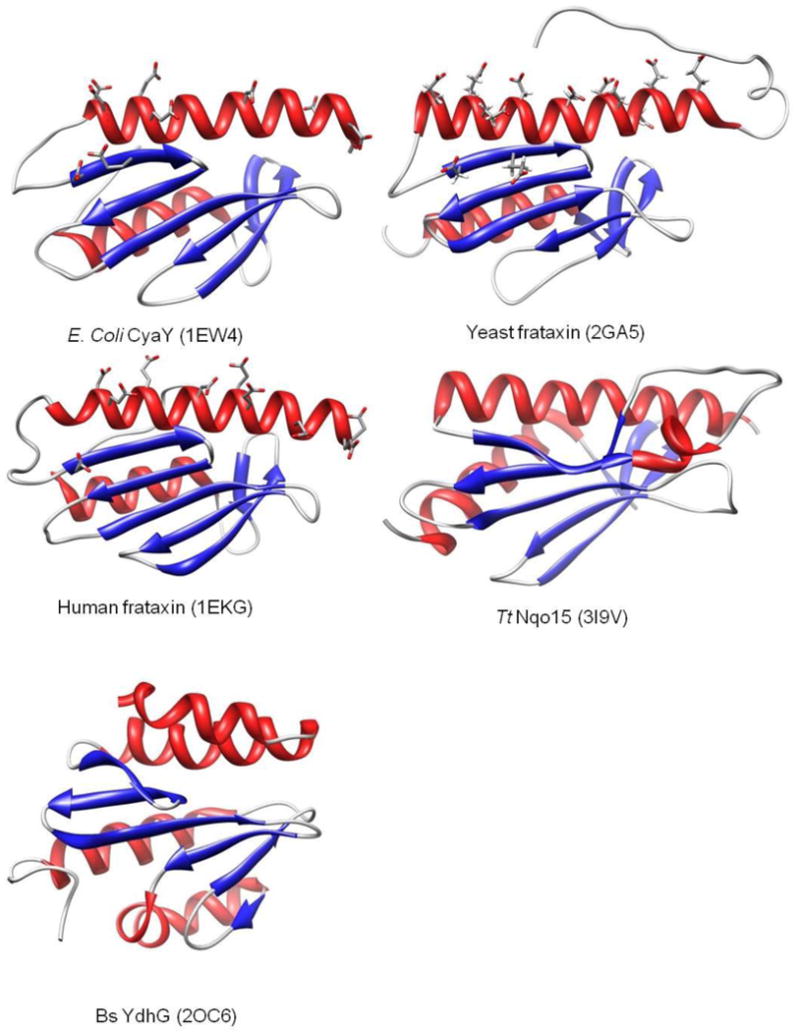
Structures of E. coli, yeast and human frataxins show an extremely similar core structure. The conserved acidic residues lying on the α1-β1 surface appear to be iron binding sites [99, 100]. Structures of the Tt Nqo15 [107, 108] and Bs YdhG proteins [89] are both observed to be very similar to the frataxin core.
3.2 Iron binding sites
Although the mechanism of iron import into mitochondria is still not well understood, it is known that mitoferrin functions as a mitochondrial iron importer that mediates iron transport through the inner mitochondrial membrane [83-86]. Controlling mitoferrin levels within the mitochondrial membrane provides a mechanism to regulate mitochondrial iron levels [85]. Frataxin deficiency in cells leads to up regulation of mitoferrin, causing increased mitochondrial iron uptake [86, 87]. Iron binding to frataxin has been confirmed in many frataxin homologs [38, 72, 74, 88-90]. ITC and fluorescence quenching experiments indicate that the N-terminal truncated form of human frataxin is able to bind six to seven ferrous or ferric ions with an apparent KD in the 10 - 50 μM range [38]. Isothermal titration calorimetry (ITC) studies have also revealed that E. coli frataxin (CyaY), yeast frataxin and Bs frataxin all bind at least two ferrous ions and Drosophila frataxin binds one ferrous ion with a KD also in the μM range [74, 89-91]. While many of these affinities appear of borderline significance in light of physiologically available concentrations, the affinities do increase dramatically in the presence of cognate partner proteins, consistent with a role in promoting delivery of iron to such partners.
The structures determined for yeast [78, 92], human [80, 93] and E. coli frataxins [94-96] are extremely similar, with an overall planar α-β sandwich structure motif [97]. Conserved acidic residues are located in the α1 and β1 region, constituting an exposed negatively-charged surface that suggests a plausible binding site for metal ions. Iron titration studies monitored by solution NMR have essentially validated the α1-β1 surface as the metal binding site (Figure. 7). Perturbation of resonances was observed for the residues lying on α1-β1 region following titration of ferrous ion to E. coli [94] or yeast [74] frataxin. XAS studies were performed on iron loaded yeast frataxin and human frataxin. In both cases ferrous ion are bound in a symmetric six-coordinate oxygen/nitrogen based ligand environment [74, 98]. This is consistent with ligation through the conserved Asp, Glu and His residues in the α1-β1 region as indicated by NMR (Figure. 8). To better define the locations of potential iron binding sites on the polyanionic surface, and those sites at the interface with human ISU, block mutants were investigated that covered the 12 conserved acidic residues. These mutants exhibit similar ITC determined binding affinities, relative to wild type frataxin, with decreased stoichiometry of iron to protein and a stoichiometry of two for derivative A(E100A/E101A/D104A/E108A/ E111A/D112A); four for derivative B (D115A/E121A/D122A/D124A); and five for derivative C (E92A/E96A/E100A/E101A), compared to seven for native protein [99]. Cellular studies show that mutation of five acidic residues in yeast frataxin D86A/E90A/E93A/D101A/E103A results in severe growth defects, while mutation of D86A/E90A or E93A/D101A/E103A as separate domains had little effect [100]. These and other reports [74, 94, 98, 100-102] suggest significant flexibility for iron and partner binding to frataxin.
3.3 Structural homologs
Solution and crystal structures have been reported for E. coli [94-96], yeast [78, 92] and human frataxin [80, 93], and each exhibit similar secondary and tertiary folds, with an overall α-β sandwich structure motif and a general topology α1β1β2β3β4β5β6α2 [97]. It was once thought that the structural similarity between frataxins derives from the high level sequence similarity among these homologs. Conserved N-terminal acidic residues in the α1-β1 domain construct a negatively-charged surface that is important for both iron and partner protein binding. The sequence identity of yeast frataxin versus E. coli frataxin and human frataxin are 28.1% and 37.8%, respectively, with the similarities been 59.8% and 65.0% [97]. However, the lack of a sequence-wise frataxin homolog in many other classes of organism, such as thermophiles and gram-positive bacteria, is inconsistent with a general role for frataxin as iron donor protein in iron-sulfur cluster assembly. In fact, sequence homologs for almost all of the other components of the ISC machinery were found in these organisms [41, 103-106]. A functionally unknown protein Nqo15 was revealed in the structure of the Thermus thermophilus (Tt) mitochondrial respiratory complex I [107, 108]. Although the sequence is very different, Nqo15 is structurally superimposable on both the E. coli CyaY frataxin (RMSD = 2.5 Å, 13% sequence identity) and truncated human frataxin (RMSD = 3.3 Å 11% sequence identity) [107] (Figure. 8). Possible iron binding sites are also present on the α1-β1 surface, and so it was proposed that Tt Nqo15 is a frataxin analog. It was subsequently demonstrated that Bacillus subtilis (Bs) YdhG also exhibits a very similar structural topology to human frataxin in spite of the very low sequence similarity [89]. In fact Bs YdhG possesses all of the functional attributes expected for a frataxin family member; including iron binding sites, binding to ISU in an iron-dependent manner, and stimulation of cluster formation in the ISU scaffold protein. All of these factors support Bs YdhG as a frataxin analog in Bacillus subtilis [89]. Homologs of Bs YdhG, though of uncharacterized function, are present in many gram-positive bacteria, but suggest that they belong to the broader frataxin family and demonstrate that frataxins are conserved structurally throughout all species.
3.4 Cellular Role
Several cellular processes related to iron metabolism have implicated iron binding roles for frataxin. These include (1) iron delivery to the iron-sulfur cluster assembly machinery [73, 75, 77] or heme biosynthetic pathway [77, 78, 98, 109, 110]; (2) iron storage [71, 111, 112]; (3) Fe-S cluster repair [76]; and (4) cellular iron sensors that regulate iron sulfur cluster biosynthesis based on the availability of cellular iron [113]. Each of these areas is discussed in turn.
There is clear evidence for a direct role for frataxin in cellular iron sulfur cluster assembly. A deficiency in the activity of the iron-sulfur cluster-containing subunits of the mitochondrial respiratory complexes and aconitase was found in FRDA patients [68]. Moreover, disruption of the YFH1 gene in yeast resulted in the loss of several iron sulfur cluster protein activities (aconitase and succinate dehydrogenase) and respiratory deficiencies [68, 114] [69], while depletion of Yfh1p resulted in deficiencies in the assembly of cytosolic iron sulfur cluster proteins as reflected by the significant decrease in iron incorporation in two reporter proteins Leu1p and Rli1p in Yfh1p-depleted yeast [115]. Cytosolic Fe-S proteins were also affected in frataxin-deleted mouse tissues [116]. Yfh1 and human frataxin interact with either the ISU1/NFS1 complex (presumably through contact to ISU1 rather than to both, although this point was not addressed in that report [73]) or to ISU. In each case binding occurred in an iron-dependent manner and stimulated cluster formation on the scaffold protein ISU [38, 73, 101, 117, 118]. A direct interaction between E. coli CyaY and IscS was also demonstrated [75], leading to an interesting interpretation of the role for frataxin in cellular iron sulfur cluster assembly, which will be discussed later. All of these in vivo and in vitro studies provide a solid indication that frataxin serves an important role in cellular iron sulfur cluster assembly as an iron donor protein.
The relationship between frataxin and cellular heme biosynthesis was first observed in a genetic study, which showed that Yfh1p functionally interacts with the yeast OCT1 gene, a metalloprotease required for maturation of ferrochelatase and other iron proteins [119]. In yeast cells lacking the YFH1 gene, a cytochrome deficiency and a low level of ferrochelatase was observed [109]. However, the observed accumulation of zinc protoporphyrin demonstrated that the cytochrome deficiency was not caused by the low level of ferrochelatase. In vitro assays using either permeabilized cells or intact mitochondria showed that protoporphyrin was produced at normal levels and was not used for heme synthesis in Δyfh1 cells, but rather the ligand was used for zinc protoporphyrin synthesis instead. These experiments demonstrated the Δyfh1 cell line to be defective in the use of iron by ferrochelatase [109]. Overexpression of Yfh1p in yeast made mitochondrial iron more available to ferrochelatase and resulted in higher rates of heme synthesis [110]. The interaction between human frataxin and ferrochelatase has been studied by ITC and binding was observed to be iron-dependent with a measured Kd ∼ 17 nM [77]. The binding interface was determined through NMR studies and a significant overlap with the frataxin iron binding surface was found, consistent both with the iron-dependent binding as well as an iron delivery role for the protein [78, 98]. Kinetic studies demonstrated the ferrochelatase activity to increase with frataxin concentration and the optimal ratio to be one frataxin per ferrochelatase dimer [77]. These results support a specific role for frataxin as an iron donor to ferrochelatase for the synthesis of heme.
Other proposed cellular functions for frataxin in cellular iron chemistry include protecting and repairing protein-bound clusters and iron storage. Frataxin is observed to bind to mitochondrial aconitase in a citrate dependent manner, protecting aconitase cluster from the oxidative disassembly [76] as well as reactivating aconitase by converting the [3Fe-4S] cluster to the native [4Fe-4S] form [76]. Yeast frataxin (Yfh1), and the bacterial (CyaY) and human frataxin homologs are all capable of binding multiple iron ions in an oligomeric form [71, 88, 111], leading to a proposed iron storage function similar to ferritin. However, the solution conditions required to form such oligomers appear non-physiological, and so the cellular relevance of this chemistry remains uncertain. Another interesting report suggests that E. coli frataxin is not only an iron chaperone but works as a gatekeeper of iron-sulfur cluster formation [113]. In vitro Fe-S reconstitution of either IscU or ferredoxin is faster in the absence of the bacterial frataxin CyaY, leading to the suggestion that CyaY might act as an inhibitor [113]. However, there is no free cellular iron under physiological conditions, since iron is always complexed by small molecule chelators or proteins, and so a direct comparison of the relative rates of cluster assembly for free iron versus frataxin-mediated iron delivery may not be physiologically relevant. In the absence of IscU the reconstitution of ferredoxin was also observed to be slower in the presence of bacterial CyaY when reconstituted via IscS/Cysteine/Fe2+, and so it was proposed that the inhibition involves IscS rather than IscU. Again, however, comparison of relative rates for free iron versus frataxin-mediated reconstitution is not physiologically relevant and is not readily interpretable in terms of an inhibitory role for frataxin. By monitoring cysteine-to-alanine conversion it was shown that the IscS desulfurase activity is not affected by CyaY. The observed inhibitory effect of CyaY on cluster reconstitution rates is greater at higher iron concentrations, and so the inhibitory effect was rationalized in terms of iron-promoted binding of frataxin to IscS. Alternatively, one might explain such observations by an iron buffering effect of frataxin with a change of free iron concentration resulting from iron binding to CyaY. NMR perturbation and immunoprecipitation assays also supported a direct interaction between CyaY and IscS [113], and led to the proposed gatekeeper role. In this model the iron-dependent binding of CyaY to IscS acts as an inhibitor when iron is in excess, preventing the unnecessary overproduction of Fe-S clusters [113]. This is certainly an interesting hypothesis for an alternative role for frataxin on the iron-sulfur cluster assembly pathway, although the conclusions are inconsistent with previous studies that show stimulation of cluster formation in the presence of frataxin and a direct interaction between frataxin and ISU [38, 73-75, 89]. More recently it has been shown that overexpression of CyaY does not interfere with sulfur transfer between IscS and IscU, favoring CyaY as a iron donor protein [47]. Some additional work is clearly needed to elaborate these details.
4 Sulfur Donor Protein-NFS/IscS
The final key component of the cellular iron sulfur cluster assembly machinery is the sulfur donor protein (NFS1 in eukaryotes) and its bacterial homologs (NifS, IscS, and SufS), all of which are pyridoxal 5′-phosphate-dependent cysteine desulfurases. Members of this enzyme family convert cysteine to alanine with the assistance of a bound PLP cofactor, resulting in a modified surface Cys in a persulfide form [120], which is later transferred to a variety of sulfur acceptors: including the iron-sulfur cluster assembly machinery, thiolation of tRNAs [47, 121, 122], and biotin synthesis [123]. Solution and crystal structures are available for Tm Nifs [105], and E. coli IscS [47, 124]. They exist as a homodimer, with each monomer divided into two domains. The larger domain contains the PLP cofactor and the smaller domain hosts the conserved active site cysteine in the middle of a highly flexible loop [105].
4.1 Sulfur donor protein
Biochemical studies have shown that NFS1 is essential for cellular iron-sulfur cluster protein biogenesis and cellular iron metabolism [22, 121, 125-128]. A deficiency of the yeast NFS1p or E. coli IscS genes resulted in a deficiency of cellular iron sulfur cluster proteins and, in the case of yeast, accumulation of mitochondrial iron [28, 125, 127, 129, 130]. While other sulfur donor proteins have been implicated in other metabolic pathways n eukaryotes, NFS has been implicated more broadly as a cellular sulfur donor rather than exclusively focused on cluster-related chemistry [47, 121-123].
Prior mass spectrometric studies and experiments with radiolabeled cysteine have been used to demonstrate direct transfer of sulfane sulfur, S0, from the cysteine persulfide on IscS to the cysteine residues on IscU [35, 54]. However, the IscS family of proteins are relatively promiscuous when studied in vitro with regard to sulfur transfer chemistry to Cys-containing proteins [131-133], with evidence for multiple sulfur transfers to form polythianes [35, 54]. Mechanistic studies of the role of IscS in cluster biosynthesis are typically carried out in vitro, where the lack of control over both selectivity and the number of sulfur centers transferred is certainly evident. In the case of sulfur delivery to apo IscU, later iron binding studies demonstrated the IscU protein product (a cysteinyl persulfide) obtained from initial sulfur transfer from IscS to have a low affinity for iron ion, and provided evidence in support of a mechanistically more reasonable pathway involving reductive cleavage of the IscS-persulfide (described in the next section, Figure 10) yielding inorganic sulfide for addition to IscU pre-loaded with bound iron centers [55].
Figure 10.
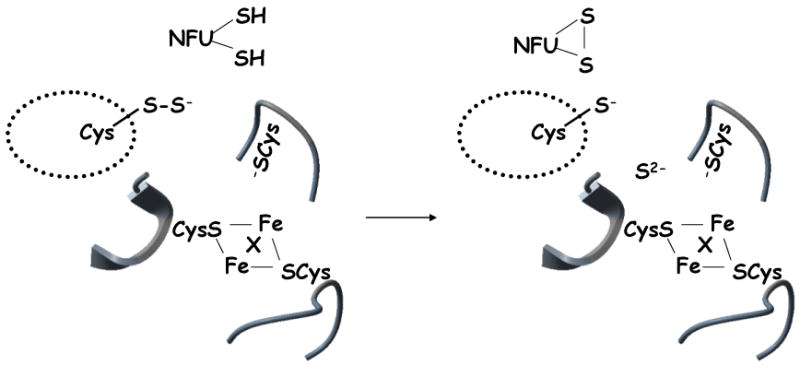
Sulfide delivery to IscU/ISU. Cleavage of the IscS persulfide bond by reduced NFU, yielding inorganic sulfide, which is positioned adjacent to the iron-loaded IscU cluster binding site, where X is a presumed stabilizing bridging ligand (possibly a conserved carboxylate [48, 57]). Details of how the iron centers are coordinated to the IscU Cys at the time of sulfide delivery remain to be established.
Tight binding between E. coli IscS and IscU, with a KD∼ 2 μM determined by ITC and SPR studies [35, 53, 54], coupled with the crystallographic definition of the IscS-IscU complex [47], are consistent with physiologically relevant complex formation and a direct transfer pathway for sulfur. The mechanism of IscS catalyzed desulfurization of cysteine includes the steps of coupling of substrate cysteine with the PLP factor, nucleophilic attack by a conserved cysteine residue to form the persulfide, and the release of alanine [120]. The crystallographically-defined structures of Tm Nifs and E. coli IscS both reveal the PLP cofactor to be anchored at Lys206 with a histidine residue, His104, lying nearby, possibly as an acid-base catalyst [105, 124]. The structure of E. coli IscS also shows the active site cysteine (C328) on another domain [124]. Using 1,5-IAEDANS- and 5-BMF-labeled Tm IscS, the KD for Tm IscS binding to Tm IscU was determined to be ∼6 μM [53]. Calculation from FRET data is consistent with this conserved cysteine as the active site cysteine [53]. Recently, it was found that in eukaryotes NFS functions as a sulfur donor only when complexed with Isd11 [134-137]. Depletion of Isd11 in yeast or human cells causes defective activity in iron-sulfur cluster containing proteins, as well as failure to form cluster in the ISU scaffold protein [134, 135]. Co-immunoprecipitation results have demonstrated human Isd11 to form a tight complex with human NFS and that the complex possesses cysteine desulfurase activity [137]. Interestingly, immunoprecipitation results also suggest Isd11 to form a complex with the iron donor protein frataxin [136]. While solution studies suggest bacterial IscS alone to be capable of sulfide delivery to the IscU scaffold protein [54, 129, 138], recent evidence suggests that eukaryotic NFS might require Isd11 to be fully functional as a sulfur donor [20].
4.2 IscU-IscS complex formation
While the crystallographically defined complex between IscS and IscU [47] is consistent with a direct functional interaction between these two proteins, the structure obtained does little to infer the mechanism of sulfur transfer, since the active Cys that promotes formation of inorganic sulfide is far removed from the cluster formation site on IscU (Figure. 9A). Possibly the reported crystallographic structure does not represent the active state required to promote transfer of reduced sulfur between the two proteins. Recent docking simulations of representative IscU and IscS proteins, and experimental FRET studies reveal critical contact surfaces at the N-terminal helix of IscU and a C-terminal loop (comprising a chaperone binding domain) that position the sulfide donor Cys on the flexible loop of IscS directly adjacent to the cluster formation site of IscU (Figure. 9B).
Figure 9.

Sulfide transfer from the IscS-IscU complex. (A) Adapted from the crystallographically defined E. coli IscS-IscU complex [47]. IscS is in light green and IscU is in gray. Residues 328-332 of IscS where Cys328 lie are not solved due to the flexibility of this region, shown as red dotted line. The three conserved cysteine residues of IscU where cluster binds are shown in yellow. Cys328 of IscS is at the position that is not directly close to cluster binding site of IscU. (B) Simulated structural model for the complex of H. influenzae IscU monomer with Tm IscS monomer [53]. Tm IscS is depicted in gray, with the blue region comprising a loop, and a portion of an α-helix adjacent to the disordered loop (residues 321-332) that contains the putative active cysteine residue C324 that mediates sulfide delivery. The position of the latter residue is illustrated with the dotted circle, but is not directly observed in the structure as a result of the flexibility of this loop. IscU is represented in yellow, with the red regions depicting the three conserved cysteine residues (C37, C63, and C106), where the asterisk represents C63, which is poised close to the catalytic C324 of Tm IscS.
4.3 NFU as a Persulfide Reductase
The final step of IscS cysteine desulfurization involves cleavage of the protein persulfide bond. A variety of reductive mechanisms have been proposed, including electrons derived from the incoming ferrous ions [139] and ferredoxin-derived electrons [140]. Human NFU, a protein with a thioredoxin-like C-terminal domain has been shown to bind to the sulfur donor protein and mediate cysteinyl persulfide bond cleavage (Figure 10) [141]. The active site within human NFU is the C-terminal domain and is homologous with the C-terminal domain of Av NFU domain[56, 142], yeast NFU [13, 143] and Synechocystis PCC6803NifU [144], all of which are important for the biogenesis of iron-sulfur clusters. The Synechocystis protein is capable of assembling a [2Fe-2S] cluster in vitro, and a preliminary report also suggest [4Fe-4S] formation on the human NFU, prompting speculation that these might be alternative scaffold proteins [61, 142, 145]. Whether these proteins serve dual functions, or if cluster formation is a physiologically relevant species remains to be elaborated.
Similarly, recent reports show that several Glutaredoxin proteins, which have CxxC or CGFS motifs in the active site, are capable of coordinating a [2Fe-2S] cluster that bridges a glutaredoxin dimer and is stabilized by glutathione coordination [146-151], resulting in speculation that in addition to thioredoxin-type activity, glutaredoxin might also serve as a cluster carrier protein [152]. As evidence for such, it has been reported that [2Fe–2S] clusters on plant chloroplast glutaredoxin can be quantitatively transferred to apo chloroplast ferredoxin [153]. Given that the NFU and glutaredoxin families of proteins are non-homologous and serve distinct cellular roles, any functional similarity or relationship must be viewed as tenuous at this time.
ITC and kinetic studies of human NFU have demonstrated binding to IscS and a facility to promote the cluster reconstitution in the human ISU scaffold protein [141, 154]. Considering the highly conserved thioredoxin-like CXXC motif present in human NFU (Figure 11), human NFU would appear to be a likely candidate to serve as a reductase for persulfide bond cleavage yielding inorganic sulfide for cluster formation [141, 154] (Figure 10).
Figure 11.

Sequences of Cys-X-X-Cys domains in NFU-type proteins. Hs, Homo sapiens; Mm, Mus musculus; Dm, Drosophila melanogaster; Ce, Caenorhabditis elegans; Av, Azotobacter vinelandii; Sys, Synechocystis PCC6803.
5 Summary and Conclusions
Iron sulfur cluster proteins are involved in a wide range of cellular processes and dysfunction in cluster biosynthesis has been related to many diseases. Much effort has been directed toward elucidation of iron sulfur cluster biosynthesis pathways, including cellular and genetic studies, and isolation and characterization of novel classes of protein. There remains a number of very interesting questions pertaining to the coordination chemistry of cluster assembly and transfer that will need to be answered to better understand the biosynthesis of iron sulfur clusters and related cofactors. The detailed mechanism of cluster assembly on the scaffold protein is not well defined. The structural and coordination chemistry underlying cluster transfer from scaffold protein to target protein has barely been addressed, including structural recognition by the partners involved. There is a clear role for redox chemistry in promoting cluster assembly and transfer, but again this is poorly understood. Finally the cellular iron chemistry of frataxin has becomes increasingly diverse and complex as it has become the focus of greater attention. While not a subject of this review, the ligand exchange and transport chemistry, underlying iron-sulfur cluster export from the mitochondrion and how all of this is regulated, is a problem where some answers might finally be emerging.
Acknowledgments
This manuscript is dedicated to Professor H. B. Gray on the occasion of his 75th birthday, and in appreciation of his many contributions and tireless efforts to advance the understanding, application, and role of inorganic chemistry in furthering chemical and biochemical science. Research support by a grant from the National Institutes of Health [AI072443] is gratefully acknowledged.
Footnotes
Contribution from Evans Laboratory of Chemistry, The Ohio State University, 100 West 18th Avenue, Columbus, Ohio 43210.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peters JW, Stowell MH, Soltis SM, Finnegan MG, Johnson MK, Rees DC. Biochemistry. 1997;36:1181–1187. doi: 10.1021/bi9626665. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DC, Dean DR, Smith AD, Johnson MK. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 3.Volbeda A, Charon MH, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. Nature. 1995;373:580–587. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 4.Iwata S, Saynovits M, Link TA, Michel H. Structure. 1996;4:567–579. doi: 10.1016/s0969-2126(96)00062-7. [DOI] [PubMed] [Google Scholar]
- 5.Robbins AH, Stout CD. Proc Natl Acad Sci U S A. 1989;86:3639–3643. doi: 10.1073/pnas.86.10.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moulis JM, Sieker LC, Wilson KS, Dauter Z. Protein Sci. 1996;5:1765–1775. doi: 10.1002/pro.5560050902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lill R. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 8.Lill R, Muhlenhoff U. Annu Rev Cell Dev Biol. 2006;22:457–486. doi: 10.1146/annurev.cellbio.22.010305.104538. [DOI] [PubMed] [Google Scholar]
- 9.Ayala-Castro C, Saini A, Outten FW. Microbiol Mol Biol Rev. 2008;72:110–125. doi: 10.1128/MMBR.00034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fontecave M, Ollagnier-de-Choudens S. Arch Biochem Biophys. 2008;474:226–237. doi: 10.1016/j.abb.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Mansy SS, Cowan JA. Acc Chem Res. 2004;37:719–725. doi: 10.1021/ar0301781. [DOI] [PubMed] [Google Scholar]
- 12.Zheng L, Cash VL, Flint DH, Dean DR. J Biol Chem. 1998;273:13264–13272. doi: 10.1074/jbc.273.21.13264. [DOI] [PubMed] [Google Scholar]
- 13.Schilke B, Voisine C, Beinert H, Craig E. Proc Natl Acad Sci U S A. 1999;96:10206–10211. doi: 10.1073/pnas.96.18.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobson MR, Cash VL, Weiss MC, Laird NF, Newton WE, Dean DR. Mol Gen Genet. 1989;219:49–57. doi: 10.1007/BF00261156. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi Y, Tokumoto U. J Biol Chem. 2002;277:28380–28383. doi: 10.1074/jbc.C200365200. [DOI] [PubMed] [Google Scholar]
- 16.Ding H, Clark RJ, Ding B. J Biol Chem. 2004;279:37499–37504. doi: 10.1074/jbc.M404533200. [DOI] [PubMed] [Google Scholar]
- 17.Ding B, Smith ES, Ding H. Biochem J. 2005;389:797–802. doi: 10.1042/BJ20050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu J, Bitoun JP, Tan G, Wang W, Min W, Ding H. Biochem J. 2010;428:125–131. doi: 10.1042/BJ20100122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. J Bacteriol. 2001;183:4562–4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lill R, Muhlenhoff U. Annu Rev Biochem. 2008;77:669–700. doi: 10.1146/annurev.biochem.76.052705.162653. [DOI] [PubMed] [Google Scholar]
- 21.Balk J, Lobreaux S. Trends Plant Sci. 2005;10:324–331. doi: 10.1016/j.tplants.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Kispal G, Csere P, Prohl C, Lill R. EMBO J. 1999;18:3981–3989. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Netz DJ, Pierik AJ, Stumpfig M, Muhlenhoff U, Lill R. Nat Chem Biol. 2007;3:278–286. doi: 10.1038/nchembio872. [DOI] [PubMed] [Google Scholar]
- 24.Roy A, Solodovnikova N, Nicholson T, Antholine W, Walden WE. EMBO J. 2003;22:4826–4835. doi: 10.1093/emboj/cdg455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hausmann A, Aguilar Netz DJ, Balk J, Pierik AJ, Muhlenhoff U, Lill R. Proc Natl Acad Sci U S A. 2005;102:3266–3271. doi: 10.1073/pnas.0406447102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balk J, Aguilar Netz DJ, Tepper K, Pierik AJ, Lill R. Mol Cell Biol. 2005;25:10833–10841. doi: 10.1128/MCB.25.24.10833-10841.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srinivasan V, Netz DJ, Webert H, Mascarenhas J, Pierik AJ, Michel H, Lill R. Structure. 2007;15:1246–1257. doi: 10.1016/j.str.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Kispal G, Csere P, Guiard B, Lill R. FEBS Lett. 1997;418:346–350. doi: 10.1016/s0014-5793(97)01414-2. [DOI] [PubMed] [Google Scholar]
- 29.Bekri S, Kispal G, Lange H, Fitzsimons E, Tolmie J, Lill R, Bishop DF. Blood. 2000;96:3256–3264. [PubMed] [Google Scholar]
- 30.Chen CA, Cowan JA. Biochim Biophys Acta. 2006;1760:1857–1865. doi: 10.1016/j.bbagen.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Kuhnke G, Neumann K, Muhlenhoff U, Lill R. Mol Membr Biol. 2006;23:173–184. doi: 10.1080/09687860500473630. [DOI] [PubMed] [Google Scholar]
- 32.Chen CA, Cowan JA. J Biol Chem. 2003;278:52681–52688. doi: 10.1074/jbc.M306472200. [DOI] [PubMed] [Google Scholar]
- 33.Lange H, Lisowsky T, Gerber J, Muhlenhoff U, Kispal G, Lill R. EMBO Rep. 2001;2:715–720. doi: 10.1093/embo-reports/kve161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Csere P, Lill R, Kispal G. FEBS Lett. 1998;441:266–270. doi: 10.1016/s0014-5793(98)01560-9. [DOI] [PubMed] [Google Scholar]
- 35.Smith AD, Agar JN, Johnson KA, Frazzon J, Amster IJ, Dean DR, Johnson MK. J Am Chem Soc. 2001;123:11103–11104. doi: 10.1021/ja016757n. [DOI] [PubMed] [Google Scholar]
- 36.Mansy SS, Wu G, Surerus KK, Cowan JA. J Biol Chem. 2002;277:21397–21404. doi: 10.1074/jbc.M201439200. [DOI] [PubMed] [Google Scholar]
- 37.Agar JN, Krebs C, Frazzon J, Huynh BH, Dean DR, Johnson MK. Biochemistry. 2000;39:7856–7862. doi: 10.1021/bi000931n. [DOI] [PubMed] [Google Scholar]
- 38.Yoon T, Cowan JA. J Am Chem Soc. 2003;125:6078–6084. doi: 10.1021/ja027967i. [DOI] [PubMed] [Google Scholar]
- 39.Ramelot TA, Cort JR, Goldsmith-Fischman S, Kornhaber GJ, Xiao R, Shastry R, Acton TB, Honig B, Montelione GT, Kennedy MA. J Mol Biol. 2004;344:567–583. doi: 10.1016/j.jmb.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 40.Shimomura Y, Wada K, Fukuyama K, Takahashi Y. J Mol Biol. 2008;383:133–143. doi: 10.1016/j.jmb.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 41.Bertini I, Cowan JA, Del Bianco C, Luchinat C, Mansy SS. J Mol Biol. 2003;331:907–924. doi: 10.1016/s0022-2836(03)00768-x. [DOI] [PubMed] [Google Scholar]
- 42.Mansy SS, Wu SP, Cowan JA. J Biol Chem. 2004;279:10469–10475. doi: 10.1074/jbc.M312051200. [DOI] [PubMed] [Google Scholar]
- 43.Cupp-Vickery JR, Peterson JC, Ta DT, Vickery LE. J Mol Biol. 2004;342:1265–1278. doi: 10.1016/j.jmb.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 44.Hoff KG, Cupp-Vickery JR, Vickery LE. J Biol Chem. 2003;278:37582–37589. doi: 10.1074/jbc.M305292200. [DOI] [PubMed] [Google Scholar]
- 45.Hoff KG, Ta DT, Tapley TL, Silberg JJ, Vickery LE. J Biol Chem. 2002;277:27353–27359. doi: 10.1074/jbc.M202814200. [DOI] [PubMed] [Google Scholar]
- 46.Kim JH, Fuzery AK, Tonelli M, Ta DT, Westler WM, Vickery LE, Markley JL. Biochemistry. 2009;48:6062–6071. doi: 10.1021/bi9002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi R, Proteau A, Villarroya M, Moukadiri I, Zhang L, Trempe JF, Matte A, Armengod ME, Cygler M. PLoS Biol. 2010;8:e1000354. doi: 10.1371/journal.pbio.1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu SP, Wu G, Surerus KK, Cowan JA. Biochemistry. 2002;41:8876–8885. doi: 10.1021/bi0256781. [DOI] [PubMed] [Google Scholar]
- 49.Bonomi F, Iametti S, Ta D, Vickery LE. J Biol Chem. 2005;280:29513–29518. doi: 10.1074/jbc.M504344200. [DOI] [PubMed] [Google Scholar]
- 50.Unciuleac MC, Chandramouli K, Naik S, Mayer S, Huynh BH, Johnson MK, Dean DR. Biochemistry. 2007;46:6812–6821. doi: 10.1021/bi6026665. [DOI] [PubMed] [Google Scholar]
- 51.Bych K, Kerscher S, Netz DJ, Pierik AJ, Zwicker K, Huynen MA, Lill R, Brandt U, Balk J. EMBO J. 2008;27:1736–1746. doi: 10.1038/emboj.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheftel AD, Stehling O, Pierik AJ, Netz DJ, Kerscher S, Elsasser HP, Wittig I, Balk J, Brandt U, Lill R. Mol Cell Biol. 2009;29:6059–6073. doi: 10.1128/MCB.00817-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nuth M, Cowan JA. J Biol Inorg Chem. 2009;14:829–839. doi: 10.1007/s00775-009-0495-7. [DOI] [PubMed] [Google Scholar]
- 54.Urbina HD, Silberg JJ, Hoff KG, Vickery LE. J Biol Chem. 2001;276:44521–44526. doi: 10.1074/jbc.M106907200. [DOI] [PubMed] [Google Scholar]
- 55.Nuth M, Yoon T, Cowan JA. J Am Chem Soc. 2002;124:8774–8775. doi: 10.1021/ja0264596. [DOI] [PubMed] [Google Scholar]
- 56.Agar JN, Yuvaniyama P, Jack RF, Cash VL, Smith AD, Dean DR, Johnson MK. J Biol Inorg Chem. 2000;5:167–177. doi: 10.1007/s007750050361. [DOI] [PubMed] [Google Scholar]
- 57.Wu G, Mansy SS, Wu Sp SP, Surerus KK, Foster MW, Cowan JA. Biochemistry. 2002;41:5024–5032. doi: 10.1021/bi016073s. [DOI] [PubMed] [Google Scholar]
- 58.Huang J, Cowan JA. Chem Commun (Camb) 2009:3071–3073. doi: 10.1039/b902676b. [DOI] [PubMed] [Google Scholar]
- 59.Morimoto K, Yamashita E, Kondou Y, Lee SJ, Arisaka F, Tsukihara T, Nakai M. J Mol Biol. 2006;360:117. doi: 10.1016/j.jmb.2006.04.067. [DOI] [PubMed] [Google Scholar]
- 60.Ollagnier-de Choudens S, Nachin L, Sanakis Y, Loiseau L, Barras F, Fontecave M. J Biol Chem. 2003;278:17993. doi: 10.1074/jbc.M300285200. [DOI] [PubMed] [Google Scholar]
- 61.Tong WH, Jameson GN, Huynh BH, Rouault TA. Proc Natl Acad Sci U S A. 2003;100:9762–9767. doi: 10.1073/pnas.1732541100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu Sp, Cowan JA. Biochemistry. 2003;42:5784–5791. doi: 10.1021/bi026939+. [DOI] [PubMed] [Google Scholar]
- 63.Babcock M, deSilva D, Oaks R, DavisKaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 64.Cavadini P, Gellera C, Patel PI, Isaya G. Human Molecular Genetics. 2000;9:2523–2530. doi: 10.1093/hmg/9.17.2523. [DOI] [PubMed] [Google Scholar]
- 65.Foury F, Cazzalini O. Febs Letters. 1997;411:373–377. doi: 10.1016/s0014-5793(97)00734-5. [DOI] [PubMed] [Google Scholar]
- 66.Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, DeMichele G, Filla A, DeFrutos R, Palau F, Patel PI, DiDonato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 67.Foury F. FEBS Lett. 1999;456:281–284. doi: 10.1016/s0014-5793(99)00961-8. [DOI] [PubMed] [Google Scholar]
- 68.Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 69.Chen OS, Hemenway S, Kaplan J. Proc Natl Acad Sci U S A. 2002;99:12321–12326. doi: 10.1073/pnas.192449599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stehling O, Elsasser HP, Bruckel B, Muhlenhoff U, Lill R. Hum Mol Genet. 2004;13:3007–3015. doi: 10.1093/hmg/ddh324. [DOI] [PubMed] [Google Scholar]
- 71.Adamec J, Rusnak F, Owen WG, Naylor S, Benson LM, Gacy AM, Isaya G. Am J Hum Genet. 2000;67:549–562. doi: 10.1086/303056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cavadini P, O'Neill HA, Benada O, Isaya G. Hum Mol Genet. 2002;11:217–227. doi: 10.1093/hmg/11.3.217. [DOI] [PubMed] [Google Scholar]
- 73.Gerber J, Muhlenhoff U, Lill R. EMBO Rep. 2003;4:906–911. doi: 10.1038/sj.embor.embor918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cook JD, Bencze KZ, Jankovic AD, Crater AK, Busch CN, Bradley PB, Stemmler AJ, Spaller MR, Stemmler TL. Biochemistry. 2006;45:7767–7777. doi: 10.1021/bi060424r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Layer G, Ollagnier-de Choudens S, Sanakis Y, Fontecave M. J Biol Chem. 2006;281:16256–16263. doi: 10.1074/jbc.M513569200. [DOI] [PubMed] [Google Scholar]
- 76.Bulteau AL, O'Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- 77.Yoon T, Cowan JA. J Biol Chem. 2004;279:25943–25946. doi: 10.1074/jbc.C400107200. [DOI] [PubMed] [Google Scholar]
- 78.He Y, Alam SL, Proteasa SV, Zhang Y, Lesuisse E, Dancis A, Stemmler TL. Biochemistry. 2004;43:16254–16262. doi: 10.1021/bi0488193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cavadini P, Adamec J, Taroni F, Gakh O, Isaya G. J Biol Chem. 2000;275:41469–41475. doi: 10.1074/jbc.M006539200. [DOI] [PubMed] [Google Scholar]
- 80.Musco G, Stier G, Kolmerer B, Adinolfi S, Martin S, Frenkiel T, Gibson T, Pastore A. Structure Fold Des. 2000;8:695–707. doi: 10.1016/s0969-2126(00)00158-1. [DOI] [PubMed] [Google Scholar]
- 81.Condo I, Ventura N, Malisan F, Rufini A, Tomassini B, Testi R. Human Molecular Genetics. 2007;16:1534–1540. doi: 10.1093/hmg/ddm102. [DOI] [PubMed] [Google Scholar]
- 82.Yoon T, Dizin E, Cowan JA. J Biol Inorg Chem. 2007;12:535–542. doi: 10.1007/s00775-007-0205-2. [DOI] [PubMed] [Google Scholar]
- 83.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 84.Froschauer EM, Schweyen RJ, Wiesenberger G. Biochim Biophys Acta. 2009;1788:1044–1050. doi: 10.1016/j.bbamem.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 85.Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J. Mol Cell Biol. 2009;29:1007–1016. doi: 10.1128/MCB.01685-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Rahmanto YS, Sheftel AD, Ponka P. Proc Natl Acad Sci U S A. 2010;107:10775–10782. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Richardson DR, Huang ML, Whitnall M, Becker EM, Ponka P, Rahmanto YS. J Mol Med. 2010;88:323–329. doi: 10.1007/s00109-009-0565-x. [DOI] [PubMed] [Google Scholar]
- 88.Adinolfi S, Trifuoggi M, Politou AS, Martin S, Pastore A. Hum Mol Genet. 2002;11:1865–1877. doi: 10.1093/hmg/11.16.1865. [DOI] [PubMed] [Google Scholar]
- 89.Qi W, Cowan JA. Chem Commun (Camb) 2010;46:719–721. doi: 10.1039/b911975b. [DOI] [PubMed] [Google Scholar]
- 90.Bou-Abdallah F, Adinolfi S, Pastore A, Laue TM, Dennis Chasteen N. J Mol Biol. 2004;341:605–615. doi: 10.1016/j.jmb.2004.05.072. [DOI] [PubMed] [Google Scholar]
- 91.Kondapalli KC, Kok NM, Dancis A, Stemmler TL. Biochemistry. 2008;47:6917–6927. doi: 10.1021/bi800366d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karlberg T, Schagerlof U, Gakh O, Park S, Ryde U, Lindahl M, Leath K, Garman E, Isaya G, Al-Karadaghi S. Structure. 2006;14:1535–1546. doi: 10.1016/j.str.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 93.Dhe-Paganon S, Shigeta R, Chi YI, Ristow M, Shoelson SE. J Biol Chem. 2000;275:30753–30756. doi: 10.1074/jbc.C000407200. [DOI] [PubMed] [Google Scholar]
- 94.Nair M, Adinolfi S, Pastore C, Kelly G, Temussi P, Pastore A. Structure. 2004;12:2037–2048. doi: 10.1016/j.str.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 95.Cho SJ, Lee MG, Yang JK, Lee JY, Song HK, Suh SW. Proc Natl Acad Sci U S A. 2000;97:8932–8937. doi: 10.1073/pnas.160270897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pastore C, Franzese M, Sica F, Temussi P, Pastore A. FEBS J. 2007;274:4199–4210. doi: 10.1111/j.1742-4658.2007.05946.x. [DOI] [PubMed] [Google Scholar]
- 97.Bencze KZ, Kondapalli KC, Cook JD, McMahon S, Millan-Pacheco C, Pastor N, Stemmler TL. Crit Rev Biochem Mol Biol. 2006;41:269–291. doi: 10.1080/10409230600846058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bencze KZ, Yoon T, Millan-Pacheco C, Bradley PB, Pastor N, Cowan JA, Stemmler TL. Chem Commun (Camb) 2007:1798–1800. doi: 10.1039/b703195e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang J, Dizin E, Cowan JA. J Biol Inorg Chem. 2008;13:825–836. doi: 10.1007/s00775-008-0369-4. [DOI] [PubMed] [Google Scholar]
- 100.Correia AR, Wang T, Craig EA, Gomes CM. Biochem J. 2010;426:197–203. doi: 10.1042/BJ20091612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang T, Craig EA. J Biol Chem. 2008;283:12674–12679. doi: 10.1074/jbc.M800399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Foury F, Pastore A, Trincal M. EMBO Rep. 2007;8:194–199. doi: 10.1038/sj.embor.7400881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zeng J, Zhao W, Liu Y, Xia L, Liu J, Qiu G. Biotechnol Lett. 2007;29:1965–1972. doi: 10.1007/s10529-007-9488-1. [DOI] [PubMed] [Google Scholar]
- 104.Albrecht AG, Netz DJ, Miethke M, Pierik AJ, Burghaus O, Peuckert F, Lill R, Marahiel MA. J Bacteriol. 2010;192:1643–1651. doi: 10.1128/JB.01536-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaiser JT, Clausen T, Bourenkow GP, Bartunik HD, Steinbacher S, Huber R. J Mol Biol. 2000;297:451–464. doi: 10.1006/jmbi.2000.3581. [DOI] [PubMed] [Google Scholar]
- 106.Wu SP, Mansy SS, Cowan JA. Biochemistry. 2005;44:4284–4293. doi: 10.1021/bi0483007. [DOI] [PubMed] [Google Scholar]
- 107.Sazanov LA, Hinchliffe P. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 108.Berrisford JM, Sazanov LA. J Biol Chem. 2009;284:29773–29783. doi: 10.1074/jbc.M109.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lesuisse E, Santos R, Matzanke BF, Knight SA, Camadro JM, Dancis A. Hum Mol Genet. 2003;12:879–889. doi: 10.1093/hmg/ddg096. [DOI] [PubMed] [Google Scholar]
- 110.Seguin A, Bayot A, Dancis A, Rogowska-Wrzesinska A, Auchere F, Camadro JM, Bulteau AL, Lesuisse E. Mitochondrion. 2009;9:130–138. doi: 10.1016/j.mito.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 111.Gakh O, Adamec J, Gacy AM, Twesten RD, Owen WG, Isaya G. Biochemistry. 2002;41:6798–6804. doi: 10.1021/bi025566+. [DOI] [PubMed] [Google Scholar]
- 112.Aloria K, Schilke B, Andrew A, Craig EA. EMBO Rep. 2004;5:1096–1101. doi: 10.1038/sj.embor.7400272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Adinolfi S, Iannuzzi C, Prischi F, Pastore C, Iametti S, Martin SR, Bonomi F, Pastore A. Nat Struct Mol Biol. 2009;16:390–396. doi: 10.1038/nsmb.1579. [DOI] [PubMed] [Google Scholar]
- 114.Wilson RB, Roof DM. Nat Genet. 1997;16:352–357. doi: 10.1038/ng0897-352. [DOI] [PubMed] [Google Scholar]
- 115.Muhlenhoff U, Richhardt N, Ristow M, Kispal G, Lill R. Hum Mol Genet. 2002;11:2025–2036. doi: 10.1093/hmg/11.17.2025. [DOI] [PubMed] [Google Scholar]
- 116.Martelli A, Wattenhofer-Donze M, Schmucker S, Bouvet S, Reutenauer L, Puccio H. Hum Mol Genet. 2007;16:2651–2658. doi: 10.1093/hmg/ddm163. [DOI] [PubMed] [Google Scholar]
- 117.Ramazzotti A, Vanmansart V, Foury F. FEBS Lett. 2004;557:215–220. doi: 10.1016/s0014-5793(03)01498-4. [DOI] [PubMed] [Google Scholar]
- 118.Leidgens S, De Smet S, Foury F. Hum Mol Genet. 2010;19:276–286. doi: 10.1093/hmg/ddp495. [DOI] [PubMed] [Google Scholar]
- 119.Branda SS, Yang ZY, Chew A, Isaya G. Hum Mol Genet. 1999;8:1099–1110. doi: 10.1093/hmg/8.6.1099. [DOI] [PubMed] [Google Scholar]
- 120.Zheng L, White RH, Cash VL, Dean DR. Biochemistry. 1994;33:4714–4720. doi: 10.1021/bi00181a031. [DOI] [PubMed] [Google Scholar]
- 121.Nakai Y, Nakai M, Hayashi H, Kagamiyama H. J Biol Chem. 2001;276:8314–8320. doi: 10.1074/jbc.M007878200. [DOI] [PubMed] [Google Scholar]
- 122.Nakai Y, Umeda N, Suzuki T, Nakai M, Hayashi H, Watanabe K, Kagamiyama H. J Biol Chem. 2004;279:12363–12368. doi: 10.1074/jbc.M312448200. [DOI] [PubMed] [Google Scholar]
- 123.Kiyasu T, Asakura A, Nagahashi Y, Hoshino T. J Bacteriol. 2000;182:2879–2885. doi: 10.1128/jb.182.10.2879-2885.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cupp-Vickery JR, Urbina H, Vickery LE. J Mol Biol. 2003;330:1049–1059. doi: 10.1016/s0022-2836(03)00690-9. [DOI] [PubMed] [Google Scholar]
- 125.Li J, Kogan M, Knight SA, Pain D, Dancis A. J Biol Chem. 1999;274:33025–33034. doi: 10.1074/jbc.274.46.33025. [DOI] [PubMed] [Google Scholar]
- 126.Muhlenhoff U, Gerber J, Richhardt N, Lill R. EMBO J. 2003;22:4815–4825. doi: 10.1093/emboj/cdg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fosset C, Chauveau MJ, Guillon B, Canal F, Drapier JC, Bouton C. J Biol Chem. 2006;281:25398–25406. doi: 10.1074/jbc.M602979200. [DOI] [PubMed] [Google Scholar]
- 128.Li K, Tong WH, Hughes RM, Rouault TA. J Biol Chem. 2006;281:12344–12351. doi: 10.1074/jbc.M600582200. [DOI] [PubMed] [Google Scholar]
- 129.Schwartz CJ, Djaman O, Imlay JA, Kiley PJ. Proc Natl Acad Sci U S A. 2000;97:9009–9014. doi: 10.1073/pnas.160261497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smid O, Horakova E, Vilimova V, Hrdy I, Cammack R, Horvath A, Lukes J, Tachezy J. J Biol Chem. 2006;281:28679–28686. doi: 10.1074/jbc.M513781200. [DOI] [PubMed] [Google Scholar]
- 131.Mihara H, Esaki N. Appl Microbiol Biotechnol. 2002;60:12–23. doi: 10.1007/s00253-002-1107-4. [DOI] [PubMed] [Google Scholar]
- 132.Mueller EG. Nat Chem Biol. 2006;2:185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]
- 133.Kessler D. FEMS Microbiol Rev. 2006;30:825–840. doi: 10.1111/j.1574-6976.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- 134.Adam AC, Bornhovd C, Prokisch H, Neupert W, Hell K. EMBO J. 2006;25:174–183. doi: 10.1038/sj.emboj.7600905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wiedemann N, Urzica E, Guiard B, Muller H, Lohaus C, Meyer HE, Ryan MT, Meisinger C, Muhlenhoff U, Lill R, Pfanner N. EMBO J. 2006;25:184–195. doi: 10.1038/sj.emboj.7600906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shan Y, Napoli E, Cortopassi G. Hum Mol Genet. 2007;16:929–941. doi: 10.1093/hmg/ddm038. [DOI] [PubMed] [Google Scholar]
- 137.Shi Y, Ghosh MC, Tong WH, Rouault TA. Hum Mol Genet. 2009;18:3014–3025. doi: 10.1093/hmg/ddp239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kurihara T, Mihara H, Kato S, Yoshimura T, Esaki N. Biochim Biophys Acta. 2003;1647:303–309. doi: 10.1016/s1570-9639(03)00078-5. [DOI] [PubMed] [Google Scholar]
- 139.Muhlenhoff U, Richhardt N, Gerber J, Lill R. J Biol Chem. 2002;277:29810–29816. doi: 10.1074/jbc.M204675200. [DOI] [PubMed] [Google Scholar]
- 140.Li J, Saxena S, Pain D, Dancis A. J Biol Chem. 2001;276:1503–1509. doi: 10.1074/jbc.M007198200. [DOI] [PubMed] [Google Scholar]
- 141.Liu Y, Cowan JA. Chem Commun (Camb) 2007:3192–3194. doi: 10.1039/b704928e. [DOI] [PubMed] [Google Scholar]
- 142.Bandyopadhyay S, Naik SG, O'Carroll IP, Huynh BH, Dean DR, Johnson MK, Dos Santos PC. J Biol Chem. 2008;283:14092–14099. doi: 10.1074/jbc.M709161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Garland SA, Hoff K, Vickery LE, Culotta VC. J Mol Biol. 1999;294:897–907. doi: 10.1006/jmbi.1999.3294. [DOI] [PubMed] [Google Scholar]
- 144.Nakamura Y, Kaneko T, Hirosawa M, Miyajima N, Tabata S. Nucleic Acids Res. 1998;26:63–67. doi: 10.1093/nar/26.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bandyopadhyay S, Chandramouli K, Johnson MK. Biochem Soc Trans. 2008;36:1112–1119. doi: 10.1042/BST0361112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Luo M, Jiang YL, Ma XX, Tang YJ, He YX, Yu J, Zhang RG, Chen Y, Zhou CZ. J Mol Biol. 2010;398:614–622. doi: 10.1016/j.jmb.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 147.Lillig CH, Berndt C, Vergnolle O, Lonn ME, Hudemann C, Bill E, Holmgren A. Proc Natl Acad Sci U S A. 2005;102:8168–8173. doi: 10.1073/pnas.0500735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Feng Y, Zhong N, Rouhier N, Hase T, Kusunoki M, Jacquot JP, Jin C, Xia B. Biochemistry. 2006;45:7998–8008. doi: 10.1021/bi060444t. [DOI] [PubMed] [Google Scholar]
- 149.Johansson C, Kavanagh KL, Gileadi O, Oppermann U. J Biol Chem. 2007;282:3077–3082. doi: 10.1074/jbc.M608179200. [DOI] [PubMed] [Google Scholar]
- 150.Rouhier N, Unno H, Bandyopadhyay S, Masip L, Kim SK, Hirasawa M, Gualberto JM, Lattard V, Kusunoki M, Knaff DB, Georgiou G, Hase T, Johnson MK, Jacquot JP. Proc Natl Acad Sci U S A. 2007;104:7379–7384. doi: 10.1073/pnas.0702268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Iwema T, Picciocchi A, Traore DA, Ferrer JL, Chauvat F, Jacquamet L. Biochemistry. 2009;48:6041–6043. doi: 10.1021/bi900440m. [DOI] [PubMed] [Google Scholar]
- 152.Rouhier N, Couturier J, Johnson MK, Jacquot JP. Trends Biochem Sci. 2010;35:43–52. doi: 10.1016/j.tibs.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bandyopadhyay S, Gama F, Molina-Navarro MM, Gualberto JM, Claxton R, Naik SG, Huynh BH, Herrero E, Jacquot JP, Johnson MK, Rouhier N. EMBO J. 2008;27:1122–1133. doi: 10.1038/emboj.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu Y, Qi W, Cowan JA. Biochemistry. 2009;48:973–980. doi: 10.1021/bi801645z. [DOI] [PubMed] [Google Scholar]